
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of biochar-based catalysts in biomass upgrading: a review
Xuefei Cao *a,
Shaoni Sunab and
Runcang Sun*a
*a,
Shaoni Sunab and
Runcang Sun*a
aBeijing Key Laboratory of Lignocellulosic Chemistry, Beijing Forestry University, Beijing 100083, China. E-mail: caoxuefei@bjfu.edu.cn; rcsun3@bjfu.edu.cn; Fax: +86-10-62336903; Tel: +86-10-62336903
bState Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510640, China
First published on 16th October 2017
Abstract
Biochar is a low-cost carbon-rich material derived from the thermochemical degradation of biomass. Due to its unique chemical structure, biochar with a large surface area and tailored surface functional groups can be easily prepared by activation and/or functionalization, and shows great potential to be used as a versatile catalyst and/or catalyst support in many chemical processes. However, the application of biochars as versatile catalysts and/or catalyst supports for biomass upgrading has not been systematically overviewed so far. In this work, the formation of pyrochar and hydrochar is proposed, and the activation and/or functionalization of biochar are also included. Subsequently, the application of biochar-based catalysts in biomass upgrading, including biochar-based solid acids for biomass hydrolysis and dehydration, biochar-based catalysts for biodiesel production, and biochars as catalyst supports for biomass pyrolysis, gasification, and bio-oil upgrading, are discussed in detail.
1. Introduction
As the most abundant and renewable resource in the world, biomass is considered as a promising alternative to fossil resources for providing sustainable fuels and chemicals.1 For decades, upgrading biomass into fuels, fine chemicals, and materials has attracted worldwide attention.2 Generally, biomass can be converted into biofuels or biochemicals using different technologies, such as thermochemical, chemical, and biochemical processes. Among these, thermochemical processing, including pyrolysis, gasification, and hydrothermal conversion, has been proposed as one of the most promising technologies for biomass upgrading due to its high efficiency.3Through the thermochemical process, various important fuels or chemicals, such as syngas, bio-oils, biochars, platform chemicals, etc., can be obtained from the degradation of biomass.3,4 However, many reactions take place during the biomass upgrading process, and a mixture containing various kinds of degradation products with high dispersion and low yield is always obtained.5–7 This complex composition of the products from biomass upgrading restricts to some degree its further utilization. Nevertheless, several previous reports showed that both the reaction rate and reaction selectivity during the biomass upgrading process could be improved to varying degrees in the presence of suitable catalysts.2,4,8 Therefore, catalytic biomass upgrading plays a crucial role in converting renewable biomass resource into a variety of commodity chemicals or liquid fuels.
Biochar is a carbonaceous material, which is a residual byproduct from the thermochemical degradation of biomass.9 Biochar can be produced from various thermochemical degradation processes, such as pyrolysis, gasification, hydrothermal carbonization, etc.10,11 As compared to carbonaceous materials from other chemical processes, biochar is an inexpensive, environmentally friendly, and easily-produced charcoal, which can be used for many purposes.10,12–14 However, the utilization of these as-prepared biochars is not ideal because of their poor physicochemical properties. In order to improve the physicochemical properties of the as-prepared biochars, various activation and functionalization approaches have been investigated to extend their applications.15,16 The physicochemical properties of biochar vary significantly according to the raw biomass material type derived, the carbonization process, and activation or functionalization methods.16–19 After activation or functionalization, the biochar shows several interesting properties, such as large surface area, multi-scale porous structure, and abundant surface functional groups. The activated biochars are widely used as activated carbon, soil amendments, carbon sequestration agents, environmental adsorbents for organic and heavy metal pollution removal, etc.16–20 More importantly, due to the large surface area and abundant surface functional groups, activated or functionalized biochars show great potential to be used as versatile catalysts and/or catalyst supports in many chemical processes.10,21–25
Cha et al.10 overviewed the production and utilization of biochars in various environmental application fields, and the application of the biochars as catalysts for syngas upgrading, biodiesel production, and air pollutant treatment were also discussed. The production of biodiesel under the catalysis of carbon-based catalysts was reviewed by Konwar et al.,23 and it was found that the catalysts from biomass were excellent catalyst support for biodiesel production because of their low cost, high surface area and thermal stability. The application of biochar-based catalysts for biomass conversion, such as polysaccharides hydrolysis, biodiesel production, bio-oil upgrading, etc., were also mentioned by Liu et al.24 and Lee et al.25 Although some reviews reported the conversion of biomass into biofuels or biochemical in the presence of biochar-based catalysts, the application of biochars as versatile catalysts and/or catalyst supports for biomass upgrading has not been systematically overviewed yet. Therefore, a comprehensive review on biochar-based catalysts for biomass upgrading is still needed.
In light of the great potential of catalytic biomass upgrading, this review is devoted to discussing the versatile applications of biochars as catalysts for biomass upgrading, including biochar-based solid acids for biomass hydrolysis and dehydration, biochar-based catalysts for biodiesel production, and biochars as catalyst supports for biomass pyrolysis, gasification, and bio-oil upgrading. Considering that the formation, activation and functionalization of biochar have been extensively reviewed by many researchers, these research topics will only be briefly discussed here.
2. Thermochemical degradation of biomass and biochar formation
Lignocellulosic biomass is mainly composed of cellulose (38–50%), hemicelluloses (23–32%), lignin (15–25%) and small amounts of extractives.26 Among these components, cellulose and hemicelluloses are chain polysaccharides, while lignin is a cross-linked three-dimensional phenolic polymer resulting from the oxidative combinatorial coupling of three monolignol monomers (p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol).27,28 The lignin units are usually linked with each other by ether and carbon–carbon bonds. The most abundant inter-unit linkage in lignin is β-O-4 (β-aryl ether), while other linkages like β-5, β-β, 5-5, α-O-4, 5-O-4, β-1, etc., are also observed with varying frequency.28 Lignin is closely intertwined with cellulose and hemicelluloses by covalent and non-covalent bonds, making up the matrix of lignocellulosic biomass.As is well known, biochar can be produced using various carbonization processes, such as pyrolysis, gasification, hydrothermal carbonization, and torrefaction. Due to the structural differences of cellulose, hemicelluloses, and lignin, the biochar yield, degradation rate, degradation mechanism and pathway of each component are different. The production of biochars has been reviewed and discussed extensively in recent years.10,11,25,29 This section focuses on explaining the degradation of biomass in the pyrolysis and hydrothermal carbonization processes, and further discussing the formation pathways of pyrochar and hydrochar.
The formation pathways of biochars (pyrochar and hydrochar) vary significantly according to the reaction medium.11 Biomass pyrolysis is a very complex process, and numerous reactions take place, in which the free radical reaction is one of the most important reactions involved.30,31 These radicals, which are very active, can further react with other compounds or radicals via free-radical substitution, free-radical addition, carbon–carbon coupling, etc., and result in the formation of syngas, bio-oils, and biochars. Meanwhile, some important intermediates and/or product, such as retroaldol products (hydroxyacetaldehyde, pyruvaldehyde, etc.), dehydrated species (furfural, 5-hydroxymethyl furfural, etc.), anhydro-monosaccharides (levoglucosan, levoglucosenone, etc.), and phenolic compounds (4-vinylguaiacol, eugenol, etc.) have also been observed among the volatile products of biomass pyrolysis, indicating that the reactions, like retroaldol condensation, dehydration, isomerization, etc., also occur during the pyrolysis process.5,7,24,32,33 Thus, a very complex product mixture is always observed.7,30,32,34 However, the morphology and the microstructure of the biomass are basically unchanged over the pyrolysis process.35 This implies that the formation of pyrochar is highly “localized”. A possible formation pathway of pyrochar is shown in Fig. 1. Throughout the whole pyrolysis process, the volatile products are gradually released from the biomass matrix, resulting in a carbonaceous pyrochar material. For more detailed information on pyrolysis of biomass components and pyrochar formation mechanisms, the readers can refer to the excellent reviews by Collard et al.5 and Liu et al.,24 and references therein.
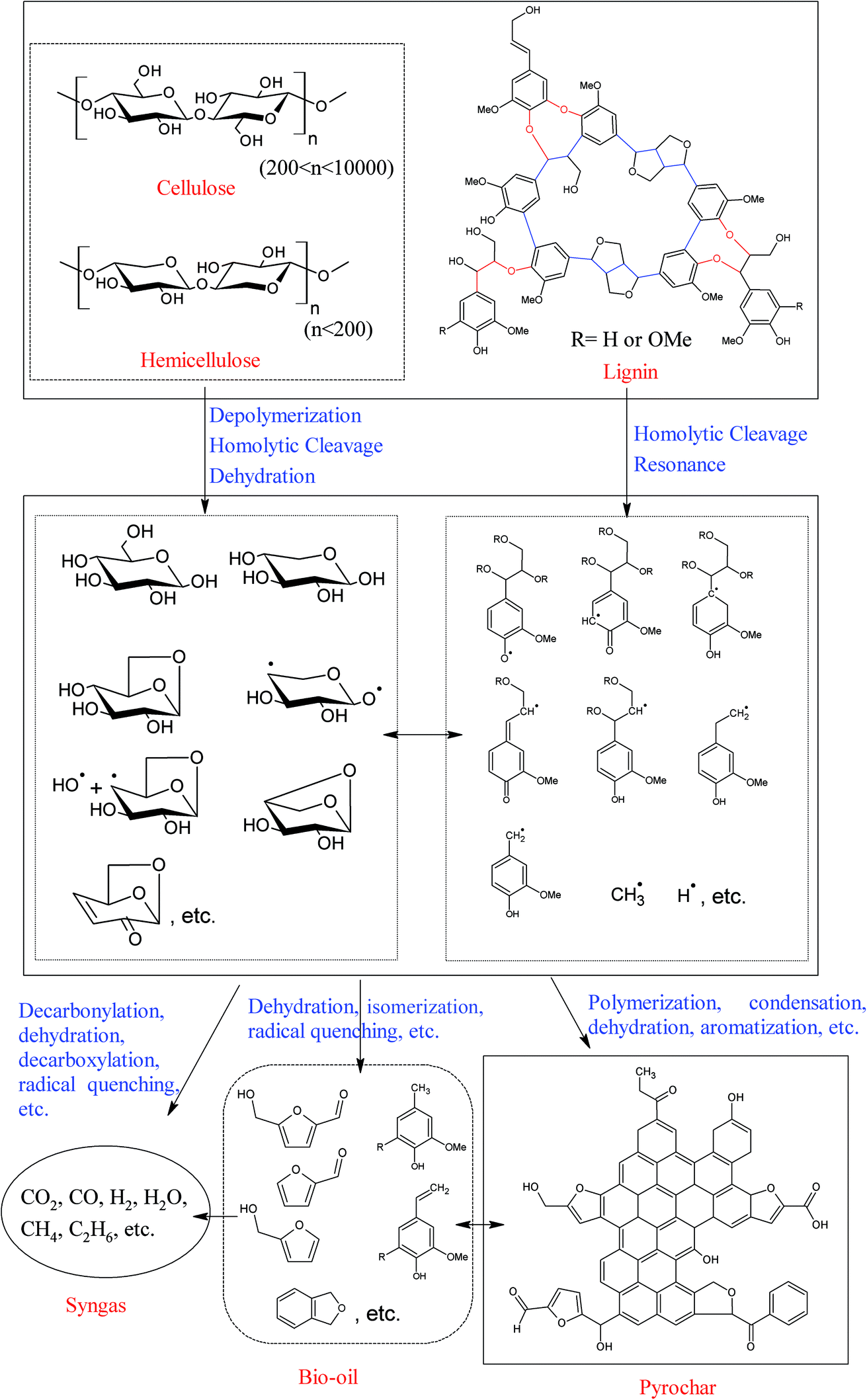 | ||
| Fig. 1 A possible formation pathway of pyrochar. | ||
As compared with pyrolysis, ionic reactions are favored in aqueous medium, and the depolymerization of cellulose, hemicelluloses, and lignin occurs at relatively low temperatures under hydrothermal conditions.11,36,37 Specifically, most of the hemicelluloses can be extracted from the lignocellulosic materials at a hydrothermal temperature below 180 °C,38,39 and the degradation of cellulose becomes obvious as the hydrothermal temperature reaches 230 °C.40 Additionally, unlike pyrochar, the hydrochar obtained from hydrothermal carbonation contains abundant functional groups, such as hydroxyl and carboxyl groups, suggesting that hydrochar is formed through a different pathway. A possible hydrochar formation pathway is illustrated in Fig. 2.
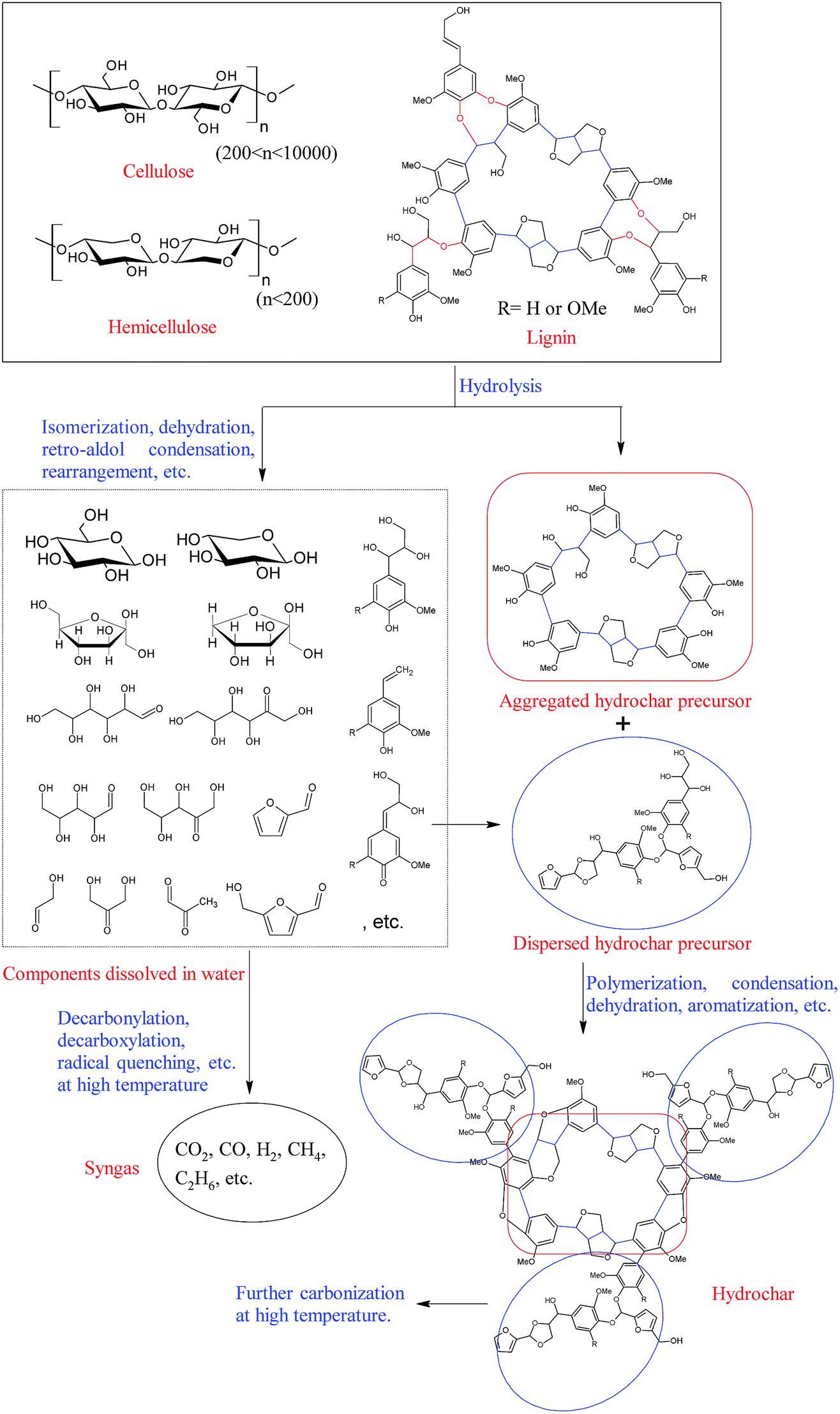 | ||
| Fig. 2 A possible formation pathway of hydrochar. | ||
During the hydrothermal carbonization process, cellulose and hemicelluloses are initially hydrolyzed into oligosaccharides and monosaccharides through the cleavage of their glycosidic bonds in hot compressed water. These water-soluble oligosaccharides and monosaccharides are further degraded into small molecule compounds, such as 5-hydroxymethyl furfural (HMF), furfural (FF), acetic acid, etc., at relatively mild conditions or decomposed into syngas at more severe conditions. Meanwhile, the ether linkages of lignin (mainly β-O-4 and α-O-4 linkages) are also unstable under hydrothermal conditions, and a large amount of lignin is converted into various water soluble phenolic compounds when the lignocellulosic material is hydrothermally treated at 240 °C.39 However, most carbon–carbon bonds in lignin (mainly β-β, β-5, β-1, and 5-5 linkages) are relatively stable, and they are left as the initial skeleton of “aggregated hydrochars”.41 These furan compounds (FF and HMF), derived from hemicelluloses and cellulose, subsequently react with the soluble phenolic compounds derived from lignin, forming “dispersed hydrochars” through polymerization.42 The reactive oxygen-containing groups on the surface of the “dispersed hydrochars” further react with the “aggregated hydrochars” and cover the surface of the “aggregated hydrochars”, resulting in a hydrochar with relatively low specific surface area and poor porosity.41,42 More detailed information on hydrothermal degradation mechanism of biomass components can be found in previous review articles.6,11
3. Activation and functionalization of biochars as catalysts or catalyst supports
Usually, the biochar directly obtained from the pyrolysis of biomass shows relatively low specific surface area, poor porosity, and limited surface functional groups.24 These inherent disadvantages hinder to some degree the application of biochars as useful catalysts or catalyst supports. Although, fortunately, these properties of the biochars can be easily tuned by the appropriate activation or functionalization processes.10,11,43 In this section, the activation and functionalization of the biochars as catalysts or catalyst supports for biomass upgrading are discussed in detail. The activation and functionalization methods of the biochars for other applications are beyond the scope of this review.3.1. Biochar activation
The main purpose of biochar activation is to increase its specific surface area and porosity prior to use. After activation, the specific surface area and porosity of the biochar can be significantly improved due to the development and opening of internal porous structure of the as-prepared biochar.11,44 As a result, the activated biochars can be further used in various applications, such as activated carbon, soil amendments, carbon sequestration agents, and environmental adsorbents.10,24 Meanwhile, the activated biochar can provide more active sites, high mass transfer capacities, and high active loading surface for catalysis.24,25 For decades, various activation methods have been explored to prepare porous biochars with special morphologies.15,43,45–47 According to the activation agents used, the activation methods can be broadly divided into the following two categories: physical activation and chemical activation.43Normally, the porosity, pore size distribution, and specific surface area of the activated biochars vary significantly depending on the biomass type, activating gas, and reaction conditions.10 For example, Lima et al.49 examined the effects of steam activation on the surface area and porosity of various biochars obtained from the fast-pyrolysis of several substrates, and further investigated their adsorptive properties for metal ion uptake. Their results showed that the surface area and micropore volume of the biochars improved dramatically from less than 5 m2 g−1 to 136–793 m2 g−1, when the biochars were steam-activated at 800 °C for 45 min. Furthermore, after activation, the metal ion adsorption performance of these biochars was improved to varying degree as a result of the improved surface area and porosity.49 Also, Koltowski et al.16 activated the biochar from the slow pyrolysis of willow using steam and CO2, separately. They found that both the steam and CO2 activation could significantly improve the surface area and porosity of the biochar. Additionally, the surface areas of the steam-activated biochar (840.6 m2 g−1) and the CO2-activated biochar (512.0 m2 g−1) were much higher than the as-prepared biochar (11.4 m2 g−1). Under the same activation program, the steam-activated biochar displayed higher specific surface area and pore size than the CO2-activated biochar.16 In contrast, Nabais et al.17 reported that the CO2-activated biochars from coffee endocarp showed higher surface areas and pore volumes than the corresponding steam activated biochars. Thus, it was deduced that this contradiction is mainly due to the different microstructure of the biomass used.
The variables during the chemical activation process, such as the activation temperature, the type and dose of activating agent, feedstock type, etc., significantly affect the porosity, pore size distribution, and specific surface area of the activated biochar.10,24 For instance, Dehkhoda et al.50 investigated the influence of the activation temperature (675 and 1000 °C) on the surface area, porosity, and electrosorption performance of the biochar impregnated with KOH solution. They found that the surface area of the biochar increased from 1.66 m2 g−1 to 614–990 m2 g−1, while the porosity increased as well from negligible to 0.6–0.9 m3 g−1. Furthermore, an increase in the activation temperature resulted in a decrease in the surface area due to a potential collapse and burn-off of the micropore walls of the biochar and/or the development of localized graphite-like structure in the biochar matrix. The total electrosorption capacitance of the biochar activated at 675 °C was more than two times higher than that of the biochar activated at 1000 °C owing to its higher contents of micropore and oxygen-containing functional groups.50
Also, Yorgun et al.18 examined the effects of the activation temperature (400–700 °C) and the impregnation ratio of ZnCl2 to biochar (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2–4:1) on the characteristics of the activated biochar prepared from Paulownia wood. Their results showed that the surface area and pore volume of the activated biochar decreased dramatically with the increase of the activation temperature, whereas the increased impregnation ratio resulted in larger surface area and pore volume. However, the results from Angin et al.19 showed that both the surface area and pore volume of the biochar from safflower seed gradually increased with the increase of the activation temperature (600–900 °C) and impregnation ratio (1:1–4:1). Therefore, despite using the same activation method, different effects may be observed with different biomass materials.
:2–4:1) on the characteristics of the activated biochar prepared from Paulownia wood. Their results showed that the surface area and pore volume of the activated biochar decreased dramatically with the increase of the activation temperature, whereas the increased impregnation ratio resulted in larger surface area and pore volume. However, the results from Angin et al.19 showed that both the surface area and pore volume of the biochar from safflower seed gradually increased with the increase of the activation temperature (600–900 °C) and impregnation ratio (1:1–4:1). Therefore, despite using the same activation method, different effects may be observed with different biomass materials.
Park and coworkers51 used HCl, H2SO4, H3PO4, KOH, MgO, ZnCl2, and K2SO4 as activation agents for chemical activation of sesame straw biochar and examined the phosphorus adsorption capacities of these activated biochars. According to their studies, ZnCl2 and MgO were more effective in improving the phosphorus adsorption characteristics of the biochar than the other activation agents. A maximum phosphorus adsorption capacity of 15 g kg−1 was achieved by the ZnCl2 activated biochar.51
Generally, short activation time and low activation temperature are required for the chemical activation, but chemical consumption, equipment corrosion, and wastewater treatment should be considered in the chemical activation process. The physical activation process is more simple and clean than the chemical activation process, but relatively high activation temperature, extended activation time, high energy consumption, and relatively low specific surface area are usually found in the physical activation process. Besides physical activation and chemical activation, the morphology, specific surface area and porosity of the formed biochar are closely related to the microstructure of the biomass. Previous studies from Zhang et al.35 showed that a pyrochar with specific surface area as high as 1610 m2 g−1 was obtained from lotus stem without any extra activation process, which was about 55% higher than that from lotus leaves.
3.2. Biochar functionalization
Biochar functionalization is mainly used to confer specific performance properties to the biochar. Due to the limited surface functional groups of the as-prepared biochars, a suitable functionalization process is indispensable for the biochars intended to be used as catalysts. Generally, biochar can gain a desirable catalytic capacity via surface modification or active substances deposition.10,43,53–56Biochar modification can be easily accomplished by introducing specific functional groups, especially acid groups, onto the surface of the organic biochar. The most commonly used biochar modification method is sulfonation with concentrated H2SO4 or its derivatives.23,24 Following sulfonation, the modified biochars can be widely used as solid acid catalysts for polysaccharide hydrolysis, sugar dehydration, biodiesel production, etc.23,56,57 Besides the –SO3H group, other weak acid groups, like the –COOH group, can also be introduced into the biochar matrix.58 These biochar-based solid acids show relatively high reaction activity and good recyclability, which are promising alternatives to mineral acid catalysts.23,25,56–61 The application of these biochar-based solid acids as effective catalysts for biomass upgrading will be discussed in detail in Sections 4.1 and 4.2.
Besides their use as catalysts, the activated biochars are also promising materials to be used as catalyst supports.24,54,55,62,63 Metals and/or metal oxides with catalytic activity can be loaded onto the biochar supports by preloading the metal precursors into the biomass matrix before pyrolysis55,64–66 or by impregnating the activated biochars with the metal precursors.62,66–68 Previous studies reported that various biochar-supported metal catalysts with high dispersion could be prepared according to the above mentioned methods.24,64,65 In this review, the application of these biochar-supported catalysts for biomass pyrolysis, gasification, and bio-oil upgrading will be discussed in detail in Section 4.3.
4. Biochar based catalysts for biomass upgrading
Carbonaceous materials have been used in heterogeneous catalysis for a long time, because of their specific physicochemical properties, such as large surface area, multi-scale porous structure, and tailoring surface functional groups.10,24,25,29,69 As compared to carbonaceous materials from other chemical processes, biochar is inexpensive, environmentally friendly, and easily-produced, which are characteristics that make biochar a promising alternative to conventional carbonaceous materials. Functionalized biochars show great potential to be used as direct catalysts or catalyst supports in many industrial applications.10,24,25,29 In this section, the utilization of biochars as versatile catalysts and/or catalyst supports for biomass upgrading is evaluated.4.1. Biochar-based solid acids for biomass hydrolysis and dehydration
Biomass has great potential to be used as a sustainable resource to provide biofuels, biochemical, and biomaterials. As the most abundant biopolymers in biomass, cellulose and hemicelluloses are the largest components of the earth's biomass. Thus, effectively converting these components into useful platform compounds, such as sugars, furfural (FF), 5-hydroxymethylfurfural (HMF), etc., is a highly desirable endeavor.4 The sugars from the hydrolysis of cellulose and hemicelluloses can be further utilized to produce alcohols. The FF and HMF from the dehydration of sugars can be converted into various corresponding furan derivatives for various applications.70,71 Therefore, the hydrolysis of biomass and its further conversion is one of the most important research topics in biomass upgrading.Many hydrolysis agents, such as enzymes, mineral acids, and solid acids, have been employed in biomass hydrolysis. However, the enzymatic hydrolysis of biomass is slow and costly, and the hydrolysis of biomass by mineral acids usually produces various environmental hazards. Recently, several studies reported that biochar-based solid acid is a promising alternative to mineral acid in biomass hydrolysis and dehydration due to its high reaction activity, recyclability, and low cost.25,56,57,59–61 The catalytic performances of various biochar-based acid catalysts in biomass hydrolysis and dehydration are summarized in Table 1.
| Biochar | Functionalization condition | Feedstock | T (°C) | t (h) | Solvent | Conv. (%) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Microwave XOS: xylooligosaccharides, TRS: total reducing sugar, FF: furfural, HMF: 5-hydroxymethylfurfural. | ||||||||
| Corn stover biochar | Sulfonated with 30% (w/v) sulfuric acid solution at 150 °C for 24 h | Corn stover, prairie cordgrass, and switch grass | 100 | 6 | Water | 18.8–21.8 (glucan) | — | 59 |
| 67.9–80.5 (xylan) | ||||||||
| Pine chip biochar | Sulfonated with sulfuric acid at 100 °C for 12–18 h | Birchwood xylan | 120 | 2 | Water | 85 | — | 60 |
| Bamboo biochar | 4 g of biochar sulfonated with 60 mL of concentrated H2SO4 at 180 °C for 12 h | Bamboo hemicellulose | 150a | 0.5 | Water | — | 55.7 (XOS) | 57 |
| Bamboo biochar | 10 g of biochar sulfonated with 50 mL of 80% sulfuric acid at 80 °C for 3 h and 50 wt% SO3 at 80 °C for 2 h | Cellulose | 90a | 1 | Water | — | 27.5 (TRS) | 72 |
| Bamboo biochar | 1.1 biochar sulfonic acid (BCSA) | Cellulose | 90a | 3 | Water | — | 34.9 (TRS) | 72 |
| 1.2 IL functionalized BCSA | ||||||||
| Bamboo biochar | 1.1 biochar sulfonic acid (BCSA) | Cellulose | 110 | 2 | Water | — | 35.6 (TRS) | 73 |
| 1.2 IL-Cu functionalized BCSA | ||||||||
| Bamboo biochar | 1.1 biochar sulfonic acid (BCSA) | Cellulose | 110 | 2 | Water | — | 58.7 (TRS) | 61 |
| 1.2 IL-Zn functionalized BCSA | ||||||||
| None | None | Cellulose | 130 | 4 | [BMIM][Cl] | 4.5 | — | 58 |
| Biochar from glucose | Containing weak acid sites | Cellulose | 130 | 3 | [BMIM][Cl] | 47 | — | 58 |
| Biochar from glucose | 20 g of glucose and 10 g of sulfosalicylic acid copolymerization at 180 °C for 4 h | Cellulose | 130 | 3 | [BMIM][Cl] | 59.4 | — | 58 |
| Biochar from glucose | 20 g of glucose and 10 g of acrylic acid copolymerization at 180 °C for 4 h | Cellulose | 130 | 3 | [BMIM][Cl] | ∼50 | — | 58 |
| Corncob biochar | Sulfonated with 0.5 M H2SO4 at room temperature for 24 h under the assistance of ultrasonic vibration | Corncob | 180 | 2.83 | Water | 62.0 (xylan) | 37.8 (FF) | 74 |
| Corncob biochar | Sulfonated with H2SO4 at 150 °C for 15 h | Perhydrolysate of corncob | 170 | 1 | Dichloromethane/water | — | 81.1 (FF) | 56 |
| SnO2–Co3O4/biochar | Loaded with SnO2–Co3O4 (Sn/Co molar ratio was 0.5–4.0) | Corncob | 180 | 3.33 | Water | — | 30.0 (FF) | 54 |
| Commercial activated carbon | Containing weak acid sites | Xylose | 200 | 3 | Water | ∼90 | 50 (FF) | 75 |
| Bamboo biochar | 1.1 biochar sulfonic acid (BCSA) | Cellulose | 100a | 2 | DMA–LiCl/water | — | 30.4 (HMF) | 61 |
| 1.2 IL-Zn functionalized BCSA | ||||||||
| Bamboo biochar | 1.1 biochar sulfonic acid (BCSA) | Cellulose | 80 | 3 | Water | — | 27.9 (HMF) | 76 |
| 1.2 IL-F1–3s functionalized BCSA | ||||||||
| Commercial activated carbon | Containing weak acid sites | Glucose | 200 | 3 | Water | ∼55 | 23 (HMF) | 75 |
Generally, the acidic active sites can be introduced into the biochar by three possible means: (i) using the weak acid groups formed during the preparation of the biochar as acidic sites; (ii) introducing the acidic sites by sulfonation or copolymerization with monomers with acid groups; (iii) loading solid acid onto the surface of the biochar. Among these methods, sulfonation is the most commonly used method to prepare biochar-based solid acids, and the –SO3H groups can be easily introduced onto the surface of the biochar using concentrated H2SO4 or its derivatives.23,24 For example, Li et al.59 reported that the sulfonation of corn stover biochar could be achieved by mixing the biochar with 30% (w/v) sulfuric acid at 150 °C for 24 h. In order to compare with H2SO4, the prepared sulfonated biochar was used to hydrolyze the hemicellulose and cellulose in corn stover, prairie cordgrass, and switch grass. Their results showed that 18.8–21.8% of glucan and 67.9–80.5% of xylan could be hydrolyzed at 100 °C in 6 h with the sulfonated biochar catalyst, and the sulfonated biochar showed higher selectivity and less soluble lignin-derived products in the hydrolysate than H2SO4.59 Similar sulfonated biochars have also been prepared to catalyze the hydrolyze of biomass to monosaccharides or oligosaccharides by many other researchers.57,60,61,72,73
Besides sulfonation, acid groups can also be introduced into the biochar matrix by copolymerization.58 For instance, Qi et al.58 prepared two kinds of biochar-based solid acid catalysts containing the –SO3H and –COOH groups, respectively, via in situ hydrothermal copolymerization of glucose with sulfosalicylic acid and acrylic acid. They found that besides the introduced acid groups, the own weak acid groups of the biochar also affect the hydrolysis of biomass. Both the as-prepared biochar and the functionalized biochars exhibited good catalytic activity for cellulose hydrolysis, and a total reducing sugar (TRS) yield around 50% was obtained at 130 °C for 3 h.58 Moreover, the content of the acid site and the catalytic activity of the catalyst were almost unchanged after five recycling experiments, indicating the stability of the catalyst for cellulose hydrolysis.58
As shown in Table 1, the biochar-based solid acids can also be used as the catalysts for the dehydration of sugars to furans under relatively severe reaction conditions.56,74,75 It was assumed that the acid groups of the biochar-based solid acid catalysts could cleave the glycosidic linkages of polysaccharides and hydrolyzed them to water-soluble sugars, which were further catalyzed by the biochar-based solid acid to produce the furan compounds.74 For example, Liu et al.74 synthesized a corncob biochar solid acid catalyst by carbonizing the mixture of corncob residue and saccharide solution from corncob and further sulfonating the as-prepared biochar. The sulfonated biochar was used to catalyze the hydrothermal degradation of corncob for furfural (FF) production. Under the optimal reaction conditions, a furfural yield of up to 37.75% and an overall corncob conversion rate of 62.00% were achieved in the presence of the sulfonated biochar catalyst.74 Also, Deng and coworkers56 used similar biochar-based solid acid to catalyze the transformation of corncob perhrydrolysate to FF in a biphasic system (dichloromethane/water system), and a much higher FF yield of 81.1% was observed at 170 °C for 1 h. Similar biochar catalysts with strong or weak acid groups have also been used to catalyze the hydrolysis and dehydration of cellulose and C6 sugars to HMF.61,75,76 Additionally, previous studies showed that the combination of Lewis and Brønsted acids is important to achieve high yields of furans from polysaccharides.77 For instance, Liu and coworkers54 prepared a series of biochar supported bimetallic oxides SnO2–Co3O4 catalysts via coprecipitation method for corncob hydrolysis into furfural. A maximum yield of furfural of 30.0% was obtained as the corncob was hydrothermally treated at 180 °C for 200 min under the catalysis of a SnO2–Co3O4/biochar catalyst.54 The Lewis acid sites from the SnO2–Co3O4 and the Brønsted acid sites from the ionization of hot-compressed water together led to the conversion of corncob hemicellulose to FF.
4.2. Biochar-based catalysts for biodiesel production
Biodiesel is a mixture of mono-alkyl esters produced from the transesterification or esterification of alcohols with vegetable oils, animal fats, or free fatty acids (FFAs), which can be used directly or in combination with petroleum diesel at any concentration in most diesel engines.23 The most commonly used alcohol is methanol because of its low cost. Other alcohols like ethanol, isopropanol and butanol can also be used to produce biodiesels with improved cold flow properties but at the expense of high cost and low transesterification efficiency.78Since the transesterification and esterification reaction can be catalyzed by either acidic or basic catalysts, numerous homogeneous and heterogeneous catalysts have been applied for biodiesel production.23,79,80 Homogeneous catalysts, especially KOH, NaOH, and NaOMe, have been widely used for biodiesel production because of their high reaction efficiency. However, these homogeneous catalysts are not reusable, and more effort is required to purify the products.23,81 Accordingly, many heterogeneous catalysts, such as CaO, MgO, Amberlyst-15, TiO2/ZrO2 and Al2O3/ZrO2, WO3/ZrO2 and various carbon-based acids or bases, have been explored as catalysts for biodiesel production.23,25,80 These catalysts can be easily separated from the products and recycled multiple times. As mentioned above, biochars have many advantages when used as catalysts or catalyst supports, such as low cost, large surface area, and tailoring surface functional groups, etc. After activation or functionalization, the biochars can be used as good heterogeneous acid or base catalysts for biodiesel production.10,23,25,53,81,82 The biodiesel productions under the catalysis of various biochar-based acid or base catalysts are summarized in Table 2.
| Biochar | Functionalization condition | Feedstock | Reaction conditions | Biodiesel yield (%) | Ref. |
|---|---|---|---|---|---|
| a FFAs: free fatty acids, FAME: fatty acid methyl ester. | |||||
| Oat hull biochar | None | Waste cooking oils | 140 °C, 30 min (microwave), MeOH/oil (10:1) |
0.5 | 84 |
| Oat hull biochar | Sulfonated with H2SO4 in microwave reactor at 100 °C for 30 min | Waste cooking oils | 140 °C, 30 min (microwave), MeOH/oil (10:1) |
28 | 84 |
| Oat hull biochar | Sulfonated with H2SO4 in microwave reactor at 140 °C for 30 min | Waste cooking oils | 140 °C, 30 min (microwave), MeOH/oil (10:1) |
72 | 84 |
| Biochar from Dynamotiv Inc. | Sulfonated with fuming H2SO4 at 150 °C for 15 h | Canola oil and oleic acid | 150 °C, 3 h, MeOH/oil (10:1) |
48 | 85 |
| Biochar from fast pyrolysis of different feedstocks | 20 g of biochar sulfonated with 200 mL of H2SO4 at 150 °C for 24 h | Vegetable oils | 60 °C, 3 h, EtOH/oil (18:1) |
77–89 | 86 |
| Biochar from peanut hulls, pine pellets, and wood chips | Sulfonated with H2SO4 at 100 °C for 12–18 h or gaseous SO3 at room temperature for six days | 5% palmitic and stearic acid | 65 °C, 3 h, MeOH/oil (20:1) |
>80 (FFA conversion) | 83 |
| Wood biochar | Sulfonated with fuming H2SO4 at 150 °C for 15 h | Canola oil | 65 °C, 12 h, MeOH/oil (15:1) |
44.2 | 87 |
| Biochar from glucose | Sulfonated with H2SO4 at 160 °C for 12 h | Palm fatty acid | 65 °C, 134 min, MeOH/oil (12.2:1) |
92.4 (FAME yield) | 88 |
| 94.5 (FFA conversion) | |||||
| Biochar from glucose | 4 g of biochar sulfonated with 100 mL of H2SO4 at 150 °C for 15 h | Palm fatty acid | 75 °C, 2 h, MeOH/oil (10:1) |
92.3 (FAME yield) | 89 |
| 95.4 (FFA conversion) | |||||
| Sulfonated starch solid acid | 5 g of biochar sulfonated with 200 mL of H2SO4 at 160 °C for 12 h | Palm fatty acid | 75 °C, 3 h, MeOH/oil (10:1) |
90.4 (FAME yield) | 90 |
| 94.6 (FFA conversion) | |||||
| Rice husk biochar | Sulfonated with H2SO4 at 90 °C for 0.5 h | Waste cooking oils | 110 °C, 15 h, MeOH/oil (20:1) |
87.6 (FAME yield) | 53 |
| >98 (FFA conversion) | |||||
| Rice husk biochar | Sulfonated with H2SO4 at 70–150 °C for 0.25–4 h | Oleic acid | 110 °C, 2 h, MeOH/oil (4:1) |
98.7 | 91 |
| Corn straw biochar | Sulfonated with fuming H2SO4 at 180 °C for 4 h | Oleic acid | 60 °C, 4 h, MeOH/oil (7:1) |
98 | 92 |
| Biochar from glucose–starch mixture | Sulfonated with H2SO4 at 150 °C for 5 h | Cotton seed oil containing 55.2 wt% of FFA | 80 °C, 12 h, MeOH/oil (20:1) |
>80 | 93 |
| Douglas fir biochar | 20 g of biochar sulfonated with 150 mL of H2SO4 at 150 °C for 24 h | Crude microalgal oil | 100 °C, 60 min, MeOH/oil (20:1) |
98.6 (FFA conversion) | 94 |
| Douglas fir biochar | 1.1 Douglas fir biochar | Crude microalgal oil | 1.1 100 °C, 60 min, MeOH/oil (20:1) |
99 | 94 |
| 1.2 CaO | 1.2 65 °C, 60 min, MeOH/oil (10:1–20:1) |
||||
| Palm kernel shell biochar | Loaded with CaO at a total basicity of 0.516 mmol g−1 | Sunflower oil | 65 °C, 300 min, MeOH/oil (9:1) |
99 | 95 |
| Palm kernel shell biochars | Loaded with 50.6% of CaO | Sunflower oil | 65 °C, 300 min, MeOH/oil (9:1) |
99 | 63 |
| Peat biochar | None | Palm oil | 65 °C, 1.5 h, MeOH/oil (8:1) |
10.5 | 81 |
| Peat biochar | Loaded with 20–40 wt% of K2CO3 | Palm oil | 65 °C, 1.5 h, MeOH/oil (8:1) |
95.2–98.6 | 81 |
| Flamboyant pods biochar | Impregnated in KOH solution | Hevea brasiliensis oil | 60 °C, 1 h, MeOH/oil (15:1) |
89.3 | 96 |
| Biochar from maize residue | None | Waste cooking oils | 300 °C, 1 h, MeOH/oil (50:1) |
∼90 | 82 |
Basically, there are two kinds of biochar catalysts that have been used for biodiesel production, namely, (a) acid-functionalized biochar catalysts and (b) base-functionalized biochar catalysts. As shown in Table 2, sulfonated biochars are the most commonly used heterogeneous catalysts for biodiesel production. The sulfonation of biochar can be easily achieved by impregnating the biochar in concentrated H2SO4 at a high temperature or by exposing the biochar to gaseous SO3, resulting in the immobilization of –SO3H groups on the surface of the biochar. These –SO3H groups can catalyze the esterification of free fatty acids (FFAs) or promote the transesterification reactions of triglycerides with alcohols to form biodiesel.23 For example, Kastner et al.83 prepared various biochar-based solid-acid catalysts by sulfonating the biochar with concentrated H2SO4 or gaseous SO3, and these catalysts were further used as effective catalysts for biodiesel production. Their results showed that H2SO4 sulfonation could increase the surface area and pore volume of the biochar matrix, whereas gaseous SO3 sulfonation generated higher –SO3H densities, as compared to H2SO4 sulfonation, without significantly altering its surface area and pore volume.83 These sulfonated biochar catalysts exhibited high catalytic activities for the esterification of fatty acids, and ∼90–100% of fatty acids could be converted into the corresponding fatty acid methyl esters (FAMEs) within 30–60 min at 55–60 °C.
Similar biochar catalysts from various biomass resources, such as wood, glucose, starch, etc., have also been prepared to be used as solid acid catalysts for biodiesel production.53,84–93 For example, Gonzalez et al.84 sulfonated the oat hull biochar with H2SO4 in a microwave reactor at different temperature, and studied the catalytic performance of these sulfonated biochars in upgrading waste cooking oils to biodiesel in a microwave reactor. In contrast to previous findings by Kastner et al.,83 it was found that the surface area of the biochar was significantly reduced during the sulfonation process, and its BET specific surface area dramatically decreased from 49.3 m2 g−1 to 30.6 and 5.4 m2 g−1 as the biochar was sulfonated with H2SO4 at 100 and 140 °C, respectively. However, the total acidity of the biochar sulfonated at 140 °C (7.0 meq. g−1) was much higher than that sulfonated at 100 °C (3.7 meq. g−1). Despite the low specific surface area, the biochar sulfonated at 140 °C showed high conversion yields to biodiesel, indicating that the total acidity of the biochar-based solid acid catalyst is an important parameter for biodiesel production.84
Recently, base-functionalized biochar catalysts, such as CaO/biochar, KOH/biochar, K2CO3/biochar, etc., have also been used for biodiesel production.94–97 For instance, McKay and coworkers63,95 prepared a CaO-based palm kernel shell biochar catalyst and further used the base-functionalized biochar to catalyze the transesterification of sunflower oil to FAME. Their results showed that a best FAME yield of 99% was achieved at the optimum reaction conditions (3 wt% catalyst loading, 65 °C, and methanol to oil molar ratio of 9:1).95 Various carbon material-supported CaO catalysts used as solid-base catalysts for biodiesel production have also been reported by Konwar and coworkers.23 Similarly, K2CO3 and KOH can also be used as the active center for transesterification reaction. Actually, Wang et al.81 synthesized a series of peat biochar-supported K2CO3 catalysts with different K2CO3 loadings (20–40 wt%) using a wet impregnation method. Also, Dhawane et al.96 prepared a series of flamboyant pods biochar-supported KOH catalysts via an excessive impregnation process. All these base-functionalized biochar catalysts were found to be efficient solid-base catalysts for the transesterification reaction of triacetin with methanol.81,96 However, the water and FFAs in the vegetable oils or animal fats would cause the leaching of the active centers in acid- or base-functionalized biochar catalysts.23 Despite the high yield of biodiesel produced from the vegetable oils or animal fats under the catalysis of the fresh acid- or base-functionalized biochar catalysts, most of these biochar-based catalysts tended to deactivate after several recycling cycles.23 Therefore, the stability of the acid- and base-functionalized biochar catalysts needs to be significantly improved to maximize their lifetime.
4.3. Biochars as catalyst supports for biomass pyrolysis, gasification, and bio-oil upgrading
As mentioned above, biochars have been used as efficient catalysts (especially biochar-based solid acids) for biomass hydrolysis, dehydration and biodiesel production due to its tailoring properties and large surface area. Besides these applications, they have many other advantages when used as catalyst supports, such as low cost, large surface area, multi-scale porous structure, etc.Previous studies showed that metal and metal oxides can be loaded onto the biochar supports with high dispersion by preloading the metal precursor into the biomass matrix before pyrolysis55,64–66 Since the abundant oxygen-containing groups in the biomass matrix can adsorb the metal cations during the preloading process, the metal precursors are highly dispersed in the biomass matrix.24,55,64,65,98 Some metal precursors like Ni2+ and Cu2+ in the biomass matrix can be reduced to metal particles via carbothermal reduction, and the metal particles simultaneously load onto the surface of the biochar, forming biochar-supported metal catalysts.24,55 Other metal precursors like Mg2+ and Fe3+ will form MgO and Fe3O4 during the biomass pyrolysis process.55,66
In addition, Richardson and coworkers64,65 found that biochar-supported nickel metal nanoparticles could be prepared by pyrolyzing the beechwood chips impregnated with aqueous Ni(NO3)2 solution. Their results showed that the Ni(II) was reduced to metallic Ni by carbothermal reduction at temperatures below 500 °C, leading to the in situ formation of metallic Ni nanoparticles on the biochar surface with high dispersion (around 4 nm).64,65 Similar results have also been observed by other researchers.55,98 For instance, a metallic Cu-anchored magnetic biochar catalyst was prepared by fast pyrolysis of fir sawdust preloaded Cu2+ and Fe3+, and metallic Cu particles were monodispersed on the biochar support with an average particle size of 21.2 nm.55 Besides the preparation of biochar-supported metal nanoparticles, they also acted as the catalysts for the generation of syngas and conversion of tar during biomass pyrolysis.64,65,98,99 The results from Richardson et al.64,65 showed that the H2 yield increased about 57–91% and the production of tar was reduced by 60–70% after the lignocellulosic biomass was impregnated with aqueous Ni(NO3)2 solution, indicating that the in situ generated metallic Ni nanoparticles exhibited significant catalytic activity for tar reforming during biomass pyrolysis. The above results show that preloading metal precursor into the biomass matrix is a very useful strategy to prepare effective biochar-supported catalysts for biomass pyrolysis or gasification.
The biochar supported metal or metal oxides can also be prepared by impregnating the biochar with metal precursor.62,66–68 Indeed, it was reported that the biochar-supported Ni catalyst was a promising catalyst for hydrogen production from biomass gasification.67 The biochars obtained from fast pyrolysis of wheat straw, rice husk, cotton stalk, and commercial active carbon were used as catalyst supports, and Ni was supported on the biochars by the impregnation method. The results revealed that the cotton stalk biochar-supported Ni catalyst showed high hydrogen production because of its high content of alkaline and alkaline earth metals and high external surface area.67 Nguyen et al.62 found that the bio-oil from microalgae could be effectively converted into hydrocarbon under the catalysis of Ni/biochar via hydrodeoxygenation (HDO) and hydrodenitrogenation (HDN) processes. Both oxygenates and nitrogenates in the bio-oil were sharply reduced after the catalytic upgrading process, indicating the high activity and selectivity of the Ni/biochar catalyst.62
Additionally, the presence of numerous inorganic compounds in the biochar matrix can also catalyze the pyrolysis or gasification of biomass.100,101 Specifically, Lei et al.100 and Ren et al.101 investigated the catalytic performance of biochar from corn stover in biomass pyrolysis and bio-oil upgrading. Their results showed that the biochar catalyst could improve the quality of syngas and bio-oil to some degree, and high-quality syngas enriched in H2, CO, and CH4 was achieved from the catalytic pyrolysis of sawdust in the presence of the biochar catalysts with a high mineral content, such as K, Ca, Mg, etc.100,101
5. Conclusions and prospects
In this review, the formation of two typical biochar (pyrochar and hydrochar) is proposed. Due to the limited surface area, porosity, and surface functional groups of the biochar, an activation and/or functionalization process is always required for the biochars intended to be used as catalysts. The physicochemical properties of the biochar, especially the surface area and porosity, can be improved to varying degrees according to the activation methods. Functional groups or substances can be further introduced into the activated biochar to endow it with specific properties, such as selective adsorption and catalysis. The activated and functionalized biochars show great potential to be used as versatile catalysts and/or catalyst supports in biomass upgrading, which have been successfully applied in carbohydrate hydrolysis and dehydration, biodiesel production, biomass pyrolysis, gasification, and bio-oil upgrading. However, the physicochemical properties of the biochars vary significantly with the biomass type, production conditions, and activation or functionalization conditions. Further investigations on the industrial scale production of biochars with stable properties should be conducted in the future. In addition, combination of the biochar production with the biomass upgrading process has not received much attention so far. More efforts should be devoted to the co-production of biochars and biofuels or biochemicals from biomass upgrading, which will be beneficial for the further utilization of biochars and the development of the biomass upgrading industry.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are extremely grateful for the financial support from the Fundamental Research Funds for the Central Universities (BLX2015-10), the National Natural Science Foundation of China (31700518), the State Key Laboratory of Pulp and Paper Engineering (201517 and 201612), and the Beijing Key Lab. Project (Z16110005016041).References
- A. Demirbaş, Energy Convers. Manage., 2001, 42, 1357–1378 CrossRef.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gartner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS PubMed.
- L. Ma, T. Wang, Q. Liu, X. Zhang, W. Ma and Q. Zhang, Biotechnol. Adv., 2012, 30, 859–873 CrossRef CAS PubMed.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- F. X. Collard and J. Blin, Renewable Sustainable Energy Rev., 2014, 38, 594–608 CrossRef CAS.
- S. S. Toor, L. Rosendahl and A. Rudolf, Energy, 2011, 36, 2328–2342 CrossRef CAS.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, Chemsuschem, 2011, 4, 636–643 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- J. Lehmann, Front. Ecol. Environ., 2007, 5, 381–387 CrossRef.
- J. S. Cha, S. H. Park, S. C. Jung, C. Ryu, J. K. Jeon, M. C. Shin and Y. K. Park, J. Ind. Eng. Chem., 2016, 40, 1–15 CrossRef CAS.
- H. S. Kambo and A. Dutta, Renewable Sustainable Energy Rev., 2015, 45, 359–378 CrossRef CAS.
- J. Marousek, M. Vochozka, J. Plachy and J. Zak, Clean Technol. Environ. Policy, 2017, 19, 311–317 CrossRef CAS.
- X. Tan, Y. Liu, Y. Gu, Y. Xu, G. Zeng, X. Hu, S. Liu, X. Wang, S. Liu and J. Li, Bioresour. Technol., 2016, 212, 318–333 CrossRef CAS PubMed.
- Y. Ding, Y. Liu, S. Liu, Z. Li, X. Tan, X. Huang, G. Zeng, L. Zhou and B. Zheng, Agron. Sustainable Dev., 2016, 36, 1–18 CrossRef CAS.
- S. Prapagdee, S. Piyatiratitivorakul and A. Petsom, Asian J. Water, Environ. Pollut., 2016, 13, 27–34 CrossRef CAS.
- M. Koltowski, B. Charmas, J. Skubiszewska-Zieba and P. Oleszczuk, Ecotoxicol. Environ. Saf., 2017, 136, 119–125 CrossRef CAS PubMed.
- J. M. V. Nabais, P. Nunes, P. J. M. Carrott, M. M. L. Ribeiro Carrott, A. M. García and M. A. Díaz-Díez, Fuel Process. Technol., 2008, 89, 262–268 CrossRef CAS.
- S. Yorgun, N. Vural and H. Demiral, Microporous Mesoporous Mater., 2009, 122, 189–194 CrossRef CAS.
- D. Angin, E. Altintig and T. E. Kose, Bioresour. Technol., 2013, 148, 542–549 CrossRef CAS PubMed.
- T. Z. Liu, B. Gao, J. N. Fang, B. Wang and X. D. Cao, RSC Adv., 2016, 6, 24314–24319 RSC.
- X. H. Lu, J. He, R. Jing, P. P. Tao, R. F. Nie, D. Zhou and Q. H. Xia, Sci. Rep., 2017, 7, 2676 CrossRef PubMed.
- M. Uchimiya, J. J. Pignatello, J. C. White, S. L. Hu and P. J. Ferreira, Sci. Rep., 2017, 7, 5027 CrossRef PubMed.
- L. J. Konwar, J. Boro and D. Deka, Renewable Sustainable Energy Rev., 2014, 29, 546–564 CrossRef CAS.
- W. J. Liu, H. Jiang and H. Q. Yu, Chem. Rev., 2015, 115, 12251–12285 CrossRef CAS PubMed.
- J. Lee, K. H. Kim and E. E. Kwon, Renewable Sustainable Energy Rev., 2017, 77, 70–79 CrossRef CAS.
- S. Du, J. A. Valla and G. M. Bollas, Green Chem., 2013, 15, 3214–3229 RSC.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- K. Qian, A. Kumar, H. Zhang, D. Bellmer and R. Huhnke, Renewable Sustainable Energy Rev., 2015, 42, 1055–1064 CrossRef CAS.
- A. Demirbaş, Energy Convers. Manage., 2000, 41, 633–646 CrossRef.
- M. L. Poutsma, J. Anal. Appl. Pyrolysis, 2000, 54, 5–35 CrossRef CAS.
- Y. C. Lin, J. Cho, G. A. Tompsett, P. R. Westmoreland and G. W. Huber, J. Phys. Chem. C, 2009, 113, 20097–20107 CAS.
- D. K. Shen, S. Gu and A. V. Bridgwater, J. Anal. Appl. Pyrolysis, 2010, 87, 199–206 CrossRef CAS.
- Q. Liu, S. Wang, Y. Zheng, Z. Luo and K. Cen, J. Anal. Appl. Pyrolysis, 2008, 82, 170–177 CrossRef CAS.
- Y. Zhang, S. Liu, X. Zheng, X. Wang, Y. Xu, H. Tang, F. Kang, Q.-H. Yang and J. Luo, Adv. Funct. Mater., 2017, 27, 1604687 CrossRef.
- D. Kim, K. Lee and K. Y. Park, J. Ind. Eng. Chem., 2016, 42, 95–100 CrossRef CAS.
- H. P. Yang, R. Yan, H. P. Chen, D. H. Lee and C. G. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- Z. Wang, X. Wang, J. Jiang, Y. Fu and M. Qin, Carbohydr. Polym., 2015, 126, 185–191 CrossRef CAS PubMed.
- X. F. Cao, X. W. Peng, S. N. Sun, L. X. Zhong and R. C. Sun, J. Agric. Food Chem., 2014, 62, 12360–12365 CrossRef CAS PubMed.
- M. Sasaki, T. Adschiri and K. Arai, Bioresour. Technol., 2003, 86, 301–304 CrossRef PubMed.
- S. Kang, X. Li, J. Fan and J. Chang, Ind. Eng. Chem. Res., 2012, 51, 9023–9031 CrossRef CAS.
- J. Ryu, Y. W. Suh, D. J. Suh and D. J. Ahn, Carbon, 2010, 48, 1990–1998 CrossRef CAS.
- M. B. Ahmed, J. L. Zhou, H. H. Ngo, W. Guo and M. Chen, Bioresour. Technol., 2016, 214, 836–851 CrossRef CAS PubMed.
- J. Li, H. Zhang, X. Tang and H. Lu, RSC Adv., 2016, 6, 100352–100360 RSC.
- U. Iriarte-Velasco, I. Sierra, L. Zudaire and J. L. Ayastuy, Food Bioprod. Process., 2016, 98, 341–353 CrossRef CAS.
- T. Shim, J. Yoo, C. Ryu, Y. K. Park and J. Jung, Bioresour. Technol., 2015, 197, 85–90 CrossRef CAS PubMed.
- H. Jin, S. Capareda, Z. Chang, J. Gao, Y. Xu and J. Zhang, Bioresour. Technol., 2014, 169, 622–629 CrossRef CAS PubMed.
- D. Jimenez-Cordero, F. Heras, N. Alonso-Morales, M. A. Gilarranz and J. J. Rodriguez, Fuel Process. Technol., 2015, 139, 42–48 CrossRef CAS.
- I. M. Lima, A. A. Boateng and K. T. Klasson, J. Chem. Technol. Biotechnol., 2010, 85, 1515–1521 CAS.
- A. M. Dehkhoda, N. Ellis and E. Gyenge, J. Appl. Electrochem., 2014, 44, 141–157 CrossRef CAS.
- J. H. Park, Y. S. Ok, S. H. Kim, J. S. Cho, J. S. Heo, R. D. Delaune and D. C. Seo, Environ. Geochem. Health, 2015, 37, 969–983 CrossRef CAS PubMed.
- R. Azargohar and A. K. Dalai, Microporous Mesoporous Mater., 2008, 110, 413–421 CrossRef CAS.
- M. Li, Y. Zheng, Y. X. Chen and X. F. Zhu, Bioresour. Technol., 2014, 154, 345–348 CrossRef CAS PubMed.
- Q.-Y. Liu, F. Yang, Z.-H. Liu and G. Li, J. Ind. Eng. Chem., 2015, 26, 46–54 CrossRef CAS.
- W.-J. Liu, K. Tian, H. Jiang and H.-Q. Yu, Green Chem., 2014, 16, 4198–4205 RSC.
- A. Deng, Q. Lin, Y. Yan, H. Li, J. Ren, C. Liu and R. Sun, Bioresour. Technol., 2016, 216, 754–760 CrossRef CAS PubMed.
- Y. Y. Bai, L. P. Xiao and R. C. Sun, Biomass Bioenergy, 2015, 75, 245–253 CrossRef CAS.
- X. Qi, Y. Lian, L. Yan and R. L. Smith, Catal. Commun., 2014, 57, 50–54 CrossRef CAS.
- S. Li, Z. Gu, B. E. Bjornson and A. Muthukumarappan, J. Environ. Chem. Eng., 2013, 1, 1174–1181 CrossRef CAS.
- R. Ormsby, J. R. Kastner and J. Miller, Catal. Today, 2012, 190, 89–97 CrossRef CAS.
- C. Zhang, Z. Fu, B. Dai, S. Zen, Y. Liu, Q. Xu, S. R. Kirk and D. Yin, Cellulose, 2014, 21, 1227–1237 CrossRef CAS.
- H. K. D. Nguyen, V. V. Pham and H. T. Do, Catal. Lett., 2016, 146, 2381–2391 CrossRef CAS.
- A. Bazargan, M. D. Kostić, O. S. Stamenković, V. B. Veljković and G. McKay, Fuel, 2015, 150, 519–525 CrossRef CAS.
- Y. Richardson, J. Blin, G. Volle, J. Motuzas and A. Julbe, Appl. Catal., A, 2010, 382, 220–230 CrossRef CAS.
- Y. Richardson, J. Motuzas, A. Julbe, G. Volle and J. Blin, J. Phys. Chem. C, 2013, 117, 23812–23831 CAS.
- W. J. Liu, H. Jiang, K. Tian, Y. W. Ding and H. Q. Yu, Environ. Sci. Technol., 2013, 47, 9397–9403 CrossRef CAS PubMed.
- D. Yao, Q. Hu, D. Wang, H. Yang, C. Wu, X. Wang and H. Chen, Bioresour. Technol., 2016, 216, 159–164 CrossRef CAS PubMed.
- J. R. Kastner, S. Mani and A. Juneja, Fuel Process. Technol., 2015, 130, 31–37 CrossRef CAS.
- F. Rodríguez-reinoso, Carbon, 1998, 36, 159–175 CrossRef.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- C. Zhang, Z. Fu, Y. C. Liu, B. Dai, Y. Zou, X. Gong, Y. Wang, X. Deng, H. Wu, Q. Xu, K. R. Steven and D. Yin, Green Chem., 2012, 14, 1928–1934 RSC.
- C. Zhang, Z. Fu, B. Dai, S. Zen, Y. Liu, Q. Xu, S. R. Kirk and D. Yin, Ind. Eng. Chem. Res., 2013, 52, 11537–11543 CrossRef CAS.
- Q. Liu, F. Yang, X. Sun, Z. Liu and G. Li, J. Mater. Cycles Waste Manage., 2017, 19, 134–143 CrossRef CAS.
- E. Sairanen, R. Karinen and J. Lehtonen, Catal. Lett., 2014, 144, 1839–1850 CrossRef CAS.
- C. Zhang, Z. Cheng, Z. Fu, Y. Liu, X. Yi, A. Zheng, S. R. Kirk and D. Yin, Cellulose, 2017, 24, 95–106 CrossRef CAS.
- V. Choudhary, S. I. Sandler and D. G. Vlachos, ACS Catal., 2012, 2, 2022–2028 CrossRef CAS.
- M. Paul Abishek, J. Patel and A. Prem Rajan, Biotechnol. Res. Int., 2014, 2014, 8 Search PubMed.
- J. M. Rafi, A. Rajashekar, M. Srinivas, B. V. S. K. Rao, R. B. N. Prasad and N. Lingaiah, RSC Adv., 2015, 5, 44550–44556 RSC.
- M. Di Serio, R. Tesser, L. Pengmei and E. Santacesaria, Energy Fuels, 2008, 22, 207–217 CrossRef CAS.
- S. Wang, C. Zhao, R. Shan, Y. Wang and H. Yuan, Energy Convers. Manage., 2017, 139, 89–96 CrossRef CAS.
- J. Lee, J.-M. Jung, J.-I. Oh, Y. S. Ok, S.-R. Lee and E. E. Kwon, Bioresour. Technol., 2017, 231, 59–64 CrossRef CAS PubMed.
- J. R. Kastner, J. Miller, D. P. Geller, J. Locklin, L. H. Keith and T. Johnson, Catal. Today, 2012, 190, 122–132 CrossRef CAS.
- M. E. Gonzalez, M. Cea, D. Reyes, L. Romero-Hermoso, P. Hidalgo, S. Meier, N. Benito and R. Navia, Energy Convers. Manage., 2017, 137, 165–173 CrossRef CAS.
- A. M. Dehkhoda and N. Ellis, Catal. Today, 2013, 207, 86–92 CrossRef CAS.
- A. M. Dehkhoda, A. H. West and N. Ellis, Appl. Catal., A, 2010, 382, 197–204 CrossRef CAS.
- J. T. Yu, A. M. Dehkhoda and N. Ellis, Energy Fuels, 2011, 25, 337–344 CrossRef CAS.
- I. M. Lokman, U. Rashid and Y. H. Taufiq-Yap, Chin. J. Chem. Eng., 2015, 23, 1857–1864 CrossRef CAS.
- I. M. Lokman, U. Rashid, Y. H. Taufiq-Yap and R. Yunus, Renewable Energy, 2015, 81, 347–354 CrossRef CAS.
- I. M. Lokman, U. Rashid and Y. H. Taufiq-Yap, Arabian J. Chem., 2016, 9, 179–189 CrossRef CAS.
- M. Li, D. Chen and X. Zhu, Chin. J. Catal., 2013, 34, 1674–1682 CrossRef CAS.
- T. Liu, Z. Li, W. Li, C. Shi and Y. Wang, Bioresour. Technol., 2013, 133, 618–621 CrossRef CAS PubMed.
- G. Chen and B. Fang, Bioresour. Technol., 2011, 102, 2635–2640 CrossRef CAS PubMed.
- T. Dong, D. F. Gao, C. Miao, X. C. Yu, C. Degan, M. Garcia-Perez, B. Rasco, S. S. Sablani and S. L. Chen, Energy Convers. Manage., 2015, 105, 1389–1396 CrossRef CAS.
- M. D. Kostic, A. Bazargan, O. S. Stamenkovic, V. B. Veljkovic and G. McKay, Fuel, 2016, 163, 304–313 CrossRef CAS.
- S. H. Dhawane, T. Kumar and G. Halder, Energy Convers. Manage., 2015, 100, 277–287 CrossRef CAS.
- S. Dai, S. Wu, N. Duan, J. Chen, Z. Zheng and Z. Wang, Biosens. Bioelectron., 2017, 91, 538–544 CrossRef CAS PubMed.
- Q. Yan, C. Wan, J. Liu, J. Gao, F. Yu, J. Zhang and Z. Cai, Green Chem., 2013, 15, 1631–1640 RSC.
- Y. Shen and K. Yoshikawa, Ind. Eng. Chem. Res., 2014, 53, 10929–10942 CrossRef CAS.
- H. Lei, S. Ren and J. Julson, Energy Fuels, 2009, 23, 3254–3261 CrossRef CAS.
- S. Ren, H. Lei, L. Wang, Q. Bu, S. Chen and J. Wu, RSC Adv., 2014, 4, 10731–10737 RSC.
| This journal is © The Royal Society of Chemistry 2017 |