
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling and tackling guanidinium peptide coupling reagent side reactions towards the development of peptide-drug conjugates†
Eirinaios I. Vrettos ,
Nisar Sayyad,
Eftychia M. Mavrogiannaki,
Evgenios Stylos,
Androniki D. Kostagianni,
Serafim Papas,
Thomas Mavromoustakos,
Vassiliki Theodorou and
Andreas G. Tzakos*
,
Nisar Sayyad,
Eftychia M. Mavrogiannaki,
Evgenios Stylos,
Androniki D. Kostagianni,
Serafim Papas,
Thomas Mavromoustakos,
Vassiliki Theodorou and
Andreas G. Tzakos*
Section of Organic Chemistry and Biochemistry, Department of Chemistry, University of Ioannina, Ioannina GR-45110, Greece. E-mail: agtzakos@gmail.com
First published on 30th October 2017
Abstract
Peptide coupling reagents and especially uronium/guanidinium salts have been extensively utilized in solid-phase peptide synthesis. However, the impact of these reagents in solution phase synthesis, normally used in the formation of peptide-drug conjugates (PDCs), has not been fully explored. Herein, we identified that when guanidinium salts are used in classical peptide coupling conditions, besides leading to the formation of amide bonds, a uronium derivative can also be installed on specific amino acid scaffolds. The formation of this side product depends on the reaction conditions, as also on the nucleophilicity of the susceptible groups. Conditions to avoid this side product formation and a putative reaction mechanism describing its formation are reported.
Introduction
An array of coupling reagents has been developed for peptide synthesis, such as phosphonium (BOP, PyBOP etc.), carbodiimide (DCC) and guanidinium (HATU, HBTU etc.). Among the most classically utilized guanidinium reagents are HATU (N-[(dimethylamino)-1H-1,2,3-triazo[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate-N-oxide) (1), and HBTU (N-[(1H-benzotriazol-1-yl)(dimethylamino)-methylene]-N-methylmethanaminium hexafluorophosphate-N-oxide) (2).1–6Based on their structure, HBTU and HATU were originally classified as O-uronium salts; later were revised to N-guanidinium salts (Fig. 1).7
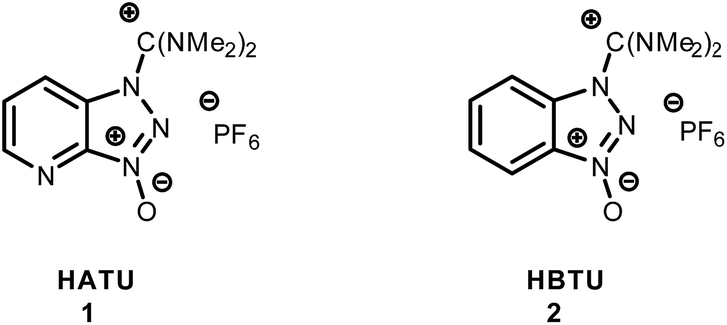 | ||
| Fig. 1 Guanidinium salts of HATU and HBTU coupling reagents. | ||
HATU8 since its innovation, has been established as a peptide coupling reagent with enhanced reactivity, devoid of side product(s) formation and racemization in the formed products compared to other commonly used coupling reagents. HATU has been applied in the macrocyclization of complicated molecules9–11 as well as in the total synthesis of the antibiotic himastatin,12,13 a symmetric cyclohexadepsipeptide dimer.
Furthermore, cyclodimerization or epimerization during cyclization of all-L-tetrapeptides or all-L-pentapeptides,10 could be surmounted by the utilization of HATU, which was found to be more effective than TBTU or BOP. During the synthesis of peptide nucleic acid (PNA) analogues, HATU was successfully used as a coupling reagent along with collidine, both in solution and solid phase.14 Furthermore, the synthetic route towards dehydrodidemnin B was improved by the use of HATU.15 Besides these, it has been reported16 that during a peptide coupling reaction, in the presence of uronium coupling reagents, a guanidino side product on the N-terminal amine group was formed. More specifically, it has been noted that in a peptide coupling reaction, aminium salts can react with the N-terminal amino moiety, leading to a guanidino side product responsible for the termination of peptide chain elongation.17,18 Guanidinium coupling reagents, like HBTU and TBTU, have been utilized to produce amine-modified analogues such as 4-aminoquinoline, which have been evaluated for their antimalarial potency,19 or peptide antagonists for the chemokine receptor CXCR4.20 Similar isothiouronium pyrimidine–curcumin compounds have been synthesized using tetramethylthiourea for Golgi localization.21
Peptide-drug conjugates (PDCs) represent a rapidly emerging class of therapeutics capable to enhance the treatment efficacy of medicinal compounds.22–24 In this frame, we evaluated the utilization of HATU/HBTU in the solution phase coupling of a PDC and specifically of the Gonadotropin Releasing Hormone (GnRH) agonist ([D-Lys]6-GnRH; Glp-His-Trp-Ser-Tyr-D-Lys-Leu-Arg-Pro-Gly-NH2) and the cytotoxic drug gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) which could be exploited for drug targeted delivery in cancer cells.24 After the construction and HPLC purification of the relevant conjugate, we observed the coexistence of a side product, bearing a mass of the expected PDC plus 99 amu. A template peptide coupling reaction was conducted to monitor the exact location of this side product using all amino acids of [D-Lys]6-GnRH sequence. The side product was assigned as a uronium moiety located on the phenol group of tyrosine. The same side product was also determined to occur on the –OH group of tyrosine of the tumor homing peptide HER2-BP1 (NH2-YWPSVTL-COOH),25 an heptapeptide that is known for its activity against erbB2, suppressing expression of the c-neu gene in breast cancer cells. Triggered by these observations we explored whether a relevant modification could be applied on other amino acids using model dipeptides. We discovered that the sulfhydryl group of cysteine is also susceptible to this modification. The relevant modification was also evident to take place in both cysteines of the C1B5141–151 subdomain peptide (RCVRSVPSLCG) of protein kinase C (PKC) γ isozymes, that is known to suppress growth of human colon cancer cells.26 Finally, a possible mechanism for the formation of the reported uronium side product is depicted and the proper conditions to surmount its formation are also reported.
Experimental
Materials and methods
Fmoc amino acids, N-[(dimethylamino)-1H-1,2,3-triazo[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HATU), N-[(1H-benzotriazol-1-yl)(dimethylamino)methylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HBTU) were purchased from Neosystem Laboratoire (Strasbourg, France). Fmoc-D-lysine was purchased from Chempep. Triisopropylsilane (TIS), N,N-diisopropylethylamine (DIPEA), trifluoroacetic acid (TFA) and phenol were purchased from Sigma-Aldrich and used without further purification. Thionyl chloride was purchased from Acros Organics. N,N-Dimethylformamide (DMF) was distillated over CaH2 and stored under molecular sieves (4 Å). High resolution ESI-MS spectra were measured on a Thermo-Fischer Scientific Orbitrap XL and/or EVOQ™ Elite ER LC-TQ system.Synthetic procedures
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. no. | Side chain (R) | HATU | HBTU | Product | ||||
| Equivalents | Products monitored with mass | Equivalents | Products monitored with mass | |||||
| a | b | a | b | |||||
| 8 | 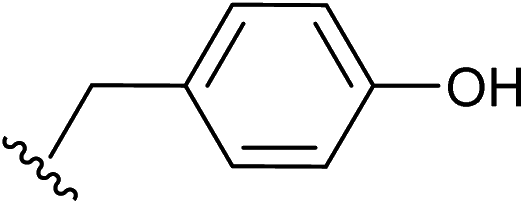 |
1.5 | ✓ | ✓ | 1.5 | ✓ | ✓ | 15 |
| 1.0 | ✓ | ✗ | 1.0 | ✓ | ✗ | |||
| 9 | 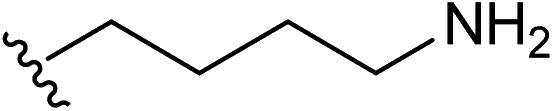 |
1.5 | ✗ | ✓ | 1.5 | ✓ | ✓ | 16 |
| 1.0 | ✓ | ✓ | 1.0 | ✓ | ✗ | |||
| 10 | 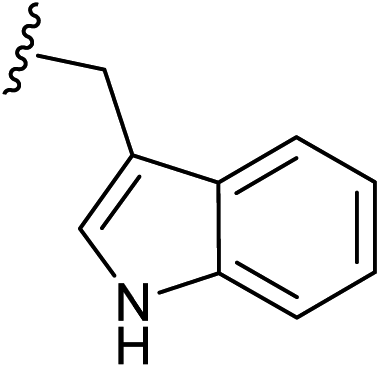 |
1.5 | ✓ | ✗ | 1.5 | ✓ | ✗ | 17 |
| 1.0 | ✓ | ✗ | 1.0 | ✓ | ✗ | |||
| 11 | 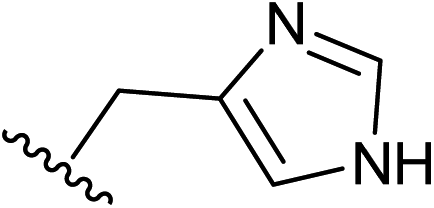 |
1.5 | ✓ | ✗ | 1.5 | ✓ | ✗ | 18 |
| 1.0 | ✓ | ✗ | 1.0 | ✓ | ✗ | |||
| 12 | 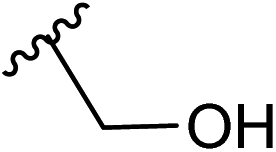 |
1.5 | ✓ | ✗ | 1.5 | ✓ | ✗ | 19 |
| 1.0 | ✓ | ✗ | 1.0 | ✓ | ✗ | |||
| 13 | 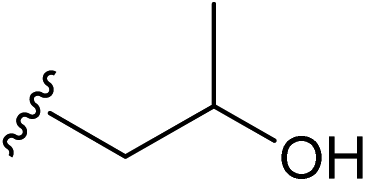 |
1.5 | ✓ | ✗ | 1.5 | ✓ | ✗ | 20 |
| 1.0 | ✓ | ✗ | 1.0 | ✓ | ✗ | |||
| 14 | 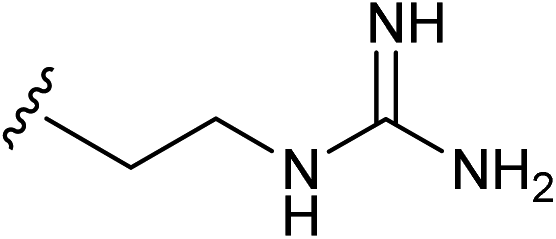 |
1.5 | ✓ | ✗ | 1.5 | ✓ | ✗ | 21 |
| 1.0 | ✓ | ✗ | 1.0 | ✓ | ✗ | |||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3:1). The cleavage of the peptides from the resin was carried out using either cocktail A: containing TFA/TIS/EDT/H2O in a percentage of 92.5/2.5/2.5/2.5 v/v (for peptides Fmoc-Cys-Tyr-NH2 and Fmoc-C1B5) or B: containing TFA/TIS/H2O in a percentage of 95/2.5/2.5 v/v (for peptides [D-Lys]6-GnRH, Fmoc-HER2-BP1 and Fmoc-Ser-Tyr-NH2).
:3:3:1). The cleavage of the peptides from the resin was carried out using either cocktail A: containing TFA/TIS/EDT/H2O in a percentage of 92.5/2.5/2.5/2.5 v/v (for peptides Fmoc-Cys-Tyr-NH2 and Fmoc-C1B5) or B: containing TFA/TIS/H2O in a percentage of 95/2.5/2.5 v/v (for peptides [D-Lys]6-GnRH, Fmoc-HER2-BP1 and Fmoc-Ser-Tyr-NH2).Results and discussion
In our former work, our group designed and reported24 the development of an innovative strategy to produce a metabolically stable analogue of gemcitabine (Scheme 1), that could be selectively delivered to prostate cancer cells (CaP). This was achieved through the formation of a gemcitabine-GnRH PDC (compound 6) that was based on the cell surface overexpression of the Gonadotropin Releasing Hormone-Receptor (GnRH-R) in cancer cells.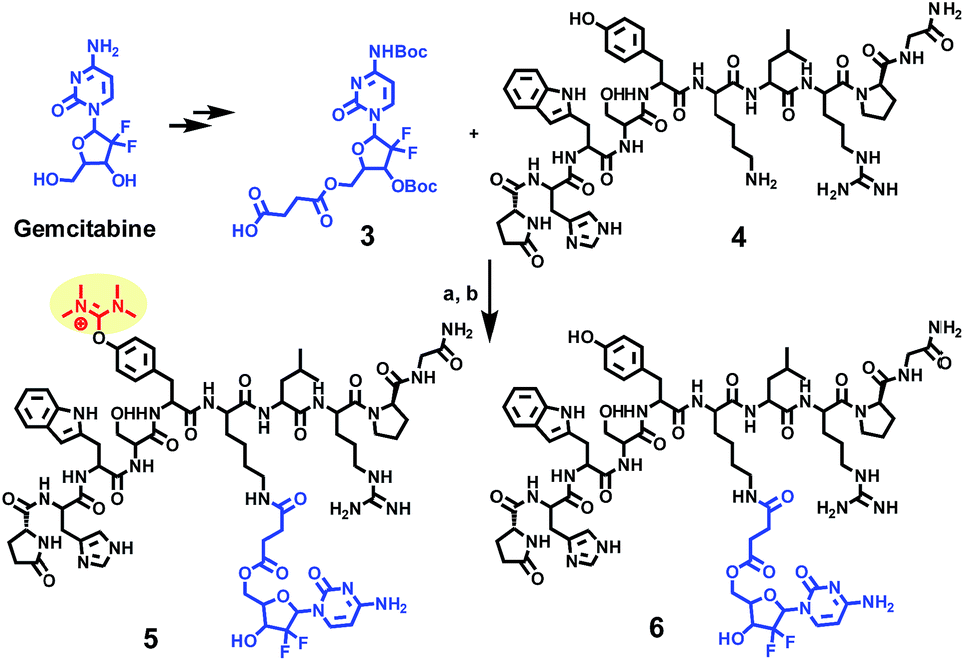 | ||
| Scheme 1 Synthesis of a gemcitabine-GnRH conjugate 6 and its uronium side product 5. (a) HATU (1.5 eq.), DIPEA (5 eq.), DMF; (b) TFA/TIS/H2O (95/2.5/2.5%). | ||
During the liquid phase synthesis of compound 6 by the reaction of Boc-protected 5′-O-gemcitabine hemisuccinate (3) and the ε-amino group of [D-Lys]6-GnRH (4), in the presence of HATU as a coupling reagent, we realized the formation of an unexpected side product (5) along with the desired product (6). Evaluation of the mass spectrum (Fig. 2) of the reaction product(s), indicated that in addition to the mass corresponding to the desired PDC (6) (molecular ions [M + 2H]2+ = 800.11 and [M + 3H]3+ = 534.11), the presence of an unexpected compound that corresponded to the mass of the desired PDC plus 99.00 amu (molecular ions [M + 2H]2+ = 849.71 and [M + 3H]3+ = 566.79).
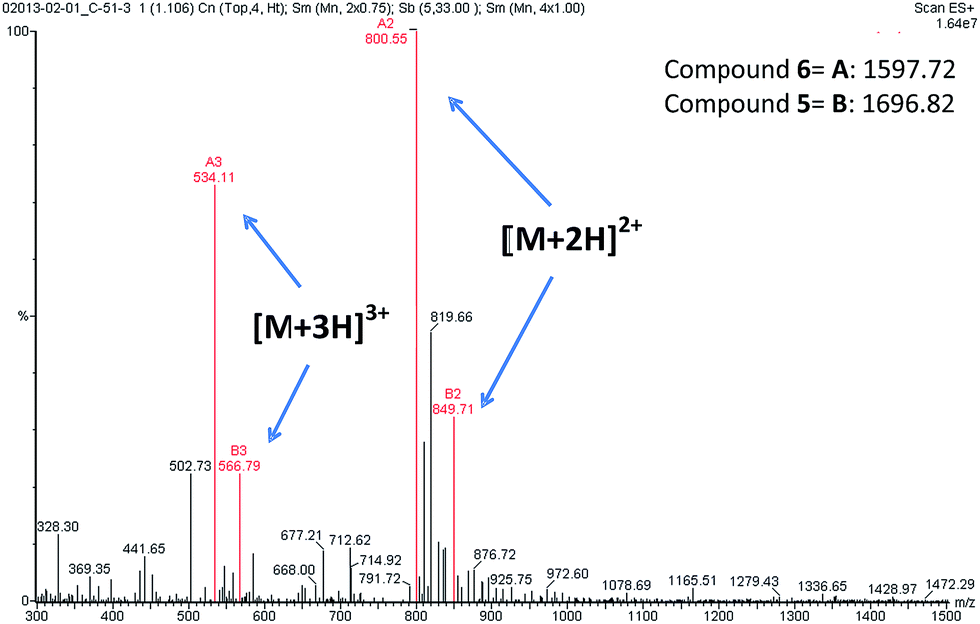 | ||
| Fig. 2 ESI-MS spectra of compounds 5 and 6. The molecular ions of compound 5 [M + 2H]2+ = 849.71 and [M + 3H]3+ = 566.79. The molecular ions of compound 6 [M + 2H]2+ = 800.55 and [M + 3H]3+ = 534.11. | ||
We hypothesized that the second observed product could correspond to the formation of a guanidinium/uronium side product due to the covalent coupling of the guanidinium group to the peptide. However, HATU-mediated guanidinylation has been reported only for free amino groups of peptides. In [D-Lys]6-GnRH the N-terminal amine is capped with pyroglutamic acid and the ε-amino group of D-Lys6 was coupled to the carboxy derivatized gemcitabine.
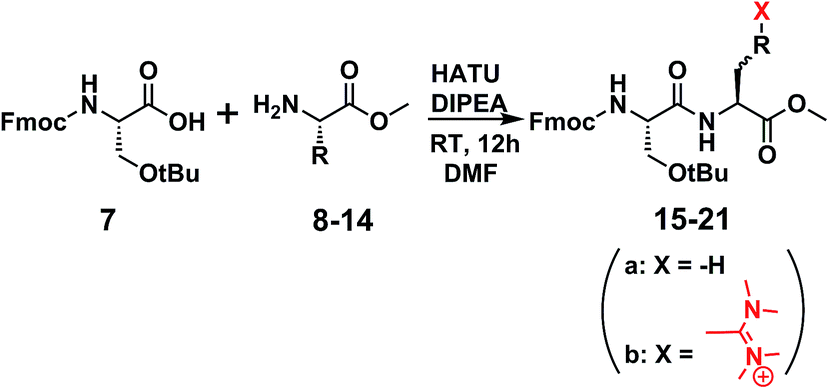
This led us to suggest that the putative guanidinium/uronium group was installed in a different location since it had not hindered the formation of the amide bond between the drug and the peptide. We thus targeted to explore the amino acid residue of the [D-Lys]6-GnRH sequence susceptible to this side product formation, having in mind that bears two potent nucleophiles (the ε-amino group of Lys6 and the hydroxyl group of Tyr5). In order to assign this, we conducted a series of reactions (Scheme 2) between the methyl ester of each individual amino acid existed in the [D-Lys]6-GnRH peptide sequence (8–14) and Fmoc-Ser(tBu)-OH (7) (Table 1, Fig. S1–S28†), using the same reaction conditions, as in Scheme 1 and we monitored the products with ESI-MS.
 | ||
| Scheme 2 Coupling of Fmoc-Ser(tBu)-OH with tyrosine-O-methyl ester: (a) HATU (1.5 eq.), DIPEA (5 eq.), DMF. | ||
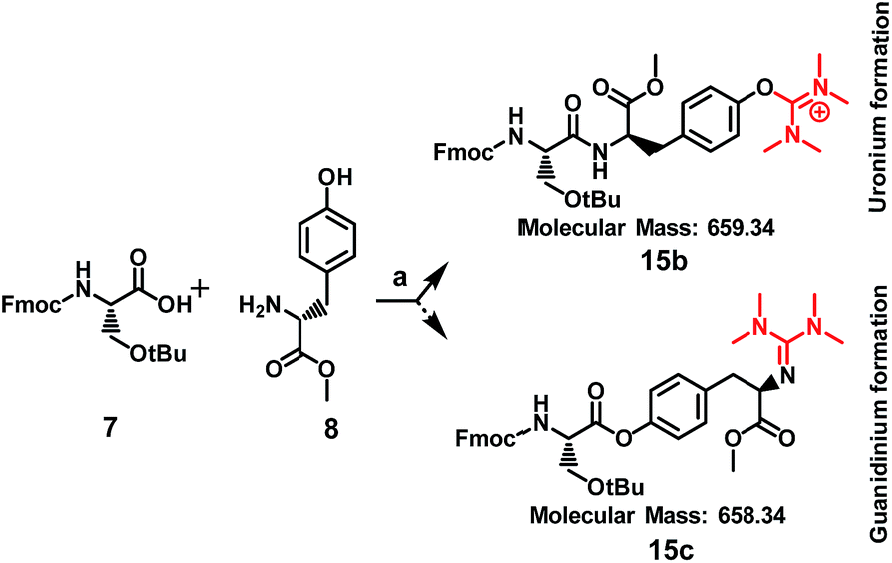
When Fmoc-Ser(tBu)-OH (7) was treated with HATU (1.5 eq.) or HBTU (1.5 eq.) and DIPEA (5 eq.) the formation of a side product was observed on the mass spectrum, but only when Tyr or Lys were used in the template reactions (data shown in Table 1). Specifically, it became evident that regarding Tyr, the formation of the side product enhanced the expected mass by M + 99 m/z, whereas for Lys by M + 98 m/z. These results indicate that compound 5, the side product of compound 6, results from the installation of an additional group to Tyr and not to Lys, since its molecular ion has been enhanced by M + 99 m/z, and not by M + 98 m/z. To further verify this finding and to exclude the possibility of an ester bond formation instead of the expected amide bond, Fmoc-glycine (1 eq.) was reacted with Tyr-OMe (1 eq.) using HATU (1.5 eq.)/DIPEA (5 eq.) in DMF, where two products could be formed: an uronium form on tyrosine (compound 28, Fig. S71†) or a guanidinium form on the amino group (compound 29, Fig. S71†).
The mass spectrum of the received product (ESI Fig. S72†) corroborates with the formation of compound 28 (m/z = 573.27 [M + H]+) and not of compound 29 (m/z = 572.26 [M + H]+).
Therefore, the possibility that the activated ester of Fmoc-Ser(tBu)-OH reacted with the hydroxyl group of Tyr, to form an ester linkage (Scheme 2), is ruled out by the MS analysis of the resulted product. As it is shown in Scheme 2, a putative compound could be 15c, bearing an ester linkage between Fmoc-Ser(tBu)-OH and Tyr-OMe and N-terminal guanidinylation has an expected molecular mass of 658.34. Compound 15b is the other alternative, with an expected molecular mass of 659.34, where the amide bond is formed between Fmoc-Ser(tBu)-OH and Tyr-OMe and an uronium group is installed on the phenol group of Tyr-OMe. ESI-MS analysis of the product corroborates to the formation of 15b due to the recorded molecular ion at m/z 660.14 (ESI Fig. S1 and S3†).
It is interesting to pinpoint that pure compound 6 bears a white color, whereas compound 5 is a pale yellow solid (dark yellow in solution phase) that is difficult to separate via HPLC from its mixture with 6 (Fig. S29†). The end point of the HATU-mediated coupling reaction is yellow-to-colourless.8 In our case, the yellow color persisted till the end of the reaction that could be an indication of the presence of the side product.
To further justify our findings in more complex compounds than template dipeptides, we reacted [D-Lys]6-GnRH (1 eq.) with HATU (1.5 eq.) and DIPEA (5 eq.) in DMF and after RP-HPLC purification we received compound 22 (Fig. 3B and S38†).
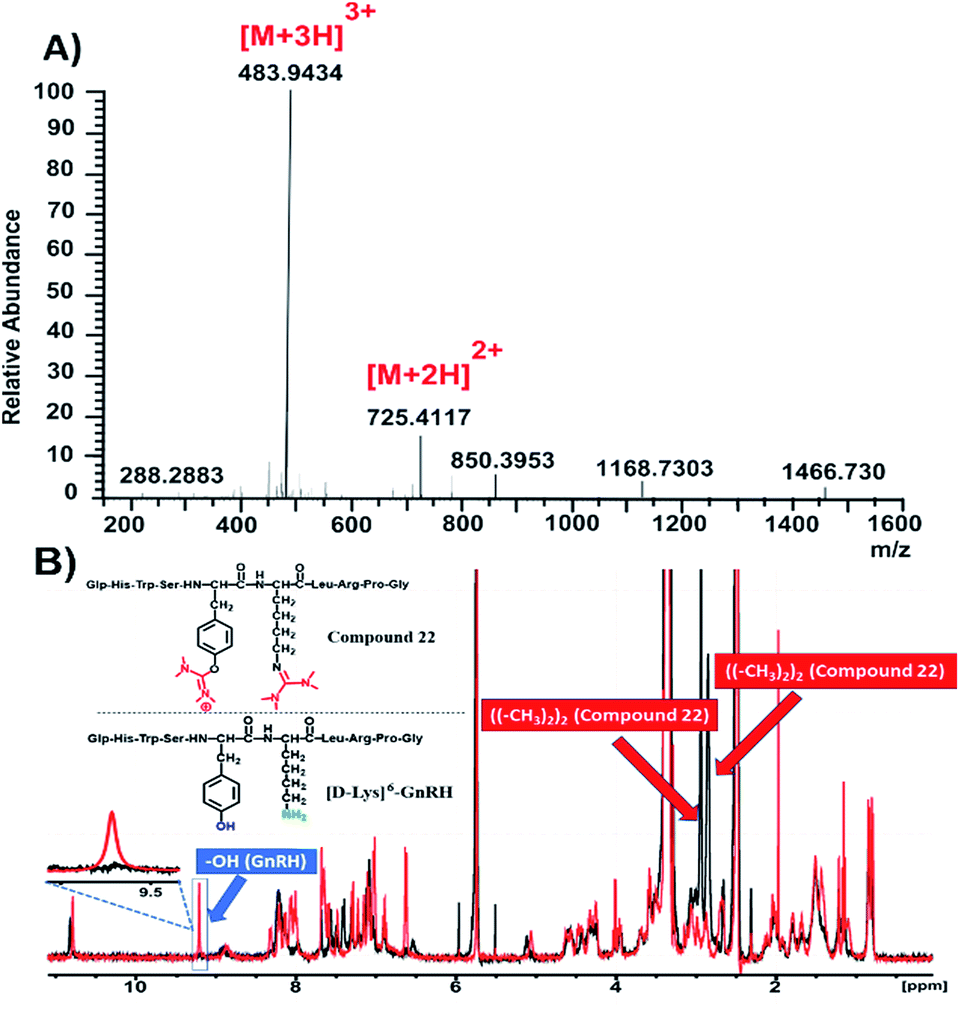 | ||
| Fig. 3 Characterization of compound 22. (A) HRMS spectrum of compound 22; (B) overlay of 1H-NMR spectra of compound 22 (black color) and [D-Lys]6-GnRH (red color) in DMSO-d6 at 298 K. | ||
The HRMS spectrum (Fig. 3A and S40†) of this compound suggests the installation of two uronium along the [D-Lys]6-GnRH sequence. On the basis of the information extracted from Table 1, it can be suggested that the side product has been installed both on the phenolic group of tyrosine and the ε-amino group of lysine. This was further verified through recording the 1H-NMR spectrum of compound 22 (Fig. 3B, black color; Fig. S41†) and overlaying it with the relevant spectrum of the [D-Lys]6-GnRH peptide (Fig. 3B, red color).
It is evident that the absorption of the phenol group has disappeared from 9.6 ppm, since it is occupied by the uronium group and also two more distinct peaks appeared (bearing an integral of 12) referring to the discrete chemical environment of the four methyl groups of the guanidinium and uronium, at 2.9 and 3 ppm, respectively (Fig. S41†).
The same methodology was applied to another tumor homing peptide widely used for peptide-drug conjugates (HER2-BP1; NH2-YWPSVTL-COOH) to afford compound 23 (Fig. 4 and S45†). The ESI-MS spectrum (Fig. 4A and S47†) of this compound suggests the installation of only one uronium group, which probably occurred on the –OH group of tyrosine. This is verified through the 1H-NMR spectrum of compound 23 (Fig. 4B, black color; Fig. S48†) overlaid with the relevant spectrum of the parent peptide (Fmoc-HER2-BP1, Fig. 4B, red color), which shows the absence of the phenol group at 9.2 ppm and the appearance of one peak at 3 ppm referring to the methyl groups of the side product with an integral of 12 (Fig. S48†).
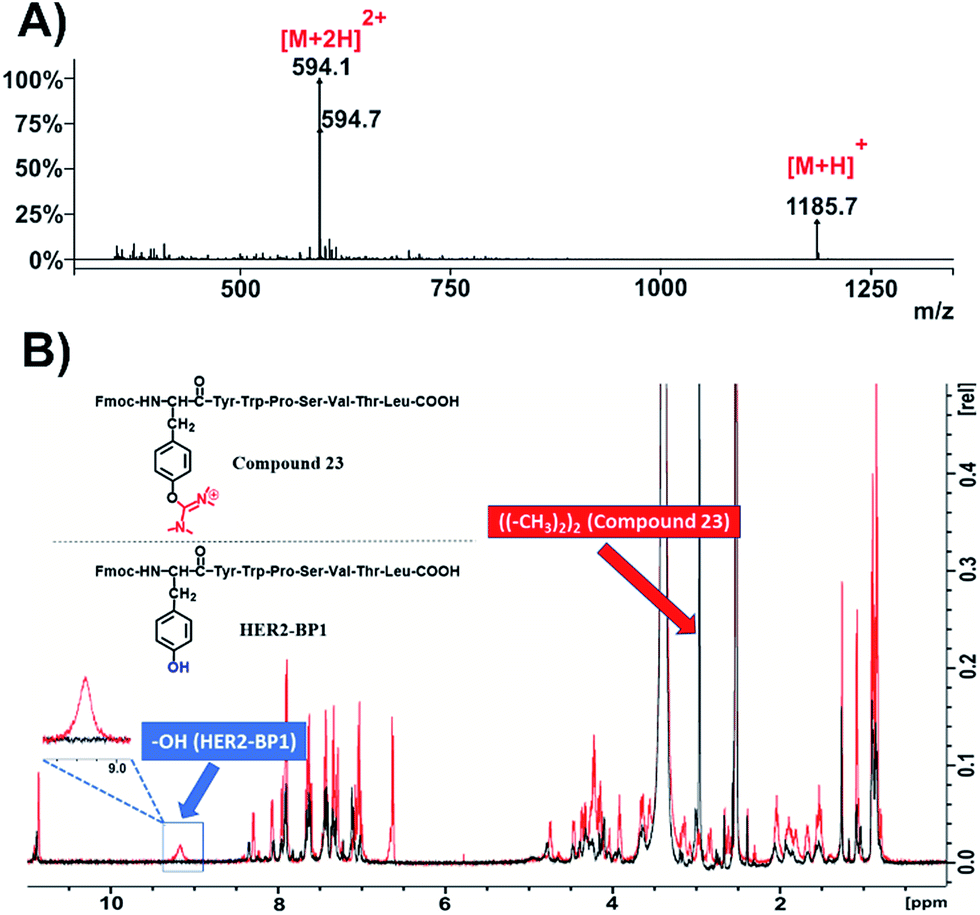 | ||
| Fig. 4 Characterization of compound 23. (A) ESI-MS spectrum of compound 23; (B) overlay of 1H-NMR spectra of compound 23 (black color) and Fmoc-HER2-BP1 (red color) in DMSO-d6 at 298 K. | ||
Due to the fact that the previously used peptides didn't contain cysteine residues in their sequences, we became interested to evaluate whether this modification is also applicable on the –SH nucleophilic group of cysteine. After preparing the dipeptide Fmoc-Cys-Tyr-NH2 and applying HATU (4 eq.) and DIPEA (4 eq.) in DMF, we recorded the ESI-MS of the product that responded to the mass of the parent compound (Fmoc-Cys-Tyr-NH2) plus 2× 99.00 amu (Fig. 5A and S57†). This indicates the installation of two aminium groups, one on cysteine and the other on tyrosine. To verify the proposed formation of the side product on both tyrosine and cysteine, we recorded 1H-NMR spectra both of the native peptide (Fig. 5B, colored in red) and compound 24 (Fig. 5B, colored in black; Fig. S59†). As it was expected, the peak referring to the phenol group of tyrosine at 9.2 ppm and the sulfuryl group of cysteine at 2.2 ppm disappeared.
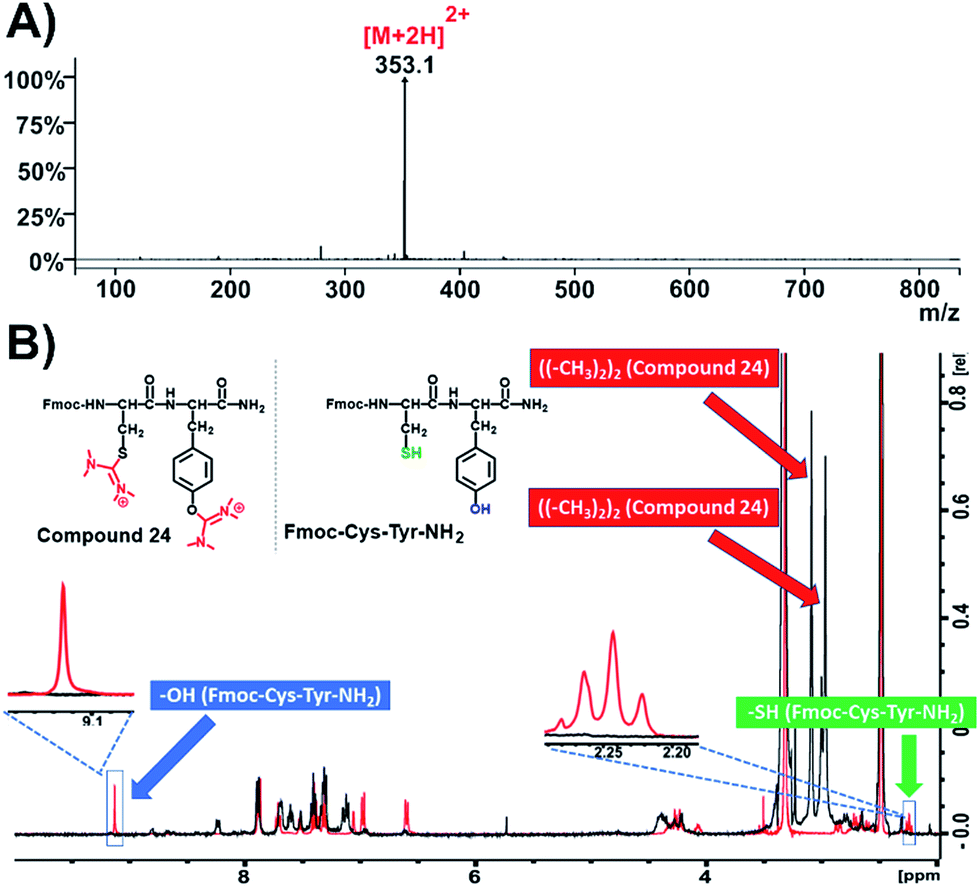 | ||
| Fig. 5 Characterization of compound 24. (A) ESI-MS spectrum of compound 24; (B) overlay of 1H-NMR spectra of compound 24 (black color) and Fmoc-Cys-Tyr-NH2 (red color) in DMSO-d6 at 298 K. | ||
This suggests that the aminium groups are installed on tyrosine and cysteine, which was further verified from the appearance of two more distinct peaks at 3 ppm derived from the methyl groups of the two aminium moieties (Fig. S59†).
In addition, we also synthesized the dipeptide Fmoc-Ser-Tyr-NH2 and post-processed it with HATU (4 eq.) and DIPEA (4 eq.) in DMF to receive compound 25 (Fig. 6B and S60†).
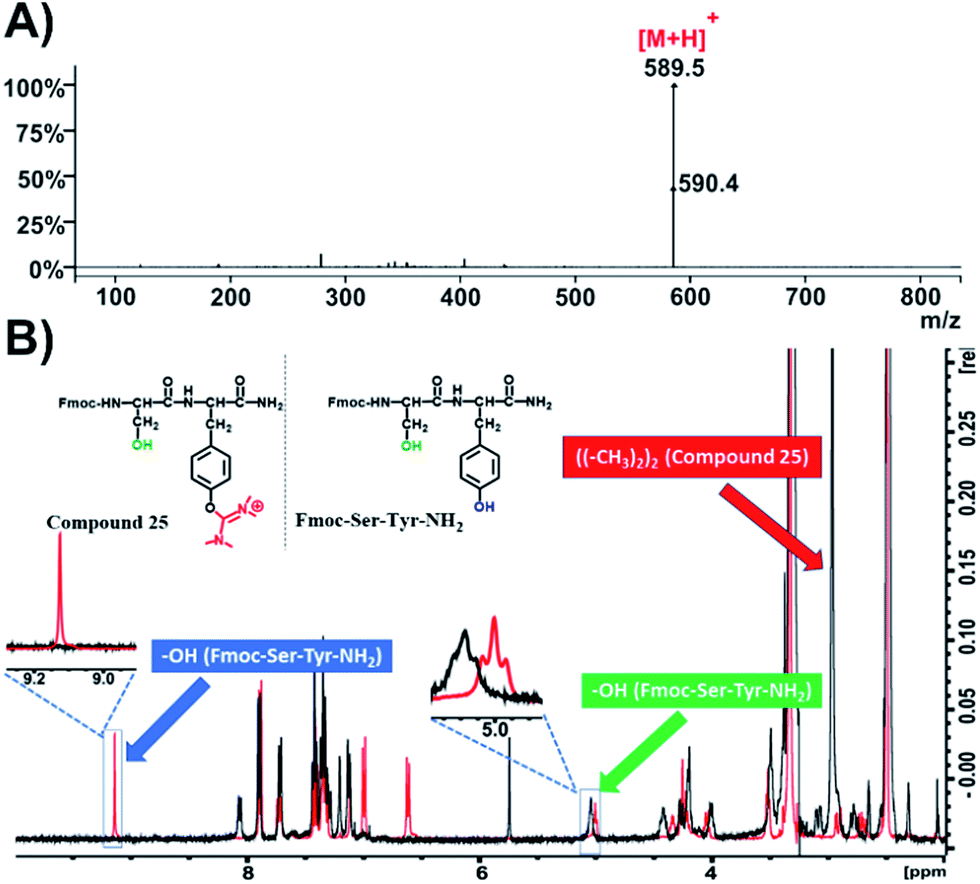 | ||
| Fig. 6 Characterization of compound 25. (A) ESI-MS spectrum of compound 25; (B) overlay of 1H-NMR spectra of compound 25 (black color) and Fmoc-Ser-Tyr-NH2 (red color) in DMSO-d6 at 298 K. | ||
We then recorded the ESI-MS of the resulted product where we observed that the mass of the parent compound has increased by 99.00 amu, which indicates the formation of a single uronium side product (Fig. 6A and S58†). 1H-NMR spectroscopy revealed that this modification is located only on the phenol group of Tyr (Fig. 6B, black color; Fig. S60†), as the peak referring to the phenol group of tyrosine at 9.1 ppm disappeared while the peak at 5 ppm, referring to the primary alcohol of serine, remained intact. Moreover, a distinct peak with an integral of 12 has appeared at 3 ppm, which is derived from the methyl groups of the uronium moiety (Fig. S60†).
These results clearly demonstrate that the modification can be applied not only to Tyr and Lys, but also to Cys.
Our next step was to explore the possibility to observe this HATU-mediated modification in bioactive peptides containing cysteine residues. We selected C1B5 subdomain peptide of protein kinase C (PKC) γ isozymes, since it has been revealed to be effective in the attenuation of the growth of colon carcinoma cells.26 C1B domain peptide of protein kinase C gamma significantly suppresses growth of human colon cancer cells in vitro and in an in vivo mouse xenograft model through induction of cell cycle arrest and apoptosis.26 The C1B5141–151 peptide has the following sequence: RCVRSVPSLCG. It contains 2 cysteines (C142 and C150) and according to our former observations we expected that the HATU-processed peptide, named compound 26, should adopt plus 2× 99 amu with respect to the MW of the parent compound (peptide Fmoc-C1B5). Indeed, the ESI-MS spectrum proved that the mass of the native peptide is 700.7 [M + 2H]2+ (Fig. 7A and S63†) and the HATU-processed peptide is 400.1 [M + 4H]4+ (Fig. 7B and S66†).
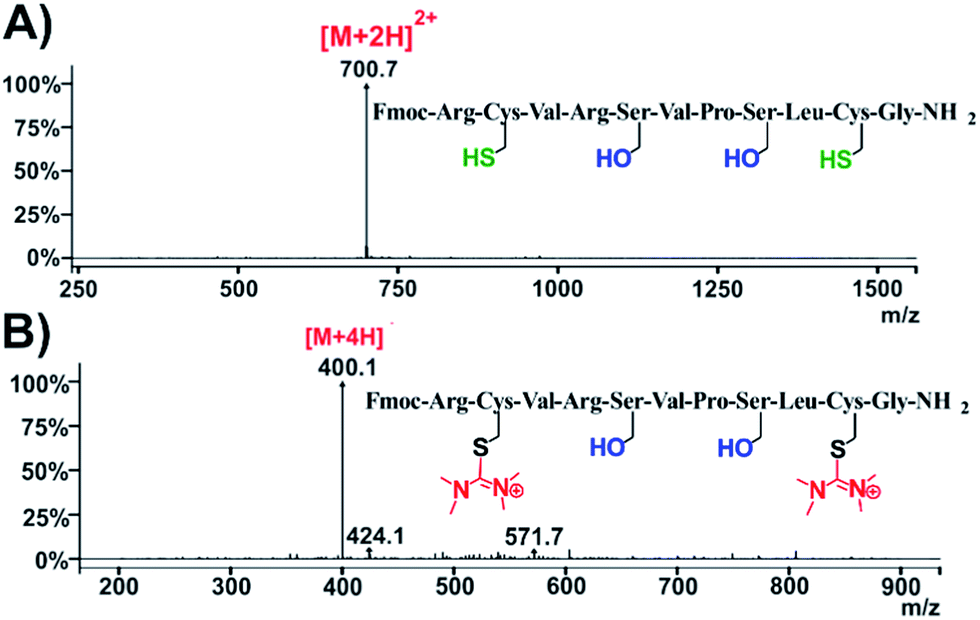 | ||
| Fig. 7 Characterization of compound 26. ESI-MS spectra of (A) native peptide C1B5141–151 and (B) HATU-processed peptide: compound 26. | ||
Afterwards, with the aim to broaden the impact of our work beyond traditional peptide chemistry, we used phenol, a simple organic molecule widely used by synthetic chemists mostly as starting material. We applied HATU (2 eq.) and DIPEA (3 eq.) in DMF to acquire the relevant HATU-modified analogue. Upon recording ESI-MS (Fig. 8A and S69†) and 1H-NMR spectrum (Fig. 8B, black color; Fig. S70†) on the resulted product (compound 27, Fig. S67†), we observed the disappearance of the –OH peak at 9.3 ppm and the formation of one new peak at 3 ppm with an integral of 12 (Fig. S70†). This fully validates the formation of the expected uronium product.
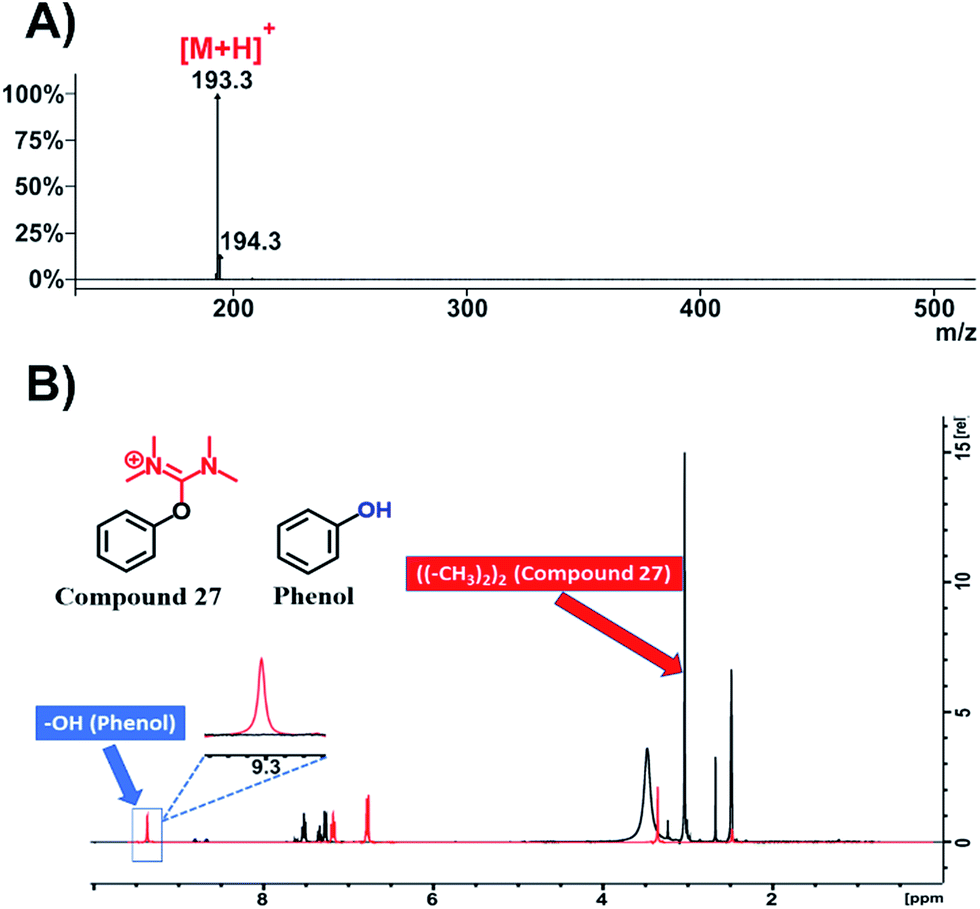 | ||
| Fig. 8 Characterization of compound 27. (A) ESI-MS spectrum of compound 27; (B) overlay of 1H-NMR spectra of compound 27 (black color) and phenol (red color) in DMSO-d6 at 298 K. | ||
Judging from the above, it can be concluded that during peptide-drug conjugation chemistry: (i) the side product can be formed without interrupting the creation of the amide bond between the drug and the peptide and (ii) the side product can be positioned on the hydroxyl group of Tyr, on the primary amine of Lys and on the sulfhydryl of Cys, which are initially ionized by DIPEA.
Moreover, during our efforts to avoid the formation of the reported side products, we conducted the reactions in Table 1, using 1 eq. of HATU/HBTU, instead of the classical and established protocols using 1.5 eq. From the ESI-MS analysis of the reaction products we discovered that by avoiding using excess of the coupling reagent, only the desired product was formed. Thus, it can be concluded that the side product is formed after the completion of the expected amide bond formation, since it is not detected with the use of equimolecular amount of the coupling reagent.
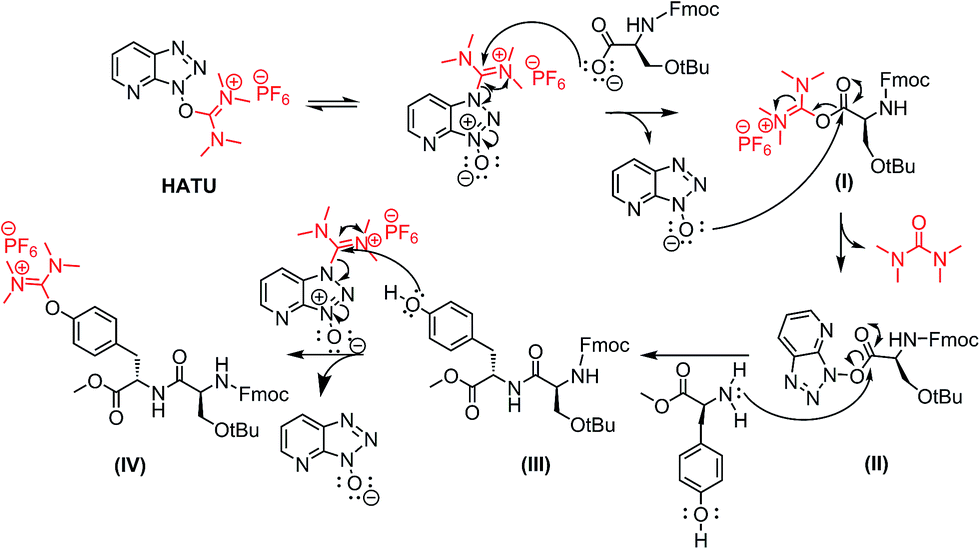
A possible mechanism for the formation of the uronium side product is depicted in Scheme 3 (similar mechanism can be applied for Lys- and Cys-mediated side products). In the first step, the deprotonated carboxylic group of the Fmoc-Ser(tBu)-OH attacks the electrophilic carbon of HATU to the formation of the activated ester (I).
 | ||
| Scheme 3 Suggested mechanism for the uronium side product formation. | ||
Then, the resulting anion HATU-N-oxide reacts further with the initially formed activated ester (I) to the production of the activated ester (II). The latter is then coupled with the primary amino group of the next amino acid to obtain the desired product (III). Excess of HATU in the reaction mixture reacts further with the nucleophilic phenol group of Tyr to the undesired product (IV), bearing the uronium group.
Conclusions
Peptide-drug conjugates have emerged as an efficient way to selectively deliver spatiotemporally drugs in complex diseases such as cancer.24 Such conjugations are frequently conducted in the solution phase, utilizing guanidinium salts as coupling reagents. Herein, we highlighted uncharted challenges that could be met during amide bond formation utilizing guanidinium salts in the solution phase. Upon usage of these reagents, besides the formation of the expected amide bond, a new aminium derivative, originated from the peptide coupling reagent, can result as a side-product on the phenol group of tyrosine (–OH), the primary amine of lysine (–NH2) and the sulfhydryl group of cysteine (–SH). Both HATU and TBTU investigated here were not devoid of the side product formation upon using classical peptide coupling conditions, i.e. 1.5 equiv. of the guanidinium coupling reagent. This observation has been monitored in template dipeptide reactions, on the synthesis of a peptide-drug conjugate and also on two peptides widely applied in PDCs. A plausible mechanism leading to the formation of this side product has been suggested. Based on this and seeking to identify conditions that could assist in surmounting the formation of this side product, different reaction conditions were screened. We identified that the optimum conditions that should be followed to avoid the formation of this side product is the usage of 1 eq. of guanidinium coupling reagent instead of the classically used 1.5 eq. This diminishes both the formation of the side product as also the associated reaction costs. The current observations can find several applications in drug discovery, where orthogonal coupling is carried out to produce drug-peptide/protein/antibody conjugates. We envisage that this side formation could be also used for constructing novel compounds. We are currently working along these lines in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr A. K. Shaikh for recording preliminary MS data. Dr N. Sayyad wishes to acknowledge the ‘Greek State Scholarships Foundation’ (IKY) for financial support for his PhD studies (2011–2015).Notes and references
- L. A. Carpino, D. Ionescu and A. El-Faham, J. Org. Chem., 1996, 61, 2460–2465 CrossRef CAS.
- J. Tulla-Puche, S. Auriemma, C. Falciani and F. Albericio, J. Med. Chem., 2013, 56, 5587–5600 CrossRef CAS PubMed.
- P. Stathopoulos, S. Papas, M. Sakka, A. G. Tzakos and V. Tsikaris, Amino Acids, 2014, 46, 1367–1376 CrossRef CAS PubMed.
- P. Stathopoulos, S. Papas, C. Pappas, V. Mousis, N. Sayyad, V. Theodorou, A. G. Tzakos and V. Tsikaris, Amino Acids, 2013, 44, 1357–1363 CrossRef CAS PubMed.
- M. Nagulapalli, G. Parigi, J. Yuan, J. Gsponer, G. Deraos, V. V. Bamm, G. Harauz, J. Matsoukas, M. R. de Planque, I. P. Gerothanassis, M. M. Babu, C. Luchinat and A. G. Tzakos, Structure, 2012, 20, 522–533 CrossRef CAS PubMed.
- I. C. Moschonas, T. F. Kellici, T. Mavromoustakos, P. Stathopoulos, V. Tsikaris, V. Magafa, A. G. Tzakos and A. D. Tselepis, Platelets, 2017, 1–10, DOI:10.1080/09537104.2017.1282607.
- L. A. Carpino, H. Imazumi, A. El-Faham, F. J. Ferrer, C. Zhang, Y. Lee, B. M. Foxman, P. Henklein, C. Hanay, C. Mugge, H. Wenschuh, J. Klose, M. Beyermann and M. Bienert, Angew. Chem., 2002, 41, 441–445 CrossRef CAS.
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef CAS.
- M. El Haddadi, F. Cavelier, E. Vives, A. Azmani, J. Verducci and J. Martinez, J. Pept. Sci., 2000, 6, 560–570 CrossRef CAS.
- A. Ehrlich, H.-U. Heyne, R. Winter, M. Beyermann, H. Haber, L. A. Carpino and M. Bienert, J. Org. Chem., 1996, 61, 8831–8838 CrossRef CAS PubMed.
- K. J. Hale and L. Lazarides, Org. Lett., 2002, 4, 1903–1906 CrossRef CAS PubMed.
- T. M. Kamenecka and S. J. Danishefsky, Chem.–Eur. J., 2001, 7, 41–63 CrossRef CAS.
- T. M. Kamenecka and S. J. Danishefsky, Angew. Chem., Int. Ed., 1998, 37, 2993–2995 CrossRef CAS.
- R. Corradini, S. Sforza, A. Dossena, G. Palla, R. Rocchi, F. Filira, F. Nastri and R. Marchelli, J. Chem. Soc., Perkin Trans. 1, 2001, 2690–2696 RSC.
- G. Jou, I. González, F. Albericio, P. Lloyd-Williams and E. Giralt, J. Org. Chem., 1997, 62, 354–366 CrossRef CAS PubMed.
- S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS.
- F. Albericio, J. M. Bofill, A. El-Faham and S. A. Kates, J. Org. Chem., 1998, 63, 9678–9683 CrossRef CAS.
- S. C. Story and J. V. Aldrich, Int. J. Pept. Protein Res., 1994, 43, 292–296 CrossRef CAS PubMed.
- V. R. Solomon, S. K. Puri, K. Srivastava and S. B. Katti, Bioorg. Med. Chem., 2005, 13, 2157–2165 CrossRef CAS PubMed.
- H. Tamamura, K. Hiramatsu, M. Mizumoto, S. Ueda, S. Kusano, S. Terakubo, M. Akamatsu, N. Yamamoto, J. O. Trent, Z. Wang, S. C. Peiper, H. Nakashima, A. Otaka and N. Fujii, Org. Biomol. Chem., 2003, 1, 3663–3669 CAS.
- S. Tong, M. Zhang, S. Wang, R. Yin, R. Yu, S. Wan, T. Jiang and L. Zhang, Eur. J. Med. Chem., 2016, 123, 849–857 CrossRef CAS PubMed.
- Y. Wang, A. G. Cheetham, G. Angacian, H. Su, L. Xie and H. Cui, Adv. Drug Delivery Rev., 2017, 110–111, 112–126 CrossRef CAS PubMed.
- O. Argyros, T. Karampelas, X. Asvos, A. Varela, N. Sayyad, A. Papakyriakou, C. H. Davos, A. G. Tzakos, D. Fokas and C. Tamvakopoulos, Cancer Res., 2016, 76, 1181–1192 CrossRef CAS PubMed.
- T. Karampelas, O. Argyros, N. Sayyad, K. Spyridaki, C. Pappas, K. Morgan, G. Kolios, R. P. Millar, G. Liapakis, A. G. Tzakos, D. Fokas and C. Tamvakopoulos, Bioconjugate Chem., 2014, 25, 813–823 CrossRef CAS PubMed.
- X. F. Wang, M. Birringer, L. F. Dong, P. Veprek, P. Low, E. Swettenham, M. Stantic, L. H. Yuan, R. Zobalova, K. Wu, M. Ledvina, S. J. Ralph and J. Neuzil, Cancer Res., 2007, 67, 3337–3344 CrossRef CAS PubMed.
- A. Kawabata, T. Matsuzuka, C. Doi, G. Seiler, J. Reischman, L. Pickel, R. Ayuzawa, T. A. Nguyen and M. Tamura, Cancer Biol. Ther., 2012, 13, 880–889 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data, 1H-NMR, 13C-NMR and MS analysis for all new compounds. See DOI: 10.1039/c7ra06655d |
| This journal is © The Royal Society of Chemistry 2017 |