
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective signalling of sialic acid in solution by circularly polarised luminescence spectroscopy using a dynamically racemic europium(III) complex
Emily R. Neil and
David Parker *
*
Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: david.parker@dur.ac.uk
First published on 16th January 2017
Abstract
A europium complex bearing a phenylboronic acid group has been created that binds reversibly in methanol to sialic acid and lactic acid, as signalled by changes in the Eu total emission spectrum and the induction of strong circularly polarised luminescence. An analogue lacking the boronate moiety displays no spectral response towards sialic acid. The CPL signature is distinctive for sialic acid, and differs from that observed with methyl sialate and N-acetyl glucosamine. A hypothetical binding model is promulgated, where binding of the amide carbonyl to the Eu centre occurs at the same time as the terminal diol group of the glycerol moiety chelates to the boron atom.
Introduction
Sialic acid (Neu-5-Ac) is an important biomolecule belonging to the family of 9-carbon amino sugars (neuraminic acids). It is present in serum as a mixture of α- (5–8%) and β-pyranose (92–95%) forms; the absence of the furanose form can be attributed to the acetamide group at the C-5 position, (Fig. 1). Sialic acid can also be found occupying the terminal positions of carbohydrate chains of glycoproteins and glycolipids of biological membranes. It is over-expressed on the surface of tumour cells1,2 and the variation in serum levels may serve as a useful biological marker, indicating the existence of a cancerous state.3 Typical concentrations in healthy adults of free sialic acid in serum lie in the range 140–200 μM.4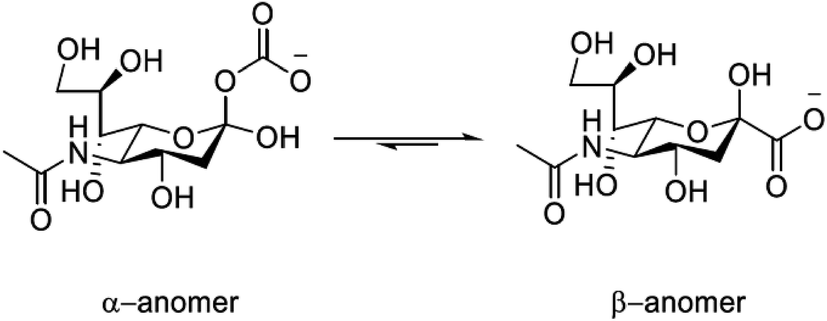 | ||
| Fig. 1 Structure of sialic acid (Neu-5-Ac) showing the α- and β-pyranose forms; pKa = 2.6 (H2O, I = 0.1, 298 K). | ||
Several lanthanide probes have been reported that target sialic acid and signal the detection using MRI5–7 and emission based techniques.8,9 Of particular importance is the use of a boronic acid moiety in the putative selective recognition mechanism of its diol functionality. The reversible formation of five- and six-membered cyclic boronate esters often results from the interaction with 1,2- and 1,3-diols, and has been extensively exploited in the design of various saccharide sensors.10 The incorporation of a phenylboronic acid (pba) moiety into the ligand framework of a lanthanide complex has also demonstrated the utility of this moiety, in the putative dynamic covalent binding to the 1,2-diol group of sialic acid.5,6
The complexes [Eu·L1]+ and [Eu·L2]+ have been shown to exhibit high quantum yield values, anion binding capability and strong induced CPL following the addition of a chiral analyte.11,12 Recently, renewed interest has been emerging in the use of CPL to allow the creation of chiral probes,13 and to permit CPL imaging and microscopy.14
Here, a heptadentate Eu(III) complex based on the bis-carboxylate ligand structure containing a phenylboronic acid moiety, [Eu·L3]+, (Fig. 2) has been designed to target sialic acid selectively. Exploiting the inherent chirality of the molecule, binding at europium was hypothesised to result in a change in emission spectral form and generation of an induced circularly polarised luminescence signal. The response of the complex is compared to the bis-carboxylate complex, [Eu·L1]+, and the N-alkylated derivative, [Eu·L2]+, which serve as controls lacking the boronate moiety.
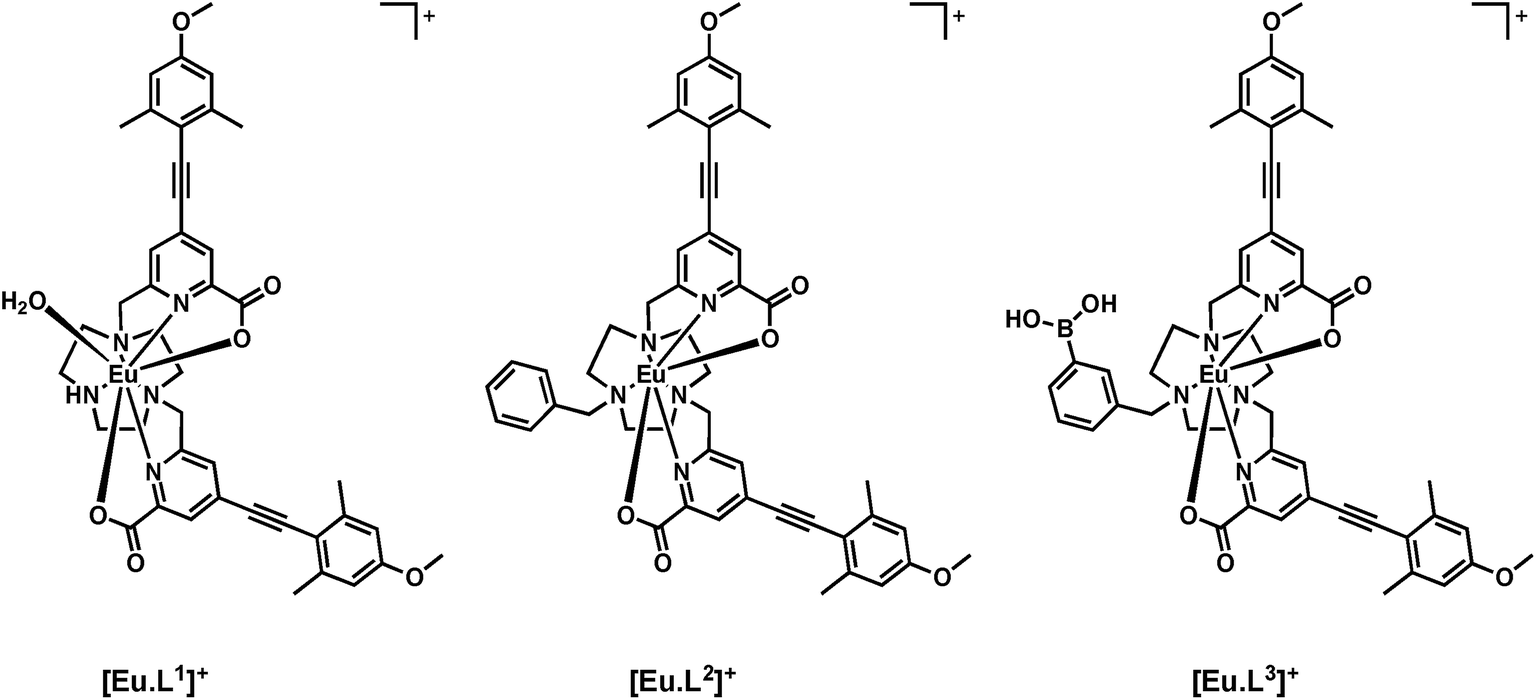 | ||
| Fig. 2 Structures of the Eu(III) complexes of L1–3. | ||
Synthesis of [Eu·L3]+
A phenylboronic acid moiety was appended to the desired ligand framework, L1, via a ring N-alkylation reaction. Initial protection of the boronic acid group as a cyclic ester was used to assist subsequent purification. The simple cyclic ester, based on ethylene glycol, was selected as the protecting group. Although rather sensitive to hydrolysis, the boronic ester was considered to be sufficiently stable towards purification using column chromatography, under carefully controlled conditions. Subsequent removal of the ester group can be achieved using mild base hydrolysis. The use of more stable hindered cyclic esters, such as a pinacol derivative was avoided, as its conversion back to the boronic acid is particularly difficult and is not compatible with the presence of other groups in the ligand framework, such as the acid-sensitive alkyne.Reaction of commercially available 3-(bromomethyl)phenylboronic acid with ethylene glycol in anhydrous pentane over 4 Å molecular sieves, yielded the protected phenylboronate in good yield, (Scheme 1). Subsequent alkylation with L1 gave the protected ligand, L3′, which was purified using flash column chromatography on silica gel. The presence of the boronic ester moiety was confirmed by a single resonance at 31.3 ppm in the 11B NMR spectrum.
 | ||
| Scheme 1 Synthesis of the Eu(III) complex of L3 containing a phenylboronic acid moiety. | ||
Base hydrolysis of both the methyl ester and the cyclic boronic ester groups was carried out using NaOH (aq.) in a MeOH–H2O mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at pH 12. After the completion of reaction was confirmed by LC-MS, the pH of the solution was lowered to 6.5 and EuCl3·6H2O was added. Formation of the complex was confirmed by ESI-MS, in 18 h.
:1, v/v) at pH 12. After the completion of reaction was confirmed by LC-MS, the pH of the solution was lowered to 6.5 and EuCl3·6H2O was added. Formation of the complex was confirmed by ESI-MS, in 18 h.
A chiral probe for sialic acid
The photophysical properties of the boronic acid derivative [Eu·L3]+ were assessed in comparison to the N-benzyl complex [Eu·L2]+. The capability of the complex to bind anions was tested using lactate in a model study, and the induced CPL behaviour was correlated to the configuration of the complex, using methods recently described.11–14 The binding affinity of sialic acid was estimated and the emission spectral changes and induced CPL signature were compared to [Eu·L1]+ and [Eu·L2]+. Investigation into the mode of binding of sialic acid was attempted, by comparison of the response with that observed following addition of the methyl-ester derivative of Neu-5-Ac.Photophysical properties of [Eu·L3]+
The key photophysical parameters for [Eu·L3]+ were determined and compared to those of the related complex, [Eu·L2]+ (Table 1). The hydration state in water of the title complex, [Eu·L3]+, was estimated to be q = 0.2.15 The absence of an inner sphere metal-bound water molecule is consistent with the reported behaviour of the N-benzyl complex, [Eu·L2]+.11,12 This observation provides further evidence that the steric bulk of the N-substituent inhibits coordination of a water molecule to the lanthanide centre.| [Eu·L2]+ | [Eu·L3]+ | |
|---|---|---|
| λ/nm | 348 | 356 |
| ε/mM−1 cm−1 | 36.0 | 35.0 |
| ϕ | 0.18 | 0.05 |
| k/ms−1 | 1.20 | 2.04 |
| k(H2O)/ms−1 | 2.00 | 2.63 |
| k(D2O)/ms−1 | 1.89 | 2.17 |
| q | 0 | 0.2 |
The europium emission quantum yield of the boronic acid complex, [Eu·L3]+, was measured to be 5%, which is significantly lower than that of the N-benzyl complex, [Eu·L2]+ (Table 1). Secondly, the radiative rate of decay of Eu3+ emission in methanol was nearly twice as fast for [Eu·L3]+ compared to [Eu·L2]+. Such behaviour suggests that the europium(III) excited state lifetime for [Eu·L3]+ is being quenched, in a manner that is not possible for [Eu·L2]+.

A likely explanation for the increase of the measured excited state decay rate (Table 1, where the measured value of k is given by kobs = k0 + knr, in which k0 is the radiative rate constant for the Eu excited state and ∑knr represent competing non-radiative processes) is that the presence of the boronic acid group simply enhances the rate of non-radiative decay in the monomer. Alternatively, it may be related to the ability of [Eu·L3]+ to undergo intermolecular association, e.g. dimer formation as shown in the X-ray crystal structure of phenylboronic acid,16 where hydrogen bonding occurs between boronic acid groups. To test this hypothesis further, a dilution study was carried out in MeOH–H2O (1:1, v/v) at pH 7.4. The luminescent lifetime of the excited state was monitored as the concentration was successively lowered from 5 μM to 0.63 μM, at which point the sample was too dilute to obtain reliable results. No significant change in the luminescent lifetime was observed, suggesting either that the intermolecular interactions are relatively strong and not disrupted over this concentration range, or that the monomer or dimer undergo additional non-radiative deactivation processes, in the latter case, for example, via intra- or intermolecular charge transfer.
Preliminary investigations into interactions with anions
The form and relative intensity of the electric-dipole allowed transitions in the europium emission spectrum is particularly sensitive to the metal coordination environment.13,14 The addition of lactate to a series of structurally related Eu(III) complexes ([Eu·L1,2]) was seen to cause significant changes in the total emission spectra, consistent with anion binding at the metal centre. With this behaviour in mind, the total emission spectral response of [Eu·L3]+ was monitored following addition of S-lactate (Fig. 3). A ten-fold enhancement in the total luminescence intensity was observed, following addition of 4000-fold excess of S-lactate. The increase in emission intensity suggested that anion binding to the metal centre results in disruption of the quenching mechanism taking place. Lactate can bind to the boronate through the carboxylate and hydroxyl group to form a 5-membered chelate,17 as well as to the lanthanide centre, consistent with the idea that the strong intermolecular association that may be occurring in the monomer, is not possible when lactate binds. | ||
| Fig. 3 Total emission spectra of [Eu·L3]+ (blue) and following addition of 2 mM S-lactate (red) (5 μM complex, λexc = 356 nm, MeOH, 295 K). | ||
The addition of R- and S-lactate to the europium complex, [Eu·L3]+, resulted in an induced CPL response, identical in nature to that of [Eu·L2]+, (Fig. 4).11 The sign and form of each transition was the same and the measured emission dissymmetry values, gem, were comparable. The binding affinity was similar to that observed earlier with [Eu·L2]+, (Table 2)11 Empirical analysis of the ΔJ = 4 transition allowed assignment of absolute configuration of the complex,11,12 as addition of R-lactate has been shown to result in Δ helicity while S-lactate gives Λ helicity in the ternary adduct.
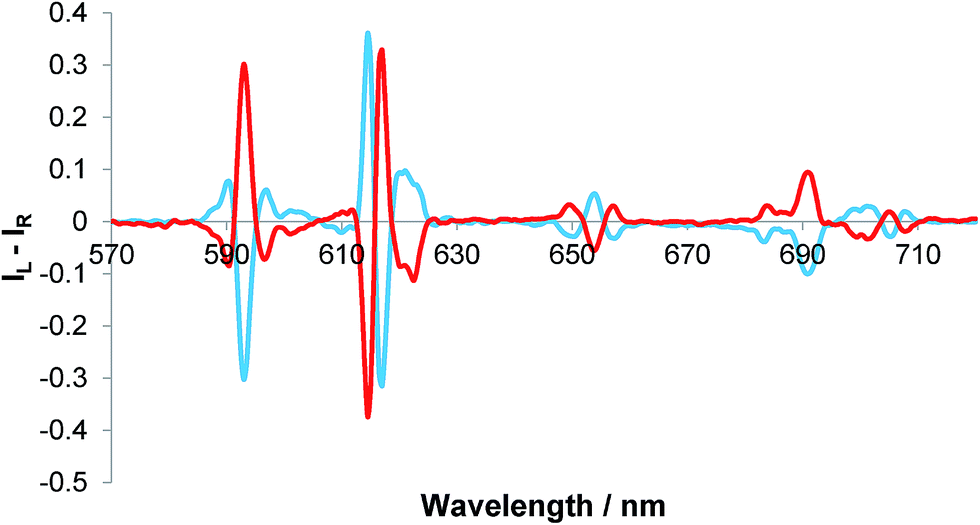 | ||
| Fig. 4 Induced CPL spectra of [Eu·L3]+ following the addition of 2 mM R- (red) and S-lactate (blue); gem (593 nm = ±0.05), gem (654 nm) = ±0.07 (5 μM complex, λexc = 356 nm, MeOH, 295 K). | ||
K, for complexation of lactate and sialic acid (295 K)
The emission and CPL spectral response of [Eu·L3]+ following addition of lactate, demonstrates that binding at the metal centre can occur, in principle, in a similar manner to that observed for [Eu·L1,2].
Investigations into the binding of sialic acid
The interaction of sialic acid with the parent bis-carboxylate europium(III) complex, [Eu·L1]+ was investigated first. An emission titration was carried out in methanol and spectral changes were monitored as a function of added concentration of sialic acid. A binding affinity in methanol of logK = 5.24 was estimated, (Table 2) which demonstrates that there is a significant interaction between the complex and the Neu-5-Ac anion in methanol, (Fig. 5). No significant interaction was observed in pure water at ambient pH; presumably the hydration energies of the complex and the sialic acid are much higher under these conditions.
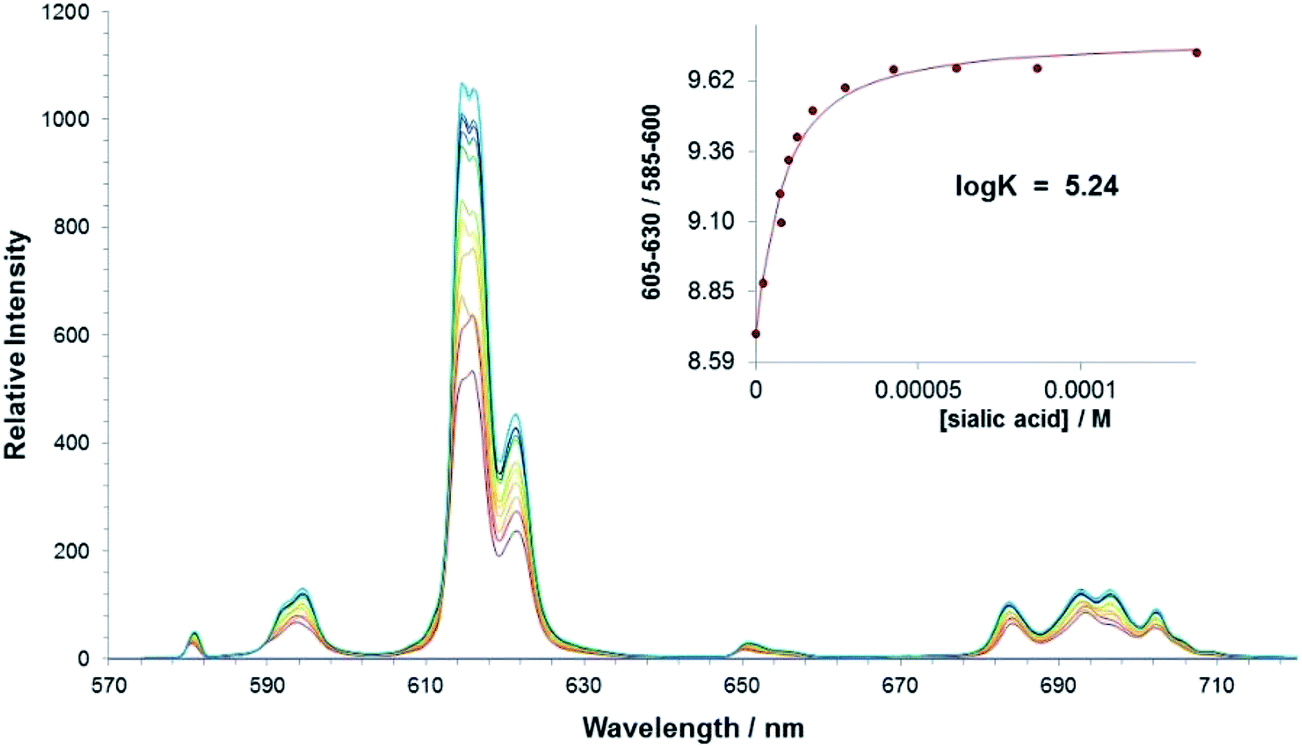 | ||
| Fig. 5 Variation of the europium(III) emission profile of [Eu·L1]+ as a function of added sialic acid. (5 μM, λexc = 352 nm, MeOH). Inset shows fit to experimental data, following iterative least squares fitting to a 1:1 binding model. | ||
The addition of sialic acid to [Eu·L1]+ also induced significant CPL. It was interesting to note that the form of the CPL spectrum, particularly in the ΔJ = 1 and ΔJ = 2 manifolds, was remarkably different to that following addition of simple carboxylates such as R-phenylpropionate, and also α-hydroxy acids, such as lactate and mandelate11 (Fig. 6). The ‘signature’ CPL response may allow simple differentiation between the detection of sialic acid and other chiral carboxylate anions in a competitive environment. It is notable, that the ΔJ = 4 region again resembles that of the R-lactate adduct of [Eu·L1]+, allowing assignment of the complex adduct configuration as Δ-[Eu·L1·Neu-5-Ac].
 | ||
| Fig. 6 CPL spectra of [Eu·L1]+ following addition of R-lactate (left) and sialic acid (right). (5 μM complex, 50 μM anion, λexc = 352 nm, MeOH). | ||
This proof of concept study demonstrated the capability of lanthanide complexes based on a heptadentate bis-carboxylate ligand, to signal the binding of sialic acid. Further development of this probe examined the behaviour of the complex, [Eu·L3]+ with incorporation of a phenylboronic acid moiety into the ligand framework.
Phenylboronic acid is a weak acid (pKa = 8.72) that is able to bind covalently and reversibly to 1,2-diols,18 such as the glycerol side-chain of sialic acid, and to α-hydroxy acids, in each case forming 5-ring chelate structures.17 The stability of esters formed by the tetragonal boronate anion is an order of magnitude greater than for the trigonal boronic acid.19 Therefore, enhanced affinity is observed under basic conditions. It was hoped that the addition of this secondary stabilising interaction would enhance the affinity of the complex for sialic acid, and permit detection in polar media.
An emission titration of [Eu·L3]+ and sialic acid in methanol was carried out to allow comparison with [Eu·L1]+. Plotting the change in ΔJ = 2 and ΔJ = 1 intensities versus concentration of anion generated a curve that did not fit to a 1:1 binding model, (Fig. 7). The plot revealed that another process perturbed the spectral signal variation process occurs before 1:1 binding between the anion and the lanthanide ion. Such behaviour indirectly supports the suggestion that there is an intermolecular association in solution between the boronate groups in two or three molecules (e.g. hydrogen bonded dimer or trimer formation has been noted before for such boronates10c). Alternatively, the interaction may occur between the boronate group acting as a ligand to the Eu centre in a second Eu complex. In each case these interactions are disrupted following addition of an anion. It is worth noting that significant changes in the emission spectrum were observed with [Eu·L3]+ at a 3-fold lower concentration of sialic acid, compared to the response observed for [Eu·L1]+. Such behaviour suggests that the addition of the phenylboronic acid moiety, significantly increases the affinity of the europium(III) complex for sialic acid. The complexity of the binding curve obtained (Fig. 7) precluded quantitative assessment of binding affinity. It is particularly interesting to note that there was no direct spectroscopic evidence of sialic acid binding to the control N-benzyl complex, [Eu·L2]+, indicating the need for the stabilising interaction provided by the boronic acid moiety.
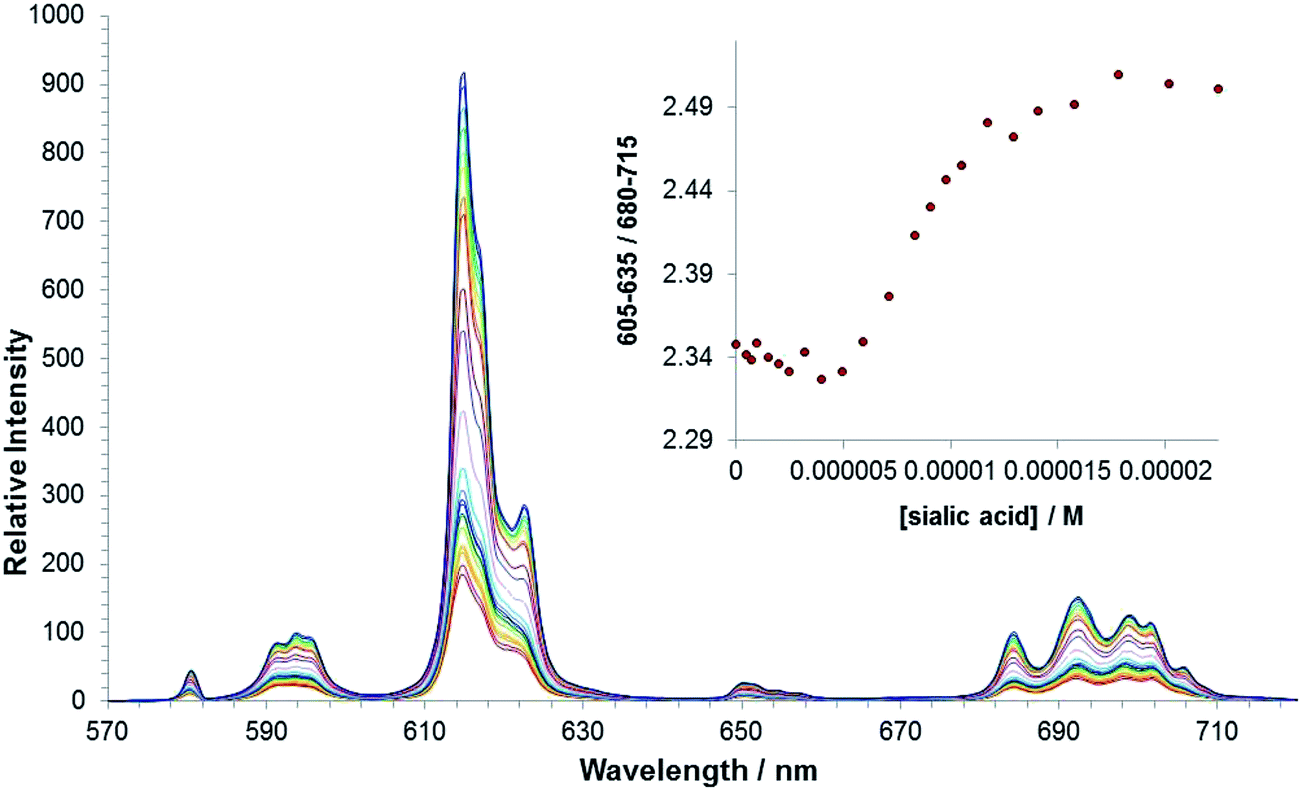 | ||
| Fig. 7 Variation of the europium(III) emission profile of [Eu·L3]+ as a function of added sialic acid. (5 μM, λexc = 356 nm, MeOH). | ||
The europium(III) total emission studies showed that there is a well-defined interaction between [Eu·L1]+, and sialic acid, (Fig. 5 above). However, no evidence for binding was observed for the N-benzyl complex, [Eu·L2]+. Such behaviour is most likely to occur as a result of the increased steric demand around the metal centre created by the N-benzyl group. Interestingly, the addition of a boronic acid group to the aromatic ring of the N-benzyl complex, [Eu·L3]+, provided an additional stabilising interaction that evidently allowed sialic acid to bind to the europium(III) complex, notwithstanding the steric hindrance imposed by the benzyl substituent.
The sign of the induced CPL in the ΔJ = 1 manifold was comparable to that for [Eu·L1]+, following addition of sialic acid. However, very little optical activity was observed in the ΔJ = 3 and ΔJ = 4 transitions, (Fig. 8). The difference in the CPL spectral form, suggests that a different coordination environment is present in these complexes. The emission dissymmetry values, gem, were larger for [Eu·L3·Neu-5-Ac]+ (gem (593 nm) = +0.03, c.f. +0.015 for [Eu·L1·Neu-5-Ac]), suggesting that enhanced binding affinity and a greater degree of conformational rigidity is present when sialic acid binds to the metal centre in the boronate complex, [Eu·L3]+. Such behaviour may be due to an additional interaction with the boronic acid group in the adduct. The CPL spectra for sialic acid and lactic acid were also very different in overall form, suggesting that the europium was not binding in the same manner.
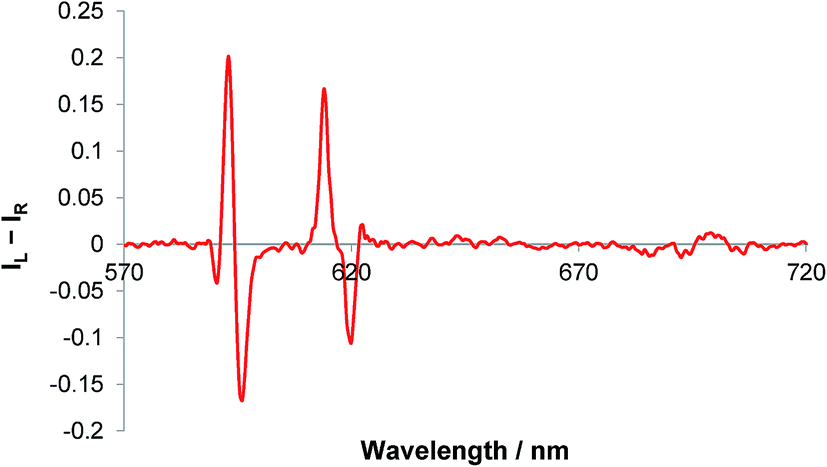 | ||
| Fig. 8 CPL spectrum of [Eu·L3]+ following addition of sialic acid (5 μM complex, 50 μM anion, λexc = 356 nm, MeOH). | ||
No evidence for binding was observed in pure water in the pH range 4–10 for [Eu·L3]+. This behaviour could be related to the fact that the conformation adopted by sialic acid in solution has been reported to be solvent dependent. 1H NMR spectra of sialic acid in MeOD and D2O were analysed at a pD of 5.5. However, no significant differences were observed in the spectra in each solvent, suggesting a minimal difference in the conformation of sialic acid.
It is likely that the difference in binding affinity of sialic acid in the two solvent systems is a direct result of the difference in solvation energy. There are five hydroxyl groups present in the molecule, so hydration of the anion in water is likely to be very high and may explain the low affinity it has for the positively charged lanthanide complex in aqueous solution. Systematic variation of the solvent composition and the effect on the emission spectral output was monitored. It was found that significant change in form and intensity was observed when 2 mM sialic acid was added to 1 mL 4:1 MeOH:H2O sample of [Eu·L3]+. However, increasing the water mole fraction any higher, resulted in no evidence for anion binding to the metal centre. The poor aqueous solubility of the complex is a key factor in the decreased affinity for sialic acid in such aqueous systems.
Investigating the mode of binding of sialic acid
The mode of binding of sialic acid to the europium(III) complexes, [Eu·L1,3]+, was investigated further by a comparative study of induced CPL following addition of sialic acid, its methyl ester and the related molecule N-acetyl-D-glucosamine, (Fig. 9). | ||
| Fig. 9 Structures of sialic acid (left), the corresponding methyl ester (centre) and N-acetyl-D-glucosamine (right). | ||
The form and sign of the CPL spectra of [Eu·L1]+ following addition of sialic acid and its methyl ester derivative was similar, strongly suggesting that the negatively charged carboxylate group of Neu-5-Ac is not involved in the primary binding interaction at the metal centre (Fig. 10A vs. B). Therefore, an alternative mechanism for the recognition of sialic acid must be considered, based on binding of the amide carbonyl oxygen to the lanthanide centre. Such a binding mode was hypothesised recently in the work of Ouchi, regarding sialic acid binding to a [Ln·(ABNOTA)] complex (Fig. 11).9 However, addition of N-acetyl-D-glucosamine, a monosaccharide containing the same acetamide moiety but lacking the glycerol moiety and a carboxylate group, resulted in no change to the total emission spectrum. Neither was any induced CPL observed, indicating that other groups in the Neu-5-Ac molecule must be cooperatively involved in the binding interaction.
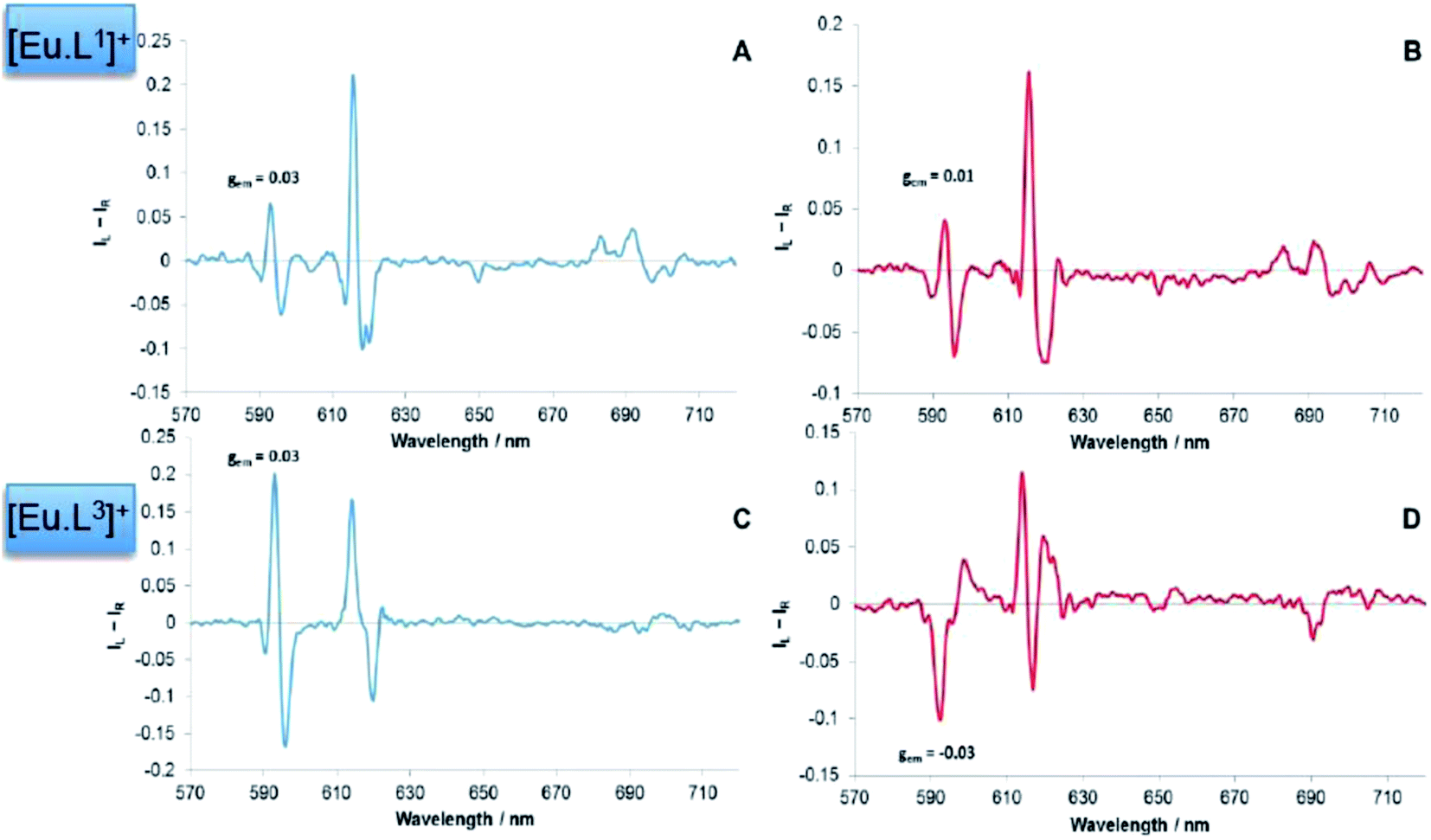 | ||
| Fig. 10 Induced CPL spectra for [Eu·L1]+ following the addition of (A) 2 mM sialic acid; (B) 2 mM sialic acid methyl ester (MeOH, λexc = 352 nm, 295 K). Induced CPL spectra for [Eu·L3]+ following the addition of (C) 2 mM sialic acid; (D) 2 mM sialic acid methyl ester (MeOH, λexc = 356 nm, 295 K). The gem values are given for the same ΔJ = 1 transition at 593 nm. | ||
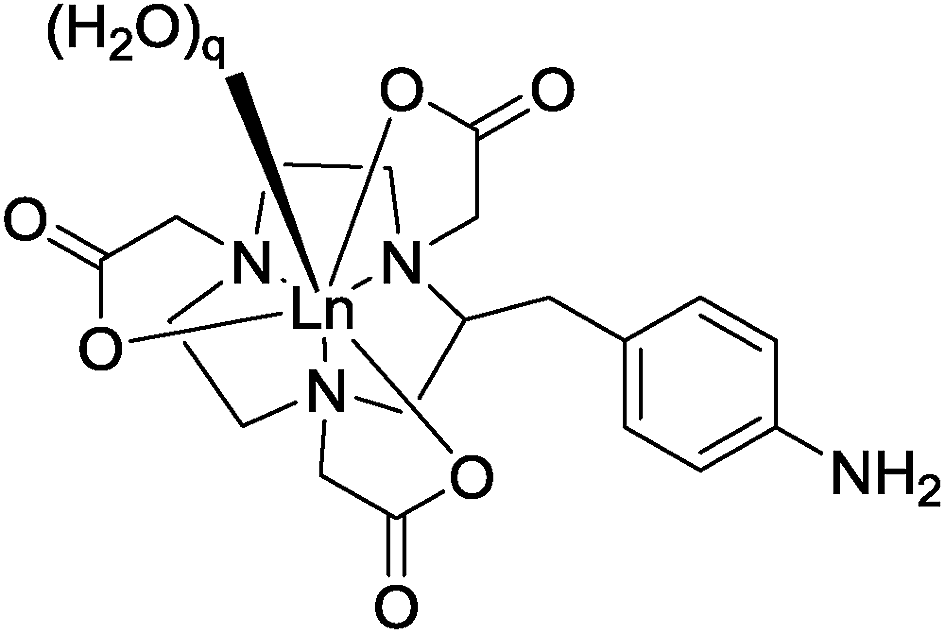 | ||
| Fig. 11 Partial structure of a [Ln·ABNOTA] complex; q = 2 or 3.9 | ||
In contrast to the behaviour of [Eu·L1]+, the induced CPL spectra of [Eu·L3]+ were rather different, following addition of sialic acid versus its methyl ester (Fig. 10C vs. D). Subtle differences in the form of the total emission spectra, particularly in the ΔJ = 1 and ΔJ = 4 transitions, were also observed. Such behaviour suggests that the carboxylate group may yet be involved in some manner, even if just via some hydrogen bonding interaction.
In work on a Gd-based MRI contrast agent for sialic acid detection, lanthanide complexes based on diethylenetriamine pentaacetic acid (DTPA) ligands incorporating a phenylboronic acid moiety have also been used, e.g. L4 and L5 (Fig. 12).5 The authors suggested that the interaction between the phenylboronic acid group and sialic acid occurred either with the glycerine moiety at C-6, or with the carboxylate and hydroxyl group at C-2, to form a five-membered cyclic ester. They did not consider any role for the amide carbonyl group in binding to the Gd3+ centre.
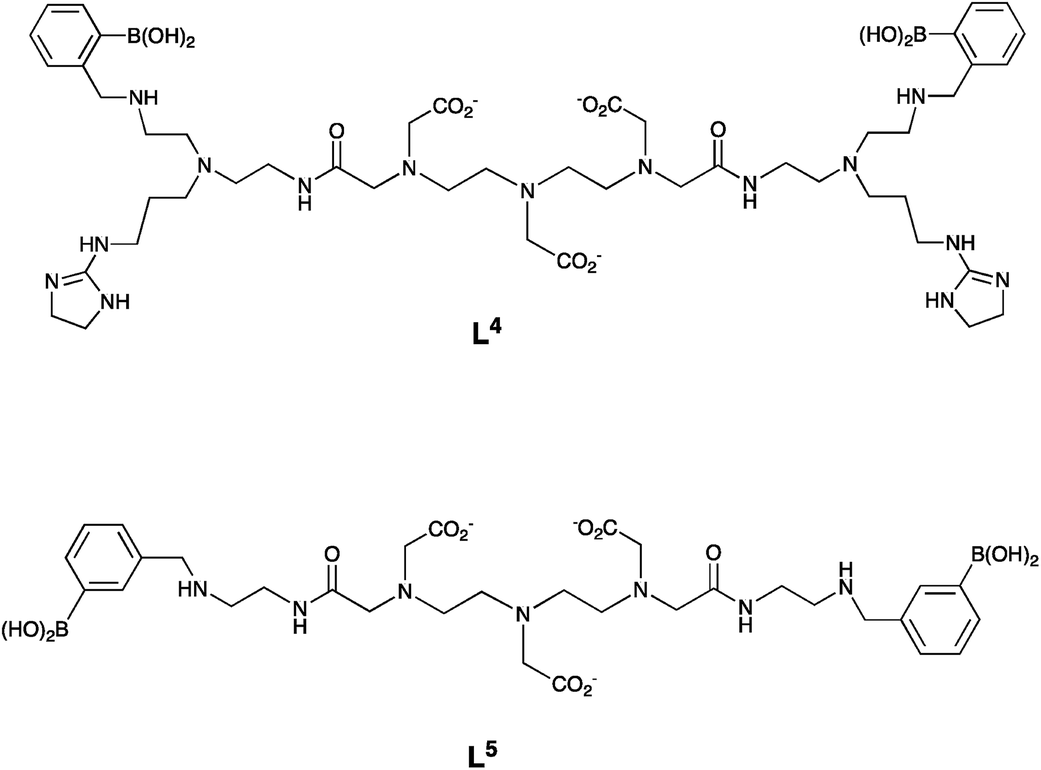 | ||
| Fig. 12 DTPA-bis amide (DTPA – diethylenetriamine pentaacetic acid) ligands, L4 and L5 used in Gd-based MRI contrast agents for sialic acid detection.5 | ||
Summary
This study has presented the first example of induced CPL to signal sialic acid binding. Evidence gathered from the induced CPL response of the phenylboronate–europium(III) complex [Eu·L3]+, allows us to hypothesise that there is a synergistic interaction involved in the binding mechanism of the europium complex and sialic acid. It is possible that this interaction involves the 2,3-hydroxyl group of the glycerol moiety, forming a cyclic ester with the boronic acid, together with an additional stabilising interaction, involving the amide carbonyl oxygen. There is indirect support for this hypothesis, based on crystal structure data for the sialic acid trisaccharide derivative, Neu-5-Ac–Gal–Glc, when bound in the active site of the serum protein complement factor H. The structure reveals a potentially favourable orientation for metal binding, involving the amide carbonyl oxygen and, in that case, the first glycerol hydroxyl group (Fig. 13).20 | ||
| Fig. 13 The sialic acid derivative binding site of the serum protein complement factor H, showing the orientation of the amide carbonyl oxygen and the first glycerol hydroxyl group to allow a cooperative binding interaction.21 | ||
With the methyl ester of sialic acid, the carbonyl oxygen atom is a much weaker lone pair donor and cannot reasonably be expected to engage in such a binding interaction. Therefore, the coordination geometry around the metal centre will be different, as observed by the emission behaviour. Indeed, the induced CPL response is distinctively different in form and sign, comparing the two chiral analytes.
Taken together, these results allow the development of tentative hypotheses for the binding mode of the sialic acid group to [Eu·L1]+ and [Eu·L3]+. Accordingly, DFT modelling studies were undertaken to assess the feasibility of putative cooperative binding models, using [Eu·L3]+. The binding to boron of the terminal diol group of the glycerol moiety, allowed the cooperative ligation of the amide carbonyl oxygen to europium, (Fig. 14). Alternative structures that were explored included one involving chelation of the alpha-hydroxyl carboxylate to boron. However, in that case no simultaneous binding to europium was geometrically feasible, and the earlier CPL work had anyway suggested very different coordination environments at Eu, in the complexation with sialic acid vs. lactic acid (vide supra).
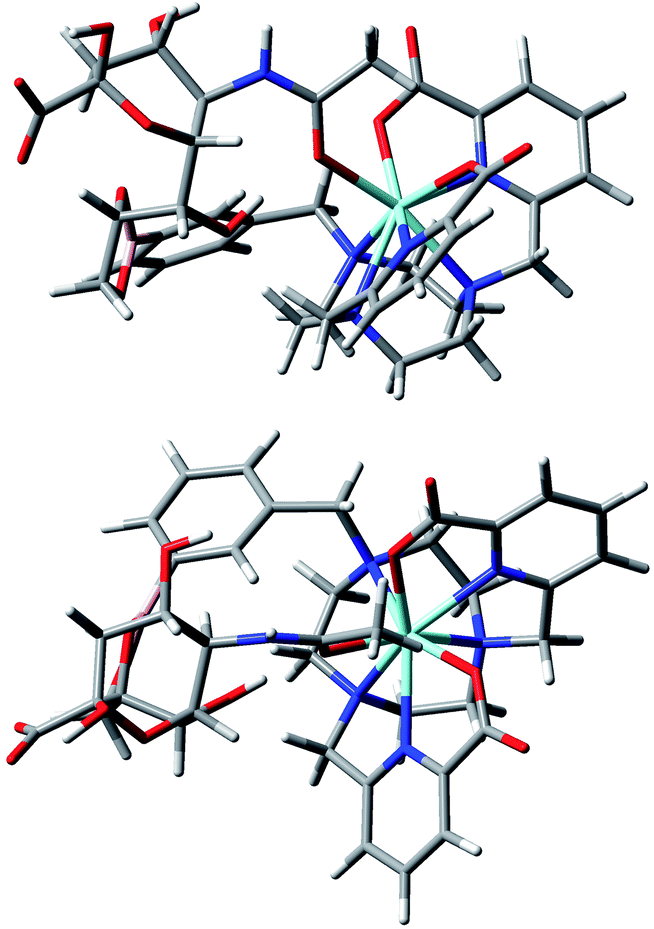 | ||
| Fig. 14 Views of the hypothetical binding of the glycerol diol group to boron and cooperative ligation of the amide carbonyl oxygen to the metal ion in an optimised model geometry for the complex [Eu·L3·sialic acid].11,12 | ||
Conclusions and future work
The utility of dynamically racemic lanthanide probes in the sensing of important chiral biomolecules, employing CPL spectroscopy as the detection technique, is a research area of growing interest. Here, the sensitivity and selectivity of this type of chirality probe has been demonstrated.The reversible binding of sialic acid by the bis-carboxylate complex, [Eu·L1]+, and the boronic acid complex [Eu·L3]+ was signalled via the induction of a ‘fingerprint’ CPL response accompanying changes in the total emission spectrum. This result demonstrates the first example of the use of CPL spectroscopy in conjunction with a lanthanide chirality probe to detect sialic acid. Other workers have examined the response of selected carbohydrates to europium, but at far higher concentrations and without any real rationale for binding selectivity.22 Further work investigated the sign and form of the CPL spectra to elucidate key structural information regarding the ternary adduct, by comparison with the methyl-ester derivative of sialic acid. A proposed binding mode was hypothesised, highlighting the role of the glycerol side chain with the boronic acid group.
It may also be possible to increase the binding affinity, and thus selectivity, of [Eu·L3]+ for sialic acid through the inclusion of an aminomethyl group in the ortho-position of the benzyl substituent. In this way, Wulff has shown that conversion of the trigonal boronic acid to the tetrahedral boronate occurs at lower pH, in the presence of this additional amino group, where the stability of the resulting ester is an order of magnitude greater.10 The presence of the tetragonal species over a greater pH range can be attributed to the B–N interaction in the phenylboronate and may aid the selective recognition of sialic acid in aqueous solution at physiological pH levels.
Experimental
Details of general experimental methods, instrumentation and purifications methods have been given elsewhere.11,12 The synthesis of L1 and L2 and their europium (III) complexes was reported in ref. 11 and 12. The methods of acquiring spectra, spectral titrations and the analysis of spectral data to estimate binding affinities are given in ref. 11.HPLC
Reverse-phase preparative HPLC was performed at 295 K using a Shimadzu system consisting of a Degassing Unit (DGU-20A5R), a Prominence Preparative Liquid Chromatograph (LC-20AP), a Prominence UV/Vis Detector (SPD-20A) and a Communications Bus Module (CBM-20A). An XBridge C18 OBD 19 × 100 mm, i.d. 5 μM column was used with a flow rate of 2 mL min−1 (analytical) or 17 mL min−1 (prep). The solvent system was H2O + 0.1% formic acid/MeOH + 0.1% formic acid (gradient elution, Table 3).| Step | Time/min | Flow (analytical/prep)/mL min−1 | % H2O (0.1% FA) | % MeOH (0.1% FA) |
|---|---|---|---|---|
| 0 | 0.0 | 2.0/17.0 | 90.0 | 10.0 |
| 1 | 10.0 | 2.0/17.0 | 5.0 | 95.0 |
| 2 | 13.0 | 2.0/17.0 | 5.0 | 95.0 |
| 3 | 13.5 | 2.0/17.0 | 90.0 | 10.0 |
| 4 | 16.5 | 2.0/17.0 | 90.0 | 10.0 |
DFT modelling and geometry optimisation
Several starting model geometries of [Eu·L3·sialic acid] with Eu replaced by Y and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC6H3Me2(OMe) groups replaced by hydrogen atoms were optimised at B3LYP/3-21G* to examine the structural feasibility of both metal-binding and boronic cycle formation, present in [Eu·L3·sialic acid]. The optimised geometry in Fig. 14 was confirmed as a true minimum by a frequency calculation. More details of the DFT modelling methodology used are given in ref. 11 and 12.
CC6H3Me2(OMe) groups replaced by hydrogen atoms were optimised at B3LYP/3-21G* to examine the structural feasibility of both metal-binding and boronic cycle formation, present in [Eu·L3·sialic acid]. The optimised geometry in Fig. 14 was confirmed as a true minimum by a frequency calculation. More details of the DFT modelling methodology used are given in ref. 11 and 12.
3-(Bromomethyl)phenylethylene boronate CAS: 1225388-28-2
3-(Bromomethyl)phenylboronic acid (100 mg, 0.465 mmol) was dissolved in anhydrous pentane (2 mL) over 4 Å molecular sieves. Ethylene glycol (30 μL, 0.465 mmol) was added and the white suspension was allowed to stir at room temperature for 1 h under an atmosphere of argon. Upon completion, CH2Cl2 (15 mL) was added to the reaction mixture and the solution was decanted from the molecular sieves. The solvent was removed under reduced pressure to give 3-(bromomethyl)phenylethylene boronate as a colourless oil which gave spectral data in accord with the literature, and was used directly without further purification (80 mg, 72%). TLC analysis Rf 0.28 (silica, 5% CH3OH in CH2Cl2); 1H NMR (295 K, 400 MHz, CDCl3) δH 7.85 (1H, s, H2), 7.75 (1H, dt, 3J 7.5, 4J 1.5, H6), 7.51 (1H, dt, 3J 7.5, 4J 1.5, H4), 7.37 (1H, t, 3J 7.5, H5), 4.50 (s, 2H, CH2Br), 4.37 (s, 4H, BO(CH2)2O); 13C NMR (295 K, 175 MHz, CDCl3) δC 137.4 (Ar–C), 135.5 (Ar–C), 134.9 (Ar–C), 132.2 (Ar–C), 128.5 (Ar–C), 66.2 (BO(CH2)2O), 33.5 (CH2Br); 11B NMR (295 K, 128 MHz, CDCl3) δB 31.41; m/z GC-EI tR = 4.68 min, 240.0 (M+), 161.1 (M+ – Br).(3-((4,7-Bis((4-((4-methoxy-2,6-dimethylphenyl)ethynyl)-6-(methoxycarbonyl)pyridin-2-yl)methyl)-1,4,7-triazonan-1-yl)methyl)phenyl)boronic acid, L3′

The bis-alkylated ligand, L1 (15 mg, 0.020 mmol) and K2CO3 (3 mg, 0.020 mmol) were dissolved in anhydrous CH3CN (2 mL) and bubbled with argon (20 minutes). 3-(Bromomethyl)phenylethylene boronate (5 mg, 0.020 mmol) was added and the mixture was stirred under argon at 55 °C and monitored by LC-MS. After 24 h the reaction was cooled and filtered to remove excess potassium salts. The solvent was removed under reduced pressure and the crude material was purified by flash column chromatography (silica, 0–5% CH3OH in CH2Cl2) to give L3 as a glassy solid (20 mg, 85%). 1H NMR (295 K, 600 MHz, CDCl3) δH 8.02 (2H, s, py-H3), 7.78–7.72 (3H, m, Ph-H), 7.53 (2H, s, py-H5), 7.39–7.34 (1H, m, Ph-H), 6.62 (4H, s, H2,2′), 4.31 (2H, s, Ph-CH2), 4.02 (4H, s, py-CH2), 3.93 (6H, s, CO2CH3), 3.80 (6H, s, OCH3), 3.53–2.95 (12H, br m, ring Hs), 2.46 (12H, s, CH3); 13C NMR (295 K, 150 MHz, CDCl3) δC 165.2 (CO2CH3), 160.3 (C1), 157.6 (py-C6), 148.1 (C4), 143.1 (Ph-C), 136.6 (Ph-C), 135.7 (Ph-C), 134.6 (C3/3′), 133.86 (Ph-C), 127.8 (py-C5), 126.0 (py-C3), 113.7 (py-C4), 112.8 (C2/2′), 94.8 (C5), 92.9 (C6), 66.2 (Ph-CH2), 61.0 (py-CH2), 55.3 (OCH3), 53.6 (ring Cs), 53.1 (CO2CH3), 51.8 (ring Cs), 46.3 (ring Cs), 21.5 (CH3); 11B NMR (295 K, 128 MHz, CDCl3) δB 31.3; m/z (HRMS+) 877.4350 [M + H]+ (C51H5710BN5O8 requires 877.4337); tR = 8.8 min.
[Eu·L3]Cl
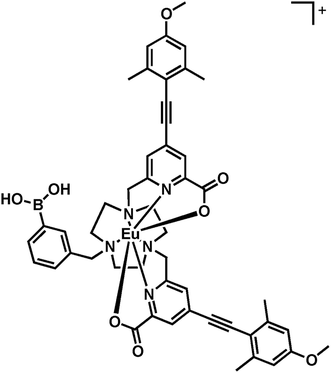
An aqueous solution of sodium hydroxide 0.1 M (0.5 mL) was added to a solution ligand L3 (5 mg, 6 μmol) in methanol (0.5 mL). The mixture was stirred at 65 °C for 4 h. The reaction was monitored by LC-MS. Upon completion, aqueous hydrochloric acid (0.1 M) was added until pH 6.5 was achieved. Europium chloride hexahydrate (3 mg, 8 μmol) was added and the pH was readjusted to 6.5 by addition of aqueous sodium hydroxide (0.1 M). The reaction was stirred at 65 °C for 24 h. The solvent was removed under reduced pressure to give the europium complex as a white solid (7 mg, 80%). m/z ESI (NH4HCO3/MeCN) 998.2887 [M + Na]+, C47H4911BN5O8155EuNa requires 998.2818; tR = 6.2 min; λexc (MeOH) = 356 nm; ϕem (MeOH) 0.05, ε (MeOH) 35000 M−1 cm−1, τ (H2O) = 0.38 ms, τ (D2O) = 0.48 ms, q = 0.2.
Acknowledgements
We thank the ERC for support (266804), Robert Pal for assistance with CPL measurements, and Mark A Fox for the DFT study.References
- G. Yogeeswaran, B. S. Stein and H. Sebastian, Cancer Res., 1978, 38, 1336–1344 CAS.
- J. Roth, C. Zuber, P. Wagner, D. J. Taatjes, C. Weisgerber, P. U. Heitz, C. Goridis and D. Bitter-Suermann, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 2999–3003 CrossRef CAS.
- F. J. Krolikowski, K. Reuter, T. P. Waalkes, S. M. Sieber and R. H. Adamson, Pharmacology, 1976, 14, 47–51 CrossRef CAS PubMed.
- E. Gruszewska, B. Cylwik, A. Panasiuk, M. Szmitkowski, R. Flisiak and L. Chrostek, BioMed Res. Int., 2014, 2014, 876096 Search PubMed.
- L. Frullano, J. Rohovec, S. Aime, T. Maschmeyer, M. I. Prata, J. J. P. de Lima, C. F. G. C. Geraldes and J. A. Peters, Chem.– Eur. J., 2004, 10, 5205–5217 CrossRef CAS PubMed.
- G. A. Lemieux, K. J. Yarema, C. L. Jacobs and C. R. Bertozzi, J. Am. Chem. Soc., 1999, 121, 4278–4279 CrossRef CAS.
- S. Geninatti Crich, D. Alberti, I. Szabo, S. Aime and K. Djanashvili, Angew. Chem., Int. Ed., 2013, 52, 1161–1164 CrossRef CAS PubMed.
- M. Regueiro-Figueroa, K. Djanashvili, D. Esteban-Gómez, T. Chauvin, É. Tóth, A. de Blas, T. Rodríguez-Blas and C. Platas-Iglesias, Inorg. Chem., 2010, 49, 4212–4223 CrossRef CAS PubMed.
- K. Ouchi, S. Saito and M. Shibukawa, Inorg. Chem., 2013, 52, 6239–6241 CrossRef CAS PubMed.
- (a) T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1996, 35, 1910–1922 CrossRef; (b) G. Wulff, Pure Appl. Chem., 1982, 54, 2093–2102 CrossRef; (c) R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1124 RSC.
- E. R. Neil, M. A. Fox, R. Pal, L.-O. Palsson, B. A. O'Sullivan and D. Parker, Dalton Trans., 2015, 44, 14937–14951 RSC.
- E. R. Neil, M. A. Fox, R. Pal and D. Parker, Dalton Trans., 2016, 45, 8355 RSC.
- For reviews addressing the scope of CPL probes: (a) F. Zinna and L. Di Bari, Chirality, 2015, 27, 1 CrossRef CAS PubMed; (b) J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1 CrossRef CAS; (c) R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673 RSC; (d) G. Muller, Dalton Trans., 2009, 9692 RSC; (e) T. Wu, X.-Z. You and P. Bour, Coord. Chem. Rev., 2015, 284, 1 CrossRef CAS.
- A. T. Frawley, R. Pal and D. Parker, Chem. Commun., 2016, 52, 13349 RSC.
- A. Beeby, I. M Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- K.-H. Chen, J.-S. Yang, C.-Y. Hwang and J.-M. Fang, Org. Lett., 2008, 10, 4401–4404 CrossRef CAS PubMed.
- O. Alptürk, O. Rusin, S. O. Fakayode, W. Wang, J. O. Escobedo, I. M. Warner, W. E. Crowe, V. Král, J. M. Pruet and R. M. Strongin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9756–9760 CrossRef PubMed.
- J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769–774 CrossRef CAS.
- S. Friedman, B. Pace and R. Pizer, J. Am. Chem. Soc., 1974, 96, 5381–5384 CrossRef CAS.
- B. S. Blaum, J. P. Hannan, A. P. Herbert, D. Kavanagh, D. Uhrín and T. Stehle, Nat. Chem. Biol., 2015, 11, 77–82 CrossRef CAS PubMed.
- R. S. Alexander, Z. F. Kanyo, L. E. Chirlian and D. W. Christianson, J. Am. Chem. Soc., 1990, 112, 933–937 CrossRef CAS.
- T. Wu, J. Průša, J. Kessler, M. Dračínský, J. Valenta and P. Bouř, Anal. Chem., 2016, 88, 8878 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |