
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive polymers and their applications
Menglian
Wei
,
Yongfeng
Gao
,
Xue
Li
and
Michael J.
Serpe
*
Department of Chemistry, University of Alberta, Edmonton, AB T6G 2G2, Canada. E-mail: michael.serpe@ualberta.ca
First published on 25th October 2016
Abstract
Responsive polymer-based materials are capable of altering their chemical and/or physical properties upon exposure to external stimuli. These materials have been intensively studied over the years for a diverse range of applications, e.g., for on-demand drug delivery, tissue generation/repair, biosensing, smart coatings, and artificial muscles. Here, we review recent advances in the areas of sensing and biosensing, drug delivery, and actuators. Specific examples are given in each of these areas, and we highlight our group's work on poly(N-isopropylacrylamide)-based microgels and assemblies.
 Menglian Wei | Menglian Wei received her B.Sc. degree from Wuhan University in 2011. She joined Prof. Michael J. Serpe's group as a Ph.D. student in 2012 and her research has focused on the development of a novel surface plasmon resonance spectrometer for studying confined polymer brushes, and for improved sensing and biosensing. |
 Yongfeng Gao | Yongfeng Gao received his B.Sc. degree in Chemistry from Tsinghua University, Beijing, China, in 2009 and obtained his M.Sc. degree in Chemical Engineering from Beijing University of Chemical Technology, Beijing, China, in 2012. He is presently a Ph.D. candidate in the Department of Chemistry at the University of Alberta, under the supervision of Prof. Michael J. Serpe. His research interests focus on stimuli-responsive microgels and microgel-based systems for controlled drug delivery. |
 Xue Li | Xue Li received her B.S. degree from Qufu Normal University in 2009, and M.Sc. degree from Nanjing University in 2011 where she studied analytical chemistry in the group of Prof. Yu Qin. In the same year she joined Prof. Michael J. Serpe's group as a Ph.D. student at the University of Alberta and received her Ph.D. in 2016. While in the Serpe group, she developed pNIPAm microgel-based etalons for biosensing applications and polymer-based materials for artificial muscles. |
 Michael J. Serpe | Michael J. Serpe received his B.S. degree from the University of Central Florida in 2000, his Ph.D. from the Georgia Institute of Technology in 2004 and completed his postdoctoral studies at Duke University in 2009. In the same year he joined the Department of Chemistry at the University of Alberta as an Assistant Professor and was promoted to Associate Professor in 2014. His group's research program is focused on developing new technologies to solve problems associated with health and the environment using fundamental and applied polymers, colloids (nano and microparticles), surface and materials chemistry. |
1. Introduction
Polymers are ubiquitous in the things we use in everyday life, and are even responsible for life itself, e.g., due to the polymers of DNA and proteins.1 For ages, polymers have been used to improve the quality of life, although the true polymeric nature of the substances (e.g., natural rubber) was not known.2 Likely driven by the economic impetus of the rubber industry in the 19th and 20th centuries more effort was spent on investigating why the materials (i.e., polymers) behave the way they do. Hence, the intense debate about the nature and structure of polymers began, and eventually Staudinger's macromolecular theory was accepted,3,4 which described polymers as being composed of many elementary units (monomers) covalently bound together. This theory was further supported by the experiments of Carothers, who first synthesized nylon.5,6 Following the work of these pioneers, a number of other researchers have made ground-breaking advances in the field, and the work has yielded many Nobel Prizes over the decades. This combined knowledge has allowed the development of polymers for nearly every application imaginable, and is single-handedly responsible for the high quality of life that many have become accustomed to.This foundational research also led to the development of a new class of polymers, which respond to their environment by changing their physical and/or chemical properties.7–9 These polymers, referred to as stimuli-responsive polymers (or smart/intelligent polymers), have been synthesized to be responsive to a variety of stimuli, e.g., pH,10 temperature,11 mechanical force,12 the presence of various small molecules and biomolecules,13 and electric/magnetic fields.14–16 Their development is oftentimes driven by the desire to mimic nature.17–19 Such intelligent polymers have found many applications in the fields of biology and medicine and can be used as sensors and biosensors,20 for controlled and triggered drug delivery,21 environmental remediation,22 chemo-mechanical actuators,23–25 and for many other applications.26–28
Of the multitude of responses to stimuli (some specifically mentioned above), by far the most well-studied and understood response is to temperature. For example, some polymers exhibit a lower critical solution temperature (LCST),29 which is the lowest temperature at which temperature induced demixing occurs. That is, below the LCST the polymer chains and solvent molecules are in one homogeneous mixed phase and above the LCST, phase separation occurs via an entropically driven process. Poly(N-isopropylacrylamide) (pNIPAm)11,30–32 is one of the most extensively studied temperature responsive polymers that exhibits a LCST at ∼32 °C, which is close to the physiological temperature. As the solution temperature rises above the LCST, pNIPAm chains undergo a transition from an extended (solvated) random coil to a compact (desolvated) globular conformation. For individual polymer chains, the coil to globule transition can be thermodynamically controlled by adjusting the polymer composition,33i.e., the LCST shifts to higher or lower temperature by copolymerization with a hydrophilic or hydrophobic monomer, respectively.34,35 There are a variety of polymers that exhibit LCSTs, such as poly[N-[2-(diethylamino)ethyl acrylamide]] (PDEAEAM),36 poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA),37–39 poly(N,N-diethylaminoethyl methacrylate) (PDEAEMA),40 poly(2-(N-morpholine)ethyl methacrylate) (PMEMA),40 poly[oligo(ethylene glycol)methacrylate]41,42 and poly(N,N-diethylacrylamide) (PDEAAM).43,44 In addition, multi-responsive polymers can be synthesized by incorporating other functional groups into the temperature responsive polymer. For example, pH responsive compounds that have ionizable functional groups capable of donating or accepting protons upon environmental pH changes could be used. Some common examples are, acrylic acid (AAc)10,45–47 and N,N-dimethylaminoethyl methacrylate (DMAEMA).48–50 Light responsive monomers can also be used to generate materials that exhibit both temperature and light responsivity; a common example is azobenzene.51–53 In most cases the response of these polymers is a result of light triggered isomerization of light sensitive molecules incorporated into the polymer, although other mechanisms are possible, including light triggered ionization. Finally, biologically responsive systems, e.g., enzyme responsive polymers,54–56 and glucose responsive polymers57,58 can also be generated, which have the ability to respond to stimuli that are inherently present in biological samples.59 Generally, the response is a result of capture biomolecules immobilized in the polymer interacting with the target, which results in network crosslinking and/or ionization.
In this review, we mainly discuss recent examples of the use of stimuli-responsive polymers for sensing and biosensing, controlled and triggered drug delivery, and artificial muscles. In these individual areas we will highlight recent work from our own group. We point out that this review will focus on applications, while many other recent reviews have focused on the synthesis and fundamental properties of stimuli responsive polymers, and those details will not be rehashed here.60–64
2. Applications of stimuli-responsive polymers
2.1 Sensing and biosensing
A sensor is a self-contained integrated device that is able to receive an input from its surroundings and convert it into an output signal that can be processed and converted to a readable result.65 Likewise, a biosensor is a device that is capable of detecting and quantifying biological species of interest. In general, a biosensor should be capable of detecting a species of interest (analyte) from a complex mixture containing a variety of interfering species, and provide accurate results in a short time. In some cases, biosensors should be able to detect biological analytes in resource-limited settings and at the point-of-care (POC) to increase efficiency of treating patients in developing parts of the world. Accomplishing this will have positive health outcomes for those people in developing parts of the world.Responsive polymer-based sensors have attracted considerable attention due to their ability to convert the presence of analytes into a physical and/or chemical change that a user can relate to the status of a system.66 In one example, Dostalek and coworkers generated a poly(N-isopropylacrylamide)-co-methacrylic acid (pNIPAm-co-MAAc) hydrogel on a surface plasmon resonance (SPR) sensor surface with indium tin oxide microheaters embedded to allow for SPR signal tuning, as shown in Fig. 1.67 Using the microheaters, rapid thermal responses of the pNIPAm-based material between swollen and de-swollen could be triggered, yielding a thermo-optical coefficient of dn/dT = 2 × 10−2 RIU K−1. Further, the hydrogel layer can serve as a 3D binding matrix for biosensor applications by engineering bio-recognition elements in the polymer network. In this example, the authors modified the hydrogel with mouse immunoglobulin G (mIgG) via 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling, which was able to capture Alexa Fluor 647 dye-labeled goat antimouse IgG (a-mIgG). Enhanced fluorescence intensity was observed at the SPR resonance angle, which was attributed to the enhanced electric field by SPR. In addition, upon a temperature increase, the binding matrix collapsed and resulted in a resonance angle shift as well as diminished fluorescence intensity. Therefore, the fluorescence signal excited by SPR at the resonance angle can be virtually switched on and off by swelling and de-swelling of the hydrogel binding matrix.
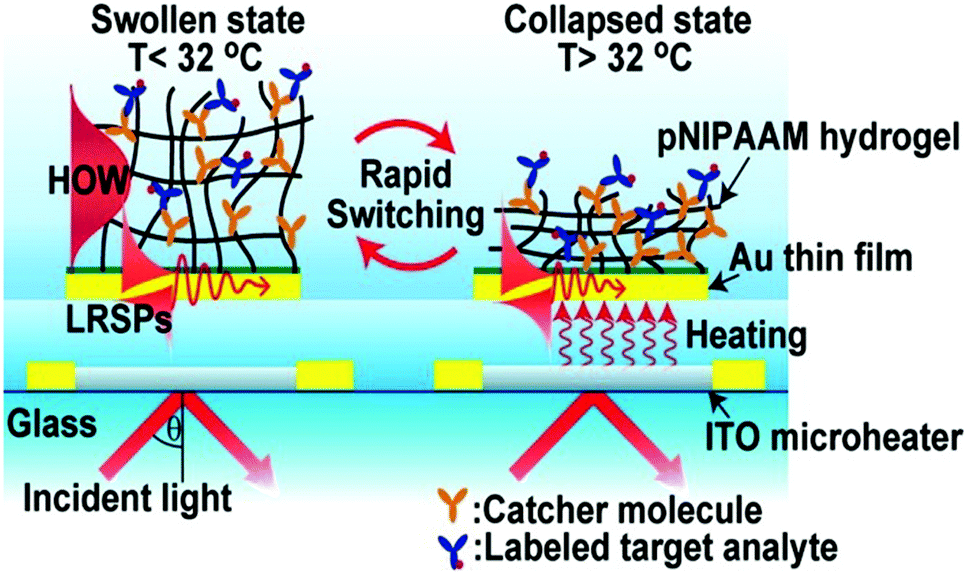 | ||
| Fig. 1 Schematic diagram of a pNIPAm-co-MAAc hydrogel modified SPR sensor device with embedded ITO microheaters enabling rapid signal tuning. Reprinted with permission from ref. 67. Copyright 2013, American Chemical Society. | ||
Stimuli-responsive polymer-modified nanoparticles have attracted significant attention in recent years for sensing and biosensing. Due to quantum effects, nanoparticles exhibit unique optical properties (e.g., bright emission with no photobleaching), which make them ideal candidates for reporters in sensors. Furthermore, the conformational change of the surface bound stimuli responsive polymers can translate into observable optical property changes, which can be used for sensing and biosensing. In one example, Hoogenboom and coworkers developed a colorimetric temperature and salt sensor by coating Au nanoparticles (AuNPs) with a thermoresponsive pNIPAm shell.68 The pNIPAm polymer was synthesized by RAFT polymerization using methyl 2-(((butylthio)carbonothioyl)thio)propanoate (MBTTC) as a chain transfer agent (CTA), and further grafted to the AuNP surface by a ligand exchange process. When exposed to solutions of different salt concentrations, the pNIPAm modified AuNPs were able to change color from red to purple or blue at elevated temperatures, as shown schematically in Fig. 2. This is due to AuNP aggregation induced electronic coupling of the SPR leading to a visible color change. In this study, the authors also found that the range of temperature sensitivity increased in the presence of NaSCN, compared to NaCl, due to charge screening and Hofmeister effects.
 | ||
| Fig. 2 Schematic illustration of the synthesis of pNIPAm brush modified AuNPs and their thermoresponsive behavior in aqueous solution and their aggregation in the presence of various concentrations of NaCl. Reprinted with permission from ref. 68. Copyright 2016, The Royal Society of Chemistry. | ||
A conceptually similar approach was used to generate CO2 sensors. CO2 has physiological significance, and abnormal concentrations have been associated with metabolism-related diseases. Ma et al. developed a CO2 sensor by coating AuNPs with the CO2 responsive polymer, poly(N-(3-amidino)-aniline) (PNAAN), as shown in Fig. 3c.69 The hybridized particles were synthesized by directly reducing HAuCl4 with the CO2 responsive monomer N-(3-amidino)-aniline (NAAN). As CO2 dissolves in solution, the amidine group of PNAAN can be protonated into hydrophilic amidinium, which induced PNAAN to swell and detach from the AuNP surface, resulting in AuNP aggregation and a color change, as shown in Fig. 3a and b. The CO2 sensor exhibited a linear range of 0.0132 to 0.1584 hPa and a limit of detection (LOD) of 0.0024 hPa by monitoring the UV absorbance change of AuNPs.
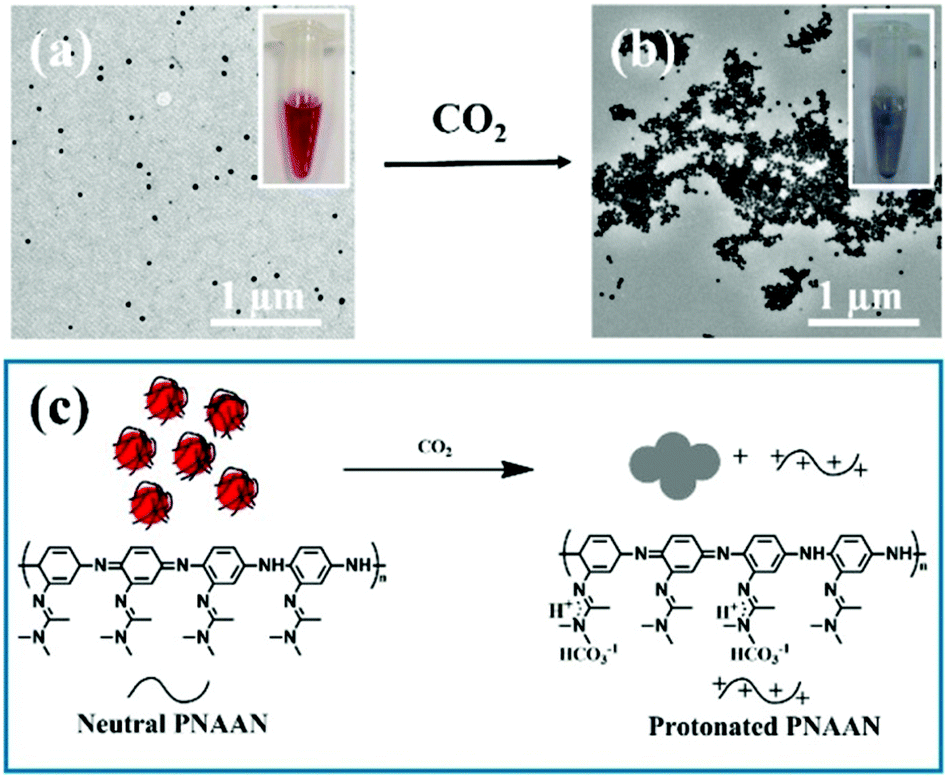 | ||
| Fig. 3 TEM images of AuNPs (a) before and (b) after being purged with CO2 gas. (c) Schematic illustration of CO2 induced AuNP aggregation. Insets of (a) and (b) show photographs of the AuNP solutions (a) before and (b) after being purged with CO2 gas. Reprinted with permission from ref. 69. Copyright 2016, American Chemical Society. | ||
In a very interesting application of nanotechnology combined with stimuli responsive polymers, Paek et al. generated a colorimetric pH sensor by anchoring “blue” and “orange” light emitting quantum dots (BQDs and OQDs) to a single graphene oxide (GO) sheet (MQD-GO) through different pH responsive linkers, as shown in Fig. 4.70 The photoluminescence emissions of the BQDs and OQDs on MQD-GO can be controlled independently through the two types of pH-responsive linkers of poly(acrylic acid) (PAA) (pKa = 4.5) and poly(2-vinylpyridine) (P2VP) (pKa = 3.0). The polymer linkers were able to change their conformation at distinct pH ranges, which ultimately tuned the Förster resonance energy transfer efficiencies from the BQDs to the GO and from the OQDs to the GO. As a result, the color of MQD-GO transitions from orange to near-white to blue over a wide range of pH values. Furthermore, the MQD-GO sensor showed excellent reversibility and high dispersion stability in pure water, all of which satisfy the critical requirements of a pH sensor.
 | ||
| Fig. 4 Structures of (a) P2VP-OQD and (b) PAA-BQD. (c) Schematic illustration of the conformation and behavior of MQD-GO at the indicated solution pH. (d) Photographs of MQD-GO in buffer solutions at the indicated solution pH under irradiation with 365 nm light. Reprinted with permission from ref. 70. Copyright 2014, American Chemical Society. | ||
Stimuli-responsive polymers can also be used as building blocks for generating photonic crystals (PCs), which exhibit structural color due to light interaction with the periodic structure of the material. These interactions ultimately lead to constructive/destructive interference of specific wavelengths of light, which yield color. Generally, PCs can be classified as one-dimensional (1-D), two-dimensional (2-D), and three-dimensional (3-D) depending on the number of dimensions the order occurs, as shown in Fig. 5.71 The wavelength of light “reflected” from the device is primarily determined by the material's lattice spacing, which also dictates the visual color of the material. If the photonic crystal is composed of responsive polymer building blocks, then the lattice spacing (and visual color) can also be tuned by applying external stimuli. Perhaps the most well-known early examples of responsive PCs come from the Asher group who pioneered the development of 3-D photonic crystal-based sensors. This was accomplished by embedding ordered crystalline colloidal arrays (CCA) in analyte-responsive hydrogel matrices. These materials were used to detect solution pH, ionic strength, temperature, and the presence of biological or chemical targets.72–78 In one example, a 3-D PC sensor for metal ions was developed by copolymerizing 4-acryloylaminobenzo-18-crown-6 (AAB18C6) into a hydrogel composed of an ordered array of polystyrene spheres.72 As metal ions bound to the crown ether of AAB18C6, the hydrogel swelled due to the charge density increase in the polymer network yielding an increase in osmotic pressure and network swelling. As a result of the swelling, the polystyrene spheres in the hydrogel network separated from one another resulting in a red shift of the “reflected” light.
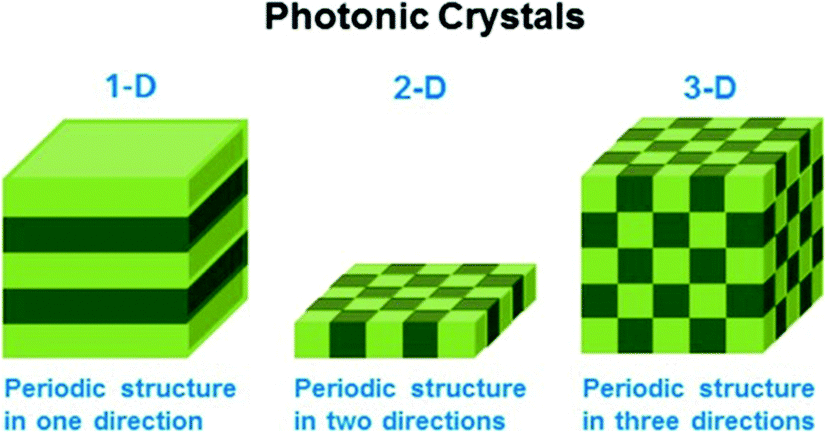 | ||
| Fig. 5 Schematic illustrating the different dimensions of order in photonic materials. Reprinted with permission from ref. 71. Copyright 2014, American Chemical Society. | ||
In another example, Hayward and coworkers developed a colorimetric temperature sensor by depositing alternating layers of high and low refractive index polymers on a surface to generate a one dimensional Bragg mirror as shown in Fig. 6b.79 The high refractive index polymer was non-responsive poly(para-methyl styrene) (PpMS), while the low refractive index polymer was temperature responsive poly(N-isopropylacrylamide)-co-acrylic acid. After immersion in water at room temperature, the polymer stack exhibited a Bragg peak at 710 nm and an orange color. Upon heating, the pNIPAm-based polymer layer shrunk, resulting in the approach of the PpMS layers and a concomitant shift in the reflected light to lower wavelengths. This also yielded a color change, as can be seen in Fig. 6a. Furthermore, the color tunability was fully reversible with temperature.
 | ||
| Fig. 6 (a) Photographs of a sensor changing color as a function of temperature—heating induces deswelling, while cooling induces swelling. (b) Schematic representation of sensor fabrication (red layers, PpMS; blue layers, pNIPAm-co-AAc). Reprinted with permission from ref. 79. Copyright 2012, Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The Serpe group has fabricated etalons (a basic 1D photonic material) that respond to a variety of stimuli, such as pH,80 light,81 electric field,82 temperature,46 nerve agents,83 and various biomolecules.84 As detailed in these publications, etalons were constructed by depositing a thin layer of Au (typically 15 nm) on top of a glass substrate by thermal evaporation followed by “painting” a layer of microgels onto the Au, and subsequent deposition of another Au layer (typically 15 nm) on top of the microgel layer. This structure allows light to enter the dielectric cavity and resonate between the two reflective Au layers. This resonating light yields constructive and destructive interference, allowing certain wavelengths of light to be reflected. The wavelength of the reflected light can be predicted using eqn (1):
λm = 2nd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ | (1) |
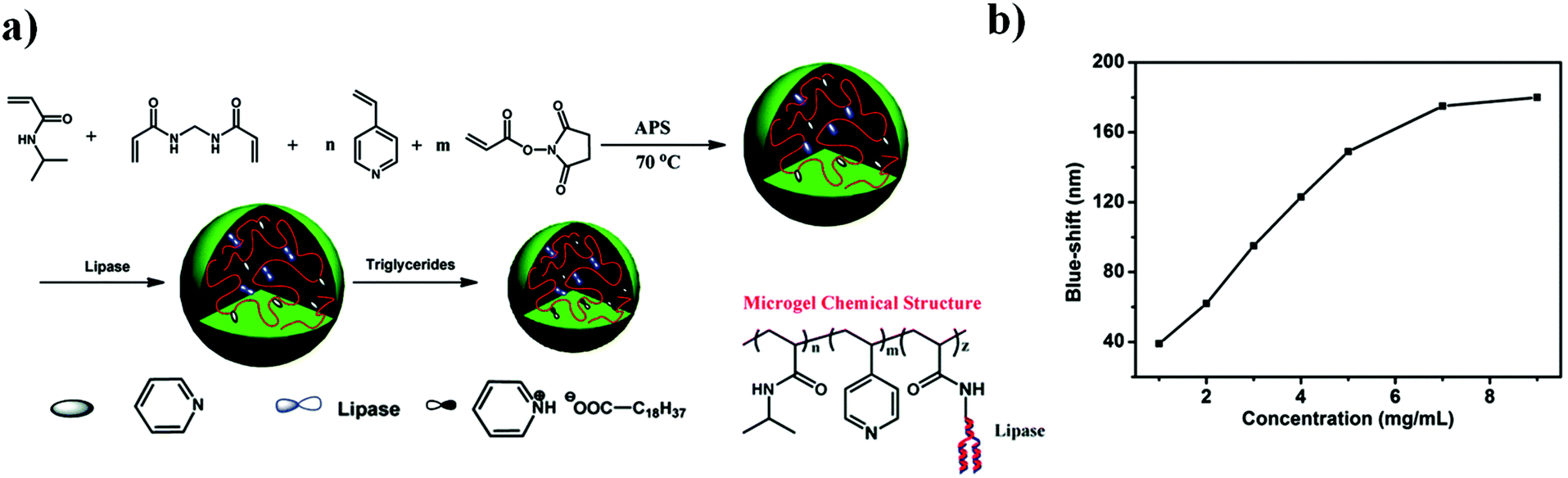 | ||
| Fig. 7 (a) Triacylglyceride responsive microgel synthesis scheme, and the resulting microgel chemical composition. (b) The magnitude of the reflectance peak blue-shift as a function of triglyceride concentration at 30 °C. Reprinted with permission from ref. 87. Copyright 2015, The Royal Society of Chemistry. | ||
Multi-responsive microgels and etalons could also be generated by incorporating a multi-responsive moiety into the pNIPAm-based microgel at the time of polymerization.83,90 Serpe and coworkers synthesized spiropyran (SP)-modified pNIPAm-based microgels and investigated their responsivity to multiple stimuli including temperature, pH, Cu2+, UV, visible, and near-infrared radiation.90 The responses were a result of the SP groups undergoing a reversible isomerization/reaction from a neutral to a charged form, as shown in Fig. 8. SP's C(spiro)–O bond can be cleaved by UV light exposure or at low pH (<4). During the process, SP isomerizes from a colorless, nonplanar, closed and neutral form to the colored, planar, open and charged merocyanine form (MC), which increases the microgel's inner osmotic pressure and results in microgel swelling. MC can also be triggered to reform SP by exposure to visible light. The C(spiro)–O bond can be cleaved by Cu2+ and subsequently bind to Cu2+ through phenoxide anions in the microgel, which result in microgel collapse. The spiropyran-modified microgels retained their responsivity after incorporation into etalons, which allows their optical properties to be tuned upon exposure to these various stimuli.
 | ||
| Fig. 8 Spiropyran modified pNIPAm-based microgel response to multiple stimuli. Reprinted with permission from ref. 90. Copyright 2015, American Chemical Society. | ||
While stimuli-responsive polymers have a great potential to be applied for sensing and biosensing, there are still many challenges that lie ahead. For example, there is always room to improve sensor sensitivity, selectivity, stability, sampling, and signal readout mechanisms. However, we believe that the benefits of stimuli-responsive-based sensors (i.e., their ease of fabrication, and versatility) far outweigh the negatives, and one will find these sensors being employed in evermore diverse environments.
2.2 Controlled drug delivery
Living systems react to external stimuli by responding and adapting to changing external conditions. Polymer scientists have been trying to mimic this behavior by creating so called smart polymers. Smart polymers have shown promise for biomedical applications, and have found use as controlled/triggered/targeted drug delivery vehicles, tissue engineering scaffolds, cell culture supports, bioseparation apparatus, sensors, and actuators/artificial muscles. The concept of stimuli-responsive polymer-based drug delivery systems was first reported in the late 1970s with the use of thermosensitive liposomes for the local release of drugs via hyperthermia.91 Subsequently, a great deal of research has been carried out on stimuli-responsive materials for drug delivery, especially on the design and application of responsive polymers.13,66,92–94 The design of new systems and approaches must meet the challenges associated with administration in the body. The systems must be: (i) simply administered; (ii) capable of delivery to the desired locations in response to a stimulus; and (iii) composed of non-toxic, biocompatible and biodegradable components. A variety of stimuli-responsive polymer-based materials have been used for this application, including crosslinked gel networks, and non-crosslinked block copolymer assemblies.One of the most important classes of stimuli-responsive polymers used for controlled drug delivery is crosslinked polymer networks, e.g., hydrogels and microgels. Hydrogels have been shown to be useful for a wide range of biomedical applications because of their porous structure and water swellability. Their porosity permits loading of drugs into the gel matrix and subsequent drug release at a rate dependent on the diffusion coefficient of the small molecule or macromolecule in the gel network. In addition, many hydrogels can change their degree of swellability in response to changes in their environment as mentioned earlier. Based on the structures and properties of hydrogels, most of them have been used for transdermal drug delivery or injectable drug delivery systems. For transdermal drug delivery, microneedles improve drug permeability into skin by providing a clear entry pathway; this improves the delivery efficiency of vaccines and pharmaceutical agents (such as small molecules, protein, DNA).95–97 Compared to the solid microneedles,98 hydrogel-forming microneedle arrays can rapidly imbibe skin interstitial fluid to form discrete in situ hydrogel bulbs to control drug administration at a rate faster than traditional patch systems used for controlled drug release. The most important feature for hydrogel-forming microneedle arrays is the swelling induced release mechanism.99 By incorporating stimuli-responsive units into microneedles, one can trigger on-demand drug release by manipulating external stimuli.100,101 In one example, the McCoy group combined hydrogel-forming microneedles and light responsive drug conjugates to generate novel devices for on-demand transdermal drug delivery.102 For this device, poly(2-hydroxyethyl methacrylate) (HEMA) was used and crosslinked by ethylene glycol dimethacrylate (EGDMA). Microneedles were generated utilizing micromolding, and exhibited desirable mechanical properties. The devices were shown to release ibuprofen over a prolonged period of time (up to 160 h) in response to light. The release could also be turned on and off by turning the light on and off, respectively. By mimicking the endoparasite Pomphorhynchus laevis that attaches to its host's intestinal wall, the researchers developed a biphase microneedle array that mechanically interlocks with tissue through swellable microneedle tips to increase adhesion strength.103 The light responsive units incorporated into the microneedles yielded patches with light triggered release properties to provide photothermal therapy and chemotherapy.104,105 The light-activated heating and releasing behavior can be precisely controlled and switched on and off on demand for several cycles.
Recently, the Gu group also developed microneedle array-based patches with responsive properties for smart insulin106,107 and controlled antibody108 delivery. A novel glucose-responsive insulin delivery device was reported based on microneedle array patches integrated with hypoxia-sensitive hyaluronic acid (HS-HA) vesicles containing insulin and glucose oxidase (GOx). By taking advantage of local generation of hypoxia, they trigger the insulin release in response to hyperglycemia. This hypoxia responsive transduction was achieved by converting hydrophobic 2-nitroimidazole to hydrophilic 2-aminoimidazoles in a hypoxic environment. The design of the microneedle array patches are shown in Fig. 9. The amphiphilic hyaluronic acid was synthesized and used to form self-assembled vesicles, which encapsulate both human insulin and GOx. When the blood glucose level is high, the dissolved oxygen is rapidly consumed due to the glucose oxidation reaction. This produces a local hypoxic environment, which reduces 2-nitroimidazole, resulting in the dissociation of vesicles and subsequent release of insulin. Furthermore, the vesicle was integrated into the microneedle array and can be used for the painless and continuous administration of insulin. This smart insulin patch with its novel triggering mechanism may offer the possibility of closed-loop delivery of insulin. It will also guide the development of a useful drug delivery platform for treating other diseases using artificial vesicles. In further studies, they also investigated the integration of microneedles with pancreatic cells and synthetic glucose-signal amplifiers for smart insulin delivery. Importantly, for the first time, a synthetic amplifier was incorporated to quickly amplify the physiological signal, in this case “glucose level,” for effective transport of the signal and sufficient stimulation of insulin secretion from the β-cells.
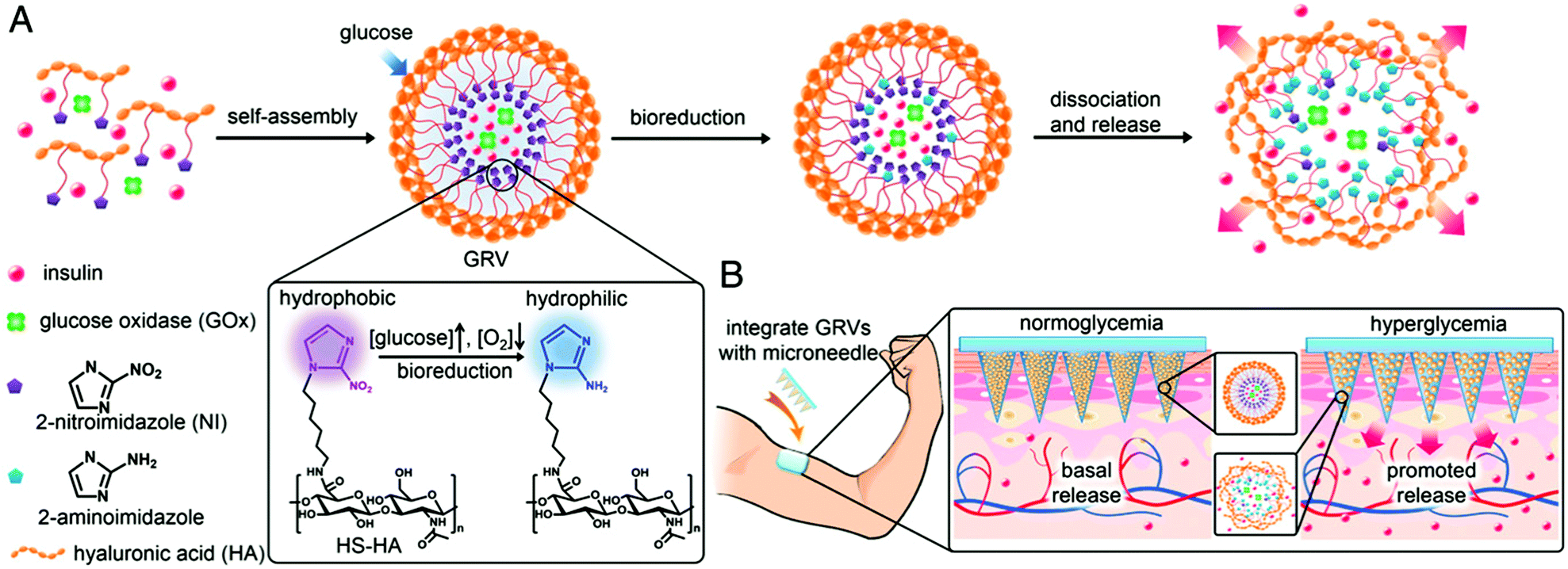 | ||
| Fig. 9 Schematic depiction of the glucose-responsive insulin delivery system using hypoxia-sensitive vesicle-loading MN-array patches. (A) Formation and mechanism of glucose responsive vesicles composed of HS-HA. (B) Schematic of the GRV-containing MN-array patch (smart insulin patch) for in vivo insulin delivery triggered by a hyperglycaemic state to release more insulin. Reprinted with permission from ref. 107. Copyright 2015, National Academy of Sciences. | ||
Similar to bulk gels, microgels can also be made biocompatible, however, due to their small size, they exhibit many advantages over bulk gels when used as biomaterials. One major advantage is that the rate of microgel response to external stimuli is much faster than bulk gels.93,109 They can also be modified chemically such that they circulate in the blood stream for long periods of time, and are biodegradable for sustained release. The degradability also allows the microgels to be quickly cleared from the body. In addition, microgels can be used as building blocks for the fabrication of biomedical devices with improved and/or new function.110 These many advantages make microgels ideal candidates for building controlled drug delivery systems.62,93,94,109–111 As a drug carrier, pNIPAm-based microgels combine the advantages of both hydrogels and nanoparticles. PNIPAm microgel particles have a sponge-like structure with interstitial spaces filled with water. Drug molecules can be loaded by equilibrium partitioning between the solution and microgel phases. Electrostatic interaction, hydrophobic interaction, and/or hydrogen-bonding may play an important role for drug loading. The Serpe group developed a novel microgel-based assembly (reservoir device) as a new platform for drug delivery.112–116 The structure of this device and the release mechanism is shown in Fig. 10. The device is usually composed of a pNIPAm-co-AAc microgel layer sandwiched between two thin Au layers (all on a glass support) and was used as a novel platform for controlled and triggered small molecule delivery. Tris(4-(dimethylamino)phenyl)methylium chloride (crystal violet, CV), which is positively charged, was loaded into the microgel layer of the device and released in a pH dependent fashion, at a rate that could be controlled by the thickness of the Au layer coating the microgel.112 The model drug could be released in an “on–off” fashion, by systematically varying the solution pH. Furthermore, by modifying the top layer Au surface, we can control the drug release with a lower thickness of Au.113
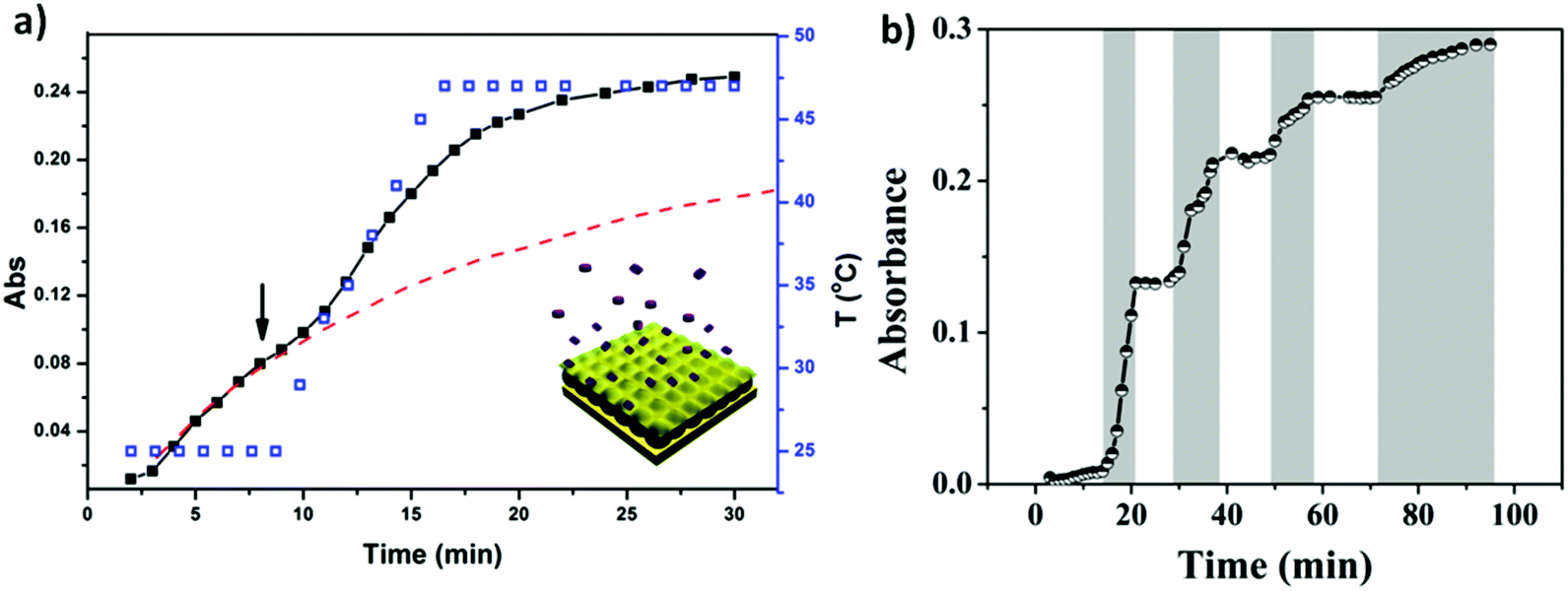 | ||
| Fig. 10 The structure of microgel-based assemblies and their (a) temperature and (b) pH triggered release profile with a 500 nm Au overlayer. For (a), the absorbance was monitored at 590 nm, (■) corresponds to the release profile (left y-axis), (□) corresponds to the temperature profile (right y-axis), while the dashed line shows the predicted release profile if the temperature wasn't increased. The shaded regions in (b) indicate where the solution pH was 3.0, while the white regions indicate where the solution pH was 6.5. The solution pH was varied by adding either HCl or NaOH. Reprinted with permission from ref. 112. Copyright 2013, American Chemical Society. | ||
In another example, the release of small molecules can be controlled by the aggregation state of microgels. For example, by combining two oppositely charged microgels, we can control the microgels’ aggregation behavior to control drug release. In this example, microgels copolymerized with acrylic acid exhibit a negative charge above pH 4.25, while the microgels copolymerized with N-[3-(dimethylamino)propyl]methacrylamide exhibit a positive charge below pH 8.4; these microgels are neutral outside of these pH ranges. We show that aggregates form when the two independent sets of microgels were exposed to one another in a solution that renders them both charged. In solutions of pH outside of this range, the microgels disaggregate because one of the microgels becomes neutralized. This behavior was exploited to load (aggregation) and release (disaggregation) a small-molecule model drug.115 This aggregate based system provides evidence of how the charged pNIPAm-based microgels are applied in controlled/triggered drug release.
Recently, we showed that a single microgel-based reservoir device could be used for sequential and controlled release of more than one small molecule to a system.114 By incorporating microgels with different chemistries that allow them to be ionized at different solution pH into a single reservoir device, the release of small molecules and their dosages can be controlled and tuned. This is shown in Fig. 11. Specifically, pNIPAm-co-AAc microgels (AAc-MG) and poly(N-isopropylacrylamide-3-(acrylamido)phenylboronic acid) (pNIPAm-co-APBA) microgels (APBA-MG) were used to construct reservoir devices and were loaded with the model drugs CV and methylene blue (MB), respectively. These two model drugs were chosen because of their strong visible light absorption and their positive charge, which allows them to interact strongly with the negatively charged microgels. At pH 10.0, CV and MB (both positively charged) exhibited strong electrostatic interactions with both the negatively charged AAc and APBA-modified microgels, respectively. At pH 7.0, the APBA groups were neutralized, allowing MB to be released from the APBA-MG only. When the solution pH was again lowered to 3.0, the AAc groups are neutralized allowing CV to be released from the AAc-MG. These systems represent a versatile approach to sequentially delivering small molecules to a system, in a triggered fashion, with tunable release kinetics. Importantly, their release behavior can be easily tuned by simply changing the microgel chemistry, e.g., by generating reservoir devices from microgels that ionize at different solution pH. This would allow one to deliver various small molecules to a system triggered by a variety of solution pH. This, combined with the tunable release kinetics and the ability to array these devices on a single substrate, make this delivery platform extremely versatile, powerful, and unique.
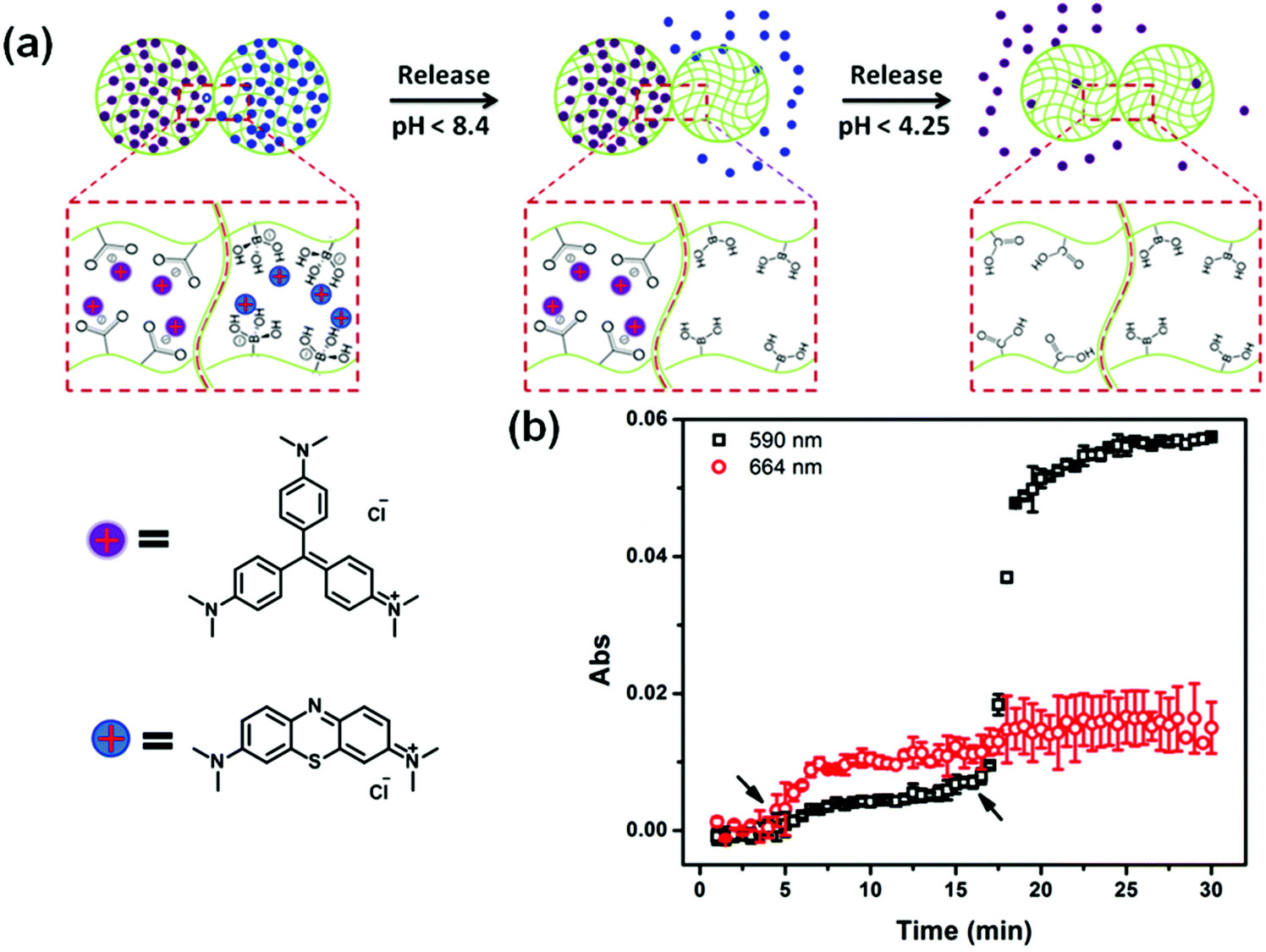 | ||
| Fig. 11 (a) Schematic illustration of pH triggered MB and CV release from APBA-MG and AAc-MG, respectively. As each microgel is neutralized, the electrostatic interactions between the microgel and the charged molecules are diminished, and the MB and CV are released from the microgel sequentially. (b) The release profile for a device made of APBA-MG and AAc-MG loaded with MB and CV, respectively. The arrows indicate the times that the solution pH was changed to 7 (at ∼5 min) and 3 (at ∼17 min). Two different wavelengths were monitored (590 nm and 664 nm, for CV and MB, respectively) that correspond to the absorbance of the two different model drugs. Reprinted with permission from ref. 114. Copyright 2016, The Royal Society of Chemistry. | ||
Another important stimuli-responsive polymer architecture used in controlled drug delivery systems are block copolymer self-assembled structures, such as liposomes, micelles, and vesicles. Temperature responsive polymeric micelles have attracted considerable attention due to their sharp change of properties in response to a small change in environmental temperature. When using a temperature responsive polymer as the hydrophobic core-forming segment, a core–shell micelle structure forms above its LCST due to the hydrophobic interaction among the dehydrated polymer chains.117–120 For example, Discher and Yang121 used poly(ethylene oxide)-block-poly(N-isopropylacrylamide) (PEO-b-pNIPAm) block copolymers to generate micelles capable of releasing small molecules as a function of temperature. The polymers become amphiphilic in water above body temperature (37 °C) and self-assemble into micelles that can encapsulate both hydrophilic and hydrophobic molecules. With a decrease in temperature, the micelles disassemble and release their contents.
Photochromic molecules have also been widely utilized in responsive materials to build light responsive polymer systems. The Liu group reported the fabrication of photochromic polymersomes exhibiting photoswitchable and reversible bilayer permeability from newly designed poly(ethylene oxide)-b-PSPA (PEO-b-PSPA) diblock copolymers, where SPA is a spiropyran (SP)-based monomer containing a unique carbamate linkage.122 Upon self-assembly into polymersomes, SP moieties within vesicle bilayers undergo reversible photo-triggered isomerization between the hydrophobic spiropyran (SP, λ2 > 450 nm irradiation) and zwitterionic merocyanine (MC, λ1 < 420 nm irradiation) states. The microstructures of both SP and MC polymersomes are synergistically stabilized due to cooperative noncovalent interactions from hydrophobic, hydrogen bonding, π–π stacking, and paired electrostatic (zwitterionic) interactions, with the latter two types being exclusive for MC polymersomes. Moreover, reversible phototriggered SP/MC polymersome transition is accompanied by membrane permeability switching from being impermeable to selectively permeable towards non-charged, charged, and zwitterionic small molecule species below critical molar masses, as shown in Fig. 12. They further demonstrate photoswitchable spatiotemporal release of 4′,6-diamidino-2-phenylindole (DAPI, cell nuclei staining dye) within living HeLa cells. UV-actuated MC polymersomes possess two types of release modules: (1) sustained release upon short UV irradiation duration by taking advantage of the unexpectedly slow spontaneous MC-to-SP transition kinetics (t1/2 > 20 h) in the dark; and (2) on-demand and switchable release under alternated UV-vis light irradiation. In another example, the Huang group incorporated azobenzene derivatives into supramolecular structures to create a supra-amphiphilic polypseudorotaxane, which can self-assemble to form vesicles in water.123 Due to the dual-responsivity of the molecular recognition motif, the reversible transformations between solid nanospheres based on the self-assembly of the polymer backbone and vesicles based on the self-assembly of the supra-amphiphilic polypseudorotaxane was achieved by adjusting the solution temperature or UV-visible light exposure. These dual-responsive aggregation behaviors were used to study the controlled release of water-soluble dye calcein molecules.
 | ||
| Fig. 12 Photochromic polymersomes exhibiting photoswitchable and reversible bilayer permeability. Reprinted with permission from ref. 122. Copyright 2015, American Chemical Society. | ||
Disulfide bonds are well known to exhibit a high propensity to be reduced into thiols, and have been proposed as a candidate to develop smart responsive systems for controlled drug delivery. Zhu and coworkers synthesized multiblock copolymers with multiple enes and disulfides in hydrophobic blocks and then encapsulated modified doxorubicin (Dox) via conjugation and core-crosslinking reactions.124 The obtained nano-prodrug micelles presented stable nano-scaled spherical particles under physiological conditions, while quickly dissociating in response to 10 mM D,L-dithiothreitol (DTT). The Oh group also recently reported a novel rosin-based block copolymer designed to self-assemble toward micellar nanocarriers with positioned disulfides at interfaces of hydrophobic rosin cores and hydrophilic poly(ethylene glycol) (PEG) coronas.125 This block copolymer-based self-assembled micellar nanocarrier exhibited glutathione (GSH)-responsivity that enhanced release of encapsulated drugs. The resulting micelles with PEG coronas had excellent colloidal stability in the presence of proteins, suggesting prolonged blood circulation in vivo. In response to 10 mM GSH as a biological reducing agent, the disulfides at the core/corona interfaces were effectively cleaved, causing the destabilization of micelles, thus leading to the enhanced release of encapsulated Dox. Due to the hydrophobicity of rosin species in micellar cores, the Dox release appeared to be slowed. These studies suggest that the designing of responsive block copolymers and their assembled nanostructures with reduction-responsive properties offers great versatility as intracellular drug-delivery nanocarriers for cancer therapy.
The wide range of stimuli that can be exploited to trigger drug release at the right place and time, and the diversity of responsive materials and other functional materials that can be assembled in different architectures, allow great flexibility in the design of stimuli-responsive drug delivery systems. As we have shown in this section, building up novel structures from stimuli-responsive polymers can yield materials with important applications to solve problems related to human health. The next section will describe their utility as artificial muscles and actuators.
2.3 Artificial muscles and actuators
Natural muscles are biological organs that transform chemical energy into mechanical energy. The process is complex, and involves an electrical pulse from the brain that triggers the liberation of ions inside the sarcomere, chemical reactions (ATP hydrolysis), and eventual conformational changes along the natural muscle fibers. For years, groups have been attempting to mimic this behavior, and Otero,126,127 Inganäs,128,129 and MacDiarmid130,131 have conducted numerous investigations on bilayers (the most common polymer-based actuator format) composed of a single actuating conducting polymer film deposited on an electromechanically inert layer. In a few examples, artificial muscles with tactile sensitivities were constructed from electrochemo-mechanical and macroscopic devices using films of PPy electrogenerated on double-sided tape to generate bilayers and trilayers.132–134 Qu and coworkers135 reported the spontaneous formation of a partially reduced graphene oxide and PPy (prGO-PPy) film via a self oxidation–reduction strategy as shown in Fig. 13. This reaction occurs by direct exposure of the GO film to pyrrole vapor at room temperature without any extra additives. This convenient method allows for the facile preparation of asymmetric GO/prGO-PPy hybrid films which can be applied in advanced actuation systems such as moisture or electrochemical actuators. PPy-based actuators and microactuators have many possible applications, particularly in cell biology and biomedicine because they can operate under a variety of conditions including various salt solutions, blood plasma, urine, and cell culture medium.136–138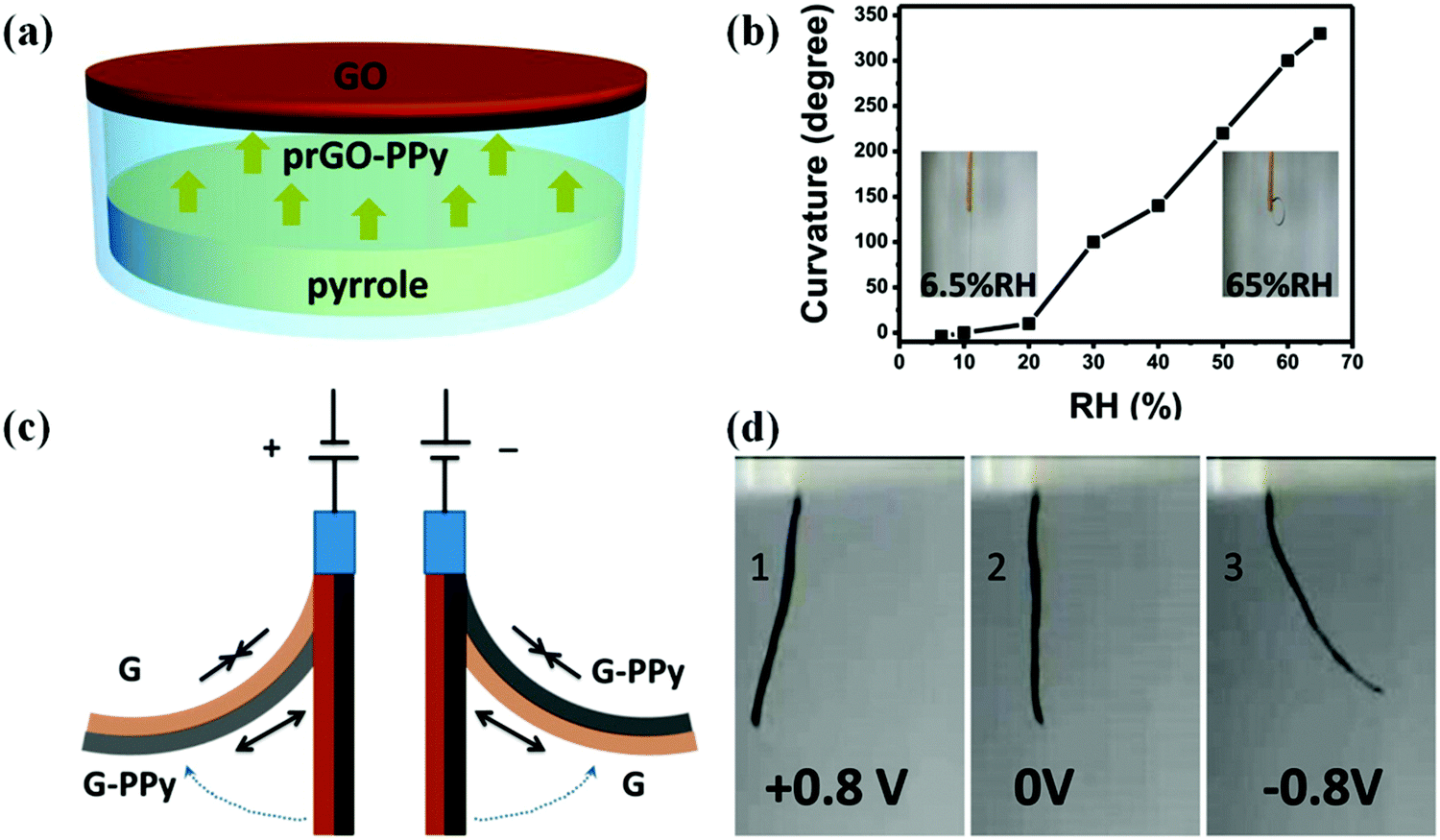 | ||
| Fig. 13 (a) Schematic illustrating the fabrication process of the humidity responsive asymmetric GO/prGO-PPy film actuator. (b) Plot of the curvature of the GO/prGO-PPy film versus relative humidity (RH). The insets are photographs of the GO/prGO-PPy film at the indicated humidities. (c) The actuation mechanism of the asymmetric G/G-PPy film. (d) Photographs of the bending for the G/G-PPy film actuator driven by the electrochemical potential within ±0.8 V. The G and G-PPy sides are placed on the left and right sides on the actuator, and the corresponding potentials of +0.8 V (1), 0 V (2), and −0.8 V (3). Reprinted with permission from ref. 135. Copyright 2016, American Chemical Society. | ||
Serpe and coworkers139,140 have demonstrated novel humidity responsive self-bending bilayer-based actuators made by depositing layers composed of poly(N-isopropylacrylamide)-based microgels and the polyelectrolyte polydiallyldimethylammonium chloride (pDADMAC) on a flexible substrate. The responsive materials bend upon drying and the degree of bending depends on the atmospheric humidity. The dried PDADMAC layer is composed of both amorphous and crystalline phases. The amorphous layer can readily absorb water, which results in actuation, while the crystalline phase templates the specific bending characteristics of the device. They worked on applying them as artificial muscles and humidity sensors based on their understanding of the bending mechanism, which is shown in Fig. 14. To investigate the humidity response, the author connected the assembly to a circuit composed of a LED and battery and collected the LED light intensity as a function of humidity; this is shown schematically in Fig. 14b, left. They also connected the assembly to a multimeter (attached onto the gold surface of the strain sensor) and placed the setup in a humidity-controlled chamber. As can be seen in Fig. 14b, right, the light intensity likewise depended on humidity. That is, as the humidity decreased the LED light intensity increased due to the decreased resistance. There was nearly no hysteresis between multiple humidity increase and decrease cycles.
 | ||
| Fig. 14 (a) A bent substrate (∼2 inches long) was hung from an arm and cycled between low and high humidity. (b, left) Schematic depiction of the experimental setup used to measure resistance and LED light intensity as a function of device bending. (b, right) Light intensity (left axis) and resistance (right axis) changes induced by the bending of the bilayers coupled to the strain sensors. Reprinted with permission from ref. 139. Copyright 2013, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim and ref. 140. Copyright 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Photoresponsive polymer-based materials can also be used to generate shape memory polymers, polymer gels, and liquid crystalline polymers (LCP).141,142 They are responsible for converting light energy into mechanical work. Generally, polymers are modified with light responsive groups, which induce photochemical reactions and visible macroscopic effects on the polymer. In a specific example, photoresponsive units were used to crosslink liquid crystalline polymers and used as actuators.143–147 Warner and coworkers introduced azobenzene moieties into a monodomain nematic LCP as crosslinkers and generated films, which were shown to exhibit significant contraction upon exposure to 365 nm radiation.148 Terentjev and coworkers also worked on incorporating various azobenzene derivatives into LCPs and examined the deformation of the system upon exposure to UV light.149 This was demonstrated in a study published by Ikeda and coworkers. They showed that light-driven bending of azobenzene LCPs could be achieved by creating an asymmetric deformation between the surface and the bulk of a film.150–152 They found that the deformation of the polymer is dependent on the manner of alignment of the LCP mesogens.141 The monodomain LCP with in-plane alignment of mesogens bent along the alignment direction towards the irradiation source. They further fabricated light driven soft actuators from azobenzene LCP/polyethylene laminated films.153 Recently, Bléger and coworkers described a liquid crystalline polymer film doped with visible light responsive fluorinated azobenzene capable of continuous “chaotic” oscillatory motion when exposed to ambient sunlight in air,154 which is shown in Fig. 15. The presence of simultaneous illumination by blue and green light is necessary for the oscillating behavior to occur, suggesting that the dynamics of continuous forward and backward switching are causing the observed effect. The motion is programmed by the molecular organization in the polymer film and is independent of the position of the light source. The unique behavior makes this material potentially useful for outdoor applications including self-cleaning coatings and surfaces. Furthermore, self-propelling/morphing soft actuators could be used to harvest and convert solar energy.
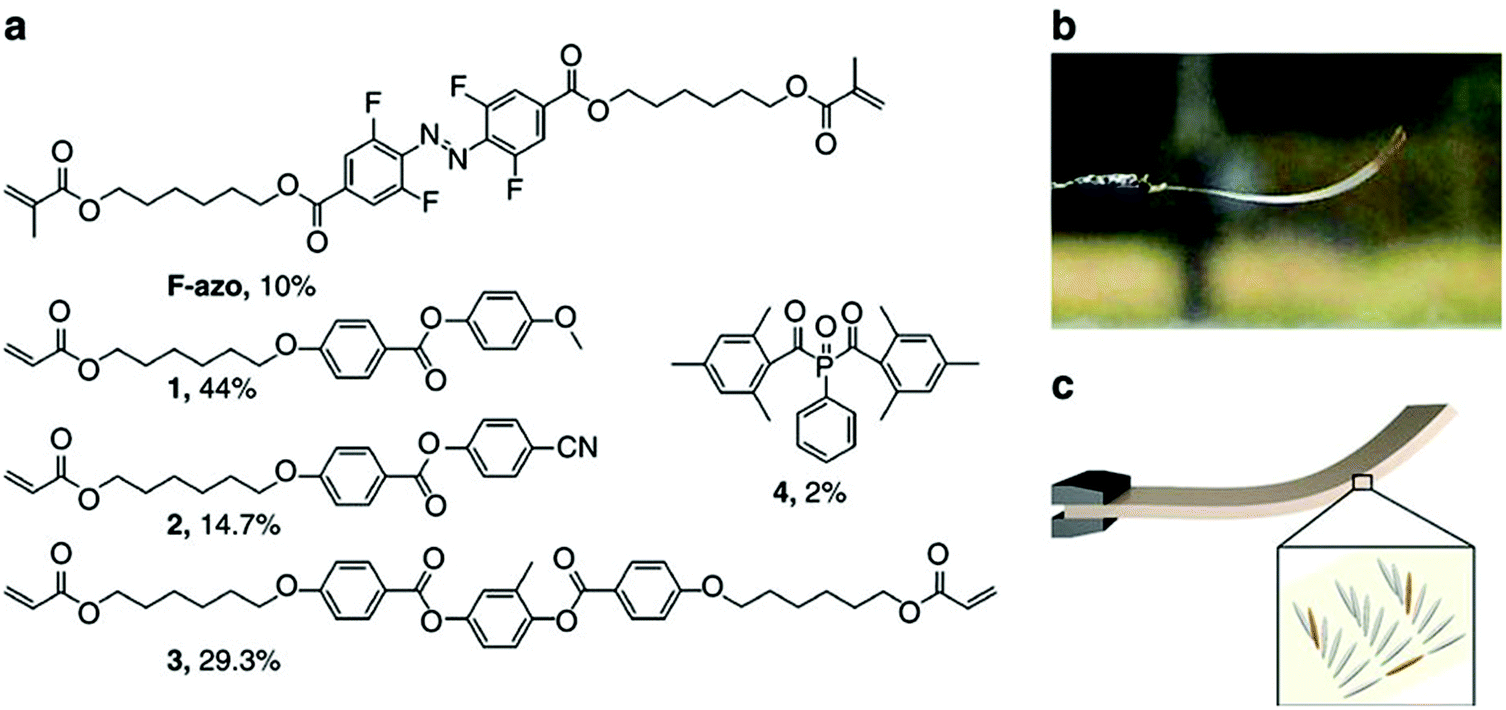 | ||
| Fig. 15 (a) Chemical structures of the components and their molar percentage used to prepare the nematic liquid crystalline network. (b) Photograph of the splay-oriented film after removal from the cell under ambient interior light (homeotropic side on top) and (c) schematic of the LC splay aligned film (grey) containing F-azo molecules (orange). Reprinted with permission from ref. 154. Copyright 2016, Nature Publication. | ||
Another very important class of actuators is composed of hydrogels that change volume in response to stimuli. Stimuli responsive hydrogels are 3D networks that can absorb water and swell and shrink in response to various external stimuli including temperature, pH, ionic strength, chemicals, electricity, and light.155,156 Hydrogel actuators produce macroscopic changes upon swelling and shrinking.157–160 In a recent example, Aida and coworkers developed a layered hydrogel consisting of cofacially oriented electrolyte nanosheets. This unusual geometry leads to significant anisotropic electrostatic repulsion in the hydrogel interior. They showed that the material could be operated by modulating its electrostatic anisotropy in response to changes of electrostatic permittivity. The electrostatic permittivity could be controlled by varying the material's solvation state, which depended on solution temperature.161 Recently, Ding and co-workers developed a computational model to demonstrate a new reversible shape-changing component design concept enabled by 3D printing of stimuli responsive polymers, shape memory polymers (SMP) and hydrogels.162 This approach uses the swelling of a hydrogel as the driving force for the shape change, and the temperature-dependent modulus of a shape memory polymer to regulate the time of such shape changes. Controlling the temperature and aqueous environment allows switching between two stable configurations. The structures are relatively stiff and can carry cargo in each state without mechanical loading for device training. By applying the design principles, they created and demonstrated the behavior of several shape-changing structures that exhibit reversible shape changes based on folding, curling, and origami concepts. Fig. 16 shows one of the examples, self-folding and unfolding flowers. The three layers were designed with different thickness ratios of layers of the elastomer, the hydrogel, and the SMP as shown in Fig. 16a. The inner layer was designed with the thinnest SMP layer of 0.3 mm, and the second layer with a 0.4 mm SMP layer, and 0.5 mm of SMP layer for the outer layer, but the total thickness of the elastomer, the hydrogel and the SMP layer were always kept the same. The corresponding curvature change profile is shown in Fig. 16b, which has the largest curvature and the fastest response speed with the thinner SMP layer. After putting this structure into low temperature water for 12 h, the inner layer bends a little bit as shown in Fig. 16d, and then on immersion into hot water, all layers bend and form the flower-like structure as shown in Fig. 16e. On taking the structure out of the hot water and letting it dry, as shown in Fig. 16g, we can see that it maintains the flower shape structure and can carry a load of 25 g. When the structure was put into hot water it became flat again as shown in Fig. 16f. The whole process can be repeated many times.
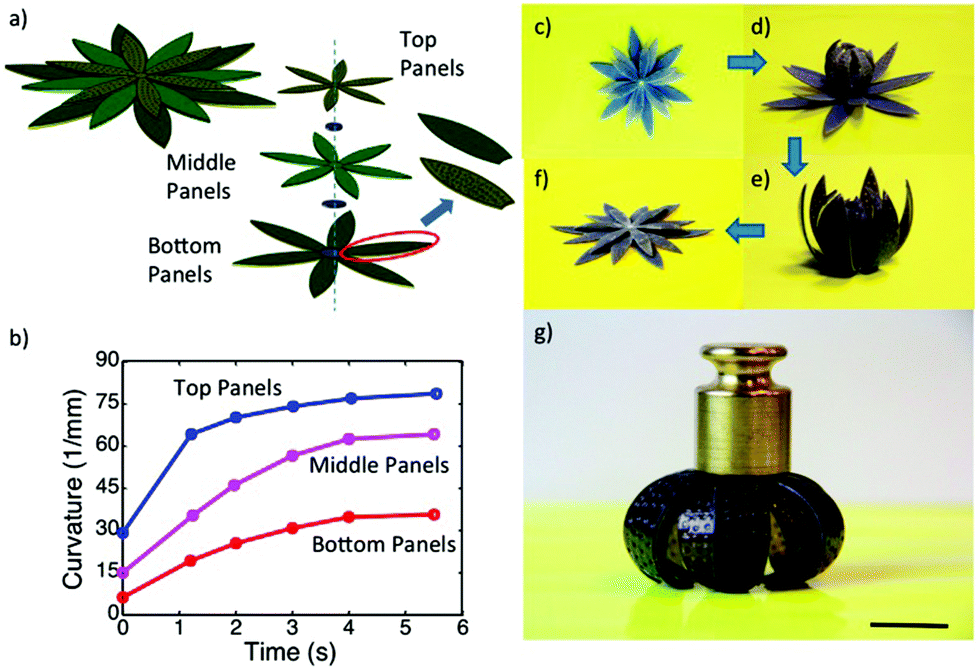 | ||
| Fig. 16 A self-folding–unfolding flower. (a) Schematic of two activated shape memory petal-like structure. (b) The curvature change under thermal activation as a function of time. (c–f) The sequence of reversible actuation. (g) The dried configuration is stiff and can carry a load of 25 g. The scale bar in (g) is 12.5 mm. Reprinted with permission from ref. 162. Copyright 2016, Nature Publication. | ||
In summary, polymer-based actuators are materials capable of converting energy from external stimuli (e.g. heat, light, and electricity) to mechanical forces, thus exhibiting shape changes. Polymer-based actuators can be generated from a variety of materials (hydrogels, liquid crystal polymers, and shape memory polymers). The material that is used depends on the intended application, e.g., soft hydrogels could be used for delicate biological applications, while hard shape memory polymers could be used for lifting/moving heavy masses. It is this versatility of materials properties, combined with the diversity of material responsivities, that makes this research area vibrant for exploration and research.
3. Conclusions and future perspective
Although stimuli-responsive polymer-based systems have been known for decades, it wasn't until relatively recently that their behavior could be understood at a level that allowed their full exploitation. Stimuli-responsive polymers have been synthesized using a variety of techniques, and employed for myriad applications; we describe only some of their applications here. In the future, combining stimuli responsive polymers with biological systems, and nanoscale materials, a variety of new functions (and properties) will be accessible. This development needs to be supported by new theories that can describe the newly found behavior such that the development of new materials can be done in a smart fashion to meet the needs of a specific application. Another challenge is to develop systems that respond to multiple external stimuli in an “intelligent” and predictable manner. These materials are required to support the development of biomimetic systems with long-term stability and durability. The concepts presented in this review encompass both the polymer history introduction, and the applications of stimuli responsive materials. A more complete picture of this broad and complex topic can be obtained from the referenced articles.Acknowledgements
MJS acknowledges funding from the University of Alberta (the Department of Chemistry and the Faculty of Science), the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Foundation for Innovation (CFI), the Alberta Advanced Education & Technology Small Equipment Grants Program (AET/SEGP), Grand Challenges Canada and IC-IMPACTS. YG and XL acknowledge Alberta Innovates Technology Futures for Graduate Student Scholarships.References
- P. J. Morris, Polymer pioneers: A popular history of the science and technology of large molecules, Chemical Heritage Foundation, Philadelphia, 2005 Search PubMed.
- M. R. Dove, Environ. Conserv., 1993, 20, 17–24 CrossRef.
- H. Staudinger, From organic chemistry to macromolecules, Wiley, New York, 1970 Search PubMed.
- R. Mülhaupt, Angew. Chem., Int. Ed., 2004, 43, 1054–1063 CrossRef PubMed.
- W. H. Carothers, US Pat, 2071251A, 1937 Search PubMed.
- W. H. Carothers, US Pat, 2130523A, 1938 Search PubMed.
- P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6677–6687 CrossRef CAS.
- A. S. Hoffman, Macromol. Symp., 1995, 98, 645–664 CrossRef CAS.
- I. Galaev and B. Mattiasson, Smart polymers: applications in biotechnology and biomedicine, CRC Press, Taylor & Francis Group, Boca Raton, USA, 2007 Search PubMed.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Chem., 1968, 2, 1441–1455 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez and S. R. White, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- Y. L. Colson and M. W. Grinstaff, Adv. Mater., 2012, 24, 3878–3886 CrossRef CAS PubMed.
- T. Tanaka, I. Nishio, S.-T. Sun and S. Ueno-Nishio, Science, 1982, 218, 467–469 CAS.
- J. Thévenot, H. Oliveira, O. Sandre and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 7099–7116 RSC.
- M. Irie, Pure Appl. Chem., 1990, 62, 1495–1502 CrossRef CAS.
- L. Zhai, Chem. Soc. Rev., 2013, 42, 7148–7160 RSC.
- A. Khan and J. Pessin, Diabetologia, 2002, 45, 1475–1483 CrossRef CAS PubMed.
- Q. M. Zhang and M. J. Serpe, in Polymer Mechanochemistry, Springer, 2015, pp. 377–424 Search PubMed.
- J. Hu and S. Liu, Macromolecules, 2010, 43, 8315–8330 CrossRef CAS.
- A. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088–1118 CrossRef CAS.
- D. Parasuraman and M. J. Serpe, ACS Appl. Mater. Interfaces, 2011, 3, 2732–2737 CAS.
- M. Ma, L. Guo, D. G. Anderson and R. Langer, Science, 2013, 339, 186–189 CrossRef CAS PubMed.
- Q. Zhao, J. W. Dunlop, X. Qiu, F. Huang, Z. Zhang, J. Heyda, J. Dzubiella, M. Antonietti and J. Yuan, Nat. Commun., 2014, 5, 4293 Search PubMed.
- Z. Hu, X. Zhang and Y. Li, Science, 1995, 269, 525 CAS.
- H. Koerner, G. Price, N. A. Pearce, M. Alexander and R. A. Vaia, Nat. Mater., 2004, 3, 115–120 CrossRef CAS PubMed.
- F. Wang, Y.-H. Lai and M.-Y. Han, Macromolecules, 2004, 37, 3222–3230 CrossRef CAS.
- F. Seker, P. R. Malenfant, M. Larsen, A. Alizadeh, K. Conway, A. M. Kulkarni, G. Goddard and R. Garaas, Adv. Mater., 2005, 17, 1941–1945 CrossRef CAS.
- G. Charlet and G. Delmas, Polymer, 1981, 22, 1181–1189 CrossRef CAS.
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS.
- Y. Okada and F. Tanaka, Macromolecules, 2005, 38, 4465–4471 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- P. Kujawa and F. M. Winnik, Macromolecules, 2001, 34, 4130–4135 CrossRef CAS.
- D. Crespy and R. M. Rossi, Polym. Int., 2007, 56, 1461–1468 CrossRef CAS.
- R. Liu, M. Fraylich and B. R. Saunders, Colloid Polym. Sci., 2009, 287, 627–643 CAS.
- Z. Song, K. Wang, C. Gao, S. Wang and W. Zhang, Macromolecules, 2016, 49, 162–171 CrossRef CAS.
- E. Karjalainen, V. Aseyev and H. Tenhu, Macromolecules, 2014, 47, 2103–2111 CrossRef CAS.
- F. A. Plamper, A. Schmalz, M. Ballauff and A. H. Müller, J. Am. Chem. Soc., 2007, 129, 14538–14539 CrossRef CAS PubMed.
- T. Thavanesan, C. Herbert and F. A. Plamper, Langmuir, 2014, 30, 5609–5619 CrossRef CAS PubMed.
- V. Bütün, S. Armes and N. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef.
- P. J. Roth, F. D. Jochum, F. R. Forst, R. Zentel and P. Theato, Macromolecules, 2010, 43, 4638–4645 CrossRef CAS.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- Y. Cao, X. Zhu, J. Luo and H. Liu, Macromolecules, 2007, 40, 6481–6488 CrossRef CAS.
- I. Idziak, D. Avoce, D. Lessard, D. Gravel and X. Zhu, Macromolecules, 1999, 32, 1260–1263 CrossRef CAS.
- L. A. Connal, Q. Li, J. F. Quinn, E. Tjipto, F. Caruso and G. G. Qiao, Macromolecules, 2008, 41, 2620–2626 CrossRef CAS.
- C. D. Sorrell, M. C. Carter and M. J. Serpe, Adv. Funct. Mater., 2011, 21, 425–433 CrossRef CAS.
- M. J. Serpe, J. Kim and L. A. Lyon, Adv. Mater., 2004, 16, 184–187 CrossRef CAS.
- F. Liu and M. W. Urban, Macromolecules, 2008, 41, 6531–6539 CrossRef CAS.
- F.-J. Xu, E.-T. Kang and K.-G. Neoh, Biomaterials, 2006, 27, 2787–2797 CrossRef CAS PubMed.
- B.-W. Liu, H. Zhou, S.-T. Zhou, H.-J. Zhang, A.-C. Feng, C.-M. Jian, J. Hu, W.-P. Gao and J.-Y. Yuan, Macromolecules, 2014, 47, 2938–2946 CrossRef CAS.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- Y.-L. Zhao and J. F. Stoddart, Langmuir, 2009, 25, 8442–8446 CrossRef CAS PubMed.
- J.-F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117–7129 RSC.
- P. D. Thornton, R. J. Mart and R. V. Ulijn, Adv. Mater., 2007, 19, 1252–1256 CrossRef.
- J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933–5949 RSC.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217–2225 RSC.
- K. Kataoka, H. Miyazaki, M. Bunya, T. Okano and Y. Sakurai, J. Am. Chem. Soc., 1998, 120, 12694–12695 CrossRef CAS.
- Z. Gu, T. T. Dang, M. Ma, B. C. Tang, H. Cheng, S. Jiang, Y. Dong, Y. Zhang and D. G. Anderson, ACS Nano, 2013, 7, 6758–6766 CrossRef CAS PubMed.
- T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766–769 CrossRef CAS PubMed.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143–159 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- M. R. Islam, Y. Gao, X. Li, Q. M. Zhang, M. Wei and M. J. Serpe, Chin. Sci. Bull., 2014, 59, 4237–4255 CrossRef CAS.
- M. A. C. Stuart, W. T. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk and M. Urban, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- M. Toma, U. Jonas, A. Mateescu, W. Knoll and J. Dostalek, J. Phys. Chem. C, 2013, 117, 11705–11712 CAS.
- S. Maji, B. Cesur, Z. Zhang, B. G. De Geest and R. Hoogenboom, Polym. Chem., 2016, 7, 1705–1710 RSC.
- Y. Ma, K. Promthaveepong and N. Li, Anal. Chem., 2016, 88, 8289–8293 CrossRef CAS PubMed.
- K. Paek, H. Yang, J. Lee, J. Park and B. J. Kim, ACS Nano, 2014, 8, 2848–2856 CrossRef CAS PubMed.
- A. Seeboth, D. Lötzsch, R. Ruhmann and O. Muehling, Chem. Rev., 2014, 114, 3037–3068 CrossRef CAS PubMed.
- J. H. Holtz and S. A. Asher, Nature, 1997, 389, 829–832 CrossRef CAS PubMed.
- S. A. Asher, K. W. Kimble and J. P. Walker, Chem. Mater., 2008, 20, 7501–7509 CrossRef CAS PubMed.
- J. H. Holtz, J. S. Holtz, C. H. Munro and S. A. Asher, Anal. Chem., 1998, 70, 780–791 CrossRef CAS.
- K. Lee and S. A. Asher, J. Am. Chem. Soc., 2000, 122, 9534–9537 CrossRef CAS.
- C. E. Reese, M. E. Baltusavich, J. P. Keim and S. A. Asher, Anal. Chem., 2001, 73, 5038–5042 CrossRef CAS PubMed.
- J. P. Walker and S. A. Asher, Anal. Chem., 2005, 77, 1596–1600 CrossRef CAS PubMed.
- J. M. Weissman, H. B. Sunkara, A. S. Tse and S. A. Asher, Thermally Switchable Periodicities and Diffraction from Novel Mesoscopically Ordered Materials, DTIC Document, 1996 Search PubMed.
- M. C. Chiappelli and R. C. Hayward, Adv. Mater., 2012, 24, 6100–6104 CrossRef CAS PubMed.
- K. C. Johnson, F. Mendez and M. J. Serpe, Anal. Chim. Acta, 2012, 739, 83–88 CrossRef CAS PubMed.
- Q. M. Zhang, X. Li, M. R. Islam, M. Wei and M. J. Serpe, J. Mater. Chem. C, 2014, 2, 6961–6965 RSC.
- W. Xu, Y. Gao and M. J. Serpe, J. Mater. Chem. C, 2014, 2, 3873–3878 RSC.
- Q. M. Zhang, W. Xu and M. J. Serpe, Angew. Chem., Int. Ed., 2014, 53, 4827–4831 CrossRef CAS PubMed.
- M. R. Islam and M. J. Serpe, Biosens. Bioelectron., 2013, 49, 133–138 CrossRef CAS PubMed.
- C. D. Sorrell and M. J. Serpe, Anal. Bioanal. Chem., 2012, 402, 2385–2393 CrossRef CAS PubMed.
- Q. M. Zhang, A. Ahiabu, Y. Gao and M. J. Serpe, J. Mater. Chem. C, 2015, 3, 495–498 RSC.
- Q. M. Zhang, D. Berg, S. M. Mugo and M. J. Serpe, Chem. Commun., 2015, 51, 9726–9728 RSC.
- M. R. Islam and M. J. Serpe, Chem. Commun., 2013, 49, 2646–2648 RSC.
- M. R. Islam and M. J. Serpe, Anal. Bioanal. Chem., 2014, 406, 4777–4783 CrossRef CAS PubMed.
- Q. M. Zhang, W. Wang, Y.-Q. Su, E. J. Hensen and M. J. Serpe, Chem. Mater., 2016, 28, 259–265 CrossRef CAS.
- M. B. Yatvin, J. N. Weinstein, W. H. Dennis and R. Blumenthal, Science, 1978, 202, 1290–1293 CAS.
- A. P. Blum, J. K. Kammeyer, A. M. Rush, C. E. Callmann, M. E. Hahn and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137, 2140–2154 CrossRef CAS PubMed.
- J. K. Oh, D. I. Lee and J. M. Park, Prog. Polym. Sci., 2009, 34, 1261–1282 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- M. Bernadete Riemma Pierre and F. Cristina Rossetti, Curr. Drug Targets, 2014, 15, 281–291 CrossRef CAS.
- X. Hong, Z. Wu, L. Chen, F. Wu, L. Wei and W. Yuan, Nano-Micro Lett., 2014, 6, 191–199 CrossRef CAS.
- R. F. Donnelly, T. R. R. Singh, M. J. Garland, K. Migalska, R. Majithiya, C. M. McCrudden, P. L. Kole, T. M. T. Mahmood, H. O. McCarthy and A. D. Woolfson, Adv. Funct. Mater., 2012, 22, 4879–4890 CrossRef CAS PubMed.
- Y. Deng, J. Chen, Y. Zhao, X. Yan, L. Zhang, K. Choy, J. Hu, H. J. Sant, B. K. Gale and T. Tang, Sci. Rep., 2016, 6, 30430 CrossRef PubMed.
- R. F. Donnelly, M. T. McCrudden, A. Z. Alkilani, E. Larrañeta, E. McAlister, A. J. Courtenay, M.-C. Kearney, T. R. R. Singh, H. O. McCarthy and V. L. Kett, PLoS One, 2014, 9, e111547 Search PubMed.
- M. J. Garland, E. Caffarel–Salvador, K. Migalska, A. D. Woolfson and R. F. Donnelly, J. Controlled Release, 2012, 159, 52–59 CrossRef CAS PubMed.
- E. M. Cahill and E. D. O'Cearbhaill, Bioconjugate Chem., 2015, 26, 1289–1296 CrossRef CAS PubMed.
- J. G. Hardy, E. Larraneta, R. F. Donnelly, N. McGoldrick, K. Migalska, M. T. McCrudden, N. J. Irwin, L. Donnelly and C. P. McCoy, Mol. Pharm., 2016, 13, 907–914 CrossRef CAS PubMed.
- S. Y. Yang, E. D. O'Cearbhaill, G. C. Sisk, K. M. Park, W. K. Cho, M. Villiger, B. E. Bouma, B. Pomahac and J. M. Karp, Nat. Commun., 2013, 4, 1702 CrossRef PubMed.
- M.-C. Chen, Z.-W. Lin and M.-H. Ling, ACS Nano, 2015, 10, 93–101 CrossRef PubMed.
- M.-C. Chen, M.-H. Ling, K.-W. Wang, Z.-W. Lin, B.-H. Lai and D.-H. Chen, Biomacromolecules, 2015, 16, 1598–1607 CrossRef CAS PubMed.
- Y. Ye, J. Yu, C. Wang, N. Y. Nguyen, G. M. Walker, J. B. Buse and Z. Gu, Adv. Mater., 2016, 28, 3115–3121 CrossRef CAS PubMed.
- J. Yu, Y. Zhang, Y. Ye, R. DiSanto, W. Sun, D. Ranson, F. S. Ligler, J. B. Buse and Z. Gu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8260–8265 CrossRef CAS PubMed.
- C. Wang, Y. Ye, G. M. Hochu, H. Sadeghifar and Z. Gu, Nano Lett., 2016, 16, 2334–2340 CrossRef CAS PubMed.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- H. Bysell, R. Månsson, P. Hansson and M. Malmsten, Adv. Drug Delivery Rev., 2011, 63, 1172–1185 CrossRef CAS PubMed.
- N. Smeets and T. Hoare, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3027–3043 CrossRef CAS.
- Y. Gao, G. P. Zago, Z. Jia and M. J. Serpe, ACS Appl. Mater. Interfaces, 2013, 5, 9803–9808 CAS.
- S. Guo, Y. Gao, M. Wei, Q. M. Zhang and M. J. Serpe, J. Mater. Chem. B, 2015, 3, 2516–2521 RSC.
- Y. Gao, K. Y. Wong, A. Ahiabu and M. J. Serpe, J. Mater. Chem. B, 2016, 4, 5144–5150 RSC.
- Y. Gao, A. Ahiabu and M. J. Serpe, ACS Appl. Mater. Interfaces, 2014, 6, 13749–13756 CAS.
- Y. Gao, W. Xu and M. J. Serpe, J. Mater. Chem. C, 2014, 2, 5878–5884 RSC.
- M. Topp, P. Dijkstra, H. Talsma and J. Feijen, Macromolecules, 1997, 30, 8518–8520 CrossRef CAS.
- J. Virtanen, S. Holappa, H. Lemmetyinen and H. Tenhu, Macromolecules, 2002, 35, 4763–4769 CrossRef CAS.
- R. Motokawa, K. Morishita, S. Koizumi, T. Nakahira and M. Annaka, Macromolecules, 2005, 38, 5748–5760 CrossRef CAS.
- W. Zhang, L. Shi, K. Wu and Y. An, Macromolecules, 2005, 38, 5743–5747 CrossRef CAS.
- S. Qin, Y. Geng, D. E. Discher and S. Yang, Adv. Mater., 2006, 18, 2905–2909 CrossRef CAS.
- X. Wang, J. Hu, G. Liu, J. Tian, H. Wang, M. Gong and S. Liu, J. Am. Chem. Soc., 2015, 137, 15262–15275 CrossRef CAS PubMed.
- X. Chi, X. Ji, D. Xia and F. Huang, J. Am. Chem. Soc., 2015, 137, 1440–1443 CrossRef CAS PubMed.
- Y. Wang, Q. Luo, W. Zhu, X. Li and Z. Shen, Polym. Chem., 2016, 7, 2665–2673 RSC.
- S. Y. An, S. H. Hong, C. Tang and J. K. Oh, Polym. Chem., 2016, 7, 4751–4760 RSC.
- T. Otero and J. Sansinena, Bioelectrochem. Bioenerg., 1997, 42, 117–122 CrossRef CAS.
- W. Takashima, M. Kaneko, K. Kaneto and A. G. MacDiarmid, Synth. Met., 1995, 71, 2265–2266 CrossRef CAS.
- Q. Pei and O. Inganläs, Adv. Mater., 1992, 4, 277–278 CrossRef CAS.
- Q. Pei and O. Inganäs, Synth. Met., 1993, 57, 3718–3723 CrossRef CAS.
- K. Kaneto, M. Kaneko, Y. Min and A. G. MacDiarmid, Synth. Met., 1995, 71, 2211–2212 CrossRef CAS.
- A. MacDiarmid, K. Kaneto, H. Saito and Y. Min, Polym. Mater. Sci. Eng., 1994, 71, 713–714 CAS.
- T. F. N. Otero and M. T. Cortes, Adv. Mater., 2003, 15, 279–282 CrossRef CAS.
- F. García-Córdova, L. Valero, Y. A. Ismail and T. F. Otero, J. Mater. Chem., 2011, 21, 17265–17272 RSC.
- T. F. Otero, J. J. Sanchez and J. G. Martinez, J. Phys. Chem. B, 2012, 116, 5279–5290 CrossRef CAS PubMed.
- Y. Jiang, C. Hu, H. Cheng, C. Li, T. Xu, Y. Zhao, H. Shao and L. Qu, ACS Nano, 2016, 10, 4735–4741 CrossRef CAS PubMed.
- K. Svennersten, M. Berggren, A. Richter-Dahlfors and E. W. Jager, Lab Chip, 2011, 11, 3287–3293 RSC.
- E. W. Jager, C. Immerstrand, K.-E. Magnusson, O. Inganas and I. Lundstrom, Biomedical applications of polypyrrole microactuators: from single-cell clinic to microrobots, in 1st Annual International Conference on Microtechnologies in Medicine and Biology, 2000 Search PubMed.
- E. W. Jager, O. Inganäs and I. Lundström, Science, 2000, 288, 2335–2338 CrossRef CAS PubMed.
- M. R. Islam, X. Li, K. Smyth and M. J. Serpe, Angew. Chem., Int. Ed., 2013, 52, 10330–10333 CrossRef CAS PubMed.
- X. Li and M. J. Serpe, Adv. Funct. Mater., 2016, 26, 3282–3290 CrossRef CAS.
- J. Wei and Y. Yu, Soft Matter, 2012, 8, 8050–8059 RSC.
- H. Jiang, S. Kelch and A. Lendlein, Adv. Mater., 2006, 18, 1471–1475 CrossRef CAS.
- J. Garcia-Amorós, A. Piñol, H. Finkelmann and D. Velasco, Org. Lett., 2011, 13, 2282–2285 CrossRef PubMed.
- T. J. White, N. V. Tabiryan, S. V. Serak, U. A. Hrozhyk, V. P. Tondiglia, H. Koerner, R. A. Vaia and T. J. Bunning, Soft Matter, 2008, 4, 1796–1798 RSC.
- T. J. White, S. V. Serak, N. V. Tabiryan, R. A. Vaia and T. J. Bunning, J. Mater. Chem., 2009, 19, 1080–1085 RSC.
- K. M. Lee, M. L. Smith, H. Koerner, N. Tabiryan, R. A. Vaia, T. J. Bunning and T. J. White, Adv. Funct. Mater., 2011, 21, 2913–2918 CrossRef CAS.
- M. Camacho-Lopez, H. Finkelmann, P. Palffy-Muhoray and M. Shelley, Nat. Mater., 2004, 3, 307–310 CrossRef CAS PubMed.
- H. Finkelmann, E. Nishikawa, G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 87, 015501 CrossRef CAS PubMed.
- P. Hogan, A. Tajbakhsh and E. Terentjev, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2002, 65, 041720 CrossRef CAS PubMed.
- T. Ikeda, M. Nakano, Y. Yu, O. Tsutsumi and A. Kanazawa, Adv. Mater., 2003, 15, 201–205 CrossRef CAS.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145–145 CrossRef CAS PubMed.
- M. Kondo, Y. Yu and T. Ikeda, Angew. Chem., Int. Ed., 2006, 118, 1406–1410 CrossRef.
- M. Yamada, M. Kondo, J. I. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS PubMed.
- K. Kumar, C. Knie, D. Bléger, M. A. Peletier, H. Friedrich, S. Hecht, D. J. Broer, M. G. Debije and A. P. Schenning, Nat. Commun., 2016, 7, 11975 CrossRef CAS PubMed.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef PubMed.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2012, 64, 49–60 CrossRef.
- N. Bassik, B. T. Abebe, K. E. Laflin and D. H. Gracias, Polymer, 2010, 51, 6093–6098 CrossRef CAS.
- R. Luo, J. Wu, N. D. Dinh and C. H. Chen, Adv. Funct. Mater., 2015, 25, 7272–7279 CrossRef CAS.
- G. H. Kwon, Y. Y. Choi, J. Y. Park, D. H. Woo, K. B. Lee, J. H. Kim and S.-H. Lee, Lab Chip, 2010, 10, 1604–1610 RSC.
- I. Tokarev and S. Minko, Soft Matter, 2009, 5, 511–524 RSC.
- Y. S. Kim, M. Liu, Y. Ishida, Y. Ebina, M. Osada, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nat. Mater., 2015, 14, 1002–1007 CrossRef CAS PubMed.
- Y. Mao, Z. Ding, C. Yuan, S. Ai, M. Isakov, J. Wu, T. Wang, M. L. Dunn and H. J. Qi, Sci. Rep., 2016, 6, 24761 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |