
Ru/ceria-catalyzed direct formylation of amines and CO to produce formamides†
Yehong
Wang
a,
Jian
Zhang
a,
Haijun
Chen
ab,
Zhixin
Zhang
a,
Chaofeng
Zhang
ab,
Mingrun
Li
a and
Feng
Wang
 *a
*a
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, 116023 Dalian, China. E-mail: wangfeng@dicp.ac.cn
bUniversity of the Chinese Academy of Sciences, 100049 Beijing, China
First published on 14th October 2016
Abstract
We herein report a new strategy of directly converting amines and CO to formamides with 100% atom utilization efficiency. It is suitable for up to 25 amine substrates with no additives. Ru/ceria is found to be an excellent catalyst for this reaction due the efficient co-activation of CO and amine on Ru species.
Formamide is a key synthetic intermediate and solvent. For decades, a corrosive and dangerous NaOCH3 catalyst has been used for industrial production of dimethylformamide (DMF),1 which is a widely-used solvent and a multipurpose building block for chemical synthesis.2 The formamide synthesis is a fundamental chemical process.3 Previous studies have been focused on testing transformylating reagents,4 such as DMF, HCOOH or in situ-generated HCOOH and derivatives. The principle of the transformylation reaction lies in the C–X bond exchange reaction between the pre-existing formyl group (R3–CHO) and the amine N–H bond (Scheme 1, Route B). This unavoidably generates byproduct R3–H, lowering atom utilization efficiency and leads to cumbersome product separation and waste disposal. Some strategies have employed oxidative formylation reactions using CO and a stoichiometric amount of oxidants,5 such as O2 and NaIO4, with alkali, or using ionic liquids combined with bases.6 Although these reactions generated a new C–N bond, they majorly produced urea instead of formamides.7 Therefore, an ideal way is to develop a heterogeneous catalyst that is capable of directly converting amines and CO to formamides with 100% atom utilization efficiency, suppressing its further conversion to urea, as well as the parallel reaction of amines to imines.
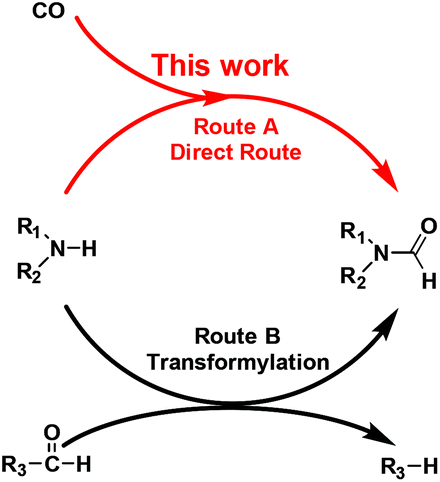 | ||
| Scheme 1 Formamides’ synthesis by the direct route (Route A, the present study) and by the transformylation route (Route B). One or two of R1 and R2 represent CmHn– groups. R3 represents a CmHn–, RO–, HO–, or RN– group. | ||
We herein report a new and direct route of converting amines and CO to formamides without using additives, hydrogen or oxygen (Scheme 1, Route A). This method is suitable for a wide range of substrates including primary and secondary amines. The Ru/ceria catalyst is found to be active for this direct formylation reaction. The excellent catalytic performance of Ru/ceria is due to the co-activation of CO and amine molecules on different Ru sites.
Due to its high oxygen storage and release capacity, and facile oxygen vacancy formation, ceria has attracted special interest as a catalyst or a support.8 Our initial test with CO and benzylamine at 180 °C and under 0.5 MPa CO over pristine ceria gave, after 4 h, merely 6% benzylamine conversion and 17% selectivity for N-benzylformamide (Fig. 1). The major product was imine. Further screening among other oxide and zeolite catalysts still failed to give satisfactory results, which made us believe that the reaction needed a metal center to activate CO.7 We then prepared several ceria-supported Ru, Rh, Pd, Pt and Au catalysts (Me/ceria) by an incipient wetness impregnation method. Catalytic results showed that the Me/ceria catalysts gave no N-benzylformamide (Fig. 1) except Ru/ceria, which afforded 56% benzylamine conversion and 93% selectivity for N-benzylformamide. On calcining Ru/ceria at 500 °C, the resulting RuO2/ceria catalyst was unreactive for the reaction. We also prepared several model nanocrystalline ceria (rod, cube and octahedron) and the corresponding Ru/ceria catalysts (2 wt% loading). As a result, Ru/ceria (rod) gave much better results (yield 46%) than that of Ru/ceria (cube) (yield 21%) and Ru/ceria (octahedron) (yield 20%). This is consistent with our previous discovery on a ceria-supported nanogold catalyst and is also found in other ceria-supported metal catalysts.9
 | ||
| Fig. 1 Catalysts’ screening using the formylation of benzylamine with CO as the model reaction. Reaction conditions: 1.5 mmol amine in 2 mL methanol, 50 mg catalyst, 0.5 MPa CO, 180 °C, 4 h. | ||
In order to test whether it was a direct formylation reaction, we examined the C source of formamide (RN–CHO) (Fig. 2). The reaction at 150 °C for 6 h over 2 wt% Ru/ceria [abbr. Ru/ceria hereinafter] afforded 61% conversion and 99% selectivity. No reaction occurred in 0.9 MPa N2 and without CO gas. We hypothesized that the methanol solvent might take part in the reaction, which was reported in the literature.10 The possibility was then excluded by using tetrahydrofuran (THF) as a solvent (conv. 47%, select. 94%). A reaction in a mixed solvent of THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13CH3OH (5:1 v/v) gave no 13C-labeled product, but a reaction with 13CO gas produced 13C-labeled N-benzylformamide (Fig. S1†), which indisputably confirmed C in RN–CHO is from CO. More interestingly, 13CO or C18O is readily accessed, as is the CO isotopologue, offering a potentially economic route to the isotopically labeled formamides. The preceding transformylation methods converted the majority of the labeled elements into byproducts.
:13CH3OH (5:1 v/v) gave no 13C-labeled product, but a reaction with 13CO gas produced 13C-labeled N-benzylformamide (Fig. S1†), which indisputably confirmed C in RN–CHO is from CO. More interestingly, 13CO or C18O is readily accessed, as is the CO isotopologue, offering a potentially economic route to the isotopically labeled formamides. The preceding transformylation methods converted the majority of the labeled elements into byproducts.
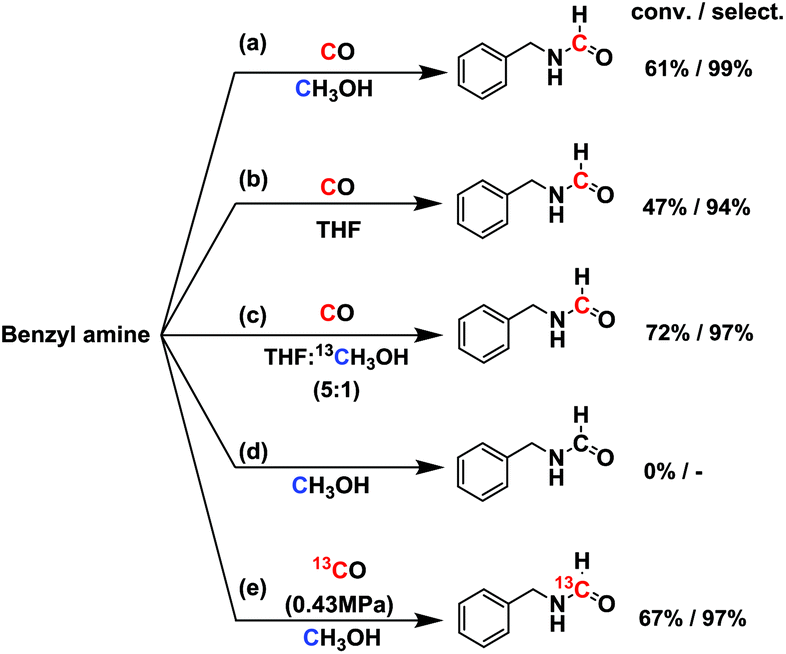 | ||
| Fig. 2 Isotopically labeling experiments. Reaction conditions: 1.5 mmol amine in 2 mL solvent, 50 mg Ru/ceria catalyst, 0.9 MPa CO, 150 °C, 6 h. | ||
We then extended this method to other amine substrates (Fig. 3). The reactions on primary aliphatic mono-amines and di-amines proceeded remarkably well (entries 1–5). In the literature, the formylation of a di-amine by CO and O2 underwent a cyclization reaction and majorly produced urea.5b In this study, N,N′-(hexane-1,6-diyl) di-formamide was obtained with >99% selectivity (entry 5). Cyclic aliphatic amines were obtained in high selectivities (entries 6 and 7). N-Benzylformamide and its derivatives were obtained with >95% selectivity (entries 8–11). 1-Phenylethanamine was also converted to the corresponding formamide with 98% selectivity (entry 12). Bulky substituents were unfavorable, and thus a long reaction time was required for β-phenyl ethylformamide (36 h) (entry 13). Amines with tetrahydrofuran, furan and thiophene were converted with 25–91% conversions and >92% selectivities (entries 14–16). The decrease in activity was possibly caused by restricting amine adsorption by an O or S atom opponent on the active sites. Secondary aliphatic amines were converted with 85–99% formamide selectivities (entries 17–21). Steric hindrance could be overcome by increasing reaction time and temperature. The presence of hydroxyl groups facilitated the reaction (entry 20). Formamides were obtained with >99% selectivities from cyclic secondary amines such as piperidine and morpholine (entries 22 and 23). The rigidity of piperazine and 1,4-diazepane majorly afforded the single-substituted formamide (entries 24 and 25). As shown above, this strategy using a heterogeneous catalyst is less efficient in synthesizing bulky formamides.
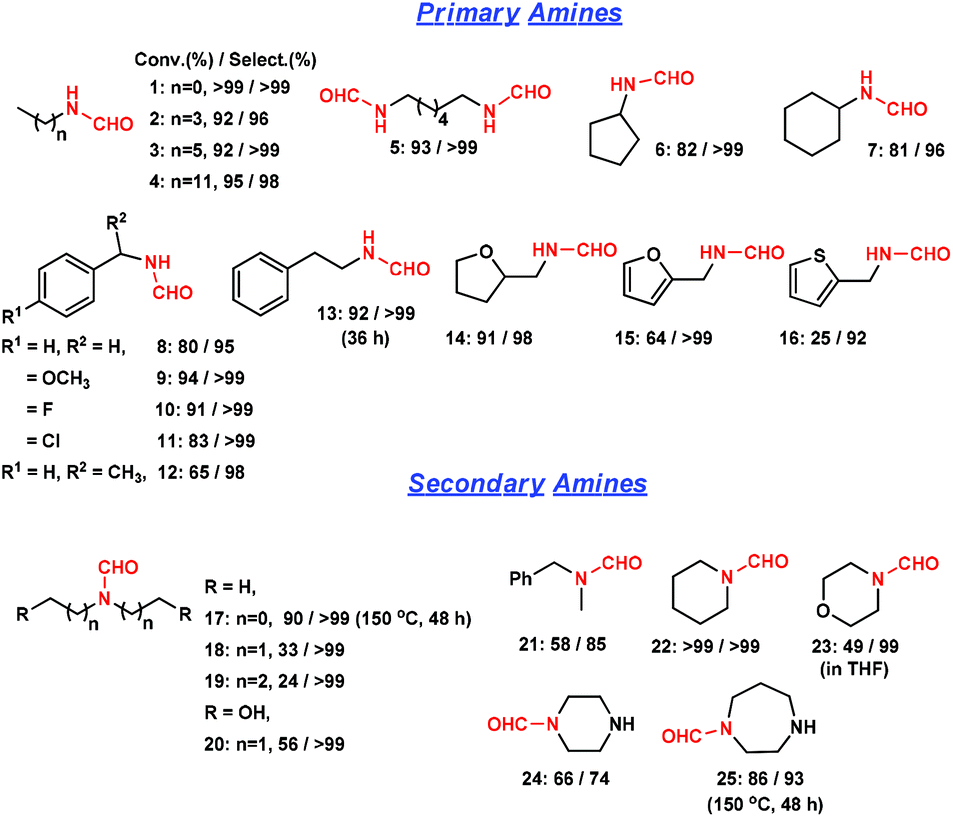 | ||
| Fig. 3 Direct formylation reaction of various amines to formamides catalyzed by Ru/ceria. Reaction conditions: 1.5 mmol amine in 2 mL methanol, 0.05 g Ru/ceria catalyst, 0.9 MPa CO, 150 °C, 24 h. | ||
Current plants use a NaOCH3 catalyst under oxidizing conditions to produce DMF, which is corrosive, potentially explosive with high-energy consumption. In comparison, using gaseous DMA and CO to produce liquid DMF is easily separated and recycled. In this study, we employed an industrially widely-used fixed-bed reactor to realize a continuous DMF synthesis (Fig. S2†). Catalytic results showed that the reaction at 180 °C and under a pressure of 2.7 MPa offered an average 40% conversion of DMA with >99% DMF selectivity in the trapped liquid product. The continuous reaction had a lower conversion than the batch reaction due to a shorter contact time, but comparably high selectivity. Unreacted CO and DMA gas were recycled upstream. The reaction can run for 288 h with a mere <5% activity loss (Fig. 4). The TON achieved 6125, which was calculated according to ref. 11, where the size of Ru particles is about 1 nm from HR-TEM (Fig. S5†) We checked the used catalyst and found a small amount of carbon deposit (Fig. S3 and S4†). The catalyst was regenerated by calcination and reduction in H2 before reuse.
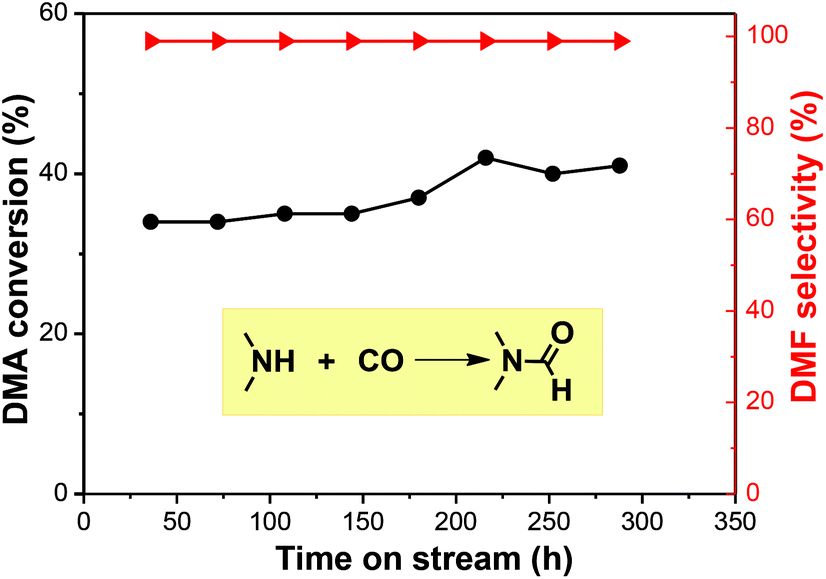 | ||
| Fig. 4 Continuous synthesis of DMF from DMA and CO in a fixed-bed reactor. Reaction conditions: reaction pressure 2.7 MPa (back valve), 180 °C, feeding gas composition: CO:N2 = 1:1 (volume ratio), DMA:CO = 1:18 (molar ratio). Ru/ceria catalyst 8 mL, GHSV 40 h−1. | ||
The CO adsorption on the Ru/ceria catalyst was investigated (Fig. 5a). There are three remarkable bands. Because of no chemical interaction between CO and ceria support, these three bands were assigned to CO adsorption on Ru species. The high-frequency bands located at 2145 cm−1 and 2080 cm−1 were assigned to multicarbonyl species on the oxidized Ru sites [Ruδ+(CO)x], although there was no consensus regarding the oxidation state of Ru (δ) and the number of carbonyl groups (x).12 The low-frequency band located at 2008 cm−1 was assigned to the stretching of CO adsorbed Ru0 related to the highly dispersed Ru atoms with defects.12a,b For comparison, when Ru is supported on SiO2 or MgO, the low-frequency band is observed at around 2030 cm−1, which is assigned to the linearly bonded CO on Ru0 sites on large Ru nanoparticles.13 These experimental results suggest that ceria support can well stabilize Ru clusters during the reduction and restrict the Ru size growth. The adsorption of amine was also investigated on Ru/ceria (Fig. 5b). n-Butylamine was used as a model molecule. We have reported that when n-butylamine was adsorbed on ceria, the bending stretch of N–H red-shifted from 1624 cm−1 of a free n-butylamine molecule to around 1560 cm−1. This shift was due to the adsorbed n-butylamine on the basic oxygen site of ceria via the Ce–Oδ−⋯Hδ+–N interaction.14 While n-butylamine was adsorbed on Ru/ceria, the bending stretch of N–H red-shifted to a much lower frequency around at 1530 cm−1, indicating a stronger interaction between amine and Ru/ceria than pristine ceria. This leads us to conclude that n-butylamine may be preferentially activated on the Ru sites. In order to further confirm the active sites for amine activation, in situ IR tests of CO and n-butylamine adsorption were conducted (Fig. 5c). Addition of n-butylamine to the CO-saturated Ru/ceria surface revealed that the peak at 2145 cm−1 assigned to CO adsorbed on Ruδ+ gradually decreased in intensity and disappeared in 30 min at room temperature. And no remarkable band related to the formamide product was observed at the same time. The IR peaks at 2080 and 2008 cm−1 remained but red-shifted. And these two bands blue-shifted to the original positions upon desorbing n-butylamine. Combining the result of amine adsorption and IR, the disappearing at 2145 cm−1 could be due to the fact that CO on Ruδ+ was taken over by n-butylamine during competitive adsorption. Thus, we proposed that amine was activated on Ruδ+. As is known that rod and nano particle ceria contain more oxygen vacancies than other ceria substrates,15 they might be beneficial for the formation of finely dispersed Ru clusters and enhanced electronic and oxygen transfer in the interface, tuning the electronic structure of noble metals supported on ceria.16 In this formylation reaction system catalyzed by Ru/ceria the transfer of electrons and oxygen might create many more Ruδ+ sites for amine activation. Thus, the rod and particle ceria with rich oxygen vacancy content showed excellent performance in the direct formylation reaction than a ceria cube or a ceria octahedron (Fig. 1). CO adsorption on Ru/ceria after calcination at 500 °C was also investigated (Fig. S6†). The result suggested that there was no chemical interaction between the Ru catalyst and CO. It could be that Ru/ceria after calcination could not catalyze the formylation reaction efficiently (Fig. 1).
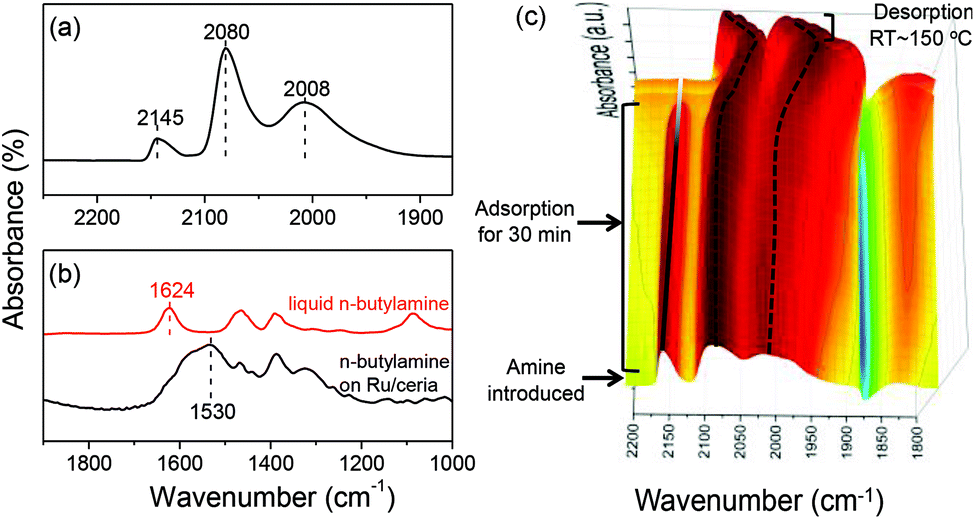 | ||
| Fig. 5 (a) IR of CO adsorption on Ru/ceria for 30 min at room temperature and then desorption at 150 °C. (b) IR of liquid n-butylamine and IR of n-butylamine adsorption at room temperature and then desorption at 150 °C. (c) In situ IR spectra of consecutive adsorption of CO followed by n-butylamine. | ||
Kinetic analysis (Fig. S7†) was conducted in a batch reactor and it showed the first order for both partial pressure of CO (PCO: 0.6–0.8 MPa) and the n-butylamine concentration (0.50–1.25 M), suggesting a possible concerted reaction mechanism.
In summary, we have reported a direct formylation reaction of amines and CO to generate formamides. The co-activation of amine and CO was achieved on different Ru species supported on ceria. The present study represents a new and convenient route for formamide synthesis, developing a new application for a supported ceria catalyst.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21422308, 21303189) and by the Strategic Priority Research Program of Chinese Academy of Sciences (XDB17020300).References
- F. Ullmann, Formamides, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2007 Search PubMed.
- S. T. Ding and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 9226–9237 CrossRef CAS PubMed.
- (a) T. M. Dine, D. Evans, J. Rouden and J. Blanchet, Chem. – Eur. J., 2016, 22, 5894–5898 CrossRef PubMed; (b) M. Lei, L. Ma and L. Hu, Tetrahedron Lett., 2010, 51, 4186–4188 CrossRef CAS; (c) Z. G. Ke, Y. Zhang, X. J. Cui and F. Shi, Green Chem., 2016, 18, 808–816 RSC; (d) O. Jacquet, C. D. Gomes, M. Ephritikhine and T. Cantat, J. Am. Chem. Soc., 2012, 134, 2934–2937 CrossRef CAS PubMed; (e) Q. Y. Bi, J. D. Lin, Y. M. Liu, S. H. Xie, H. Y. He and Y. Cao, Chem. Commun., 2014, 50, 9138–9140 RSC; (f) X. N. Li, K. Liu, X. L. Xu, L. Ma, H. Wang, D. H. Jiang, Q. F. Zhang and C. S. Lu, Chem. Commun., 2011, 47, 7860–7862 RSC; (g) V. K. Das, R. R. Devi, P. K. Raul and A. J. Thakur, Green Chem., 2012, 14, 847–854 RSC; (h) T. V. Q. Nguyen, W. J. Yoo and S. Kobayashi, Angew. Chem., Int. Ed., 2015, 54, 9209–9212 CrossRef CAS PubMed.
- (a) B. A. Aleiwi, K. Mitachi and M. Kurosu, Tetrahedron Lett., 2013, 54, 2077–2081 CrossRef CAS PubMed; (b) L. Zhang, Z. B. Han, X. Y. Zhao, Z. Wang and K. L. Ding, Angew. Chem., Int. Ed., 2015, 54, 6186–6189 CrossRef CAS PubMed; (c) C. L. Allen, B. N. Atkinson and J. M. J. Williams, Angew. Chem., Int. Ed., 2012, 51, 1383–1386 CrossRef CAS PubMed; (d) O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC; (e) P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1994, 116, 8851–8852 CrossRef CAS; (f) W. F. Li and X. F. Wu, Chem. – Eur. J., 2015, 21, 14943–14948 CrossRef CAS PubMed; (g) S. Tanaka, T. Minato, E. Ito, M. Hara, Y. Kim, Y. Yamamoto and N. Asao, Chem. – Eur. J., 2013, 19, 11832–11836 CrossRef CAS PubMed; (h) K. Motokura, N. Takahashi, D. Kashiwame, S. Yamaguchi, A. Miyaji and T. Baba, Catal. Sci. Technol., 2013, 3, 2392–2396 RSC; (i) S. P. Pathare, A. K. H. Jain and K. G. Akamanchi, RSC Adv., 2013, 3, 7697–7703 RSC; (j) S. N. Rao, D. C. Mohan and S. Adimurthy, Org. Lett., 2013, 15, 1496–1499 CrossRef CAS PubMed.
- (a) C. J. Gerack and L. McElwee-White, Chem. Commun., 2012, 48, 11310–11312 RSC; (b) J. H. Park, J. C. Yoon and Y. K. Chung, Adv. Synth. Catal., 2009, 351, 1233–1237 CrossRef CAS; (c) P. A. Shelton, Y. Zhang, T. H. H. Nguyen and L. McElwee-White, Chem. Commun., 2009, 947–949 RSC; (d) P. G. Reddy, G. D. K. Kumar and S. Baskaran, Tetrahedron Lett., 2000, 41, 9149–9151 CrossRef.
- Y. S. Choi, Y. N. Shim, J. Lee, J. H. Yoon, C. S. Hong, M. Cheong, H. S. Kim, H. G. Jang and J. S. Lee, Appl. Catal., A, 2011, 404, 87–92 CrossRef CAS.
- M. Beller, Catalytic carbonylation reactions, Springer, 2006, vol. 978 Search PubMed.
- (a) E. N. Ntainjua, M. Piccinini, J. C. Pritchard, J. K. Edwards, A. F. Carley, C. J. Kiely and G. J. Hutchings, Catal. Today, 2011, 178, 47–50 CrossRef CAS; (b) Y. Pan, Y. Cui, C. Stiehler, N. Nilius and H. J. Freund, J. Phys. Chem. C, 2013, 117, 21879–21885 CrossRef CAS; (c) N. J. Divins, I. Angurell, C. Escudero, V. Perez-Dieste and J. Llorca, Science, 2014, 346, 620–623 CrossRef CAS PubMed; (d) J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed; (e) M. Cargnello, V. V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS PubMed; (f) Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed; (g) L. Vivier and D. Duprez, ChemSusChem, 2010, 3, 654–678 CrossRef CAS PubMed; (h) M. C. Ribeiro, M. K. Gnanamani, I. R. Azevedo, R. C. Rabelo-Neto, G. Jacobs, B. H. Davis and F. B. Noronha, Top. Catal., 2014, 57, 550–560 CrossRef CAS; (i) M. Tamura, T. Kitanaka, Y. Nakagawa and K. Tomishige, ACS Catal., 2016, 6, 376–380 CrossRef CAS; (j) J. Carrasco, D. Lopez-Duran, Z. Y. Liu, T. Duchon, J. Evans, S. D. Senanayake, E. J. Crumlin, V. Matolin, J. A. Rodriguez and M. V. Ganduglia-Pirovano, Angew. Chem., Int. Ed., 2015, 54, 3917–3921 CrossRef CAS PubMed; (k) M. Tamura, A. Satsuma and K. Shimizu, Catal. Sci. Technol., 2013, 3, 1386–1393 RSC; (l) A. Trovarelli, C. de Leitenburg, M. Boaro and G. Dolcetti, Catal. Today, 1999, 50, 353–367 CrossRef CAS; (m) G. Vile, B. Bridier, J. Wichert and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2012, 51, 8620–8623 CrossRef CAS PubMed; (n) J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed; (o) N. Barrabés, K. Föttinger, A. Dafinov, F. Medina, G. Rupprechter, J. Llorca, J. E. Sueiras and K. A. Abboud, Appl. Catal., B, 2009, 87, 84–91 CrossRef.
- (a) M. Wang, F. Wang, J. P. Ma, M. R. Li, Z. Zhang, Y. H. Wang, X. C. Zhang and J. Xu, Chem. Commun., 2014, 50, 292–294 RSC; (b) N. Wang, W. Z. Qian, W. Chu and F. Wei, Catal. Sci. Technol., 2016, 6, 3594–3605 RSC; (c) W. Q. Li, S. Goverapet Srinivasan, D. R. Salahub and T. Heine, Phys. Chem. Chem. Phys., 2016, 18 CAS; (d) Z. A. Qiao, Z. L. Wu and S. Dai, ChemSusChem, 2013, 6, 1821–1833 CrossRef CAS PubMed.
- N. Ortega, C. Richter and F. Glorius, Org. Lett., 2013, 15, 1776–1779 CrossRef CAS PubMed.
- T. Abe, M. Tanizawa, K. Watanabe and A. Taguchi, Energy Environ. Sci., 2009, 2, 315–321 CAS.
- (a) S. Y. Chin, C. T. Williams and M. D. Amiridis, J. Phys. Chem. B, 2006, 110, 871–882 CrossRef CAS PubMed; (b) M. Kantcheva and S. Sayan, Catal. Lett., 1999, 60, 27–38 CrossRef CAS; (c) K. Hadjiivanov, J. C. Lavalley, J. Lamotte, F. Mauge, J. Saint-Just and M. Che, J. Catal., 1998, 176, 415–425 CrossRef CAS; (d) Z. Opre, D. Ferri, F. Krumeich, T. Mallat and A. Baiker, J. Catal., 2007, 251, 48–58 CrossRef CAS.
- C. Crisafulli, R. Maggiore, S. Scire and S. Galvagno, J. Chem. Soc., Faraday Trans., 1994, 90, 2809–2813 RSC.
- Y. Wang, F. Wang, C. Zhang, J. Zhang, M. Li and J. Xu, Chem. Commun., 2014, 50, 2438–2441 RSC.
- (a) A. Leyva-Perez, D. Combita-Merchan, J. R. Cabrero-Antonino, S. I. Al-Resayes and A. Corma, ACS Catal., 2013, 3, 250–258 CrossRef CAS; (b) Z. L. Wu, M. J. Li, J. Howe, H. M. Meyer and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skala, A. Bruix, F. Illas, K. C. Prince, V. Matolin, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02603f |
| This journal is © The Royal Society of Chemistry 2017 |