
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereospecific functionalizations and transformations of secondary and tertiary boronic esters
Christopher
Sandford
and
Varinder K.
Aggarwal
 *
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: v.aggarwal@bristol.ac.uk
First published on 29th March 2017
Abstract
The formation of highly enantioenriched boronic esters through both stoichiometric and catalytic methods has received much attention over the past decade. Accordingly, the transformations of the boronic ester moiety into other functional groups is of considerable interest in synthesis. Specifically, transformations which retain the high enantioenrichment of the starting boronic ester, either through a stereoretentive or a stereoinvertive pathway, lead to the formation of new C–C, C–O, C–N, C–X, or C–H bonds at stereogenic centres. This feature article summarises the current state of the art in stereospecific transformations of both secondary and tertiary boronic esters into other functionalities and groups, whilst considering critically the transformations that are currently unattainable and would represent future advances to the field.
 Christopher Sandford | Christopher Sandford was born in Bristol, UK in 1990. He obtained his MChem from the University of Oxford in 2013, conducting research under the supervision of Prof. Darren Dixon. Chris is currently pursuing his PhD at the University of Bristol under the direction of Prof. Varinder Aggarwal, where he is investigating the reactivity of boronate complexes. |
 Varinder K. Aggarwal | Varinder K. Aggarwal studied chemistry at Cambridge University and received his PhD in 1986 under the guidance of Dr Stuart Warren. After postdoctoral studies (1986–1988) under Prof. Gilbert Stork, Columbia University, he returned to the UK as a Lecturer at Bath University. In 1991 he moved to Sheffield University, where he was promoted to Professor in 1997. In 2000 he moved to Bristol University where he holds the Chair in Synthetic Chemistry. He was elected Fellow of the Royal Society in 2012. |
1. Introduction
Organoboron compounds are of significant utility in asymmetric synthesis. Accompanied with a large expansion in routes to obtain enantioenriched organoboron compounds, their subsequent transformations into a range of functional groups provide access to a broad array of diverse molecules with high enantioselectivity.Asymmetric hydroboration, reported in 1961 by H. C. Brown,1 heralded the birth of modern asymmetric synthesis because, for the first time, it was shown that small molecules (Ipc2BH) were capable of providing high levels of enantioselectivity, levels that had previously been the sole preserve of enzymes. Stereospecific oxidation of the chiral alkylboranes (R3B) formed through hydroboration gave enantioenriched alcohols. Subsequently, further stereospecific transformations of the alkylboranes were elucidated, enabling the C–B bond to be converted into a wide range of other functional groups.2
However, despite the range of reactions that can be carried out using alkylboranes, their synthetic utility is tempered by their air and moisture sensitivity. Changing the organoborane into a boronic ester (RB[OR′]2) enables easier purification, especially in the case of boronic acid pinacol esters (RBpin).3 This characteristic facilitates the use of boronic esters in synthesis and as a tool to create new bonds. Perhaps most notably, the Suzuki–Miyaura reaction has been applied extensively because of operational simplicity and the range of arylboronic esters and acids that are commercially available.4
As a tool for chiral synthesis, the development of novel methods to obtain enantioenriched alkylboronic esters has received much attention over the past decade. Methods to introduce the boronic ester moiety into organic compounds with high levels of asymmetric induction include both stoichiometric methods (such as homologation5 and lithiation–borylation6) and catalytic methods (such as hydroboration,7 conjugate addition8 and diboration9). Having established routes to the synthesis of these molecules, attention has also turned to their manipulation and subsequent stereospecific transformations into a wide range of functional groups,10 in a similar vein to the range of transformations open to alkylboranes.
In this feature article, we review the range of stereospecific transformations that are currently available to secondary and tertiary boronic esters, operating through either a stereoretentive or a stereoinvertive pathway (i.e. not a radical pathway). The transformations covered (Fig. 1) include oxidations (C–O bond formation), conversions of boron to other heteroatoms (C–N, C–X), protodeboronation (C–H), as well as the formation of new C–C bonds (resulting from homologations, olefinations, alkynylations and coupling reactions). These transformations have been selected for their applicability to any general alkylboronic ester, and so reactions that are dependent on the presence of specific functional groups (such as allylic boronic esters, or coupling reactions activated by neighbouring directing groups), except for benzylic boronic esters, have been omitted. With this review, we aim to showcase the work that we, and others, have conducted to expand the synthetic utility of enantioenriched boronic esters, whilst also identifying transformations that remain elusive and would represent significant future advances to the field.
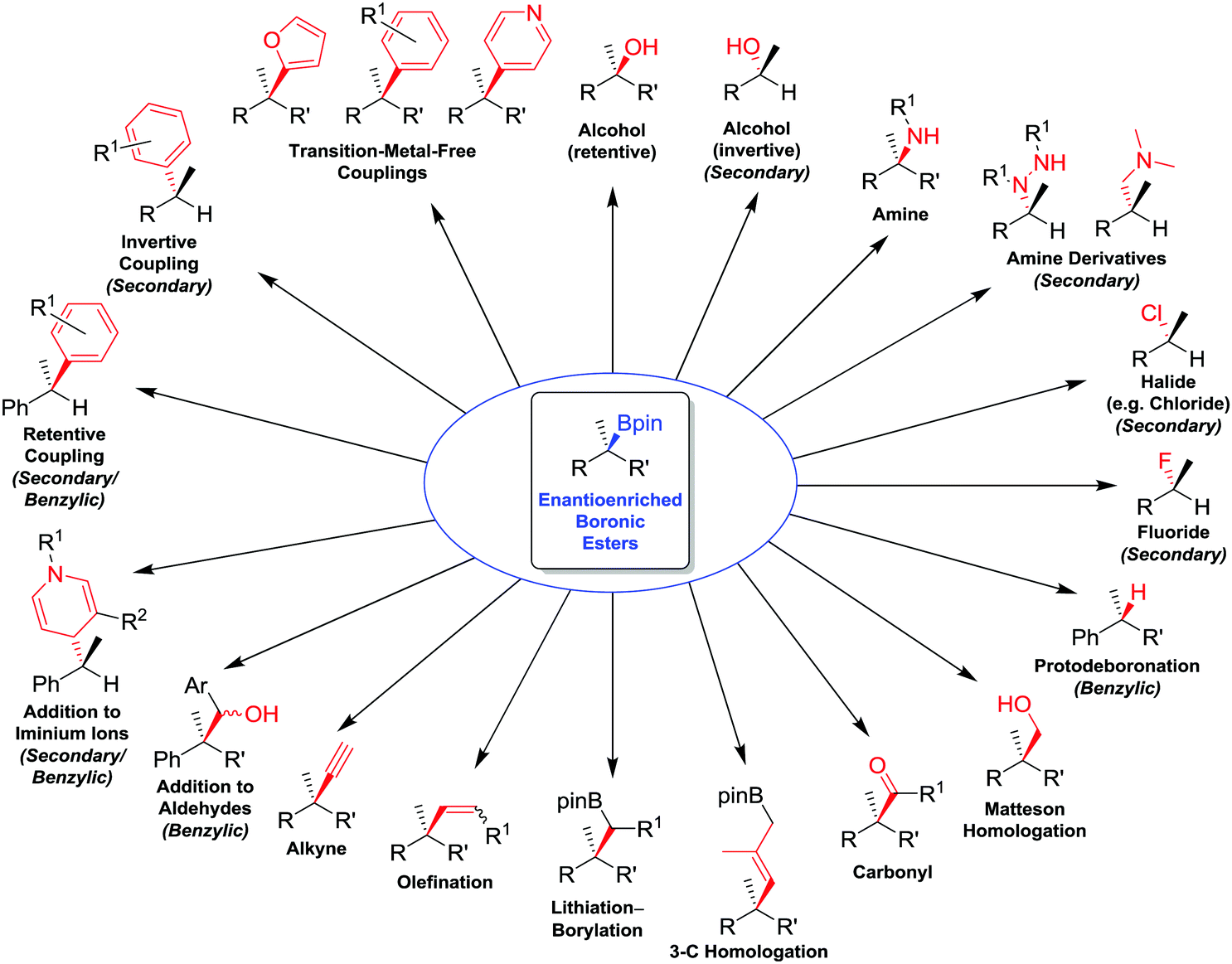 | ||
| Fig. 1 Summary of stereospecific functional group transformations of secondary and tertiary boronic esters. | ||
2. Carbon–heteroatom bond formation
2.1 Oxidation of boronic esters to alcohols
The stereospecific oxidation of boronic esters to the corresponding alcohol is the most versatile and widely used functionalization reaction of boronic esters. Building upon the oxidation of organoboranes through the use of basic hydrogen peroxide, developed by Brown and co-workers in 1961,11 the reaction of an enantioenriched boronic ester under these conditions affords the alcohol with complete retention of configuration. Addition of the peroxide anion to the empty p-orbital of the boron atom forms boronate complex 1 (Scheme 1). The boronate complex then undergoes a 1,2-metallate rearrangement – migration of the C–B σ-bond onto the adjacent oxygen atom, with loss of hydroxide. The nature of this 1,2-metallate rearrangement step is such that the stereoconfiguration of the carbon initially attached to boron is retained, a feature that is common to many of the stereospecific transformations discussed throughout this review. Finally, hydrolysis cleaves the O–B bond to afford alcohol 3. The oxidation of a boronic ester is slower than for analogous boranes owing to donation of the oxygen lone pairs into the empty p-orbital of boron, an attribute that decreases the electrophilicity of the boron.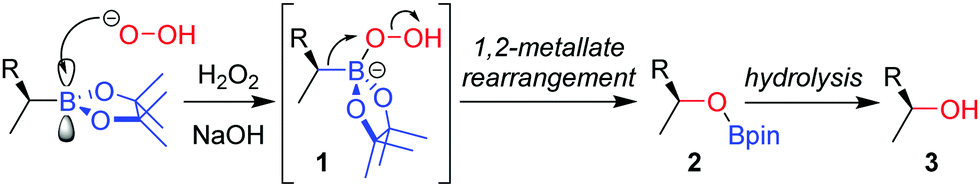 | ||
| Scheme 1 Mechanism of the oxidation of boronic esters by basic hydrogen peroxide. | ||
Although numerous synthetic routes are available to produce primary and secondary alcohols, the formation of stereodefined tertiary alcohols is significantly less developed. We found that oxidation can also be applied to tertiary boronic esters (Scheme 2),12 which can be prepared by lithiation–borylation (see Section 4.3). During the course of our studies on the oxidation of tertiary boronic esters, we have found that it is sometimes advantageous to add dibutylhydroxytoluene (BHT) to prevent the formation of peroxy radicals, and ethylenediaminetetraacetic acid (EDTA) to minimize the decomposition of hydrogen peroxide by trace metals.12,13 Furthermore, we find that THF is an ideal solvent for improving miscibility with the aqueous phase.
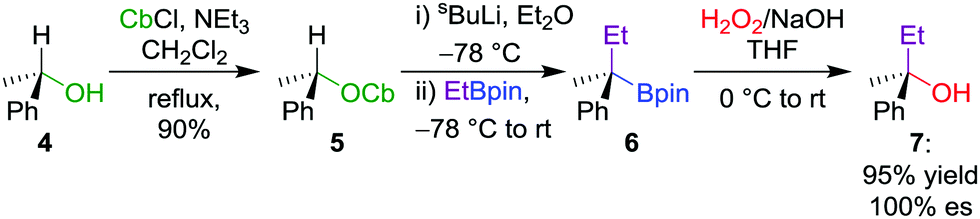 | ||
| Scheme 2 Conversion of chiral secondary alcohols into tertiary alcohols by lithiation–borylation (see Section 4.3) and the stereospecific oxidation of boronic esters. OCb = N,N-diisopropylcarbamate. | ||
As a milder alternative to the hydrogen peroxide conditions, sodium perborate effects the oxidation with higher functional-group tolerance.14 For example, Fontani et al. reported that the oxidation of diastereomerically pure cyclopropylboronic acid pinacol ester 8 with hydrogen peroxide led to a poor conversion along with the formation of ring-opened carbonyl compounds.15 Conversely, the oxidation with sodium perborate was found to afford the desired alcohol 9 in 81% yield (Scheme 3). Furthermore, sodium perborate is also often the reagent of choice for oxidations where the boron moiety is situated β to an electron-withdrawing group (such as a sulfone or a carbonyl group).16 Household bleach (aq. NaOCl) has also been used to oxidise a tertiary β-keto boronic ester with complete retention of configuration.17
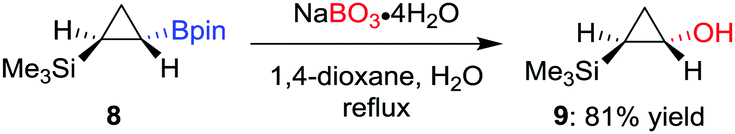 | ||
| Scheme 3 Oxidation of cyclopropylboronic esters using sodium perborate by Fontani et al. | ||
2.2 Amination of boronic esters
Although trialkylboranes can be readily converted into the corresponding amines by reaction with reagents such as chloramine18 or alkyl azides,19 the reduced Lewis acidity of boronic esters prevents reactivity with these weakly Lewis basic reagents due to ineffective association. To facilitate the stereospecific amination of boronic acid glycol esters 10, Brown and co-workers (Scheme 4a) reported the initial conversion into the more Lewis acidic borinic ester 11 with methyllithium and subsequent amination by using hydroxylamine-O-sulfonic acid (NH2OSO3H).20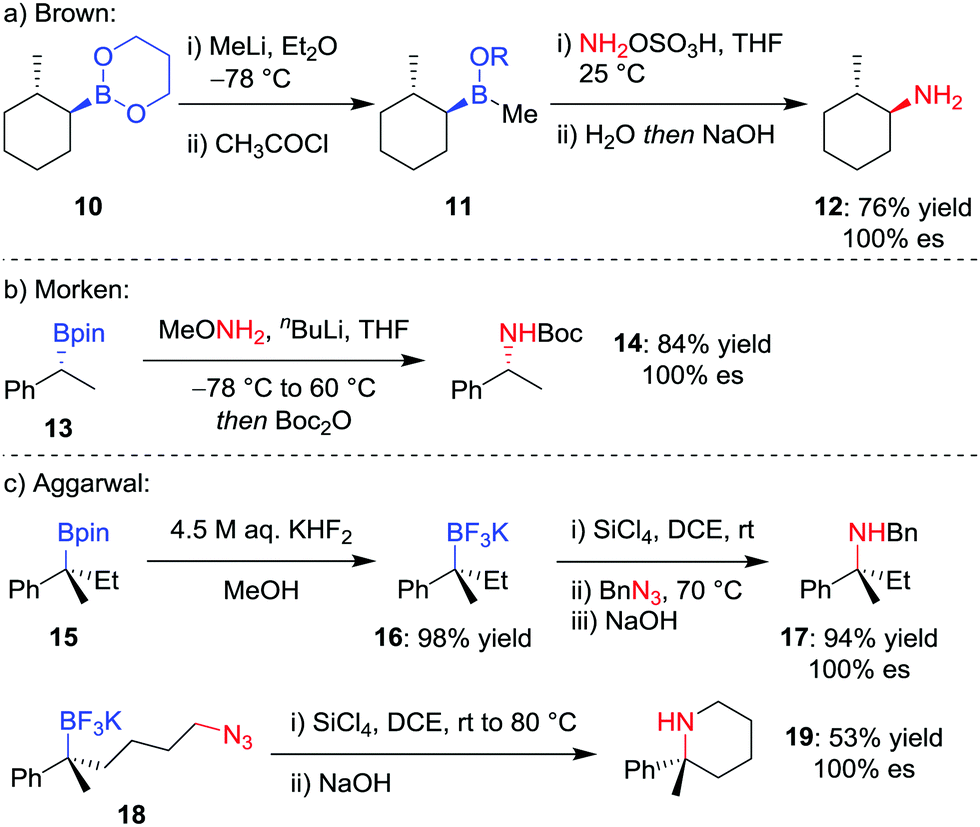 | ||
| Scheme 4 Procedures for the stereospecific amination of secondary and tertiary boronic esters. R = CH2(CH2)2OC(O)CH3. | ||
Seeking a direct stereospecific amination protocol for alkylboronic acid pinacol esters, Morken and co-workers21 (Scheme 4b) proposed increasing the nucleophilicity of the amination reagent by using lithiated alkoxy amines. Reaction of enantioenriched boronic ester 13 with 3.0 equivalents of methoxyamine and n-butyllithium, followed by Boc protection, was found to afford Boc-protected amine 14 in 84% yield. This transformation was found to proceed with complete retention of stereoconfiguration for secondary boronic esters, an outcome that is consistent with a 1,2-metallate rearrangement involving migration of the C–B σ-bond onto the nitrogen atom with loss of methoxide. However, these conditions were not amenable for the amination of tertiary alkylboronic esters.
Although a method for the direct amination of tertiary boronic esters has yet to be reported, we sought to provide the desired tertiary amines by first converting the boronic ester 15 into the trifluoroborate salt 16 (Scheme 4c),22 which can be achieved in essentially quantitative yields using KHF2 in MeOH.23 Pleasingly, we found that a modification of Matteson's amination conditions for trifluoroborate salts,24 using SiCl4 to form the alkyldichloroborane25 and then reaction with an alkylazide,19b led to the corresponding amine 17 through a retentive pathway without loss in stereochemical integrity. The reaction can occur intramolecularly to obtain piperidines (19), and we subsequently discovered that the methodology can also be applied to non-benzylic tertiary boronic esters.26
2.3 Boronate complexes as chiral organometallic-type reagents
In addition to the functionalization of boronic esters via the stereospecific migration of the C–B bond, we envisaged that enantioenriched boronic esters could serve as precursors to chiral organometallic-type nucleophilic reagents.27 We proposed that the boronic ester could be activated by addition of an electron-rich aryllithium to form a nucleophilic boronate complex. The complex is configurationally stable and should thus confer the chirality of the starting material into the desired functionalized compound.Upon addition of p-MeOC6H4Li 21 to boronic ester 20 (Scheme 5a),28 followed by reaction with I2 as the electrophile, the desired C–I bond was formed in 80% yield and 97% es. It was determined that the reaction is invertive at the stereogenic carbon (a polar, SE2inv pathway). However, high enantiospecificity was not observed when using diisopropyl azodicarboxylate (DIAD) as the electrophile: hydrazine 25 was formed in 82% yield and only 13% es (Scheme 5b). We postulated that the low enantiospecificity resulted from a competing single-electron transfer (SET) pathway (Fig. 2), a mechanism that is supported by radical-clock experiments. Nonetheless, the SET pathway could be reduced by adjusting the electronics of the aryllithium used to form the boronate complex; by using the electron-deficient aryllithium 24, the enantiospecificity of the reaction was found to increase to 66%.
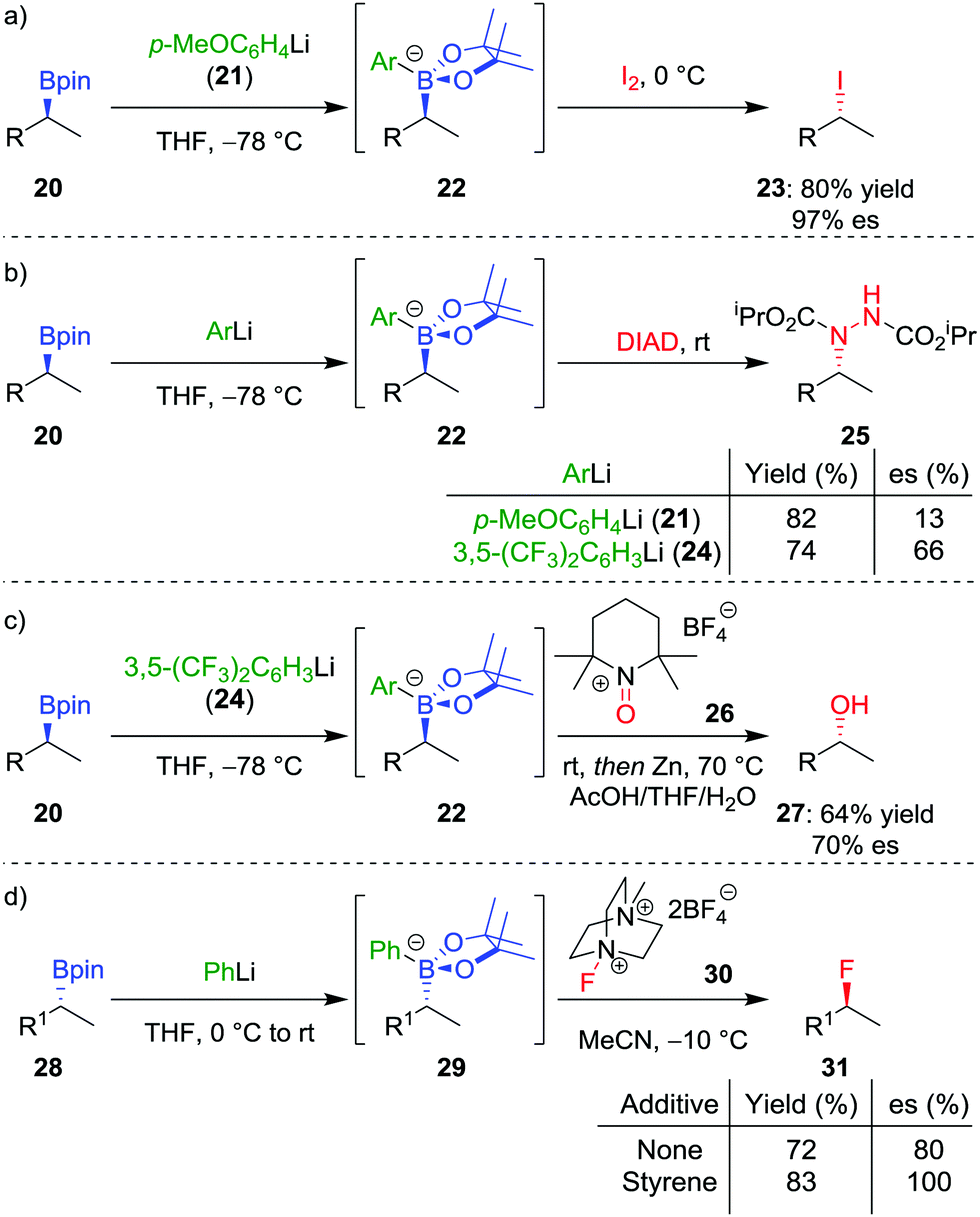 | ||
Scheme 5 Stereospecific conversions of secondary boronic esters into other heteroatoms by the reactions of nucleophilic boronate complexes with electrophiles. R = CH2CH2Ph, R1 = CH2CH2C6H4OMe, DIAD = iPrO2C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–CO2iPr. N–CO2iPr. | ||
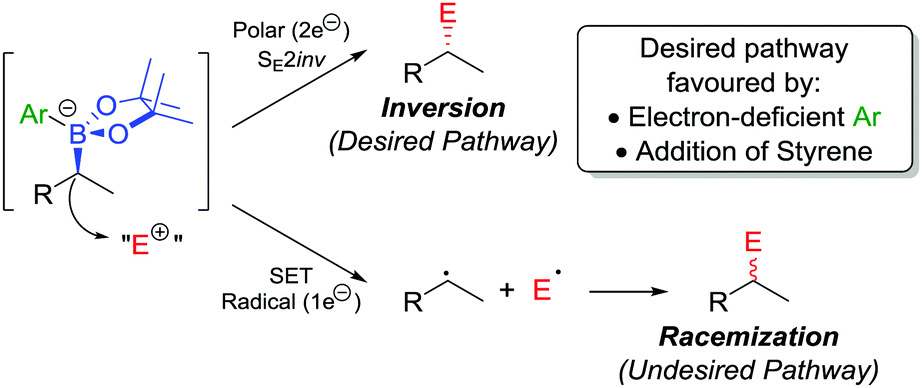 | ||
| Fig. 2 Reaction pathways of chiral boronate complexes. E+ = electrophile, SET = single-electron transfer. | ||
With the optimal nucleophilic boronate complex in hand, we were able to functionalize the C–B bond of enantioenriched secondary boronic esters with a range of different heteroatom electrophiles with up to complete stereoinversion – enabling iodination, chlorination, bromination, as well as the formation of C–N bonds. C–O bonds can also be formed using this stereoinvertive method, thus being complementary to the standard oxidation conditions which are stereoretentive (see Section 2.1). For example, reaction of the boronate complex with 2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (26) and subsequent N–O bond cleavage affords the corresponding alcohol 27 in 64% yield and 70% es (Scheme 5c). The reaction was found to be applicable to both benzylic and non-benzylic substrates, with the former exhibiting both enhanced reactivity and enantiospecificity. These reactions are related to the halogenation of boronate complexes derived from chiral boranes which also occurred with inversion.29
The range of heteroatoms that can be introduced using a boronate complex was further expanded to include fluorine,30 providing access to enantioenriched alkylfluorides (Scheme 5d). Boronate complex 29 was formed by reacting boronic ester 28 with phenyllithium, and was then reacted with Selectfluor II (30) in acetonitrile, resulting in the formation of alkylfluoride 31 in 72% yield, but only 80% es. We discovered that the addition of styrene as a cheap and readily-available additive enhanced the enantiospecificity, enabling the product to be formed in 83% yield and complete enantiospecificity. Interestingly, the use of styrene led to no measurable decrease in yield in the reaction, suggesting that it was not merely acting as a radical trap for the SET pathway. Instead, we proposed that a fast radical propagation mechanism is inhibited by styrene, which acts as a radical scavenger (Fig. 3), thus allowing the desired SE2inv pathway to dominate.
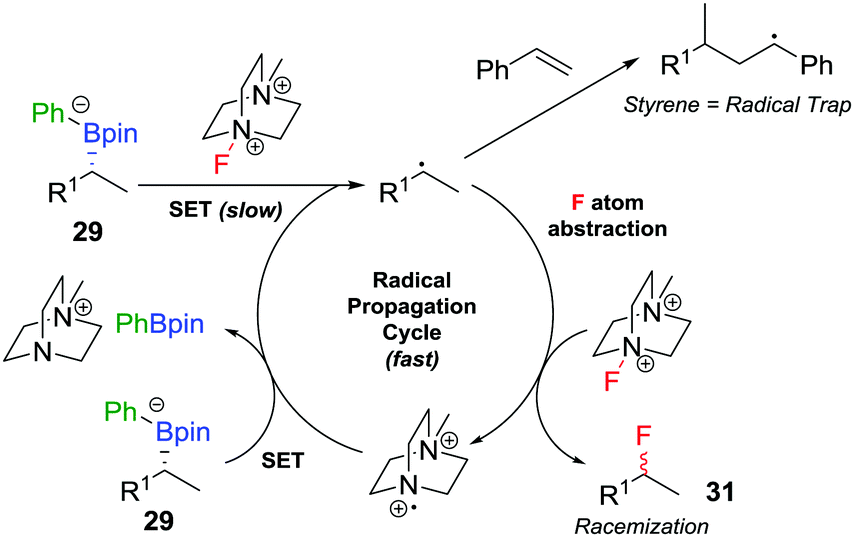 | ||
| Fig. 3 Undesired radical pathways for the fluorination of boronate complexes, leading to the formation of racemic product, unless prevented by styrene. | ||
In order to determine the range of electrophiles that might be suitable for reactions with boronate complexes, we sought to determine the nucleophilicity parameter (N) of the complexes to compare with other nucleophiles.31 In collaboration with Mayr and co-workers, we discovered that the aryl group has a significant effect on reactivity: the boronate complex derived from phenyllithium is 100 times more reactive than that derived from 3,5-(CF3)2C6H3Li (24).32 Furthermore, the order of the nucleophilicity parameter follows the trend: secondary benzylic > primary alkyl > primary benzylic > secondary alkyl. With these relative levels of reactivity known, we aim to explore other electrophiles that should participate in this SE2inv pathway (see also Section 4.5).
3. Carbon–hydrogen bond formation
3.1 Protodeboronation of tertiary benzylic boronic esters
The stereospecific conversion of the C–B bond of a tertiary boronic ester into a C–H bond represents an efficient synthesis of tertiary alkyl stereogenic centres, including 1,1-diarylalkanes. Tertiary boranes are known to undergo protodeboronation by using propionic acid as the reagent of choice,33 owing to strong B–O bond formation driving the reaction. We discovered that this reagent is not efficient for the protodeboronation of tertiary benzylic boronic esters,34 leading to slow conversion predominantly towards elimination products. We reasoned that the reaction could be facilitated if the driving force was changed to the formation of stronger B–F bonds. Consequently, we found that the addition of CsF and H2O in dioxane at 30 °C leads to the protodeboronation of diarylalkyl boronic ester 32 in 91% yield and 99% es (Scheme 6a), via a stereoretentive pathway. The methodology also provides access to enantioenriched deuterated tertiary alkanes by simply substituting the H2O with D2O.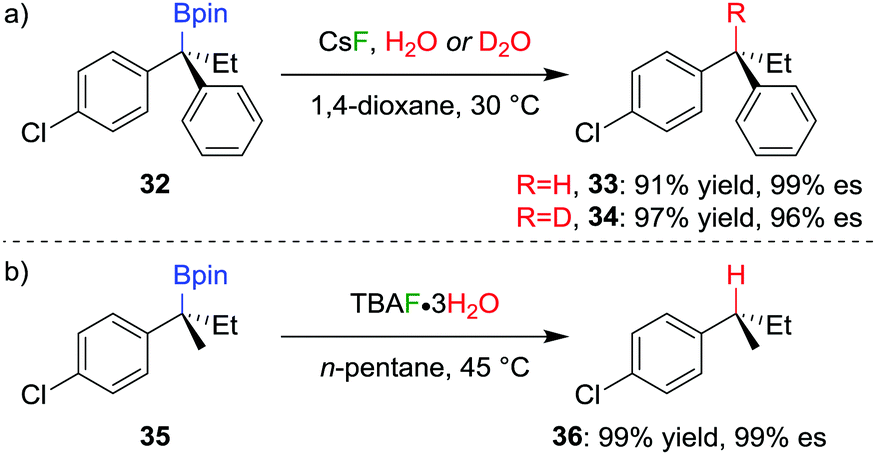 | ||
| Scheme 6 Protodeboronation of tertiary dibenzylic and monobenzylic boronic esters. | ||
In contrast with diarylalkyl boronic esters, aryldialkyl boronic esters were found to be significantly more difficult to protodeboronate. Nevertheless, using the more reactive TBAF·3H2O enabled the protodeboronation of 35 in 99% yield and 99% es when conducted in n-pentane (Scheme 6b). Notably, the protodeboronation conditions require activated substrates, such as a benzylic (or allylic35) boronic ester. However, our research group found that non-benzylic boronic esters could undergo protodeboronation by employing oxidative conditions, leading to homolytic cleavage of the C–B bond,36 the resulting C–H bond being formed without stereospecificity owing to the radical nature of the intermediates.
The TBAF·3H2O reaction is believed to occur via a hydrogen-bonded boronate complex, which delivers the proton to the same side as the boron atom (Scheme 7). In more polar solvents, solvation of the water molecules is proposed to cause a competing invertive pathway and subsequent erosion in enantiospecificity.
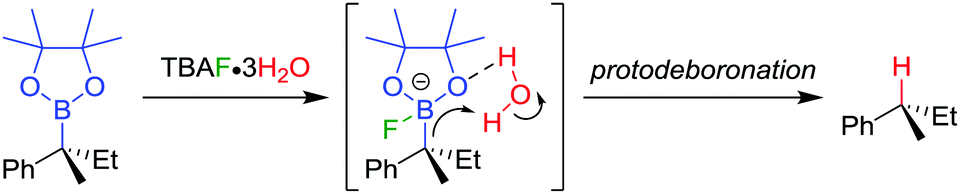 | ||
| Scheme 7 Proposed mechanism for the stereoretentive protodeboronation of tertiary benzylic boronic esters. | ||
This protodeboronation strategy provides ready access to enantioenriched alkanes that are otherwise difficult to access, since the newly formed stereogenic centre can be placed distal to other functional groups. Indeed, we have applied the methodology to the synthesis of a wide range of biologically and therapeutically active molecules (Fig. 4).34,37
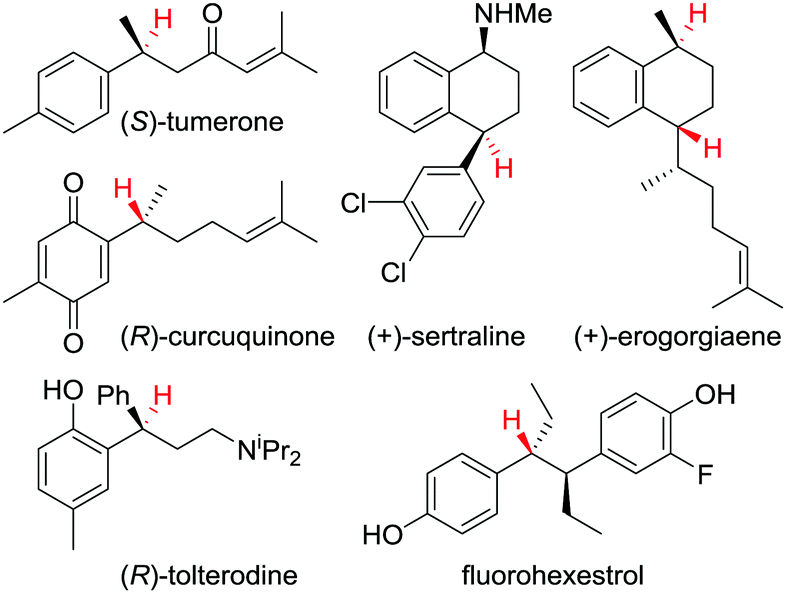 | ||
| Fig. 4 Examples of biologically and therapeutically active molecules synthesised using a protodeboronation step to introduce the displayed hydrogen(s) enantiospecifically. | ||
4. Carbon–carbon bond formation
4.1 One-carbon homologations
The homologation of an enantioenriched boronic ester38 by a one-carbon unit allows facile chain extension whilst retaining the valuable boronic ester group for future functionalization. The simplest such homologation is the insertion of a methylene (CH2) unit into the C–B bond, a reaction that was initially reported by Sadhu and Matteson in 1985.39 In their seminal work, it was found that addition of n-butyllithium to a mixture of boronic ester 37 and iodochloromethane in THF at −78 °C affords the homologated boronic ester 39 in 95% yield, a transformation that proceeds through a stereoretentive 1,2-metallate rearrangement (Scheme 8a). Initially, iodine–lithium exchange forms LiCH2Cl, which adds to the empty boron p-orbital to form boronate complex 38, thus triggering the 1,2-metallate rearrangement with loss of chloride. A number of alternative protocols were subsequently investigated by the groups of Matteson and Brown,40 including in situ reduction of α-chloroboronic esters,41 with bromochloromethane becoming the reagent of choice for the Matteson homologation procedure.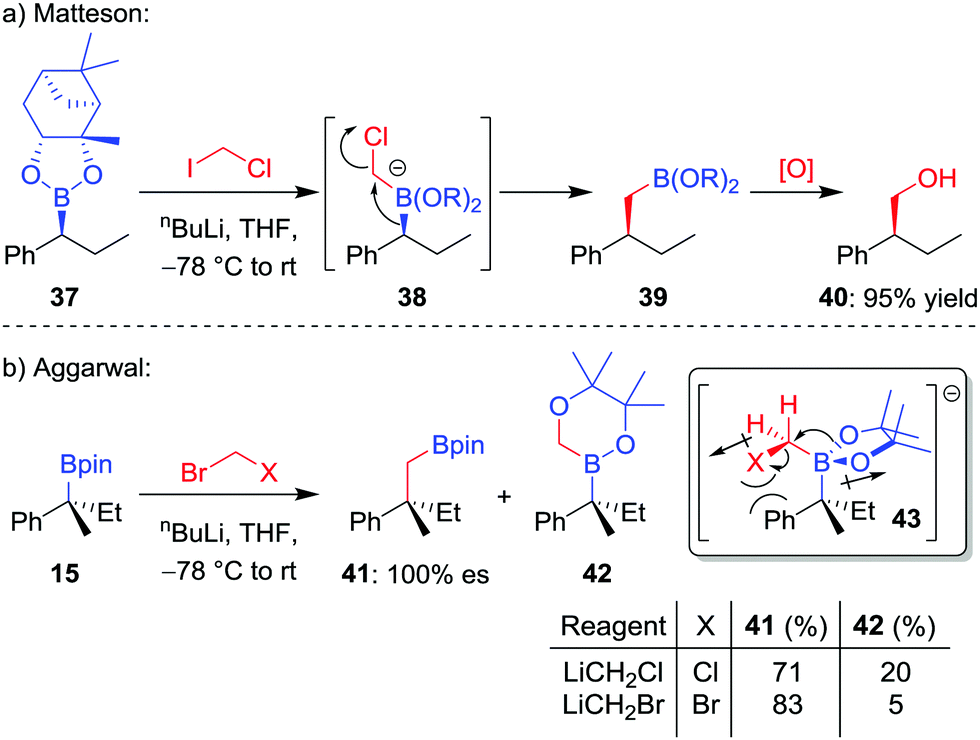 | ||
| Scheme 8 Stereospecific Matteson homologations of enantioenriched boronic esters. Yield and enantiospecificity of 41 recorded after oxidation; yield of 42 determined by 11B NMR analysis of the crude material. | ||
When we investigated the homologation of tertiary boronic esters,26,42 we discovered that the use of bromochloromethane led to competitive oxygen migration, forming borinic ester 42 (Scheme 8b). However, through analysis of conformation 43, we reasoned that using bromide as a bulkier and less-polar leaving group would disfavour oxygen migration; indeed, when using in situ formed bromomethyllithium, the carbon-migration product 41, which was subsequently oxidized to the alcohol, was formed in 83% yield and with complete stereoretention.
Computational studies on the effect of sterics in this migration conducted by Elliott et al. have shown that the energy difference between the barriers to carbon- and oxygen-migrations are significantly reduced for sterically bulky tertiary boronic esters, and that decomplexation and/or undesired reactions of the boronate complex can take place in these cases.43 Consequently, greater yields were found by decreasing steric hindrance around the boron atom through replacing the pinacol ligand with propane-1,3-diol.
Manipulation of the oxidation state of the carbon atom introduced from the organolithium allows access to alternative homologated functional groups. Using LiCHCl2 (derived from the deprotonation of CH2Cl2) provides access to the homologated aldehyde 4544 after oxidation of the intermediate α-chloroboronic ester 44 (Scheme 9a).45 For a tertiary boronic ester, we have found the homologation reaction to proceed in 68% yield with complete stereoretention (Scheme 9b).42 Fandrick et al. have subsequently applied this homologation on a decagram scale to install a quaternary stereogenic centre in their synthesis of 5-lipoxygenase-activating protein (FLAP) inhibitor 50 (Scheme 9c).46
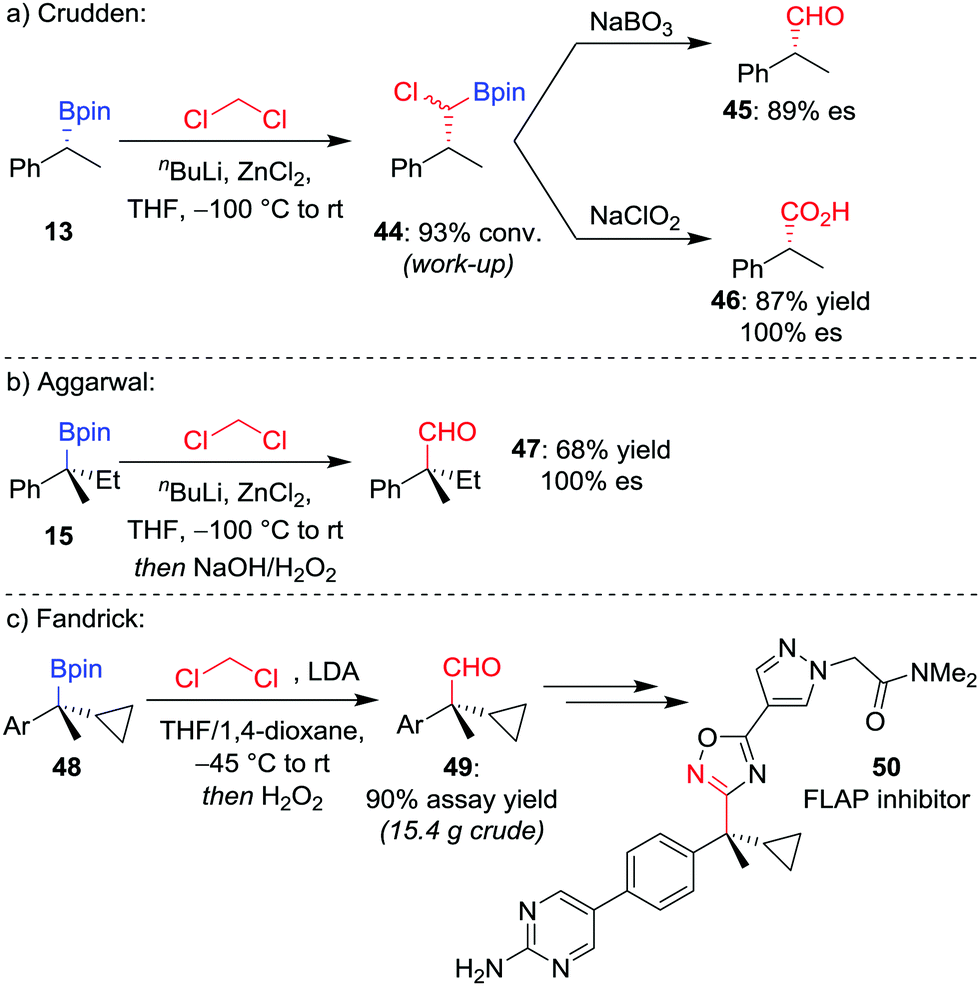 | ||
| Scheme 9 One-carbon homologations of boronic esters to aldehydes and carboxylic acids. Ar = p-BrC6H4. | ||
For the homologation to the corresponding carboxylic acid, Crudden and co-workers found the direct homologation to be challenging,44 but can be achieved via a two-step procedure involving oxidation of the α-chloroboronic ester 44 with NaClO2 (Scheme 9a). This two-step strategy had previously been applied by Matteson and Beedle to oxidise α-azido boronic esters into the corresponding carboxylic acids in their synthesis of enantioenriched α-amino acids.47 As an alternative route, we have also applied a two-step procedure involving oxidation of the homologated alcohol with pyridinium dichromate.48
4.2 Three-carbon homologations
As an alternative to one-carbon homologation processes where the 1,2-metallate rearrangement proceeds by an SN2-type displacement of a leaving group (see Section 4.1), an SN2′-type process can be achieved by using a vinylboronate complex with an allylic leaving group, thereby introducing a three-carbon motif and forming an allylic organoboron compound. This process works well for boranes,49 and the homologation was applied to achiral tertiary boronic esters by Smith et al.50 When our research group applied this technique, homologation of enantioenriched tertiary boronic ester 51 with (3-chloroprop-1-en-1-yl)-lithium 52 afforded, after oxidation, allylic alcohol 53 in 68% yield and 100% es, albeit in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr (Scheme 10a).26 To obtain higher diastereoselectivity, we have found an SN2-type displacement of chloride to be preferential since the configurationally labile organolithium is then able to undergo a dynamic kinetic resolution. In this case, the homologation of 15 with 1-chloro-allyllithium 54 afforded the allylic boronic ester 55 in 73% yield, 100% es and 24:1 dr (Scheme 10b).42
:1 dr (Scheme 10a).26 To obtain higher diastereoselectivity, we have found an SN2-type displacement of chloride to be preferential since the configurationally labile organolithium is then able to undergo a dynamic kinetic resolution. In this case, the homologation of 15 with 1-chloro-allyllithium 54 afforded the allylic boronic ester 55 in 73% yield, 100% es and 24:1 dr (Scheme 10b).42
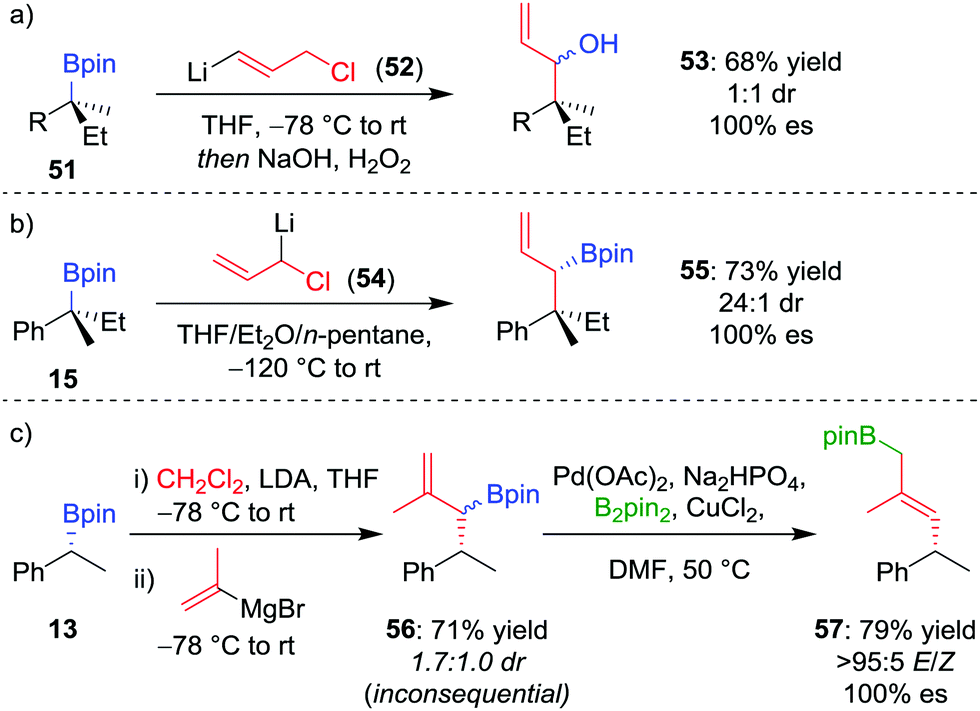 | ||
| Scheme 10 Three-carbon homologations of enantioenriched boronic esters, and the palladium-catalyzed 1,3-borotropic shift. R = CH2CH2Ph. | ||
We envisaged that if we could identify a method for promoting a 1,3-borotropic rearrangement of the products, we would have access to terminal allylic boronic esters for further functionalization.48 However, this posed two challenges: firstly, the homologation of a chiral secondary boronic ester by using 1-chloro-allyllithium leads to a mixture of under- and over-homologated products; secondly, allylic pinacol boronic ester products are stable to 1,3-borotropic rearrangements, unlike their propylene glycol analogues.50b,51
Consequently, to address the first challenge, we adopted a two-step, one-pot protocol involving a one-carbon Matteson homologation followed by in situ treatment with vinyl magnesium bromide (Scheme 10c).48 Next, to facilitate the 1,3-rearrangement, we discovered that palladium catalysis in the presence of base and B2pin2 gave access to a Bpin-bound π-allyl-PdII complex 60 (Fig. 5) which, upon reductive elimination, affords the desired product with excellent E selectivity. The three-carbon homologation of secondary boronic ester 13 was found to afford 57 in 61% yield (over 2 steps), E/Z >95:5 and 100% es (Scheme 10c). To demonstrate the synthetic utility of the three-carbon homologation, we applied our methodology to the synthesis of a fragment of (+)-jasplakinolide.
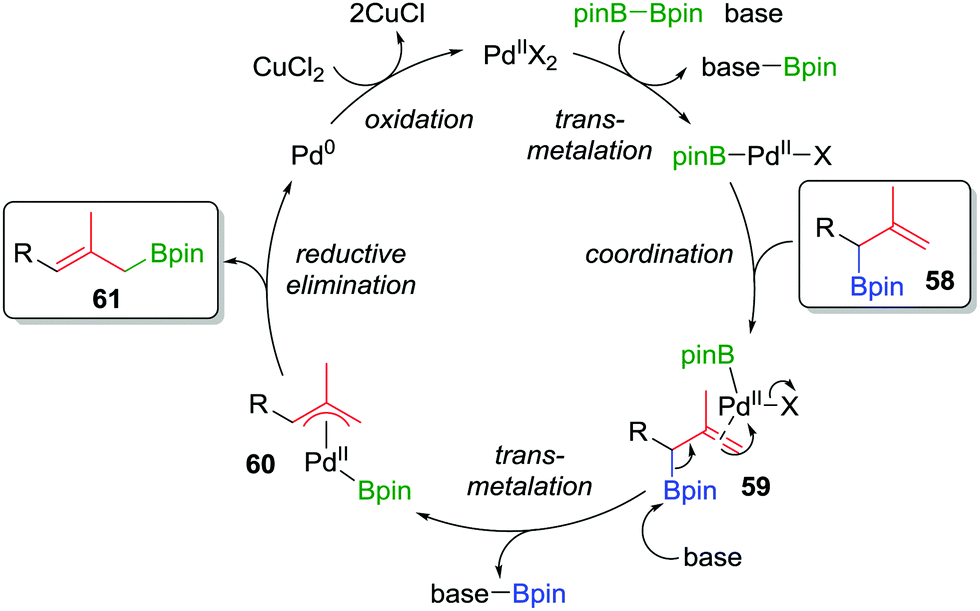 | ||
| Fig. 5 Proposed mechanism of the palladium-catalyzed 1,3-rearrangement of allylic boronic esters used in a two-step three-carbon homologation. | ||
4.3 Lithiation–borylation homologations
Owing to the stereospecific nature of the 1,2-metallate rearrangement, there is the opportunity to create two or more configurationally well-defined contiguous stereocentres with high fidelity through a stereoselective homologation. We have realised this through the application of our lithiation–borylation methodology,6,12,13d,52 in which a configurationally stable carbenoid, possessing a suitable leaving group, is added to a boronic ester to ultimately form a new carbon–carbon bond at a stereogenic centre. Lithiated carbamate 63 can be formed by the deprotonation of the parent carbamate using sec-butyllithium in the presence of (−)-sparteine (Scheme 11a).53 The addition of enantioenriched boronic ester 62 leads to the formation of boronate complex 64 which, following 1,2-metallate rearrangement and oxidation of the boronic ester, affords alcohol 66 in 82% yield, with >98:2 er and 96:4 dr.52a Simply changing the enantiomer of sparteine (or using O'Brien's (+)-sparteine surrogate54) during the lithiation gives access to the other diastereomer of the product (70). We have subsequently applied this methodology to install two contiguous stereocentres in the synthesis of a range of natural products.37a,55
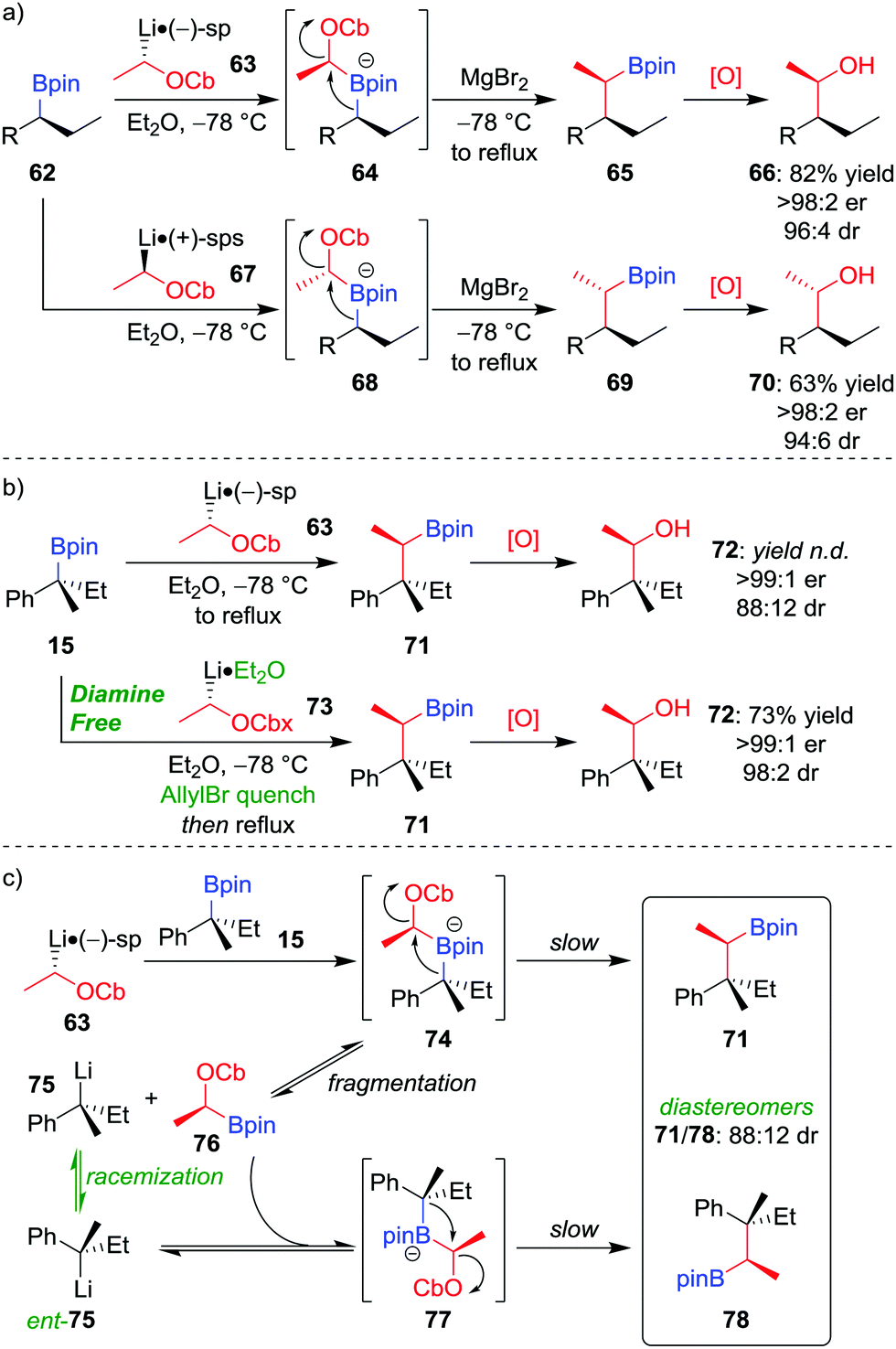 | ||
| Scheme 11 Lithiation–borylation reactions of enantioenriched boronic esters to form (a) tertiary–tertiary and (b) quaternary–tertiary contiguous stereogenic centres. (c) Proposed mechanism for the generation of diastereomers in the formation of quaternary–tertiary contiguous stereogenic centres by lithiation–borylation. OCb = N,N-diisopropylcarbamate, OCbx = 3,3-dimethyl-1-oxa-4-azaspiro[4.5]decane-4-carboxylate, sp = sparteine, sps = sparteine surrogate, R = CH2CH2Ph, n.d. = not determined. | ||
A modified method can be used to incorporate quaternary stereocentres. Application of the standard method, described above, to the homologation of tertiary boronic ester 15 (99:1 er) with lithiated carbamate 63 (98:2 er), gives alcohol 72 with a surprisingly low dr (88:12; Scheme 11b).56 It was discovered that epimerization had occurred at the quaternary stereogenic centre derived from the boronic ester. Consequently, the origin of minor diastereomer 78 was proposed to result from fragmentation of the sterically crowded boronate complex, forming a new organolithium (75) which can racemize and recombine with boronic ester 76 (Scheme 11c). To suppress this racemization pathway, the procedure was adapted as follows: firstly, adding allyl bromide after formation of boronate complex 74 traps any subsequently formed fragmentation anion 75; secondly, using diamine-free conditions (73 obtained by tin–lithium exchange from the corresponding stannane) minimises steric hindrance of the lithiated carbamate, thus enabling boronate complex formation. Application of these conditions affords alcohol 72 in 73% yield, >99:1 er and 98:2 dr (Scheme 11b).
Extending these conditions further to the synthesis of contiguous quaternary–quaternary stereogenic centres was found to be even more challenging, with no 1,2-metallate rearrangement observed upon the addition of a tertiary boronic ester to a lithiated secondary carbamate.57 However, the reaction does proceed with mixed borane 80 (derived from in situ addition of MeMgBr to neopentyl glycol boronic ester 79) to afford, after oxidation, alcohol 82 in 70% yield, >99:1 er and 98:2 dr (Scheme 12). Notably, these mixed boranes complex with the benzylic lithium carbenoids with inversion of configuration at the carbenoid carbon atom.12 In addition to oxidation, amination of the borane intermediate using chloramine also worked well.
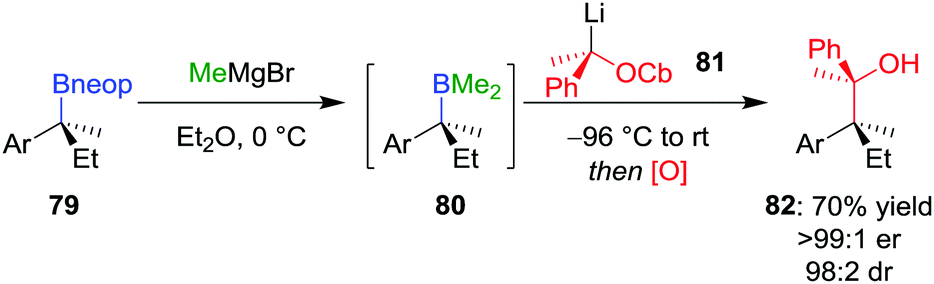 | ||
| Scheme 12 Lithiation–borylation reactions of enantioenriched boronic esters to form quaternary–quaternary contiguous stereogenic centres. OCb = N,N-diisopropylcarbamate, Ar = p-OMeC6H4, neop = neopentyl glycol. | ||
Using lithiation–borylation methodology, we have been able to develop a route to multiple contiguous stereocentres through an assembly-line process, without the requirement for multiple purifications. Nine iterative homologations, starting from boronic ester 83 and using lithiated benzoate ester 84 as the carbenoid (the OTIB group acts as a better leaving group when compared to OCb52b), afforded boronic ester 85 in 58% yield with only two aqueous work-up steps and one purification (Scheme 13a).58 The all-anti isomer was found to adopt an irregular conformation, but by varying the enantiomer of 84 during the assembly-line process, the all-syn (86) and alternating syn–anti (87) isomers could be obtained, molecules that were found to adopt helical and linear conformations respectively. Assembly-line synthesis has also been applied using combinations of different types of homologation reactions. For example, incorporating sequential lithiation–borylation and Matteson homologation steps (see Section 4.1) followed by Morken's amination protocol (see Section 2.2) afforded 89 in 52% yield (Scheme 13b), an intermediate in the synthesis of (+)-kalkitoxin (90).59 Sequential homologations were also utilised in the synthesis of (+)-hydroxyphthioceranic acid (92) from boronic ester 91 in 12.4% overall yield (Scheme 13c).
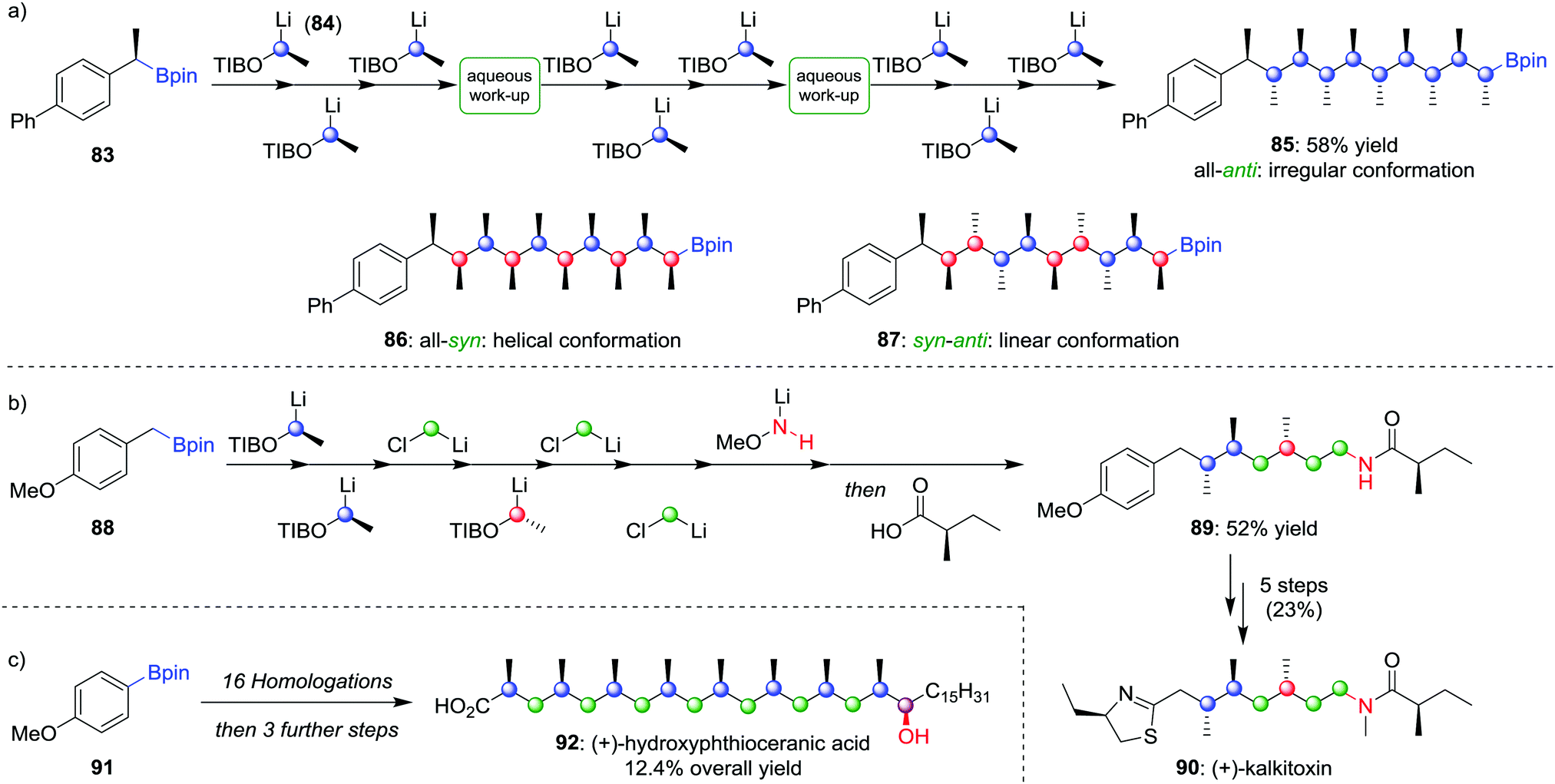 | ||
| Scheme 13 Assembly-line synthesis used to form molecules with contiguous stereocentres by using multiple homologations. OTIB = 2,4,6-triisopropylbenzoate. | ||
4.4 Olefinations and alkynylations
The selective formation of a single alkene isomer from a borane starting material was first reported by Zweifel and co-workers in 1967.60 In their seminal study, it was found that the addition of iodine to alkenyldialkylborane 93 (formed by hydroboration across an alkyne) results in the formation of iodonium ion 94, which, upon migration of an alkyl substituent of the boron, is opened to afford β-iodo-organoborane 95 (Scheme 14a). Deboronoiodination of this intermediate (usually facilitated by hydroxide or methoxide as a base) leads to cis-alkene 96. The reaction was subsequently expanded to the coupling of a boronic ester (98) with a vinyllithium (97) by Evans and co-workers (Scheme 14b).61 Owing to the stereospecific nature of the 1,2-metallate rearrangement, the reaction can be used to couple enantioenriched boronic esters with olefinic functional groups without loss of stereochemical integrity.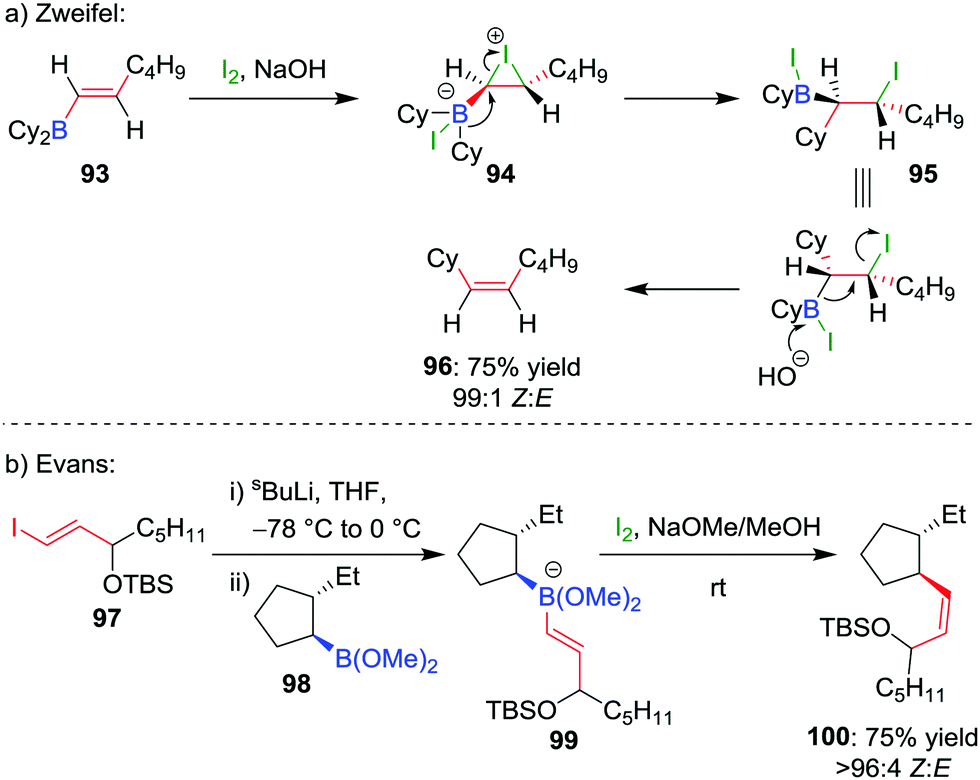 | ||
| Scheme 14 Early examples of the Zweifel olefination reaction applied to boranes and boronic esters. | ||
Our research group has shown that upon subjection of tertiary benzylic boronic esters to Zweifel olefination conditions, vinylmagnesium bromide adds not once but three times to the boronic ester, to form boronate complex 101 (Scheme 15a).42 The addition of four equivalents of vinylmagnesium bromide leads to complete conversion to this boronate complex, which, upon addition of iodine and NaOMe/MeOH, affords the olefination product 102 in 79% yield and 100% es.
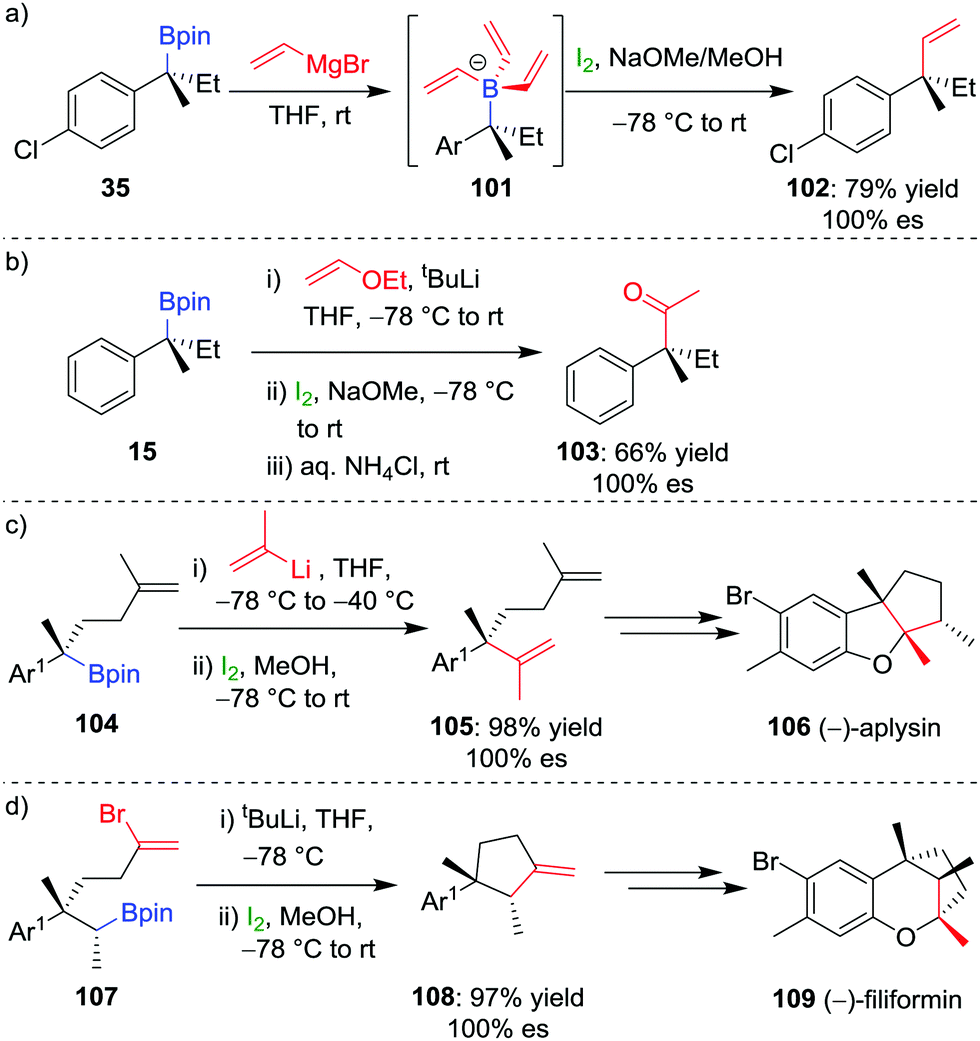 | ||
| Scheme 15 Application of the Zweifel olefination reaction to the functionalization of secondary and tertiary boronic esters. Ar = p-ClC6H4, Ar1 = 2-MeO-4-Me-C6H3. | ||
In further studies within our group, we have identified that Zweifel olefination conditions can be applied to the homologation of a boronic ester to the corresponding methyl ketone using ethoxy vinyllithium (Scheme 15b).26,42 We have also applied this versatile reaction in synthesis – propenylation of 104 affords compound 105 as an intermediate in the synthesis of (−)-aplysin 106 (Scheme 15c),62 and an intramolecular Zweifel reaction of compound 107 was used in the synthesis of (−)-filiformin 109 (Scheme 15d).56
The deboronoiodination of intermediate 95 occurs through an anti elimination process and gives access to the cis alkene. We reasoned that access to the alternative trans isomer would be facilitated through a syn elimination step.63 Replacing iodine with PhSeCl as the electrophile, followed by chemoselective oxidation of the β-selenoboronate intermediate 111 to the corresponding selenoxide 112, initiates syn elimination of selenium and boron (Scheme 16a). This protocol allowed the coupling of vinyllithium 114 (derived by lithium–halogen exchange from the parent vinylbromide) with boronic ester 28 to afford trans alkene 115 in 74% yield, >98:2 E/Z selectivity and 100% es (Scheme 16b).64 Taken in combination with the Zweifel cis olefination, the two procedures provide stereodivergent access to either alkene isomer.65
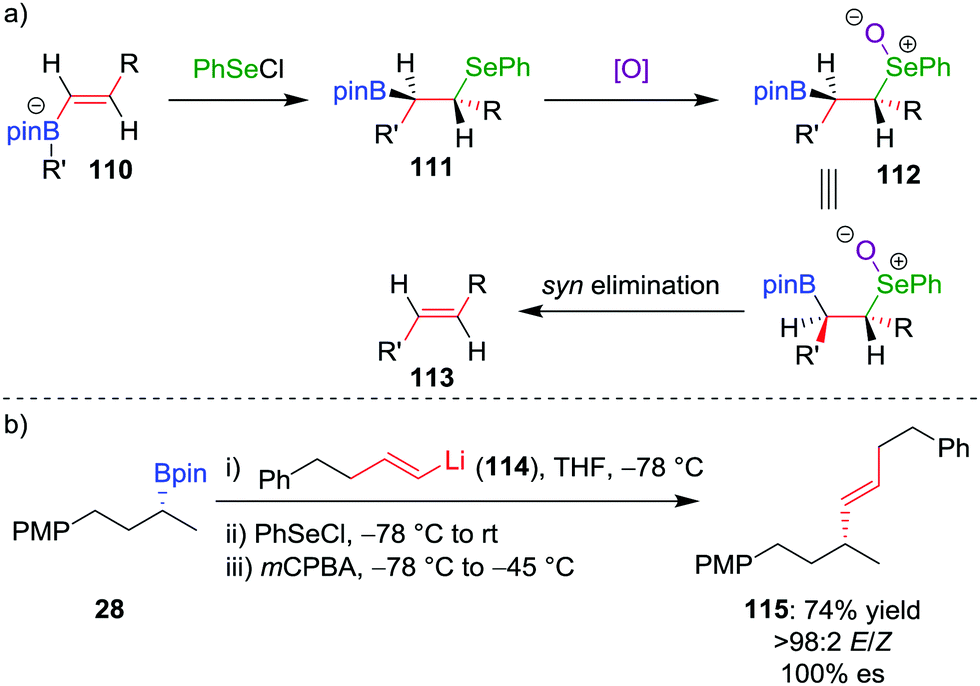 | ||
| Scheme 16 Trans selective variant of the Zweifel olefination reaction. PMP = para-methoxyphenyl. | ||
Extension of olefination techniques to alkynylation is more challenging. The reaction of a metallated alkyne with a boronic ester is reversible,66 and, consequently, addition of iodine affords an iodoalkyne rather than the desired π-electrophilic activation of the alkynyl boronate. However, our group has shown that suitably functionalized alkenes, which can be coupled to boronic esters by using the Zweifel olefination, can give an alkyne through a subsequent 1,2-elimination.67 Deprotonation of vinylbromide by LDA and addition to enantioenriched secondary alkylboronic ester 116, followed by the addition of iodine in methanol, was found to afford 1,1-bromoalkyl-alkene 117 (Scheme 17a). A solution of crude vinyl bromide 117 was then treated with TBAF to achieve elimination and afford the desired alkyne 118 in 81% overall yield and complete enantiospecificity.
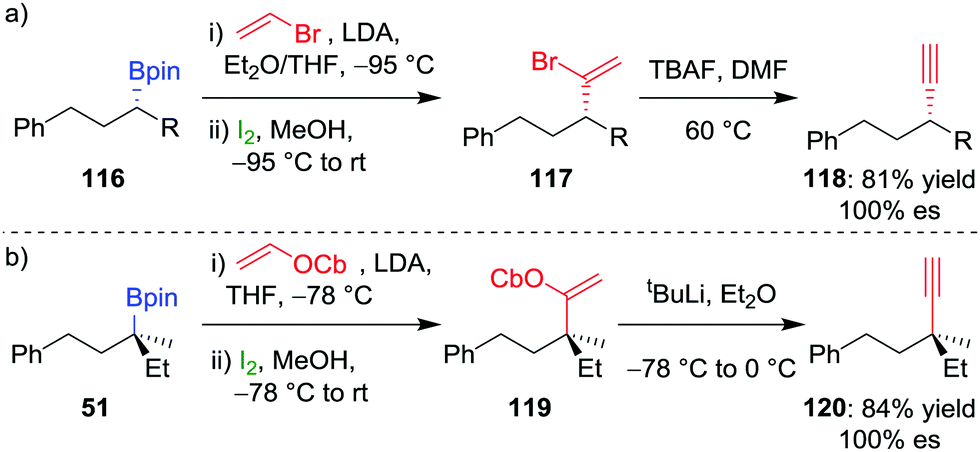 | ||
| Scheme 17 Stereospecific alkynylation of secondary and tertiary boronic esters. R = CH2CH(CH3)2, OCb = N,N-diisopropylcarbamate. | ||
For the alkynylation of tertiary boronic esters, we found that vinyl carbamate is a superior reagent, with the intermediate carbamoylalkylalkene 119 easily undergoing elimination by using tert-butyllithium to afford alkyne 120 in 84% yield and 100% es (Scheme 17b). Secondary benzylic boronic esters also undergo alkynylation by using vinyl carbamate, but the stoichiometry of the tert-butyllithium must be carefully controlled to prevent deprotonation at the benzylic carbon centre and a concomitant reduction in enantiospecificity.
4.5 Addition to aldehydes and iminium ions
During our studies into the reactivity of boronic esters, we observed that benzylic boronic esters react with aldehydes in the presence of CsF, albeit with only 43% es.68 Switching to a benzylic trifluoroborate salt 121 (obtained from the parent boronic ester in excellent yields and stereospecificity)23 and using a catalytic amount of [{RhCl(cod)}2] enabled the addition to aldehyde 122 to be conducted in 86% yield and 100% es, through a stereoretentive pathway (Scheme 18a). The retention of configuration was explained by invoking rhodium acting as a Lewis acid to simultaneously coordinate both the aldehyde and boron compound and facilitate carbon–carbon bond formation (Fig. 6). Both secondary and tertiary benzylic trifluoroborate salts participate in this reaction, and TEMPO-mediated oxidation of the products led to the corresponding ketone 124 without stereoerosion. Yun and co-workers extended this methodology to form γ-butyrolactones 127, using KHF2 as an additive to enable the reaction of benzylic boronic esters, albeit with moderate diastereocontrol (Scheme 18b).69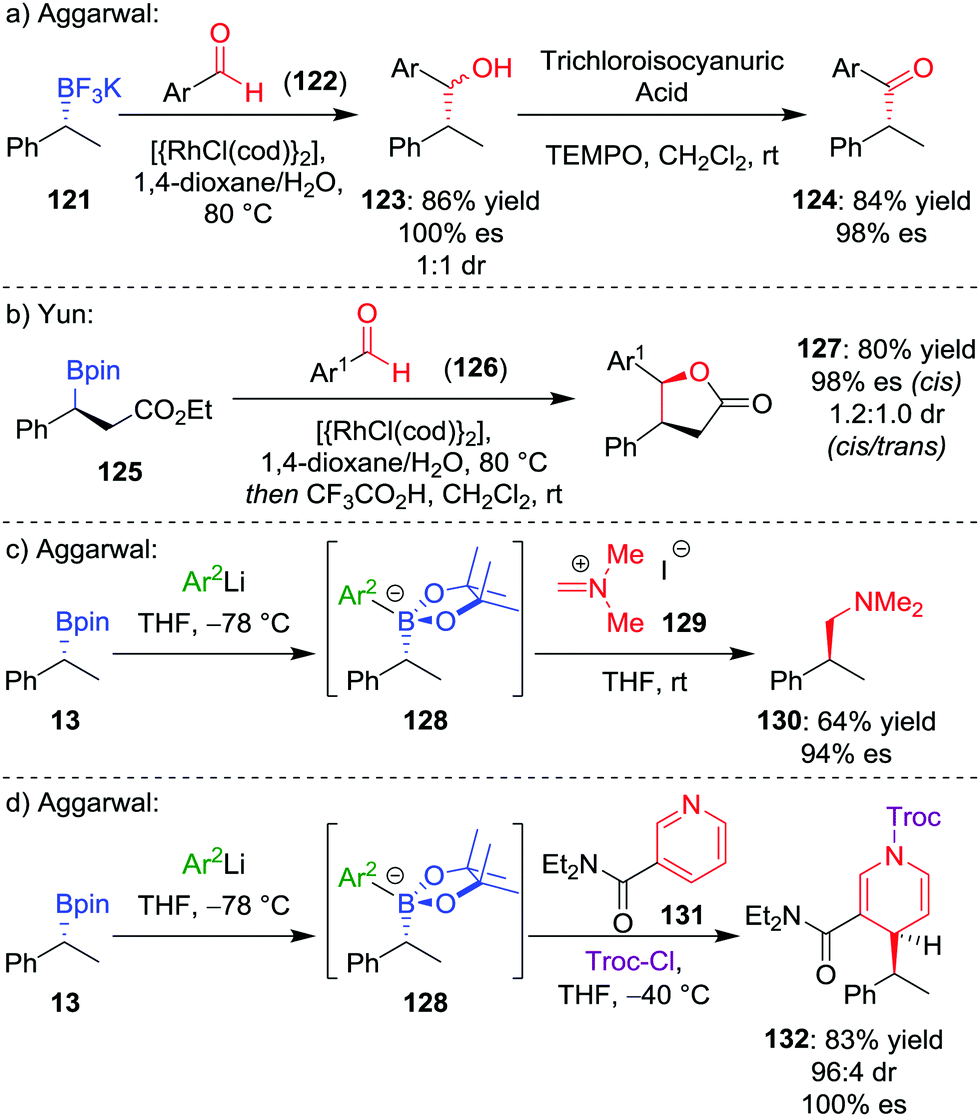 | ||
| Scheme 18 Additions of trifluoroborate salts and boronate complexes to aldehydes and iminium ions. Ar = p-CF3C6H4, Ar1 = p-NO2C6H4, Ar2 = 3,5-(CF3)2C6H3, Troc-Cl = 2,2,2-trichloroethyl chloroformate. | ||
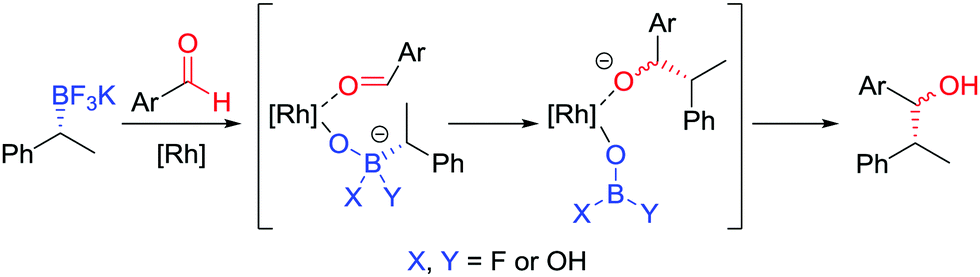 | ||
| Fig. 6 Proposed mechanism for the stereospecific addition of trifluoroborate salts to aldehydes, catalysed by [{RhCl(cod)}2]. | ||
In addition to the reaction of trifluoroborate salts, our group has also shown that boronate complexes (see Section 2.3) react with iminium ions to form C–C bonds without loss of stereochemical integrity. Reaction of boronate 128 with commercially available Eschenmoser's salt 129 gives tertiary amine 130 in 64% yield and 94% es (Scheme 18c).28 Benzylic boronate complexes also react with cyclic iminium ions – the addition of boronate 128 to pyridine 131 (activated using Troc-Cl) affords dihydropyridine 132 in 83% yield, 96:4 dr and 100% es (Scheme 18d).70 Activated quinolines and dihydroisoquinolines can also be used as electrophiles.
4.6 sp2–sp3 coupling reactions
The formation of key C–C bonds by cross-coupling reactions, such as the Suzuki–Miyaura reaction, is one of the most established methods in modern synthesis.4,71 However, the Suzuki–Miyaura reaction is largely limited to the formation of sp2–sp2 bonds, owing to problems of slow transmetalation and competing β-hydride elimination when extended to coupling of sp3 carbon centres.72 Specifically, β-hydride elimination and subsequent hydropalladation leads to racemization of stereogenic centres and loss of stereochemical information. Accordingly, significant research has focussed on the coupling of sp3 organometallic nucleophiles within recent years.73The first general stereospecific coupling of an enantioenriched secondary benzylic boronic ester was achieved by Crudden and co-workers in 2009;74 using [Pd2(dba)3]/PPh3 as the catalyst, boronic ester ent-13 was coupled in a stereoretentive manner with aryl iodide 133 in 62% yield and 91% es (Scheme 19a). Crucially, the use of Ag2O as the base, which increases the rate of the transmetalation step, was found to be critical to obtaining high yields and high levels of enantiospecificity.75 Crudden and co-workers have further extended their methodology to enable the coupling of dibenzylic boronic esters; in this case, neopentyl glycol boronic ester 135 was coupled to give triarylmethane 136 in high yield and enantiospecificity (Scheme 19b).76 Liao and co-workers found that although dibenzylic boronic esters coupled with retention of configuration, the corresponding trifluoroborates coupled with inversion of configuration (Scheme 19c).77
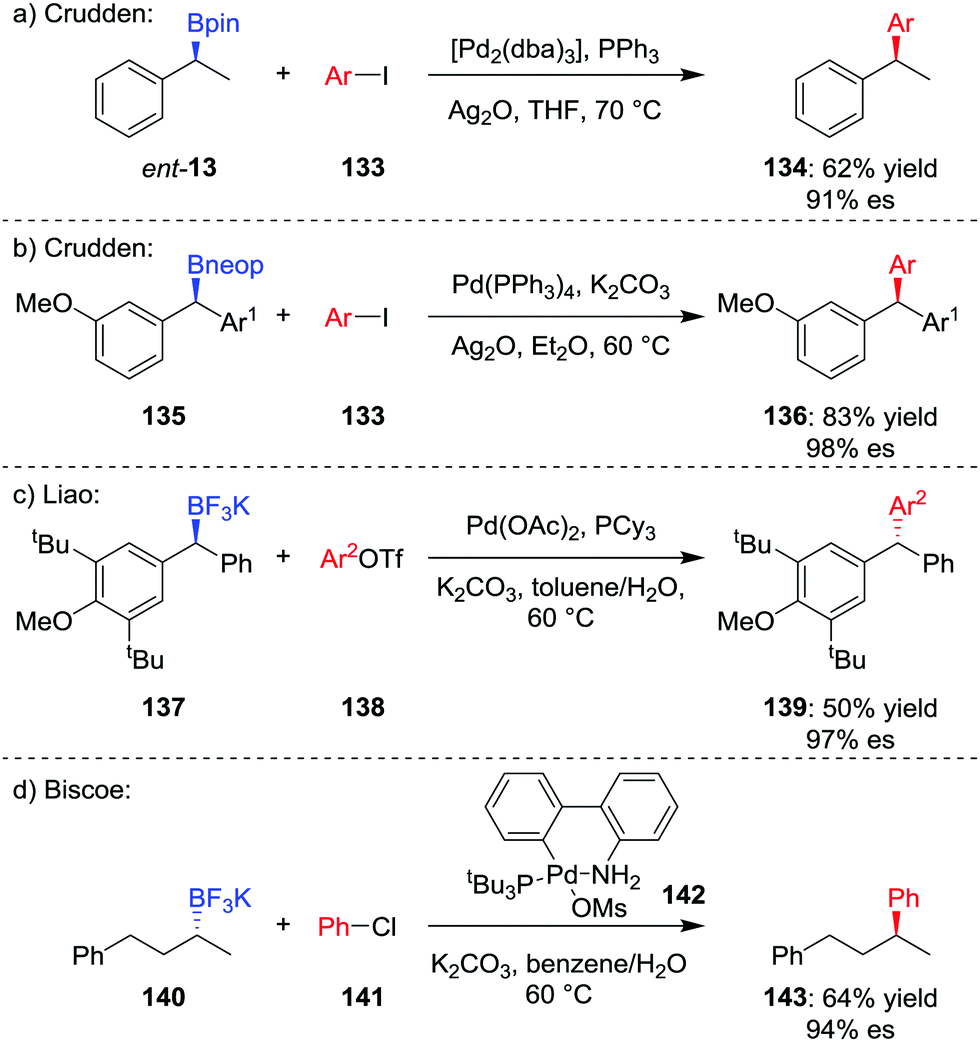 | ||
| Scheme 19 Enantiospecific transition-metal-mediated sp2–sp3 cross-coupling of unactivated secondary boronic esters and trifluoroborate salts. Ar = p-ClC6H4, Ar1 = p-CH3CH2C6H4, Ar2 = p-CH3C6H4, Ms = CH3SO2, neop = neopentyl glycol. | ||
The first report of an enantiospecific Suzuki–Miyaura coupling of an unactivated dialkylboron compound was reported by Biscoe and co-workers in 2014.78 Stereoinvertive coupling of enantioenriched trifluoroborate salt 140 with chlorobenzene 141 was achieved using preformed PtBu3-ligated palladium precatalyst 142,79 affording 143 in 64% yield and 94% es (Scheme 19d). The bulky, electron-rich phosphine ligand is used to suppress β-hydride elimination and subsequent isomerization, as reported by the groups of Dreher and Molander.80
As a consequence of the difficulties associated with transition-metal-mediated sp2–sp3 coupling reactions, particularly the inability to couple tertiary boronic esters under current protocols, we reasoned that the desired C–C bond could be formed using an alternative transition-metal-free route. In our approach, we proposed that activation of the aromatic ring of boronate complex 144 by an electrophile (in an analogous fashion to the activation of the alkene in a Zweifel reaction; see Section 4.4) would facilitate the desired 1,2-metallate rearrangement (Fig. 7).81 Upon nucleophilic attack at the boron centre, the compound would re-aromatize with the elimination of the electrophile, affording the desired coupled product 146 in a stereoretentive fashion.
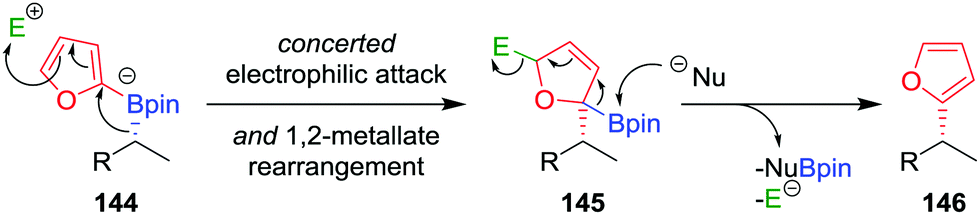 | ||
| Fig. 7 Mechanism for the stereospecific transition-metal-free coupling of boronic esters with furan. E+ = electrophile, Nu− = nucleophile. | ||
We initially tested this reaction manifold on the coupling of 2-lithiofuran (formed by deprotonation of furan by n-butyllithium) with enantioenriched boronic ester ent-20 (Scheme 20a).81a,b Following formation of boronate complex 147, addition of N-bromosuccinimide as the electrophile facilitates the desired coupling in 91% yield and 100% es. Significantly, the procedure can be applied to tertiary boronic esters (Scheme 20b), which are not good substrates for the alternative transition-metal-mediated coupling reactions discussed above. The reaction was also found to be applicable to coupling six-membered aromatics: the reaction of lithiated meta-methoxybenzene with boronic ester ent-20 gave the coupled product in 72% yield and 100% es (Scheme 20c). It was found that an electron-donating group at the meta position of the aromatic ring was essential to prevent reaction of the boronate complex with the electrophile at the sp3 carbon–boron bond (cf. Section 2.3).
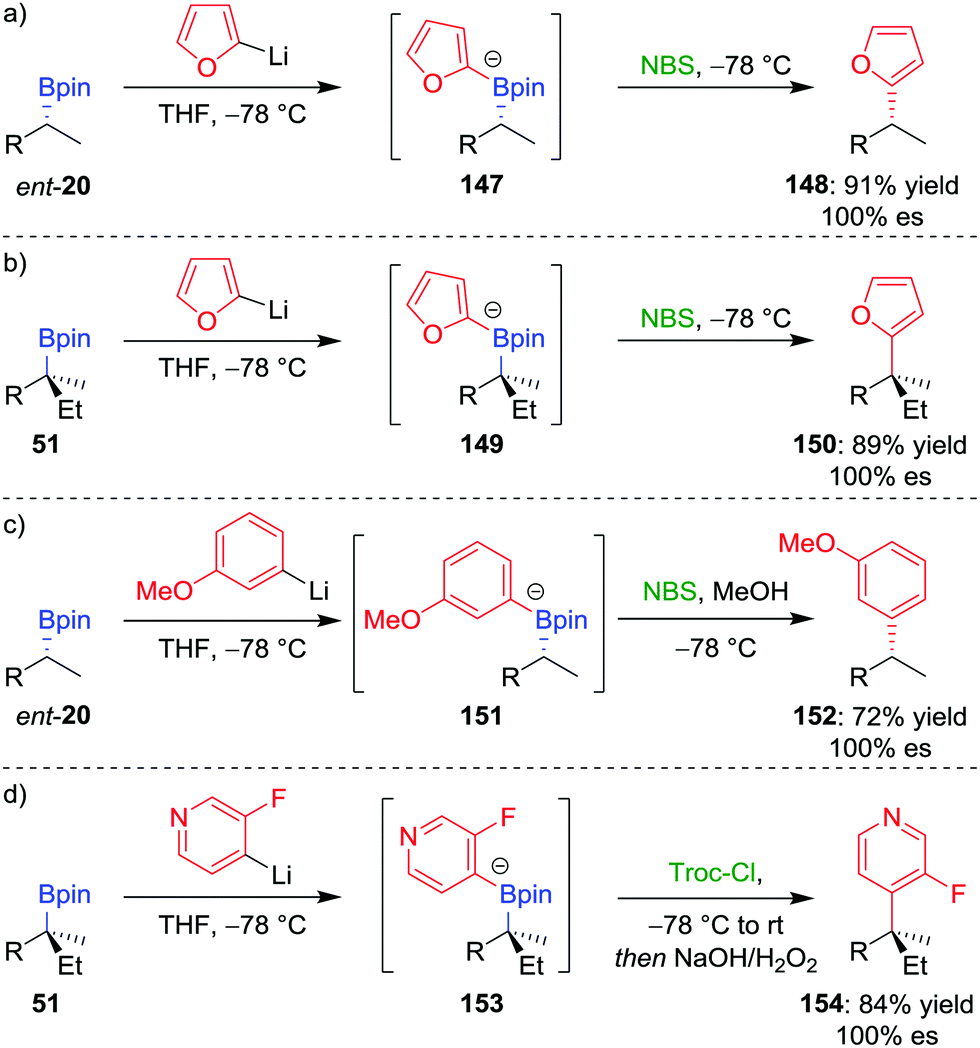 | ||
| Scheme 20 Transition-metal-free cross-coupling reactions of secondary and tertiary boronic esters. R = CH2CH2Ph, NBS = N-bromosuccinimide, Troc-Cl = 2,2,2-trichloroethyl chloroformate. | ||
The scope of this coupling reaction has been extended to include N-heteroaromatic compounds.82 Reminiscent of the reactions involving the addition of nucleophilic boronate complexes to activated cyclic iminium ions (see Section 4.5),70 we proposed that activation of the nitrogen atom of boronate complex 153, through acylation with Troc-Cl, would facilitate 1,2-metallate rearrangement (Scheme 20d). Oxidation of the resulting boronic ester facilitated rearomatization to afford coupled product 154 in 84% yield and with complete retention of configuration.
In a further extension to this methodology, we reasoned that using a carbon-based electrophile would afford intermediate 145 (Fig. 7), but this intermediate would not be susceptible to elimination. Oxidation of the boronic ester and subsequent rearomatization would instead yield a three-component coupling product. To test this strategy,83 we applied electrophilic trifluoromethylation to obtain 2,5-disubstituted furans containing a key CF3 group. For example, trifluoromethylation of boronate complex 155, through the addition of Umemoto's reagent, afforded intermediate 156, which could be oxidised using iodine to give the three-component coupled product 157 in 72% yield and 100% es (Scheme 21a). Tertiary boronic esters could also be used, but the oxidation step was found to be more challenging: only one diastereomer of 160 could be oxidised to the three-component coupled product 159 under the reaction conditions (Scheme 21b). Mechanistic studies determined that the reaction proceeds through a radical propagation cycle; the trifluoromethyl radical was observed by EPR spectroscopy using a spin trapping experiment.
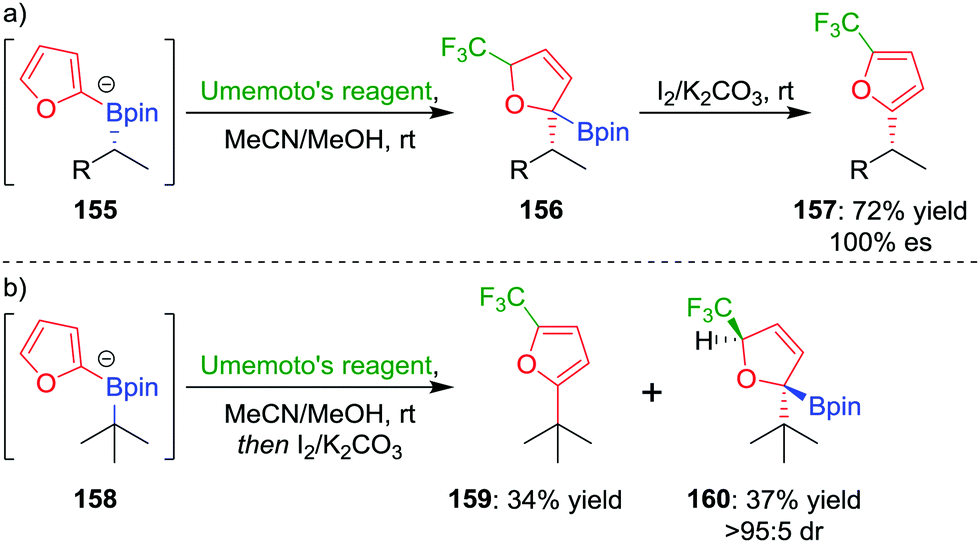 | ||
| Scheme 21 Transition-metal-free three-component couplings of secondary and tertiary boronic esters with trifluoromethylated furans. R = CH2CH2C6H4OMe, Umemoto's reagent = 5-(trifluoromethyl)dibenzothiophenium trifluoromethanesulfonate. | ||
5. Conclusions and outlook
The capability of boronic esters to undergo stereospecific transformations facilitates access to a wide range of enantioenriched building blocks for synthesis. Consequently, new asymmetric methods to create chiral boronic esters represent an efficient route towards diverse functionality. These two research areas are currently developing synergistically to form a major pillar in modern asymmetric synthesis.26,42,84For the main part, the stereospecificity of these reactions results from a 1,2-metallate rearrangement of a boronate complex, in which the carbon–boron σ-bond migrates with retention of configuration. This 1,2-metallate rearrangement has been manipulated to enable a range of new bond-forming reactions, most notably, oxidation, amination, homologation, olefination, alkynylation and arylation reactions. However, in some cases new reactivity pathways for boronic esters have been uncovered: the reaction of nucleophilic boronate complexes with electrophiles, a transformation that occurs with inversion of configuration; complexation to water molecules in protodeboronation; boron–palladium transmetalation without significant β-hydride elimination.
Broad though these methods are, access to certain functional groups remains elusive. Considering homologation reactions, if the desired nucleophile for attacking the boronic ester is too stabilised, it simply decomplexes from the boron centre rather than undergo 1,2-migration. This reluctance to form stable boronate complexes has thwarted some attempts at developing novel functionalization reactions of boronic esters, such as direct alkynylation by using alkynyl anions (see Section 4.4). We anticipate that new strategies to circumvent these problems will provide access to new functionalities, notable current remaining challenges being stereospecific transformations to thiols, nitriles, carboxylic acids, nitro compounds, trifluoromethyl groups, and phosphorus functional groups.
Finally, the stereospecific coupling of secondary and tertiary boronic esters to aromatic groups is a field that has received considerable interest over the last decade, owing to the synthetic utility of the products. Some progress has been made, but further developments to expand its generality are still required. One significant transformation that remains to be achieved is a general traceless sp3–sp3 coupling reaction between an enantioenriched boronic ester and a suitable coupling partner (such as an alkyl halide). The development of such a method would further extend the broad array of transformations available to enantioenriched boronic esters and would be a major step forward for the field of asymmetric synthesis.
Acknowledgements
We acknowledge financial support from EPSRC (EP/I038071/1) the European Research Council (FP7, ERC grant no. 670668) and the University of Bristol. We thank Dr Eddie L. Myers for valuable discussions.References
- (a) H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1961, 83, 486–487 Search PubMed ; see also: ; (b) G. Zweifel and H. C. Brown, J. Am. Chem. Soc., 1964, 86, 393–397 CrossRef CAS; (c) H. C. Brown, M. C. Desai and P. K. Jadhav, J. Org. Chem., 1982, 47, 5065–5069 CrossRef CAS.
- H. C. Brown and B. Singaram, Acc. Chem. Res., 1988, 21, 287–293 CrossRef CAS.
- (a) A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC; (b) D. S. Matteson, J. Organomet. Chem., 1999, 581, 51–65 CrossRef CAS.
- (a) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS; (b) J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- D. S. Matteson, Chem. Rev., 1989, 89, 1535–1551 CrossRef CAS.
- (a) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174–3183 CrossRef CAS PubMed; (b) D. Leonori and V. K. Aggarwal, in Synthesis and Application of Organoboron Compounds, ed. E. Fernández and A. Whiting, Springer International Publishing, Cham, 2015, pp. 271–295 Search PubMed.
- (a) C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695–4712 CrossRef CAS; (b) A.-M. Carroll, T. P. O'Sullivan and P. J. Guiry, Adv. Synth. Catal., 2005, 347, 609–631 CrossRef CAS.
- (a) S. Lee and J. Yun, in Synthesis and Application of Organoboron Compounds, ed. E. Fernández and A. Whiting, Springer International Publishing, Cham, 2015, pp. 73–92 Search PubMed; (b) J. A. Schiffner, K. Müther and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 1194–1196 CrossRef CAS PubMed.
- (a) J. R. Coombs and J. P. Morken, Angew. Chem., Int. Ed., 2016, 55, 2636–2649 CrossRef CAS PubMed; (b) J. Takaya and N. Iwasawa, ACS Catal., 2012, 2, 1993–2006 CrossRef CAS.
- (a) H. K. Scott and V. K. Aggarwal, Chem. – Eur. J., 2011, 17, 13124–13132 CrossRef CAS PubMed; (b) T. Chinnusamy, K. Feeney, C. G. Watson, D. Leonori and V. K. Aggarwal, in Comprehensive Organic Synthesis, ed. G. A. Molander and P. Knochel, Elsevier, Oxford, 2nd edn, 2014, vol. 7, pp. 692–718 Search PubMed.
- H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1961, 83, 2544–2551 CrossRef CAS.
- J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778–782 CrossRef CAS PubMed.
- (a) A. G. Davies and B. P. Roberts, J. Chem. Soc. B, 1967, 17–22 RSC; (b) G. W. Burton and K. U. Ingold, J. Am. Chem. Soc., 1981, 103, 6472–6477 CrossRef CAS; (c) L. Shu and Y. Shi, J. Org. Chem., 2000, 65, 8807–8810 CrossRef CAS PubMed; (d) V. Bagutski, R. M. French and V. K. Aggarwal, Angew. Chem., Int. Ed., 2010, 49, 5142–5145 CrossRef CAS PubMed.
- G. W. Kabalka, T. M. Shoup and N. M. Goudgaon, Tetrahedron Lett., 1989, 30, 1483–1486 CrossRef CAS.
- (a) P. Fontani, B. Carboni, M. Vaultier and G. Maas, Synthesis, 1991, 605–609 CrossRef CAS ; see also: ; (b) M. M. Hussain, H. Li, N. Hussain, M. Ureña, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2009, 131, 6516–6524 CrossRef CAS PubMed.
- (a) C. N. Farthing and S. P. Marsden, Tetrahedron Lett., 2000, 41, 4235–4238 CrossRef CAS; (b) H.-S. Sim, X. Feng and J. Yun, Chem. – Eur. J., 2009, 15, 1939–1943 CrossRef CAS PubMed.
- S. Radomkit and A. H. Hoveyda, Angew. Chem., Int. Ed., 2014, 53, 3387–3391 CrossRef CAS PubMed.
- H. C. Brown, K.-W. Kim, M. Srebnik and S. Bakthan, Tetrahedron, 1987, 43, 4071–4078 CrossRef CAS.
- (a) H. C. Brown, A. Suzui, S. Sonao, M. Itoh and M. M. Midland, J. Am. Chem. Soc., 1971, 93, 4329–4330 CrossRef CAS; (b) H. C. Brown, M. M. Midland and A. B. Levy, J. Am. Chem. Soc., 1972, 94, 2114–2115 CrossRef CAS.
- H. C. Brown, K. W. Kim, T. E. Cole and B. Singaram, J. Am. Chem. Soc., 1986, 108, 6761–6764 CrossRef CAS.
- S. N. Mlynarski, A. S. Karns and J. P. Morken, J. Am. Chem. Soc., 2012, 134, 16449–16451 CrossRef CAS PubMed.
- V. Bagutski, T. G. Elford and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 1080–1083 CrossRef CAS PubMed.
- V. Bagutski, A. Ros and V. K. Aggarwal, Tetrahedron, 2009, 65, 9956–9960 CrossRef CAS.
- D. S. Matteson and G. Y. Kim, Org. Lett., 2002, 4, 2153–2155 CrossRef CAS PubMed.
- B. J. Kim and D. S. Matteson, Angew. Chem., Int. Ed., 2004, 43, 3056–3058 CrossRef CAS PubMed.
- A. P. Pulis, D. J. Blair, E. Torres and V. K. Aggarwal, J. Am. Chem. Soc., 2013, 135, 16054–16057 CrossRef CAS PubMed.
- For other examples of chiral organometallic nucleophiles, see: (a) A. Boudier and P. Knochel, Tetrahedron Lett., 1999, 40, 687–690 CrossRef CAS; (b) R. W. Hoffmann, B. Hölzer, O. Knopff and K. Harms, Angew. Chem., Int. Ed., 2000, 39, 3072–3074 CrossRef CAS; (c) T. Thaler, B. Haag, A. Gavryushin, K. Schober, E. Hartmann, R. M. Gschwind, H. Zipse, P. Mayer and P. Knochel, Nat. Chem., 2010, 2, 125–130 CrossRef CAS PubMed.
- R. Larouche-Gauthier, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2011, 133, 16794–16797 CrossRef CAS PubMed.
- (a) H. C. Brown and C. F. Lane, J. Chem. Soc. D, 1971, 521–522 RSC; (b) H. C. Brown, N. R. De Lue, G. W. Kabalka and H. C. Hedgecock, J. Am. Chem. Soc., 1976, 98, 1290–1291 CrossRef CAS; (c) H. C. Brown and C. F. Lane, Tetrahedron, 1988, 44, 2763–2772 CAS.
- C. Sandford, R. Rasappan and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10100–10103 CrossRef CAS PubMed.
- (a) H. Mayr and M. Patz, Angew. Chem., Int. Ed. Engl., 1994, 33, 938–957 CrossRef; (b) H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS PubMed; (c) G. Berionni, A. I. Leonov, P. Mayer, A. R. Ofial and H. Mayr, Angew. Chem., Int. Ed., 2015, 54, 2780–2783 CrossRef CAS PubMed.
- K. Feeney, G. Berionni, H. Mayr and V. K. Aggarwal, Org. Lett., 2015, 17, 2614–2617 CrossRef CAS PubMed.
- H. C. Brown and K. J. Murray, Tetrahedron, 1986, 42, 5497–5504 CrossRef CAS.
- S. Nave, R. P. Sonawane, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 17096–17098 CrossRef CAS PubMed.
- M. J. Hesse, C. P. Butts, C. L. Willis and V. K. Aggarwal, Angew. Chem., Int. Ed., 2012, 51, 12444–12448 CrossRef CAS PubMed.
- (a) R. Rasappan and V. K. Aggarwal, Nat. Chem., 2014, 6, 810–814 CrossRef CAS PubMed ; see also: ; (b) D. Pozzi, E. M. Scanlan and P. Renaud, J. Am. Chem. Soc., 2005, 127, 14204–14205 CrossRef CAS PubMed; (c) G. Povie, G. Villa, L. Ford, D. Pozzi, C. H. Schiesser and P. Renaud, Chem. Commun., 2010, 46, 803–805 RSC.
- (a) T. G. Elford, S. Nave, R. P. Sonawane and V. K. Aggarwal, J. Am. Chem. Soc., 2011, 133, 16798–16801 CrossRef CAS PubMed; (b) S. Roesner, J. M. Casatejada, T. G. Elford, R. P. Sonawane and V. K. Aggarwal, Org. Lett., 2011, 13, 5740–5743 CrossRef CAS PubMed; (c) V. K. Aggarwal, L. T. Ball, S. Carobene, R. L. Connelly, M. J. Hesse, B. M. Partridge, P. Roth, S. P. Thomas and M. P. Webster, Chem. Commun., 2012, 48, 9230–9232 RSC; (d) S. Roesner and V. K. Aggarwal, Can. J. Chem., 2012, 90, 965–974 CrossRef CAS PubMed; (e) S. Roesner, D. J. Blair and V. K. Aggarwal, Chem. Sci., 2015, 6, 3718–3723 RSC.
- S. P. Thomas, R. M. French, V. Jheengut and V. K. Aggarwal, Chem. Rec., 2009, 9, 24–39 CrossRef CAS PubMed.
- K. M. Sadhu and D. S. Matteson, Organometallics, 1985, 4, 1687–1689 CrossRef CAS.
- (a) H. C. Brown, S. M. Singh and M. V. Rangaishenvi, J. Org. Chem., 1986, 51, 3150–3155 CrossRef CAS; (b) T. J. Michnick and D. S. Matteson, Synlett, 1991, 631–632 CrossRef CAS; (c) R. Soundararajan, G. Li and H. C. Brown, Tetrahedron Lett., 1994, 35, 8957–8960 CrossRef CAS.
- (a) D. S. Matteson and D. Majumdar, Organometallics, 1983, 2, 1529–1535 CrossRef CAS; (b) H. C. Brown, T. Imai, P. T. Perumal and B. Singaram, J. Org. Chem., 1985, 50, 4032–4036 CrossRef CAS; (c) D. S. Matteson, Tetrahedron, 1998, 54, 10555–10607 CrossRef CAS.
- R. P. Sonawane, V. Jheengut, C. Rabalakos, R. Larouche-Gauthier, H. K. Scott and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 3760–3763 CrossRef CAS PubMed.
- (a) M. C. Elliott, K. Smith, D. H. Jones, A. Hussain and B. A. Saleh, J. Org. Chem., 2013, 78, 3057–3064 CrossRef CAS PubMed; (b) M. C. Elliott and K. Smith, Organometallics, 2013, 32, 4878–4881 CrossRef CAS ; see also: ; (c) A. Bottoni, M. Lombardo, A. Neri and C. Trombini, J. Org. Chem., 2003, 68, 3397–3405 CrossRef CAS PubMed.
- A. Chen, L. Ren and C. M. Crudden, J. Org. Chem., 1999, 64, 9704–9710 CrossRef CAS.
- (a) M. W. Rathke, E. Chao and G. Wu, J. Organomet. Chem., 1976, 122, 145–149 CrossRef CAS; (b) D. S. Matteson, H.-W. Man and O. C. Ho, J. Am. Chem. Soc., 1996, 118, 4560–4566 CrossRef CAS.
- K. R. Fandrick, J. A. Mulder, N. D. Patel, J. Gao, M. Konrad, E. Archer, F. G. Buono, A. Duran, R. Schmid, J. Daeubler, J.-N. Desrosiers, X. Zeng, S. Rodriguez, S. Ma, B. Qu, Z. Li, D. R. Fandrick, N. Grinberg, H. Lee, T. Bosanac, H. Takahashi, Z. Chen, A. Bartolozzi, P. Nemoto, C. A. Busacca, J. J. Song, N. K. Yee, P. E. Mahaney and C. H. Senanayake, J. Org. Chem., 2015, 80, 1651–1660 CrossRef CAS PubMed.
- D. S. Matteson and E. C. Beedle, Tetrahedron Lett., 1987, 28, 4499–4502 CrossRef CAS.
- P. J. Unsworth, D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 9846–9850 CrossRef CAS PubMed.
- (a) G. Zweifel, A. Horng and J. T. Snow, J. Am. Chem. Soc., 1970, 92, 1427–1429 CrossRef CAS; (b) G. Zweifel and A. Horng, Synthesis, 1973, 672–674 CrossRef.
- (a) K. Smith, M. C. Elliott and D. H. Jones, J. Org. Chem., 2013, 78, 9526–9531 CrossRef CAS PubMed for examples starting from a vinylboronic ester, see: ; (b) M. Lombardo, S. Morganti, M. Tozzi and C. Trombini, Eur. J. Org. Chem., 2002, 2823–2830 CrossRef CAS; (c) F. Possémé, M. Deligny, F. Carreaux and B. Carboni, J. Org. Chem., 2007, 72, 984–989 CrossRef PubMed for an example of a direct enantioselective SN2′-allylic alkylation without formation of the boronate complex, see: ; (d) L. Carosi and D. G. Hall, Angew. Chem., Int. Ed., 2007, 46, 5913–5915 CrossRef CAS PubMed.
- H. C. Brown, M. V. Rangaishenvi and S. Jayaraman, Organometallics, 1992, 11, 1948–1954 CrossRef CAS.
- (a) J. L. Stymiest, G. Dutheuil, A. Mahmood and V. K. Aggarwal, Angew. Chem., Int. Ed., 2007, 46, 7491–7494 CrossRef CAS PubMed; (b) R. Larouche-Gauthier, C. J. Fletcher, I. Couto and V. K. Aggarwal, Chem. Commun., 2011, 47, 12592–12594 RSC for an alternative approach to the generation of contiguous stereocentres using α-chlorosulfoxides as enantioenriched chiral carbenoid sources, see: ; (c) P. R. Blakemore and M. S. Burge, J. Am. Chem. Soc., 2007, 129, 3068–3069 CrossRef CAS PubMed; (d) C. R. Emerson, L. N. Zakharov and P. R. Blakemore, Org. Lett., 2011, 13, 1318–1321 CrossRef CAS PubMed; (e) X. Sun and P. R. Blakemore, Org. Lett., 2013, 15, 4500–4503 CrossRef CAS PubMed.
- D. Hoppe, F. Hintze and P. Tebben, Angew. Chem., Int. Ed. Engl., 1990, 12, 1422–1424 CrossRef.
- M. J. Dearden, C. R. Firkin, J.-P. R. Hermet and P. O'Brien, J. Am. Chem. Soc., 2002, 124, 11870–11871 CrossRef CAS PubMed.
- (a) G. Dutheuil, M. P. Webster, P. A. Worthington and V. K. Aggarwal, Angew. Chem., Int. Ed., 2009, 48, 6317–6319 CrossRef CAS PubMed; (b) C. A. Brown and V. K. Aggarwal, Chem. – Eur. J., 2015, 21, 13900–13903 CrossRef CAS PubMed; (c) A. Millán, J. R. Smith, J. L. Y. Chen and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 2498–2502 CrossRef PubMed; (d) A. Noble, S. Roesner and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 15920–15924 CrossRef CAS PubMed; (e) A. Varela, L. K. B. Garve, D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 2127–2131 CrossRef CAS PubMed.
- D. J. Blair, C. J. Fletcher, K. M. P. Wheelhouse and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 5552–5555 CrossRef CAS PubMed.
- C. G. Watson, A. Balanta, T. G. Elford, S. Essafi, J. N. Harvey and V. K. Aggarwal, J. Am. Chem. Soc., 2014, 136, 17370–17373 CrossRef CAS PubMed.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183–188 CrossRef CAS PubMed.
- S. Balieu, G. E. Hallett, M. Burns, T. Bootwicha, J. Studley and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 4398–4403 CrossRef CAS PubMed.
- G. Zweifel, H. Arzoumanian and C. C. Whitney, J. Am. Chem. Soc., 1967, 89, 3652–3653 CrossRef CAS.
- D. A. Evans, T. C. Crawford, R. C. Thomas and J. A. Walker, J. Org. Chem., 1976, 41, 3947–3953 CrossRef CAS PubMed.
- C. J. Fletcher, D. J. Blair, K. M. P. Wheelhouse and V. K. Aggarwal, Tetrahedron, 2012, 68, 7598–7604 CrossRef CAS.
- For an example of E olefin synthesis by syn elimination of a borane, see: G. Zweifel, R. P. Fisher, J. T. Snow and C. C. Whitney, J. Am. Chem. Soc., 1972, 94, 6560–6561 CrossRef CAS.
- R. J. Armstrong, C. García-Ruiz, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 786–790 CrossRef CAS PubMed.
- For an alternative recent approach employing an enantioenriched boronic ester to provide access to either alkene isomer, see: Z. Wu, X. Sun, K. Potter, Y. Cao, L. N. Zakharov and P. R. Blakemore, Angew. Chem., Int. Ed., 2016, 55, 12285–12289 CrossRef CAS PubMed.
- H. C. Brown and M. Srebnik, Organometallics, 1987, 6, 629–631 CrossRef CAS.
- Y. Wang, A. Noble, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 4270–4274 CrossRef CAS PubMed.
- A. Ros and V. K. Aggarwal, Angew. Chem., Int. Ed., 2009, 48, 6289–6292 CrossRef CAS PubMed.
- (a) C. Zhang and J. Yun, Org. Lett., 2013, 15, 3416–3419 CrossRef CAS PubMed for a comparable example to form lactams from trifluoroborate salts, see: ; (b) S. Lee, W. M. Lee and J. Yun, Adv. Synth. Catal., 2015, 357, 2219–2222 CrossRef CAS.
- M. Mohiti, C. Rampalakos, K. Feeney, D. Leonori and V. K. Aggarwal, Chem. Sci., 2014, 5, 602–607 RSC.
- (a) V. F. Slagt, A. H. M. de Vries, J. G. de Vries and R. M. Kellogg, Org. Process Res. Dev., 2010, 14, 30–47 CrossRef CAS; (b) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- (a) K. Matos and J. A. Soderquist, J. Org. Chem., 1998, 63, 461–470 CrossRef CAS PubMed; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed.
- For selected recent reviews, see: (a) D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2015, 54, 1082–1096 CrossRef CAS PubMed; (b) C.-Y. Wang, J. Derosa and M. R. Biscoe, Chem. Sci., 2015, 6, 5105–5113 RSC; (c) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed.
- (a) D. Imao, B. W. Glasspole, V. S. Laberge and C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024–5025 CrossRef CAS PubMed ; see also: ; (b) C. M. Crudden, C. Ziebenhaus, J. P. G. Rygus, K. Ghozati, P. J. Unsworth, M. Nambo, S. Voth, M. Hutchinson, V. S. Laberge, Y. Maekawa and D. Imao, Nat. Commun., 2016, 7, 11065 CrossRef PubMed.
- J. Uenishi, J. M. Beau, R. W. Armstrong and Y. Kishi, J. Am. Chem. Soc., 1987, 109, 4756–4758 CrossRef CAS.
- D. C. Matthew, B. W. Glasspole, P. Eisenberger and C. M. Crudden, J. Am. Chem. Soc., 2014, 136, 5828–5831 CrossRef PubMed.
- Y. Lou, P. Cao, T. Jia, Y. Zhang, M. Wang and J. Liao, Angew. Chem., Int. Ed., 2015, 54, 12134–12138 CrossRef CAS PubMed.
- L. Li, S. Zhao, A. Joshi-Pangu, M. Diane and M. R. Biscoe, J. Am. Chem. Soc., 2014, 136, 14027–14030 CrossRef CAS PubMed.
- M. R. Biscoe, B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 6686–6687 CrossRef CAS PubMed.
- S. D. Dreher, P. G. Dormer, D. L. Sandrock and G. A. Molander, J. Am. Chem. Soc., 2008, 130, 9257–9259 CrossRef CAS PubMed.
- (a) A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584–589 CrossRef CAS PubMed; (b) M. Odachowski, A. Bonet, S. Essafi, P. Conti-Ramsden, J. N. Harvey, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2016, 138, 9521–9532 CrossRef CAS PubMed for examples with boranes, see: ; (c) E. R. Marinelli and A. B. Levy, Tetrahedron Lett., 1979, 25, 2313–2316 CrossRef; (d) I. Akimoto and A. Suzuki, Synthesis, 1979, 146–147 CrossRef CAS; (e) M. Ishikura, W. Ida and K. Yanada, Tetrahedron, 2006, 62, 1015–1024 CrossRef CAS.
- J. Llaveria, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10958–10961 CrossRef CAS PubMed.
- Y. Wang, A. Noble, C. Sandford and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 1810–1814 CrossRef CAS PubMed.
- (a) K. Kubota, E. Yamamoto and H. Ito, J. Am. Chem. Soc., 2015, 137, 420–424 CrossRef CAS PubMed; (b) N. Hu, G. Zhao, Y. Zhang, X. Liu, G. Li and W. Tang, J. Am. Chem. Soc., 2015, 137, 6746–6749 CrossRef CAS PubMed; (c) V. M. Shoba, N. C. Thacker, A. J. Bochat and J. M. Takacs, Angew. Chem., Int. Ed., 2016, 55, 1465–1469 CrossRef CAS PubMed; (d) Y. Xi and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 6703–6706 CrossRef CAS PubMed; (e) G. Casoni, E. L. Myers and V. K. Aggarwal, Synthesis, 2016, 3241–3253 CAS; (f) J. Schmidt, J. Choi, A. Tianxiang Liu, M. Slusarczyk and G. C. Fu, Science, 2016, 354, 1265–1269 CrossRef CAS PubMed; (g) D. J. Blair, D. Tanini, J. M. Bateman, H. K. Scott, E. L. Myers and V. K. Aggarwal, Chem. Sci., 2017, 8, 2898–2903 RSC.
| This journal is © The Royal Society of Chemistry 2017 |