
Cobalt nitrides as a class of metallic electrocatalysts for the oxygen evolution reaction†
Pengzuo
Chen‡
a,
Kun
Xu‡
a,
Yun
Tong
a,
Xiuling
Li
ac,
Shi
Tao
b,
Zhiwei
Fang
a,
Wangsheng
Chu
b,
Xiaojun
Wu
ac and
Changzheng
Wu
*a
aHefei National Laboratory for Physical Science at the Microscale, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), Hefei Science Center (CAS) and CAS Key Laboratory of Mechanical Behavior and Design of Materials, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: czwu@ustc.edu.cn
bNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230029, PR China
cCAS Key Laboratory of Materials for Energy Conversion and Department of Material Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
First published on 20th November 2015
Abstract
The development of highly-efficient, stable and cost-effective electrocatalysts for the oxygen evolution reaction (OER) is critical for a range of renewable-energy technologies, including metal–air batteries, fuel cells and water-splitting reactions. However, most of the well-developed electrocatalysts are semiconductors or insulators with poor conductivity, which has profoundly inhibited their overall OER efficiency. In this study, metallic cobalt nitrides (Co2N, Co3N and Co4N) arising from electron delocalization modulation have been investigated for OER electrocatalysts in alkaline solution for the first time. Benefiting from the synergistical engineering of the electrical conductivity and nitrogen content, the simple metallic Co4N catalyst without modifications exhibits a stable current density of 10 mA cm−2 at a small overpotential of 330 mV for OER with a Tafel slope as low as 58 mV dec−1 in alkaline medium, which is superior to most of the unmodified metal oxide electrocatalysts reported to date. Our finding introduces new possibilities for the design of highly active electrocatalysts using synergistical electrical conductivity regulation and composition modulation.
 Changzheng Wu | Changzheng Wu obtained his B.S. degree (2002) and his Ph.D. degree (2007) in the Department of Chemistry at the University of Science and Technology of China (USTC). He then worked as a postdoctoral fellow in the Hefei National Laboratory for Physical Sciences at Microscale. Since 2012, he is a full professor at the Department of Chemistry, USTC. Prof. Wu's current research interests focus on the synthesis and characterization of inorganic 2D nanomaterials, as well as the regulation of their intrinsic physical properties for wide applications in energy storage or energy conversion. He has received a number of prestigious awards, including Top-notch Young Talents of Chinese Organization Department (2015), the Chinese Chemical Society Young Chemist Award (2014), the National Prize for Natural Sciences of China (Second Place) and so on. |
Introduction
Oxygen electrochemistry plays a critical role in the development of efficient energy conversion and storage devices, such as rechargeable metal–air batteries, regenerative fuel cells and water-splitting.1 The oxygen evolution reaction (OER), as one of the most important processes for producing molecular oxygen via the electrochemical oxidation of water, has sparked intensive research in recent years.2 However, the kinetics of the OER is sluggish and suffers from multistep proton-coupled electron transfer, which imposes considerable overpotential requirements.3 Although precious-metal oxides such as RuO2 and IrO2 exhibit superior OER catalytic activity, their low abundance and high cost render them unsuitable for use in large-scale commercial applications.4 Therefore, the development of alternative OER electrocatalysts that can expedite the reaction and reduce the overpotential, thereby enhancing the energy conversion efficiency, is highly desired.Over the past few years, numerous alternative OER electrocatalysts have been exploited, such as 3d-transition-metal oxides,5 hydro(oxy)oxides,6 perovskites,7 transition-metal sulfides,8 and phosphates,9 along with various molecular catalysts.10 Among these well-developed catalysts, 3d-transition-metal compounds, especially cobalt-based compounds, have attracted tremendous attention and have exhibited superior OER electrocatalytic activity because of their exceptional 3d electronic configurations.11 The modulation of electrical behavior is one of the most important methods to promote the OER catalytic activities of the cobalt-based material system. For example, gold-supported cobalt oxide has exhibited a superior OER catalytic performance because of the enhanced electron transport of the semiconducting cobalt oxide mediated by the metallic Au support.12 Moreover, an N-doped graphene/Co3O4 hybrid has also been reported to exhibit high OER activity arising from the synergistic electrical coupling effects between the wide-bandgap semiconducting Co3O4 and the conductive graphene.13 Most of the cobalt-based electrocatalysts are semiconductors or insulators with poor intrinsic conductivity, which has profoundly inhibited their overall water splitting efficiency. Tremendous progress has been achieved through doping certain heteroatoms or incorporating other functional materials.14 However, there is currently a lack of in-depth research into the intrinsic conductivity of the cobalt-based frameworks to further accelerate the electron transport and to facilitate the transition from the surface low oxidation state to higher states, thereby enhancing their catalytic performance.15
Cobalt nitride, as one of the binary nitride systems of 3d metals, has been regarded as a metallic interstitial compound.16 The nitrogen atoms incorporating into the interstices of the cobalt-based framework are bonded covalently to the cobalt atoms combined with the cobalt–cobalt interactions, giving metal-like properties.17 Currently, metallic metal nitrides have been exploited as highly efficient catalysts for the hydrogen evolution reaction, but have rarely been investigated as OER catalysts.18 In this regard, cobalt nitride will be an ideal platform for investigations into the OER electrocatalytic performance of metallic cobalt-based compounds with superior intrinsic conductivity. Notably, although cobalt nitrides hold significant promise as OER catalysts, the relative study has been ignored over the past years. Herein, we report a category of metallic cobalt-based material systems (Co2N, Co3N, and Co4N), with tunable electrical conductivity arising from electron delocalization, representing new metallic OER electrocatalysts. Metallic Co4N without any modification brings synergic advantages of ultrahigh electrical conductivity and modulated nitrogen content, achieving the best OER catalytic activity and excellent stability among the serial cobalt nitrides in alkaline solution.
Experimental section
Preparation of the α-Co(OH)2 and β-Co(OH)2 precursors
The α-Co(OH)2 and β-Co(OH)2 precursors were prepared using a modification of the method reported in the literature.19 In a typical procedure for the preparation of the α-Co(OH)2 precursor, 4 mmol of CoCl2·6H2O, 5 mmol of NaCl, and 8 mmol of hexamethylenetetramine (HMT) were added to a solution of 20 ml of ethanol and 180 ml of distilled water under vigorous stirring. The solution was heated at 90 °C for 1 h and then allowed to cool to room temperature. The final product was collected by centrifugation, washed with water and ethanol several times and then dried under vacuum overnight for further characterization. For the β-Co(OH)2 precursor, the synthetic procedure was similar to that for α-Co(OH)2 with the adjustment of the concentrations of CoCl2·6H2O and HMT to 1.5 mmol and 8 mmol, respectively, as the raw materials.Synthesis of the Co3O4 precursor
In a typical procedure, 200 mg of the as-obtained β-Co(OH)2 precursor was heated at 400 °C for 3 h in air and then cooled to room temperature. The final Co3O4 precursors were obtained in black colour.Synthesis of the metallic Co2N sample
The Co2N sample was obtained from the β-Co(OH)2 precursor by a simple nitridation reaction. In a typical procedure, 50 mg of the β-Co(OH)2 precursor was placed in a tube furnace. The furnace was heated to 380 °C at 8 °C min−1 under a flowing NH3 atmosphere and was maintained at this temperature for 2 h. The furnace was then allowed to cool to room temperature, and the final product was collected for further characterization.Synthesis of the metallic Co3N sample
The Co3N sample was synthesized through a similar nitridation reaction: 50 mg of the α-Co(OH)2 precursor was nitrided with the heating rate of 8 °C min−1 at 380 °C for 2 h to produce the black Co3N sample.Synthesis of the metallic Co4N sample
The Co4N sample was obtained from the Co3O4 precursor via a similar nitridation reaction: 50 mg of the Co3O4 precursor was heated with the heating rate of 8 °C min−1 at 420 °C for 2 h under a flowing NH3 atmosphere and was then allowed to cool to room temperature.Structural characterization
X-ray powder diffraction (XRD) was performed using a Philips X'Pert Pro Super diffractometer with Cu-Kα radiation (λ = 1.54178 Å). Transmission electron microscopy (TEM) and high-resolution TEM (HR-TEM) were performed on a JEM-2100F field-emission electron microscope operated at an acceleration voltage of 200 kV. X-ray photoelectron spectra (XPS) were obtained on an ESCALAB MK II X-ray photoelectron spectrometer with Mg Kα as the excitation source. The binding energies achieved in the XPS spectral analysis were corrected for specimen charging by referencing C 1s to 284.5 eV. The scanning transmission electron microscopy (STEM) images were obtained on a JEM 2100F (field-emission) transmission electron microscope equipped with an Oxford INCA X-sight EDS Si (Li) detector: the microscope was operated at an acceleration voltage of 200 kV. The electrical transport property measurements were carried out using a Keithley 4200-SCS semiconductor characterization system and the two-point probe method.Electrochemical tests
All of the electrochemical measurements were performed in a three-electrode system on an electrochemical workstation (CHI660B). In a typical procedure, 4 mg catalysts (Co2N, Co3N and Co4N) and 40 μl of 5 wt% Nafion solutions (Sigma Aldrich) were dispersed in 1 ml of water–isopropanol solution with a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by sonication for 30 min to form a homogeneous ink. Next, 5 μl of the prepared dispersion was loaded onto a glassy carbon electrode with a diameter of 3 mm (loading: 0.285 mg cm−2). Linear-sweep voltammetry was conducted at a scan rate of 5 mV s−1 in 0.1 M and 1 M KOH solution (purged with oxygen for 30 min) using a Ag/AgCl (3.3 M KCl) electrode as the reference electrode, Pt gauze as the counter electrode, and the glassy carbon electrode with one of the catalysts. All of the potentials in this study are reported vs. Ag/AgCl. The polarization curves were obtained by sweeping the potential from 0 V to 1.0 V at a sweep rate of 5 mV s−1 in an alkaline solution. The stability of the as-obtained catalyst was carried out in an O2-saturated 0.1 M KOH solution at room temperature by applying a static overpotential of 437 mV.
:1 by sonication for 30 min to form a homogeneous ink. Next, 5 μl of the prepared dispersion was loaded onto a glassy carbon electrode with a diameter of 3 mm (loading: 0.285 mg cm−2). Linear-sweep voltammetry was conducted at a scan rate of 5 mV s−1 in 0.1 M and 1 M KOH solution (purged with oxygen for 30 min) using a Ag/AgCl (3.3 M KCl) electrode as the reference electrode, Pt gauze as the counter electrode, and the glassy carbon electrode with one of the catalysts. All of the potentials in this study are reported vs. Ag/AgCl. The polarization curves were obtained by sweeping the potential from 0 V to 1.0 V at a sweep rate of 5 mV s−1 in an alkaline solution. The stability of the as-obtained catalyst was carried out in an O2-saturated 0.1 M KOH solution at room temperature by applying a static overpotential of 437 mV.
Results and discussion
The serial cobalt nitrides were prepared through a series of simple precursor nitridation reactions. Briefly, the corresponding XRD patterns of the serial cobalt nitrides in Fig. 1 demonstrate that the peaks match well with the standard XRD profiles and that no impurities are detected. Fig. 1a shows that the metallic Co2N has an orthorhombic crystal structure (JCPDS card no. 651458), which belongs to the space group Pnnm (58). After the nitrogen content was further decreased, Co3N (space group P63/mmm (182)) with a hexagonal crystal structure is obtained, which is isostructural to Ni3N (space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (221)). Further nitrogen-depletion led to Co4N, which is packed in an fcc arrangement with a smaller nitrogen atom situated at the centre of the unit cell. Moreover, the corresponding crystal structures of the serial cobalt nitrides indicate that the electron cloud overlapping of neighbouring cobalt elements gradually multiplied from Co2N to Co4N (Fig. 1d). This observation led us to expect an obvious enhancement of the electron delocalization and an increase of the electrical conductivity in Co4N, which contains more metal Co atoms in each unit cell. In order to further demonstrate the metallic properties of cobalt nitrides based on experimental evidence, analysis of the temperature-dependent resistivity of the cobalt nitrides was carried out. As shown in Fig. 1e, the corresponding resistivity values of the cobalt nitrides slightly increase with increasing temperature from 100 K to 320 K, indicating a typical metal-like character. More importantly, the electrical conductivity of the cobalt nitrides was obviously enhanced with decreasing nitrogen content in the Co2N, Co3N, and Co4N samples. In this case, the as-obtained Co4N with the most nitrogen-depleted crystal structure exhibits an extremely low resistivity of 6.0 × 10−6 Ω m at room temperature, which is 6.3 times lower than the measured resistivity of Co3N and 7.7 times lower than that of Co2N. This result reflects the higher intrinsic conductivity of the Co4N sample resulting from the lower N content in the Co-based framework.
m (221)). Further nitrogen-depletion led to Co4N, which is packed in an fcc arrangement with a smaller nitrogen atom situated at the centre of the unit cell. Moreover, the corresponding crystal structures of the serial cobalt nitrides indicate that the electron cloud overlapping of neighbouring cobalt elements gradually multiplied from Co2N to Co4N (Fig. 1d). This observation led us to expect an obvious enhancement of the electron delocalization and an increase of the electrical conductivity in Co4N, which contains more metal Co atoms in each unit cell. In order to further demonstrate the metallic properties of cobalt nitrides based on experimental evidence, analysis of the temperature-dependent resistivity of the cobalt nitrides was carried out. As shown in Fig. 1e, the corresponding resistivity values of the cobalt nitrides slightly increase with increasing temperature from 100 K to 320 K, indicating a typical metal-like character. More importantly, the electrical conductivity of the cobalt nitrides was obviously enhanced with decreasing nitrogen content in the Co2N, Co3N, and Co4N samples. In this case, the as-obtained Co4N with the most nitrogen-depleted crystal structure exhibits an extremely low resistivity of 6.0 × 10−6 Ω m at room temperature, which is 6.3 times lower than the measured resistivity of Co3N and 7.7 times lower than that of Co2N. This result reflects the higher intrinsic conductivity of the Co4N sample resulting from the lower N content in the Co-based framework.
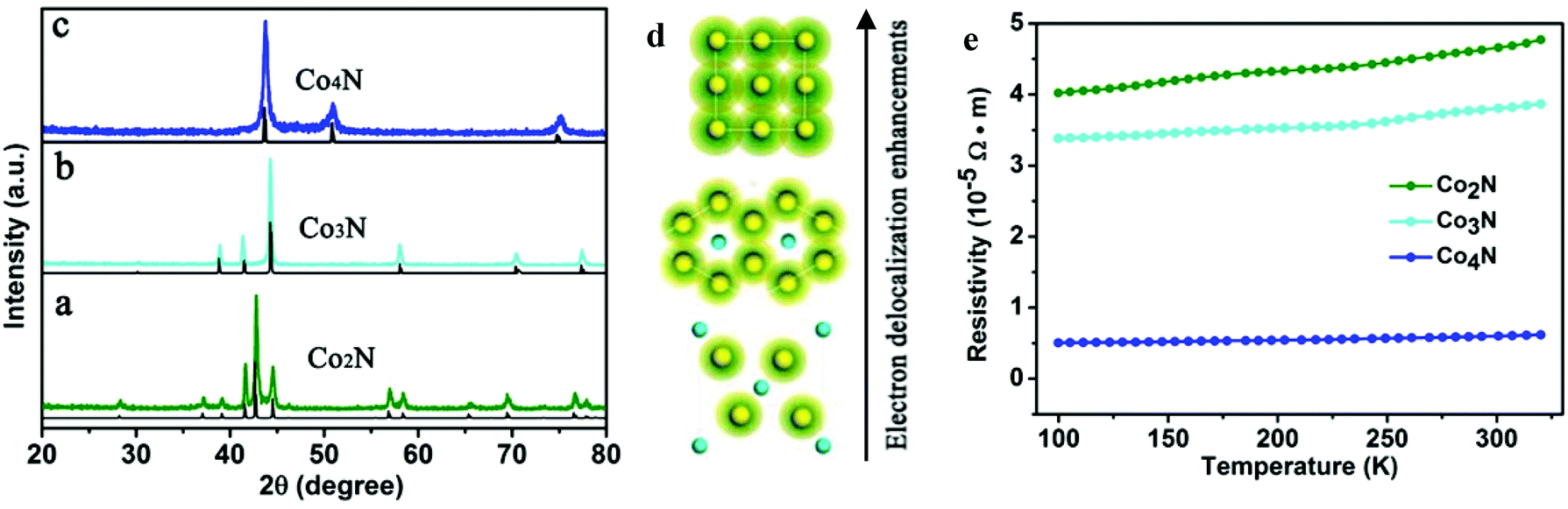 | ||
| Fig. 1 X-ray diffraction (XRD) patterns of (a) Co2N, (b) Co3N and (c) Co4N. (d) Schematic of the electron delocalization for the serial cobalt nitrides viewed from the c-orientation. (e) Temperature-dependent electrical resistivity of serial cobalt nitrides. | ||
Microscopic characterization was also performed to further analyse the phases of all of the as-obtained products. The high resolution transmission electron microscopy (HRTEM) images are shown in Fig. 2a–c, which confirm that lattice fringes of all serial cobalt nitrides match with their lattice fringes well, verifying the crystalline cobalt nitrides. Moreover, electron energy loss spectroscopy (EELS) was performed to analyse the Co and N elements in our obtained cobalt nitrides. The presence of Co and N in the as-prepared cobalt nitride catalysts was confirmed by the EELS analysis of the K-edges of N on the Co2N, Co3N and Co4N products, as shown in Fig. S4, S5a† and Fig. 2d. All of the EELS spectra exhibit ionization edges at ca. 398.3 eV, which is consistent with the K-shell of the N element, and the peak located at 780.2 eV corresponds to the edges of Co–L.20 Moreover, EELS line-scanning profiles and the mapping images were collected to further analyse the elemental distribution in our cobalt nitrides. Fig. S4, S5b† and Fig. 2e confirm that the compositions of the as-prepared products consist of Co and N, with no other obvious contaminants detected in the cobalt nitrides. Finally, the EELS element mapping images of the serial cobalt nitrides in Fig. S4, S5(c–f)† and Fig. 2f, where the Co (indicated by green) and N (indicated by red) exhibit homogeneous spatial distributions, indicate that the serial cobalt nitrides were successfully prepared. Therefore, these results clearly reveal that the obtained cobalt nitrides are composed of Co and N. As previously described, with the incorporation of N into the Co framework, serial metallic cobalt nitrides with a wide range of continuously adjustable components were developed. Therefore, both excellent conductivity and modulated nitrogen content coexisting in these metallic cobalt nitrides are expected to bring enhancement of the OER catalytic activity.
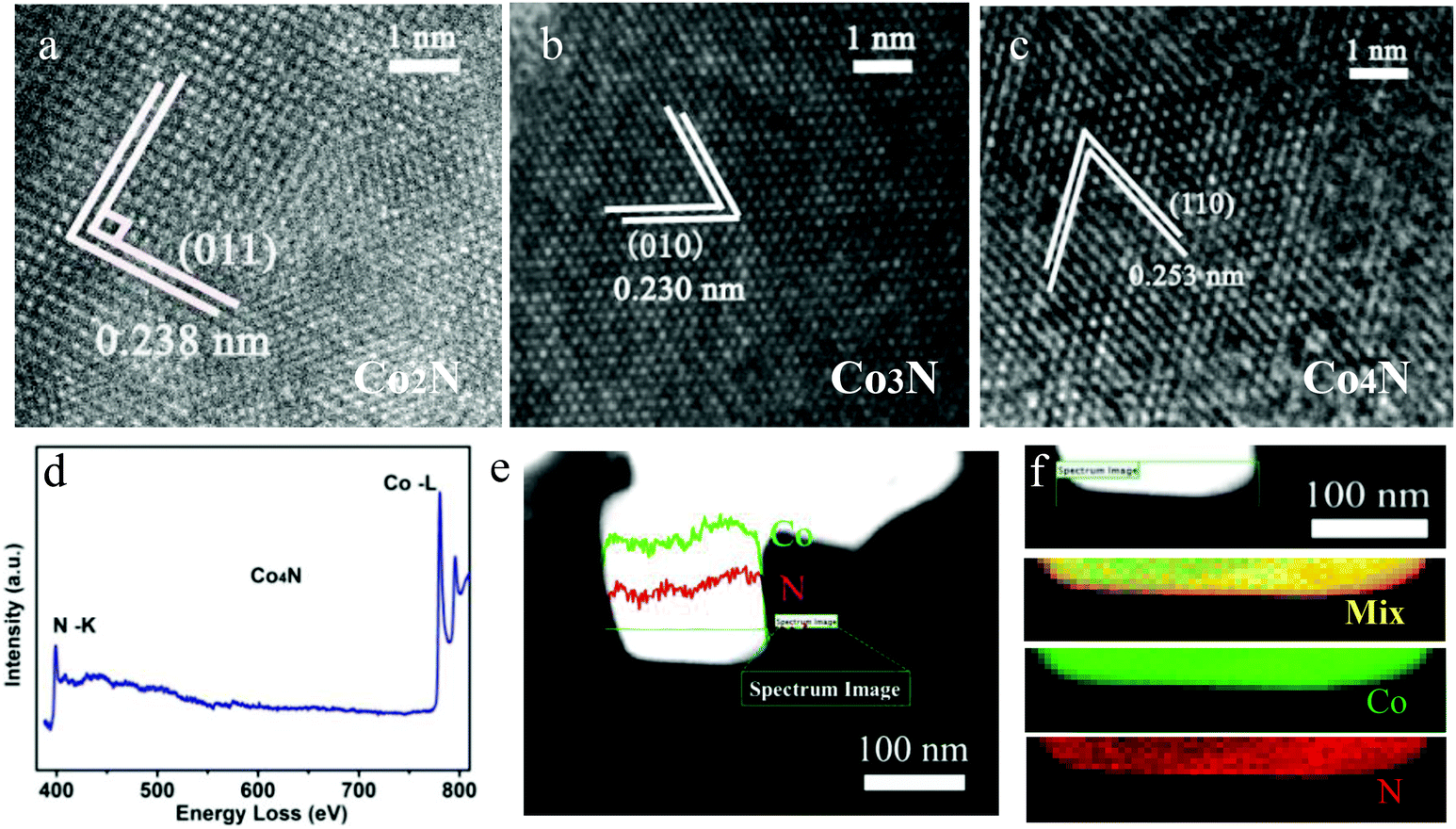 | ||
| Fig. 2 HRTEM images of the (a) Co2N, (b) Co3N and (c) Co4N catalysts. (d) Electron energy loss spectroscopy (EELS), (e) EELS line scanning profiles of the Co4N sample. (f) The typical HAADF-STEM and EELS element mapping images of the obtained Co4N product. | ||
To demonstrate the role of the intrinsic conductivity and the modulated nitrogen content in cobalt nitrides, the OER catalytic properties of the serial cobalt nitrides were initially evaluated by linear-sweep voltammetry (LSV) in a 0.1 M KOH solution. As shown in Fig. 3a, Co4N exhibits a higher current density than that of other catalysts at the same applied potential and achieves a smaller overpotential of 370 mV compared with that of the Co2N (510 mV) and Co3N (470 mV) catalysts at a current density of 10 mA cm−2. This result is comparable to the commercial RuO2 catalyst of the previous reported work (Table S2†). In addition, the corresponding IR-corrected Tafel plots [η vs. log(j) ] in Fig. 3b exhibit a Tafel slope of 80 mV dec−1 in the case of the Co4N electrode, which is significantly smaller than those of Co2N (145 mV dec−1) and Co3N (130 mV dec−1). It is noted that metallic Co4N has a further decrease in the slope value compared to the other catalysts (Fig. 3b). These phenomena were related to the resistance discrepancy for materials and charge transfer on the surface state, both of which have influence upon their Tafel slopes. Moreover, the measured currents were further normalized by BET surface areas to obtain the normalized current density (Fig. S6†). As shown in Fig. S8,† the Co4N also exhibits the best OER catalytic performance among the investigated catalysts, and the varying pattern of the OER catalytic activities for each catalyst is consistent with the analysis results prior to normalization, thereby indicating the intrinsic OER catalytic performance of all of the as-prepared catalysts.
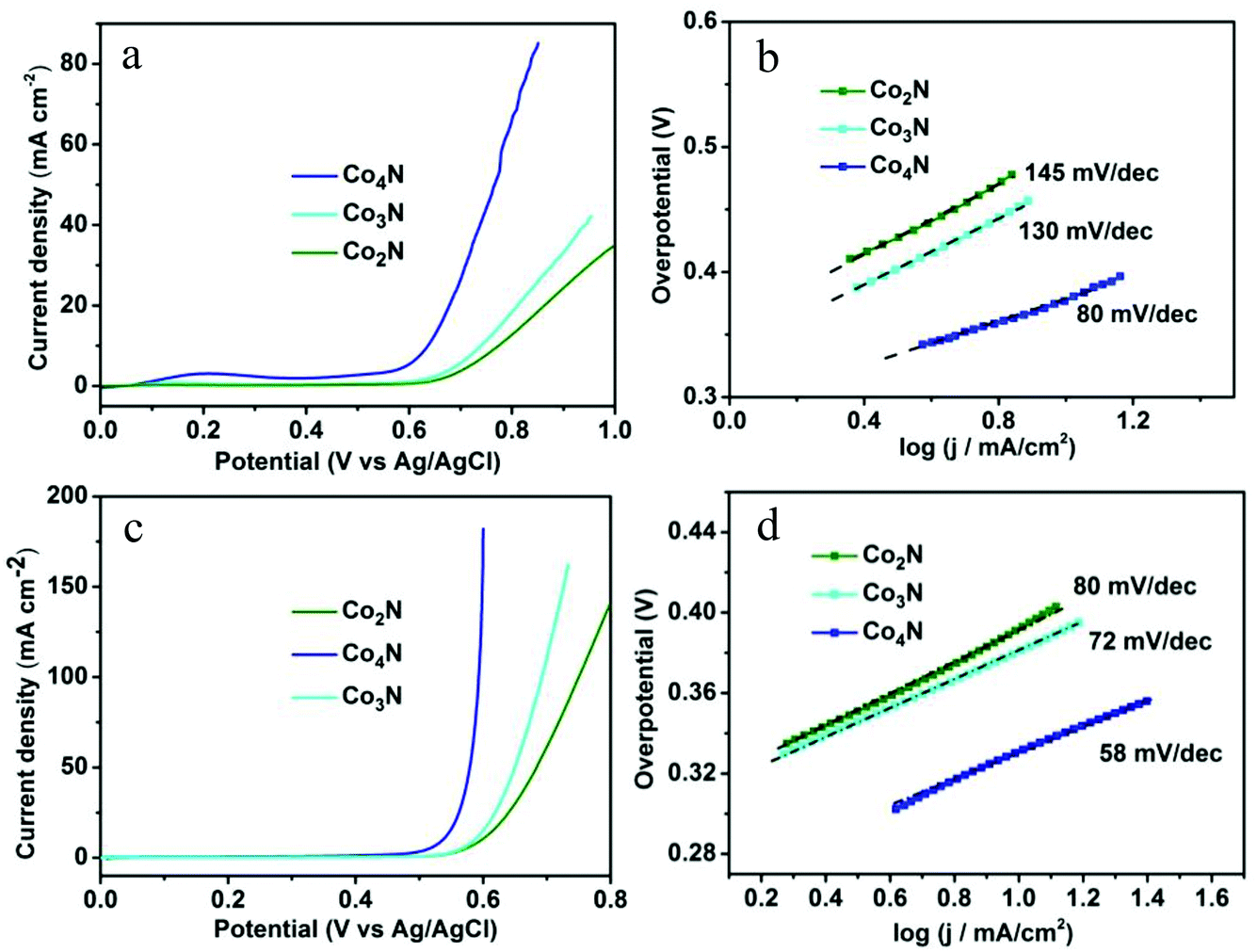 | ||
| Fig. 3 (a) IR-corrected polarization curves for the serial cobalt nitrides. (b) The corresponding Tafel plots in 0.1 M KOH solution. (c) IR-corrected polarization curves for all catalysts. (d) The corresponding Tafel plots in 1 M KOH solution. | ||
Given the significance of being able to control the electrical conductivity and the composition in the design of the OER catalysts, the catalytic activities of all of the catalysts were further investigated in a 1 M KOH solution. As shown in Fig. 3c, the nitrogen-depleted Co4N exhibited a higher current density of 10 mA cm−2 at a smaller overpotential of 330 mV in the 1 M solution due to the greater ionic conduction in the higher-concentration oxygen-saturated electrolyte. This low overpotential value is much better than most of the metal oxide electrocatalysts without modifications reported to date.5b Meanwhile, the nitrogen-depleted Co4N also exhibits the lowest Tafel slope of 58 mV dec−1 among the serial cobalt nitrides, further confirming the significance of the intrinsic conductivity and the modulated nitrogen content in metallic Co4N catalyst (Fig. 3d). In addition, the varying pattern of the OER electrocatalytic performance for serial cobalt nitrides is in accordance with the measured results achieved in 0.1 M KOH solution.
Stability is another significant criterion by which to evaluate the catalysts. The stability of the metallic Co4N in the OER process was evaluated via long-term cycling tests performed in a 0.1 M KOH solution. After the 1000 cycle test, the Co4N catalyst maintained the same performance exhibited in the initial cycle (Fig. 4a), indicating that the metallic Co4N exhibits superior stability in a long-term electrochemical process. Moreover, a continuous OER test under a static potential was performed to further verify the stability of the cobalt nitrides under alkaline conditions. As shown in Fig. 4b, when an overpotential of 437 mV was applied, the current density of Co4N exhibited only a slight degradation (4.5%) after a long period of 13000 seconds, revealing its excellent stability under the OER conditions. By contrast, the current densities of the Co2N and Co3N decreased gradually with time (only maintaining 65.9% and 58.5% of their initial current densities after 13000 seconds, respectively, as shown in Fig. 4b). The amount of oxygen evolved from water with increasing electrocatalytic time and the Faradaic efficiency of the metallic Co4N catalyst were measured using a fluorescence-based oxygen sensor (Fig. 4c). The bulk electrolysis tests were performed in a 0.1 M KOH solution in a gas-tight electrochemical cell under an inert atmosphere and under an applied continuous overpotential of 437 mV. In general, the theoretical amount of produced O2 molecule was calculated by assuming that all of the charge was generated from the 4e− oxidation of water. As shown in Fig. 4c, the amount of oxygen matches the theoretical amount of oxygen during the process of electrolysis well, corresponding to a Faradaic efficiency of >98% in 60 min. All the above results demonstrate that the metallic Co4N could serve as a highly effective OER electrocatalyst.
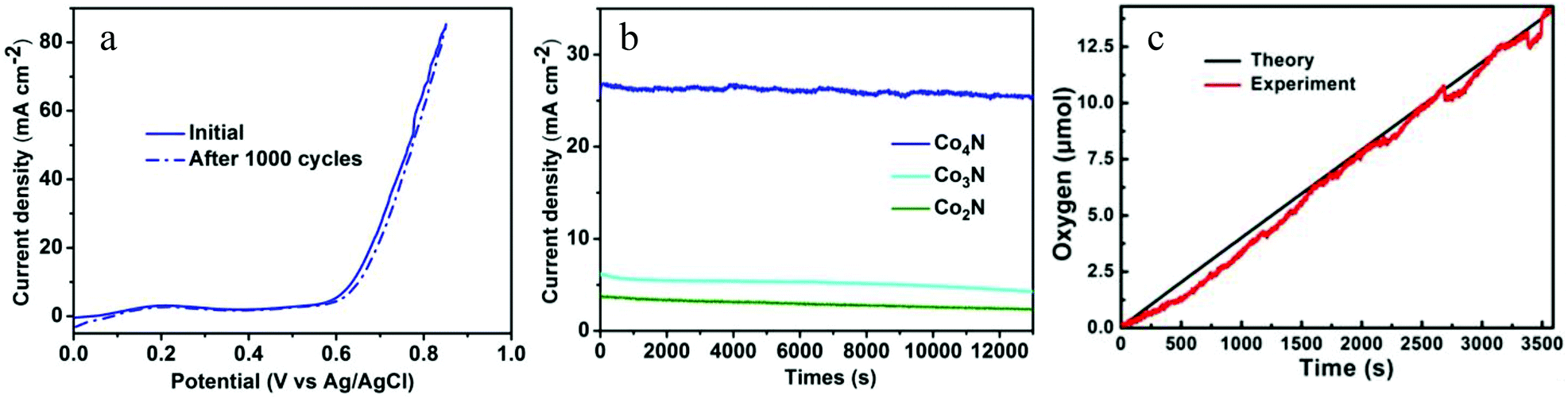 | ||
| Fig. 4 (a) Stability of Co4N with an initial polarization curve and after 1000 cycles in a 0.1 M KOH solution. (b) Time dependence of the current density under a static overpotential of 437 mV in 0.1 M KOH solution. (c) Theoretical (black plot) and experimental (red plot) amounts of O2 produced for the Co4N catalyst measured using a fluorescence-based oxygen sensor. | ||
Currently, a possible mechanism for Co-based OER electrocatalysts has already been proposed in an alkaline medium.15,21 For example, in our case, a part of Co atoms on the surface of cobalt nitrides are firstly oxidized and transformed into CoOOH to form the CoOOH/CoxN (x = 2, 3, 4) complex species as the OER catalytically active sites, which then promote the oxidation of OH− into O2. The CoOOH is then further oxidized to CoO2 to form the CoO2/CoxN (x = 2, 3, 4) complex, which is a more efficient species for the water oxidation process.22 Therefore, the effective nitrogen content modulation brings about the enhancement of intrinsic metallic characteristics in the Co4N sample, enabling its more facile oxidation reactions to overcome the kinetically sluggish OER process, and thereby largely enhancing its electrocatalytic activity. Considering the excellent performance of the simple, unmodified metallic Co4N as the only catalyst, it is expected that further improvement of the electrocatalytic activity would be realized by doping certain heteroatoms, morphology modification or incorporating other functional materials.
Conclusions
In conclusion, by utilization of electron delocalization modulation, a category of metallic electrocatalyst systems i.e., the serial cobalt nitrides (Co2N, Co3N, and Co4N), with tunable conductivity and continuously adjustable components, have been reported for efficient OER electrocatalysts. Due to the synergic advantages of ultrahigh electrical conductivity and modulated nitrogen content, metallic Co4N catalyst without any modification shows the best OER catalytic activity among the serial cobalt nitrides in alkaline medium. Briefly, this work not only offers a facile and scalable strategy for fabricating metallic cobalt nitrides as highly-efficient OER catalysts but also experimentally demonstrates that the synergistic effects of the electrical transport and the modulated composition in cobalt nitrides play a critical role in optimizing water oxidation. This approach therefore holds great promise for improving the activity of various electrocatalysts.Acknowledgements
This work was financially supported by the National Basic Research Program of China (2015CB932302), the National Natural Science Foundation of China (21222101, U1432133, 11132009, 21331005, 11321503, U1532265, J1030412), the Chinese Academy of Sciences (XDB01020300), the Fok Ying-Tong Education Foundation, China (Grant No. 141042), the Fundamental Research Funds for the Central Universities (WK2060190027) and the foundation of China Postdoctoral Science Foundation (No. 2015M580539).Notes and references
- (a) J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed; (b) H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed; (c) T. E. Mallouk, Nat. Chem., 2013, 5, 362 CrossRef CAS PubMed.
- (a) E. Mirzakulova, R. Khatmullin, J. Walpita, T. Corrigan, N. M. Vargas-Barbosa, S. Vyas, S. Oottikkal, S. F. Manzer, C. M. Hadad and K. D. Glusac, Nat. Chem., 2012, 4, 794 CrossRef CAS PubMed; (b) K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119 CrossRef CAS PubMed.
- (a) Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 Search PubMed; (b) T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2014, 53, 7281 CrossRef CAS PubMed; (c) Y. Hou, Z. Wen, S. Cui, S. Ci, S. Mao and J. Chen, Adv. Funct. Mater., 2015, 25, 872 CrossRef CAS.
- (a) F. Li, B. Zhang, X. Li, Y. Jiang, L. Chen, Y. Li and L. Sun, Angew. Chem., Int. Ed., 2011, 50, 12276 CrossRef CAS PubMed; (b) V. Petrykin, K. Macounova, O. A. Shlyakhtin and P. Krtil, Angew. Chem., Int. Ed., 2010, 49, 4813 CrossRef CAS PubMed.
- (a) Z. Lu, H. Wang, D. Kong, K. Yan, P.-C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CAS; (b) C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed; (c) C. Zhang, M. Antonietti and T.-P. Fellinger, Adv. Funct. Mater., 2014, 24, 7655 CrossRef CAS; (d) Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976–3982 RSC.
- (a) M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077 CrossRef CAS PubMed; (b) F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CAS; (c) Z. Zhao, H. Wu, H. He, X. Xu and Y. Jin, Adv. Funct. Mater., 2014, 24, 4698 CrossRef CAS.
- (a) J.-I. Jung, H. Y. Jeong, J.-S. Lee, M. G. Kim and J. Cho, Angew. Chem., Int. Ed., 2014, 53, 4582 CrossRef CAS PubMed; (b) M. Risch, K. A. Stoerzinger, S. Maruyama, W. T. Hong, I. Takeuchi and Y. Shao-Horn, J. Am. Chem. Soc., 2014, 136, 5229 CrossRef CAS PubMed; (c) Y. Guo, Y. Tong, P. Chen, K. Xu, J. Zhao, Y. Lin, W. Chu, Z. Peng, C. Wu and Y. Xie, Adv. Mater., 2015, 27, 5989 CrossRef CAS PubMed.
- (a) W. Zhou, X.-J. Wu, X. Cao, X. Huang, C. Tan, J. Tian, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921 RSC; (b) J. Xie, R. Wang, J. Bao, X. Zhang, H. Zhang, S. Li and Y. Xie, Inorg. Chem. Front., 2014, 1, 751–756 RSC.
- K. Jin, J. Park, J. Lee, K. D. Yang, G. K. Pradhan, U. Sim, D. Jeong, H. L. Jang, S. Park, D. Kim, N.-E. Sung, S. H. Kim, S. Han and K. T. Nam, J. Am. Chem. Soc., 2014, 136, 7435 CrossRef CAS PubMed.
- (a) Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342 CrossRef CAS PubMed; (b) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238 CrossRef CAS PubMed.
- (a) X. Long, J. Li, S. Xiao, K. Yan, Z. Wang, H. Chen and S. Yang, Angew. Chem., Int. Ed., 2014, 53, 7584–7588 CrossRef CAS PubMed; (b) J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS; (c) J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat Mater., 2011, 10, 780 CrossRef CAS PubMed.
- (a) D. Hou, W. Zhou, K. Zhou, Y. Zhou, J. Zhong, L. Yang, J. Lu, G. Li and S. Chen, J. Mater. Chem. A, 2015, 3, 15962 RSC; (b) W. Zhou, Y. Zhou, L. Yang, J. Huang, Y. Ke, K. Zhou, L. Li and S. Chen, J. Mater. Chem. A, 2015, 3, 1915 RSC; (c) W. Zhou, J. Zhou, Y. Zhou, J. Lu, K. Zhou, L. Yang, Z. Tang, L. Li and S. Chen, Chem. Mater., 2015, 27, 2026 CrossRef CAS.
- N. H. Chou, P. N. Ross, A. T. Bell and T. D. Tilley, ChemSusChem, 2011, 4, 1566–1569 CrossRef CAS PubMed.
- R. Juza and W. Sachsze, Z. Anorg. Chem., 1945, 253, 95 CrossRef CAS.
- K. Oda, T. Yoshio and K. Oda, J. Mater. Sci., 1987, 22, 2729 CrossRef CAS.
- (a) J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615 RSC; (b) W.-F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed, 2012, 51, 6131 CrossRef CAS PubMed; (c) B. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186 CrossRef CAS PubMed; (d) P. Chen, K. Xu, Z. Fang, Y. Tong, J. Wu, X. Lu, X. Peng, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 1470 Search PubMed.
- Z. Liu, R. Ma, M. Osada, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2005, 127, 13869 CrossRef CAS PubMed.
- N. Fontaíña-Troitiño, S. Liébana-Viñas, B. Rodríguez-González, Z.-A. Li, M. Spasova, M. Farle and V. Salgueiriño, Nano Lett., 2014, 14, 640 CrossRef PubMed.
- Z. Zhao, H. Wu, H. He, X. Xu and Y. Jin, Adv. Funct. Mater., 2014, 24, 4698 CrossRef CAS.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: XRD, TEM, EELS mapping and additional electrochemical data. See DOI: 10.1039/c5qi00197h |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |