
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron bis(oxazoline) complexes in asymmetric catalysis
Thierry
Ollevier
Département de chimie, Pavillon Alexandre-Vachon, Université Laval, 1045 avenue de la Médecine, Québec (Qc) G1V 0A6, Canada. E-mail: thierry.ollevier@chm.ulaval.ca
First published on 23rd October 2015
Abstract
Asymmetric reactions catalyzed by iron complexes have attracted considerable attention because iron is a ubiquitous, inexpensive, and environmentally benign metal. Various chiral iron complexes can be prepared from bis(oxazoline) ligands and be used in asymmetric reactions. This overview charts the development and application of chiral iron bis(oxazoline) and pyridine-2,6-bis(oxazoline) catalysts through their most prominent and innovative uses in asymmetric catalysis, especially in Lewis acid and oxidation catalysis.
 Thierry Ollevier | Born in Brussels, Thierry Ollevier obtained his B.Sc. (1991) and Ph.D. (1997) at the Université de Namur, Belgium, and was postdoctorate fellow at the Université catholique de Louvain, Belgium under István E. Markó (1997), NATO postdoctorate fellow at Stanford University under Barry M. Trost (1998–2000), then postdoctorate fellow at the Université de Montréal under André B. Charette (2000–2001). After an Assistant Professor appointment (2001) at Université Laval, he became Associate (2006) and is currently Full Professor. Current research in his group aims at designing novel catalysts, developing catalytic reactions and applying these methods to chemical synthesis. He is active in the areas of Lewis acids, asymmetric catalysis, organic chemistry in aqueous conditions, and green synthetic chemistry. |
1. Introduction
Main chiral metal catalysts are key elements in the toolbox of organic chemists. Iron is one of the most abundant metals on earth; it is inexpensive, environmentally benign, and relatively nontoxic in comparison with other metals. From a green chemistry aspect, it is interesting to develop new iron-catalyzed methods.1 Indeed, many catalysts used in asymmetric synthesis are derived from noble and rare metals and their price or toxicity prevent their use on an industrial scale. Iron, which is ubiquitous, is thus becoming one of the most promising transition metals. Various reviews have been published in the field of asymmetric catalysis using iron.2 This article aims at reviewing the catalytic applications of FeII and FeIII complexes derived from oxazoline ligands. Both bis(oxazoline) (box) and pyridine-2,6-bis(oxazoline) (pybox) have been used as chiral ligands with various metals.3 Their use, conjointly with iron salts, is gaining increasing attention. The present review covers the most prominent uses of bis(oxazoline) and pyridine-2,6-bis(oxazoline) as chiral ligands for enantioselective iron catalysis and is primarily organized according to the type of chiral ligand used. Various box and pybox will be presented, ranging from bidentate to tetradentate oxazoline-based ligands for both FeII and FeIII. Polydentate oxazolines have been used as highly effective chiral ligands for asymmetric catalysis. Nitrogen donor atoms with the in-plane lone pair combined with additional binding sites result in highly stereodirecting ligands. The resulting chiral iron complexes have been used in a broad range of catalytic transformations, particularly in Lewis acid catalysis. This account is an introduction to the most significant developments on the use of iron bis(oxazoline) complexes, including elements about the geometry of selected complexes and applications of these catalysts as Lewis acids.2. Iron bis(oxazoline) catalysts
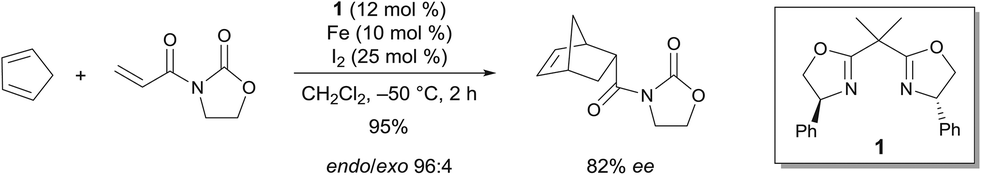
Seminal work by Corey demonstrated the efficiency of a C2-symmetric chiral bis(oxazoline) FeIII complex for the enantioselective Diels–Alder reaction.4 The reaction of 3-acryloyl-1,3-oxazolidin-2-one and cyclopentadiene in the presence of 10 mol% of the catalyst, which was prepared from a chiral bis(oxazoline), Fe and I2, led to an excellent yield of the endo product (endo/exo 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) with a very good enantioselectivity (82% ee) for the endo product (Scheme 1). The Lewis acid can either be prepared from Fe and I2 or from FeCl2 and I2. In both cases, after complexation with 1, the catalytic species is believed to be 1·FeX2+. Reasonable models of the dienophile bound to this one were proposed based on the knowledge of the metal geometry to rationalize the stereoselectivity of the process. The geometry of 1·FeX2+ is believed to be square-planar. This would lead to the predominant formation of the axial–equatorial chelate of the dienophile.
:4) with a very good enantioselectivity (82% ee) for the endo product (Scheme 1). The Lewis acid can either be prepared from Fe and I2 or from FeCl2 and I2. In both cases, after complexation with 1, the catalytic species is believed to be 1·FeX2+. Reasonable models of the dienophile bound to this one were proposed based on the knowledge of the metal geometry to rationalize the stereoselectivity of the process. The geometry of 1·FeX2+ is believed to be square-planar. This would lead to the predominant formation of the axial–equatorial chelate of the dienophile.
 | ||
| Scheme 1 FeIII-catalyzed enantioselective Diels–Alder reaction. | ||
The concept of chiral relay was disclosed by Sibi in 2006. Achiral fluxional additives containing multiple sites for modification were reported to amplify the enantioselectivity of the above-mentioned Diels–Alder reaction in the presence of 1 and Fe(ClO4)2.5 However, the origin of ee enhancements from these fluxional additives is not completely clear.
Fe catalyzed cycloisomerization reactions have been reported using an Fe catalyst obtained in situ via the reduction of Fe(acac)3 with Et3Al (3 equivalents) in the presence of a bis(oxazoline) ligand.6 Only moderate levels of stereoinduction were disclosed though.
Pfaltz reported the preparation of chiral diamino-bis(oxazoline) ligands and their use as FeII complexes.7 When the ligands were derived from N,N′-dimethylcyclohexane-1,2-diamine, the resulting complexes showed different coordination modes depending on the ligand diastereoisomer used: the Fe complex adopted either a pentacoordinate (2a) or an hexacoordinate (2b) geometry (Scheme 2a). The ionic versions of the Fe complexes with SbF6− as weakly coordinating anion were also prepared to generate two vacant coordination sites. Ligands prepared from N,N′-dimethylethane-1,2-diamine reacted with FeCl2 in a stereoselective manner to give an octahedral mononuclear complex 3, after subsequent removal of the chloride anions using 2 equiv. of AgSbF6 (Scheme 2b). Preliminary catalytic tests for the epoxidation of trans-β-methyl-styrene showed that the Fe complexes were less active and less enantioselective than analogous Mn-based catalysts.
 | ||
| Scheme 2 Chiral FeII diamino-bis(oxazoline) complexes prepared by Pfaltz. | ||
Fe complexes of spiro-bis(oxazoline) ligands 4 and 5a were highly efficient catalysts for asymmetric O–H bond insertion reactions.8 Based on the lower enantioselectivities obtained with other chiral ligands in the O–H insertion reaction, the rigid spiro scaffold of ligands 4 and 5 appeared to be essential for a high level of chiral induction. The complexes catalyzed insertions into the O–H bond of a wide variety of alcohols with excellent enantioselectivities under mild reaction conditions (Scheme 3). Interestingly, allylic alcohols could also be used in these conditions and no competitive cyclopropanation was detected. The Fe complex also catalyzed the asymmetric O–H insertion reaction between water and methyl α-diazophenylacetate with outstanding enantioselectivities. Higher enantioselectivites were obtained when using Fe as compared to other transition metals, including Cu and Rh, used under the same reaction conditions.
 | ||
| Scheme 3 FeII catalyzed O–H bond insertion reactions. | ||
The same authors also demonstrated that the complexes prepared in situ from chiral spiro bis(oxazoline) 5b and an FeII salt was competent to catalyze the C–H functionalization of indoles with α-aryl-α-diazoesters.9 α-Aryl α-indolyl-acetate derivatives were isolated as the reaction products in high yields and high enantioselectivities (up to 78% ee) (Scheme 4). Other chiral bis(oxazoline) and pyridine-2,6-bis(oxazoline) ligands were less efficient in this reaction. The α-aryl-α-diazoester is believed to be decomposed by the FeII catalyst into an FeII carbene. The enantioselective proton migration on an FeII zwitterionic intermediate is most likely the rate-determining step, as demonstrated by kinetic isotope effect experiments. Alternative mechanisms cannot be ruled out though. The efficiency of this FeII catalytic system was also exploited in the asymmetric intramolecular cyclopropropanation of α-diazoesters.10 Using chiral ligand 5b, Fe(ClO4)2·4H2O in the presence of NaBArF (sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate), [3.1.0]bicycloalkane lactone derivatives were obtained in high yields and excellent enantioselectivities (up to 97% ee) (Scheme 4). Based on the stereochemistry of the products, the authors suggested that the cyclopropanation undergoes a concerted process. According to various reaction features (higher reactivity of FeIIvs. FeIII, higher reactivity of electron-rich alkenes and observation of fumarates and maleates as by-products), electrophilic FeII carbenoids are most likely involved in the process.
 | ||
| Scheme 4 FeII catalyzed reactions with α-diazoesters. | ||
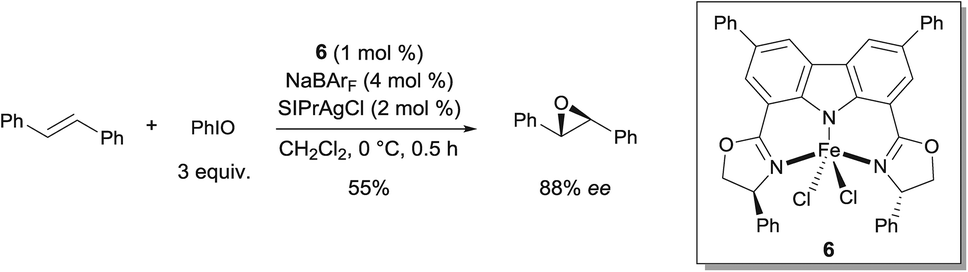
In 2012, Niwa and Nakada developed a FeII complex bearing a carbazole-based tridentate ligand that catalyzes the asymmetric epoxidation of (E)-alkenes with excellent enantioselectivity (Scheme 5).11 They found that complex 6, which was prepared from FeCl2·4H2O and a tridentate carbazole ligand, in the presence of NaBArF, catalyzed the asymmetric epoxidation of trans-stilbene to afford the corresponding chiral epoxide in moderate yield and enantioselectivity. The use of 2 mol% of SIPrAgCl as an additive improved both yield and enantioselectivity of the reaction (55% and 88% ee). The key intermediate in the epoxidation was demonstrated to be an FeIV-oxo complex bearing a π-cation radical. The Fe complex bearing the carbazole ligand behaves in a similar way as Fe porphyrins in this oxidation reaction, as demonstrated by UV-vis and EPR analysis. When an FeIII complex derived from tridentate bis(oxazolinylphenyl)amine (bopa) ligand 7 (Scheme 6), which lacks the C–C bond between the two phenyl rings, such as in the carbazole-based tridentate system 6, was used, no epoxidation occurred.
 | ||
| Scheme 5 Asymmetric epoxidation of an alkene with a FeIII carbazole–bis(oxazoline) complex. | ||
 | ||
| Scheme 6 Asymmetric hydrosilylation of aryl ketones catalysed by Fe bopa complexes. | ||
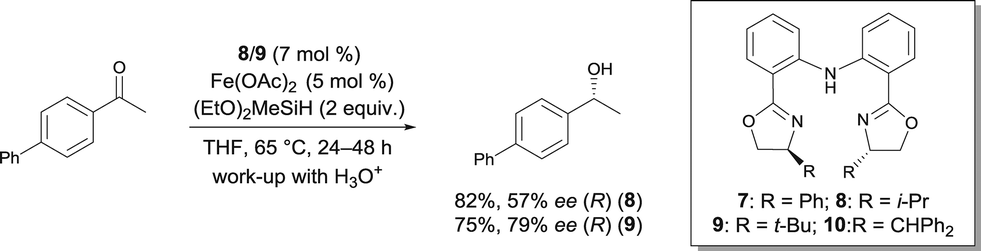
In 2007, Nishiyama investigated the asymmetric hydrosilylation of ketones using iron catalysis.12 The addition of chiral tridentate bis(oxazoline-phenyl)amine (bopa) 8 and 9 to Fe(OAc)2 in THF at 65 °C formed in situ a catalytically active species to mediate the hydrosilylation of ketones with (EtO)2MeSiH (Scheme 6). The isolated yields were up to 82% and the enantioselectivity of the obtained alcohols were 57 and 79% ee respectively. Interestingly, the use of a chiral pyridine-2,6-bis(oxazoline) (pybox) led to a lower enantioselectivity (37% ee, Scheme 17).
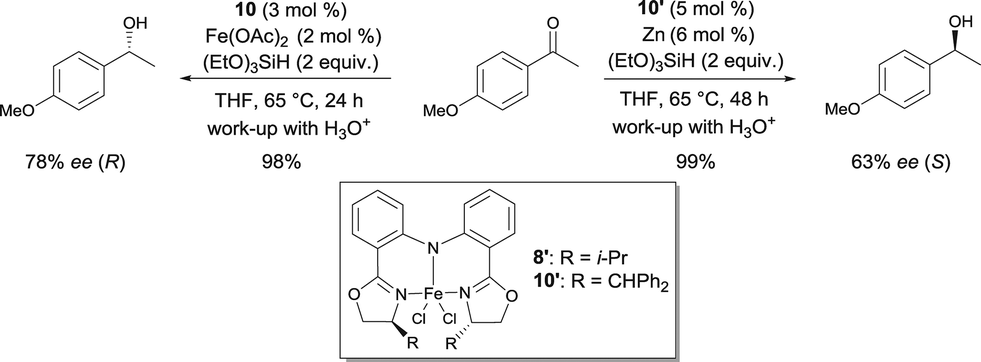
Nishiyama was able to improve the asymmetric hydrosilylation of ketones by ligand design. Bulky substituents on the oxazoline ring led to higher enantioselectivity (up to 88% ee).13 To elucidate the mechanism of the reaction, the differences between well-defined complexes and in situ prepared catalytic systems were investigated.14 Interestingly, more hindered ligand 10 (Scheme 6, R = CHPh2) can afford both enantiomers of the product either used as in situ formed catalyst (Fe(OAc)2/10) or as well-defined iron complex 10′, conjointly with zinc as an activator (Scheme 7). The authors suggest that zinc serves as a reducing agent for the reduction of FeIII to FeII. Control experiments ruled out the possibility that only a Zn bopa complex was responsible for the asymmetric induction.
 | ||
| Scheme 7 Asymmetric hydrosilylation of aryl ketones catalysed by in situ or well-defined Fe bopa complexes. | ||
Other trans-chelating tridentate ligands were reported to be efficient ligands for various metals. The resulting complexes have been characterized as cationic aqua complexes.15 In particular, (R,R)-4,6-dibenzofurandiyl-2,2′(4-phenyloxazoline) 11 was reported to be an appropriate ligand for FeII.15,16 The obtained complex with Fe(ClO4)2 was an efficient catalyst for the Diels–Alder reaction of cyclopentadiene with 3-acryloyl-2-oxazolidinone, leading to high yield and excellent stereoselectivity (Scheme 8). The absolute configuration of the product can be easily deduced from the structure of the complex coordinated to the substrate. Structural evidence was provided from the analogous Ni complex, which appeared to be a square bipyramidal structure containing an octahedral NiII. Although the complex of Fe(ClO4)2 gave an excellent selectivity, Fe(ClO4)3·nH2O (n = 6–9) was poorly selective (−40 °C, 67% ee).
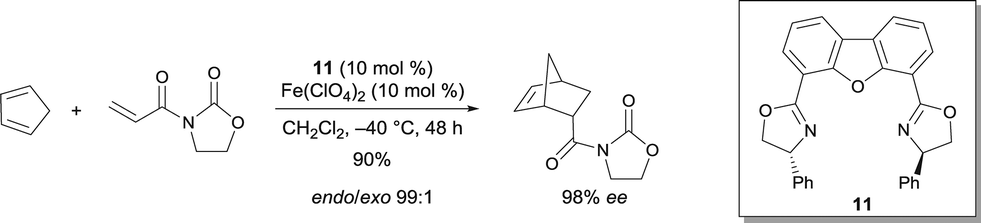 | ||
| Scheme 8 FeIII/dibenzofuran ligand 11-catalyzed enantioselective Diels–Alder reaction. | ||
Yoon reported the asymmetric oxyamination reaction of alkenes using FeII bis(oxazoline) 12.17 The process was highly stereoselective and regioselective using N-sulfonyl oxaziridines (Scheme 9). The oxyamination products were obtained with high cis diastereoselectivity and excellent enantioselectivity. The authors suggested that the high diastereoselectivity observed in the reaction is a consequence of a kinetic resolution process occuring with the racemic N-sulfonyl oxaziridines (Ns = nosyl). 1,2-Amino alcohols were prepared with high regio- and stereochemical control after clean removal of the aminal functionality in standard acid-catalyzed hydrolysis conditions.
 | ||
| Scheme 9 FeII bis(oxazoline) catalyzed asymmetric oxyamination of olefins. | ||
N-sulfonyl oxaziridines have also been rearranged into the corresponding N-sulfonyl imides.18 A highly selective kinetic resolution of N-sulfonyl oxaziridines was promoted by FeII bis(oxazoline) complexes (Scheme 10). Among other ligands, cyclopropane-bridged indanyl ligand 13 gave the best yields and selectivity. A variety of substituted N-benzenesulfonyl groups were tolerated, and similar selectivity s factor values were obtained. The chiral catalyst promotes the efficient rearrangement of oxaziridines to the corresponding N-sulfonyl imides. Interestingly, the process was scaled up to gram quantities, which is an attractive practical method for the synthesis of enantio-enriched N-sulfonyl oxaziridines. The results obtained on large scale nicely demonstrated the opportunity to offset the lower intrinsic stereoselectivity of the reaction by increasing the ee at the expense of the yield by running the resolution to higher conversion.
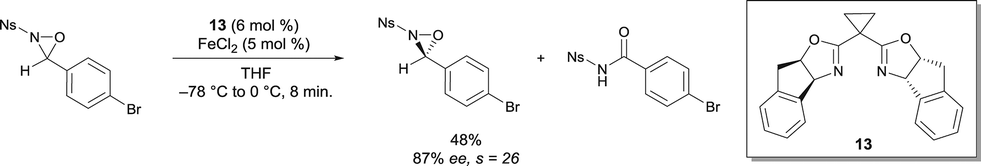 | ||
| Scheme 10 FeII catalyzed kinetic resolution of N-sulfonyl oxaziridines. | ||
In 2013, Xu discovered a new FeII bis(oxazoline) 14 complex catalyzed intramolecular olefin aminohydroxylation with functionalized hydroxylamines, where both the N and O functional groups are efficiently transferred (Scheme 11).19a Mechanistic studies revealed that an Fe nitrenoid is a possible intermediate, arising from the reductive cleavage of the N–O σ bond by the FeII complex. A stepwise cycloamination would then presumably occur, giving a carbo-radical species that would next undergo a ligand (OR) transfer, occurring faster than the competing olefin aziridination. An enantioselective intramolecular indole aminohydroxylation reaction was also reported to be catalyzed by FeII and bis(oxazoline) 14.19b Xu also observed that the FeII catalyzed asymmetric olefin aminofluorination is stereoconvergent: isomeric olefins are converted to fluoro oxazolidinones with essentially the same ee (81%) and dr (Scheme 11).20 Taking into account that the aminohydroxylation and aminofluorination are competing pathways, the authors suggested that, in the presence of Et3N·3HF and XtalFluor-E, a facile anion metathesis process can happen to convert an Fe nitrenoid intermediate into an Fe fluoride-based nitrenoid. After subsequent cycloamination, a fluoride transfer would occur at the formed radical – or possibly cationic – center. The same authors recently reported an FeII-catalyzed enantioselective and diastereoselective intramolecular olefin aminochlorination reaction.21 The reaction proceeds very efficiently with high stereoselectivities (up to 92% ee, up to 15:1 dr).
 | ||
| Scheme 11 FeII bis(oxazoline) catalyzed asymmetric aminohydroxylation and aminofluorination of olefins. | ||
Enantioselective azidations of β-keto esters and oxindoles using complex 15′ conjointly with a readily available azidoiodinane as an N3-transfer reagent were reported by Gade in 2013.22 A number of α-azido-β-keto esters were prepared with up to 93% ee. 3-Azido-3-aryloxindoles were obtained with up to 94% ee using the catalyst prepared from iron(II) propionate and bis(oxazolinyl-methylidene)isoindoline boxmi ligand 15in situ (Scheme 12).
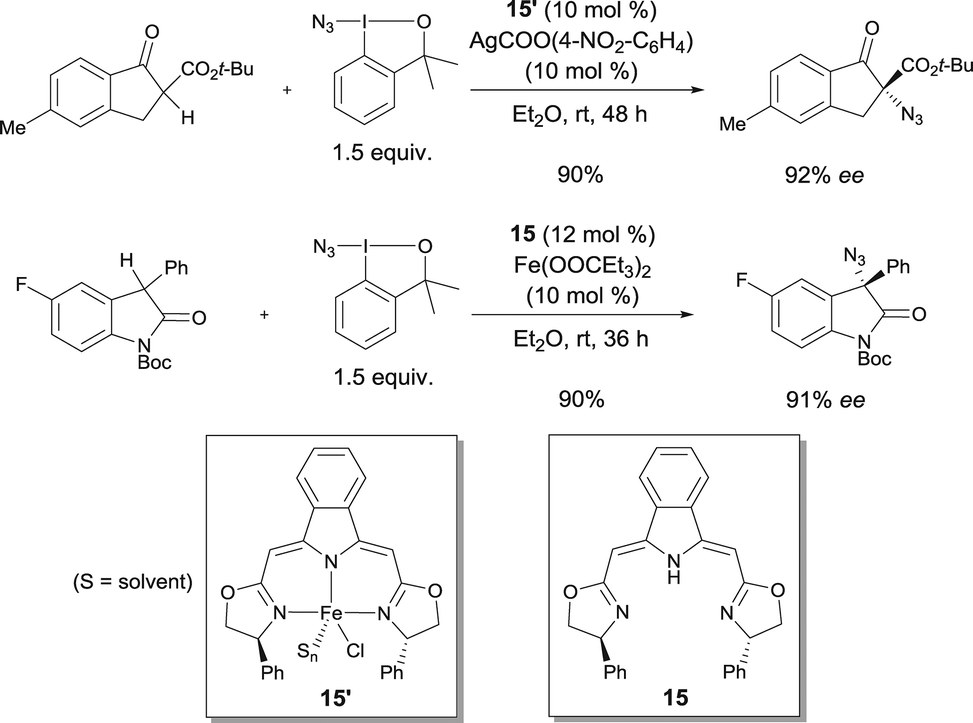 | ||
| Scheme 12 Enantioselective azidation of β-keto esters and oxindoles. | ||
Chiral Fe alkyl and Fe alkoxide complexes prepared from boxmi N3 ligands have been disclosed as catalysts for enantioselective hydrosilylation reactions with unprecendented activity and selectivity (up to 99% ee for alkyl aryl ketones), which match the performance of previously established noble-metal-derived catalysts (Scheme 13).23 Highly reactive Fe alkyl pre-catalyst 16 was transformed into an alkoxido Fe complex 17, which was obtained by clean alcoholytic transformation using (S)-1-phenyl-1-ethanol at ambient temperature. Since this species was imagined to be an important intermediate in the catalytic cycle of the studied reaction, it was believed to be an active catalyst to develop. Both of the isolated pyridine adducts of 16 and 17 were found to be as active as the in situ generated catalysts. The isolated complexes show a trigonal-bipyramidal coordination sphere with the ligand forming the equatorial plane of the bipyramid.
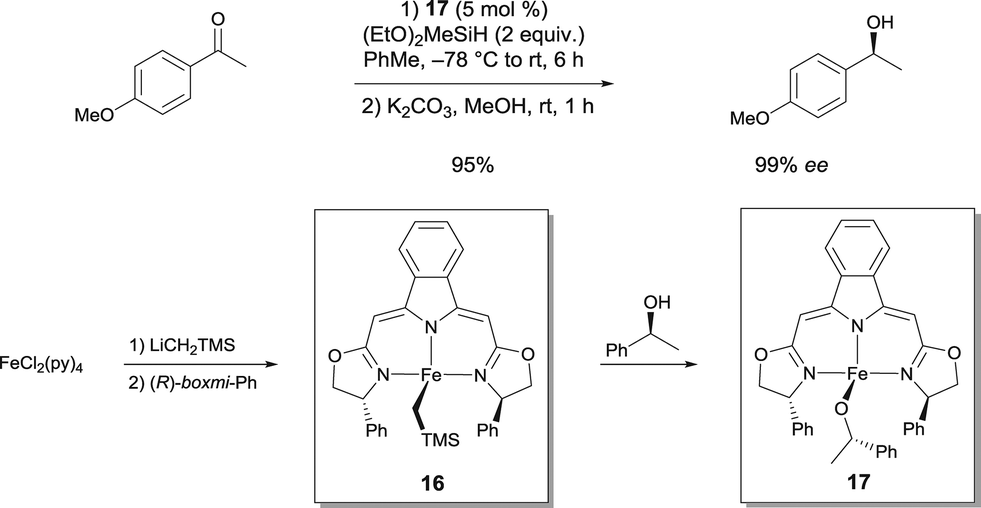 | ||
| Scheme 13 Porphyrin-inspired Fe catalyzed asymmetric epoxidation of electron-deficient olefins. | ||
A porphyrin-inspired N4 ligand bearing chiral oxazolines was reported for the asymmetric epoxidation of di- and trisubstituted enones (Scheme 14).24 The epoxidation of various enones occurred in excellent yields and high enantioselectivities (up to 99% ee). The practical utility of the catalyst was demonstrated on the gram-scale preparation of an enantio-enriched epoxide. Based on Hammett analysis for the epoxidation of para-substituted cyclic enones, it was suggested that the transition state of the reaction is electron-demanding and the active Fe complex an electrophilic oxidant.
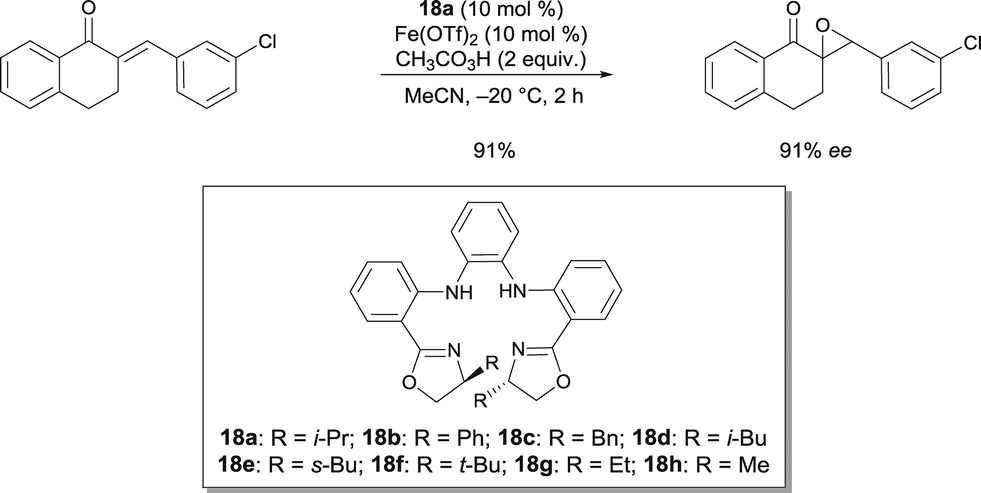 | ||
| Scheme 14 Porphyrin-inspired FeII catalyzed asymmetric epoxidation of electron-deficient olefins. | ||
3. Iron pyridine-bis(oxazoline) catalysts
In 2004, Shibasaki developed a catalytic enantioselective Diels–Alder reaction using a cationic FeIII Ar-pybox19 complex as catalyst.25 This reaction is the first catalytic enantioselective Diels–Alder reaction of acylic 4,4-disubstituted 1,3-dienes. It allowed the efficient and rapid synthesis of chiral polysubstituted cyclohexanones, which are difficult to access using other methods (Scheme 15). The use of other pybox ligands, such as i-Pr-pybox and t-Bu-pybox, afforded the product in low yield and enantioselectivity. Interestingly, other Lewis acid metal salts such as Sc(OTf)3, Cu(OTf)2, Zn(OTf)2 and cationic FeII salts gave much worse results. This methodology was used elegantly in the catalytic asymmetric total synthesis of ent-hyperforin.26 | ||
| Scheme 15 FeIIIpybox catalyzed Diels–Alder reaction. | ||
FeCl2 used with pyridine-2,6-bis(oxazoline) ligands was reported to be an effective catalyst for the asymmetric Mukaiyama aldol in aqueous media.27 The aldols were obtained in good yields and syn diastereoselectivities (Scheme 16). Moderate enantioselectivities (up to 75% ee) were afforded with ligand 20. Using bulkier ligand 21 with O-t-butyldiphenylsilyl groups,28 the enantioselectivity of the process was increased (up to 92% ee). It is believed that, using 21, the resulting FeII complex is more stable in the reaction conditions, affording better reproducibility of the reaction yield and ee. The bulky silyl groups in 21 are assumed to better shield the FeII cation against oxidation.
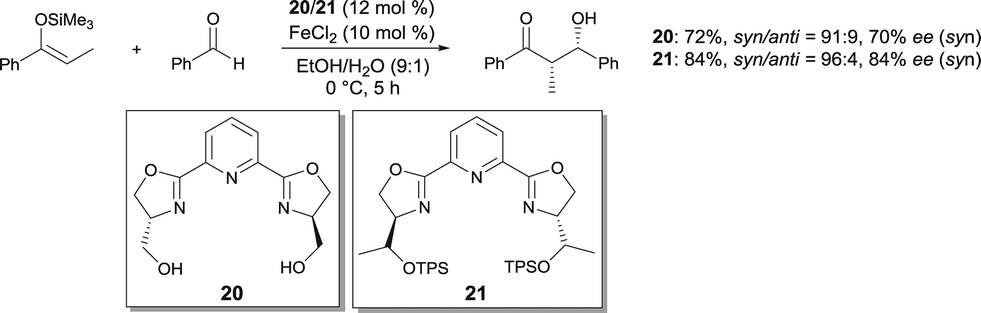 | ||
| Scheme 16 Enantioselective Mukaiyama aldol reaction. | ||
The use of pyridine-bis(oxazoline) was part of Nishiyama's study of the asymmetric hydrosilylation of ketones (Scheme 17).12 The addition of pybox22 to Fe(OAc)2 in THF at 65 °C allowed the preparation of enantiomerically enriched alcohols.
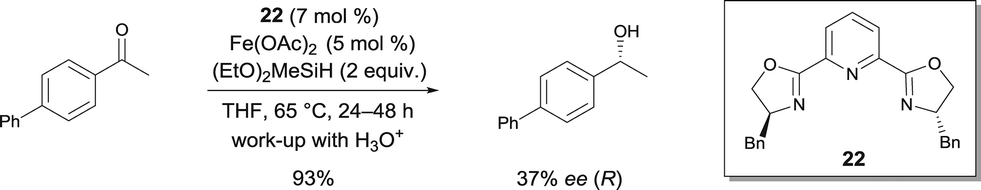 | ||
| Scheme 17 FeII pyridine-bis(oxazoline) catalyzed hydrosilylation. | ||
In connection with his work with pybox22, Nishiyama reported chiral iron bis(oxazolinyl)phenyl (phebox) complex 23 obtained by oxidative addition of Fe0 and an aryl bromide ligand.29 Interestingly, FeII-complex 23 showed up to 66% ee with full conversion of methyl(4-phenylphenyl)ketone using (EtO)2MeSiH (Scheme 18). Therefore this report by Nishiyama detailing the synthesis and structural characterization of chiral iron complexes with bis(oxazolinyl)phenyl ligands resulting from the oxidative addition of Fe2(CO)9 to phebox-Br represents a major breakthrough in the field. The authors suggest that an active Fe–H intermediate can be generated by the initial reaction of 23 with Na(acac) and the hydrosilane.
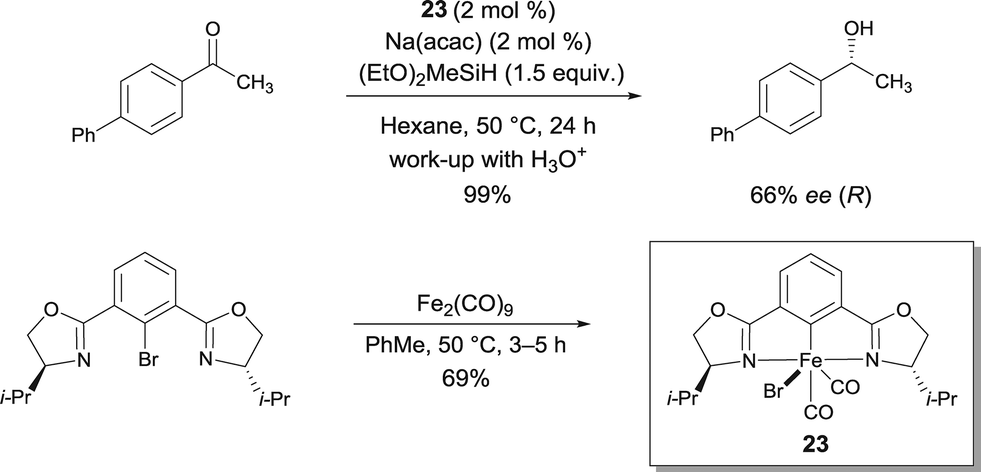 | ||
| Scheme 18 FeII bis(oxazolinyl)phenyl catalyzed hydrosilylation. | ||
The enantioselective conjugate addition of thiols to (E)-3-crotonoyloxazolidin-2-one was catalyzed by the complex prepared from pyridine-bis(oxazoline) 24 and Fe(BF4)2.30 1,4-Addition products were obtained in very good enantioselectivities (up to 95% ee) (Scheme 19). However, this catalytic system was not effective when using alkyl thiols. Using 24, the corresponding Co(ClO4)2·6H2O complex also catalyzed the same reaction with a high enantioselectivity.
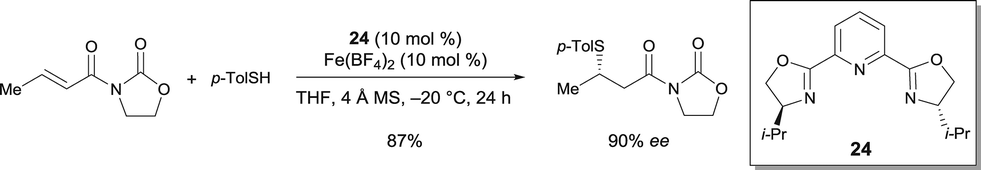 | ||
| Scheme 19 FeII catalyzed conjugate addition of thiols. | ||
Chirik studied FeII complexes prepared from pyridine-2,6-bis(oxazoline) and bis(oxazoline) ligands in order to develop an enantioselective hydrosilylation reaction of ketones.31 These ligands are commercially available or easily synthesized from available enantiopure amino alcohols. Following the same synthetic protocol as for the bis(imino)pyridine Fe-dialkyl derivatives, the corresponding pybox and box Fe-dialkyl complexes have been isolated by using the alkylation of the appropriate Fe dihalide precursor (Scheme 20). Even though high conversions were reported for the hydrosilylation of various ketones, the chiral induction of these systems was rather poor. When the hydrosilylation reactions were run for longer times, a competing catalyst deactivation pathway, i.e. the formation of bis(chelate) [(S,S)-i-Pr-pybox]2Fe, was observed. Activation of these Fe-dialkyl complexes with B(C6F5)3 resulted in catalysts able to reduce quantitatively acetophenones and α-tetralone with ee in the 34–54% range. Chirik also demonstrated that sodium amalgam reduction of (S,S)-i-Pr-pybox-FeCl2 under 1 atm of CO afforded the desired Fe dicarbonyl complex (S,S)-i-Pr-pybox-Fe(CO)2.
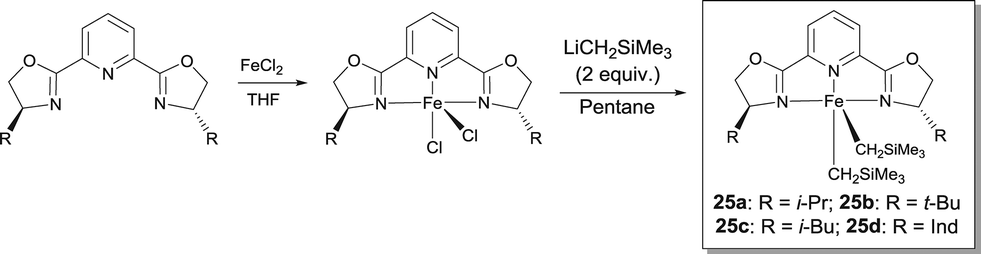 | ||
| Scheme 20 Preparation of FeII pyridine-bis(oxazoline) catalysts for asymmetric hydrosilylation. | ||
FeII pyridine-bis(oxazoline) complexes were also used in the catalytic asymmetric aziridine forming reaction of N-benzylideneaniline and ethyl diazoacetate.32 In these conditions (complex 24′ used together with an excess of ligand 24 and AgSbF6), the corresponding cis-aziridine was obtained in poor performance – moderate yield and enantioselectivity (up to 49% ee) (Scheme 21).
 | ||
| Scheme 21 Catalytic asymmetric aziridination reaction of arylimines. | ||
Asymmetric aziridination reaction of styrene was studied using various FeII derived catalysts.33 Among various tridentate ligands tested with Fe(OTf)2 and N-(p-tolyl-sulfonyl)imino phenyliodinane (Scheme 22), ligand 24 was found to be the most effective leading to the product with up to 40% ee in 72% yield. When studying the aziridination of cis-stilbene using Fe(OTf)2 as a catalyst, Bolm suggested that a concerted mechanism was unlikely, and that the formation of a radical intermediate during the nitrogen transfer process can be invoked.
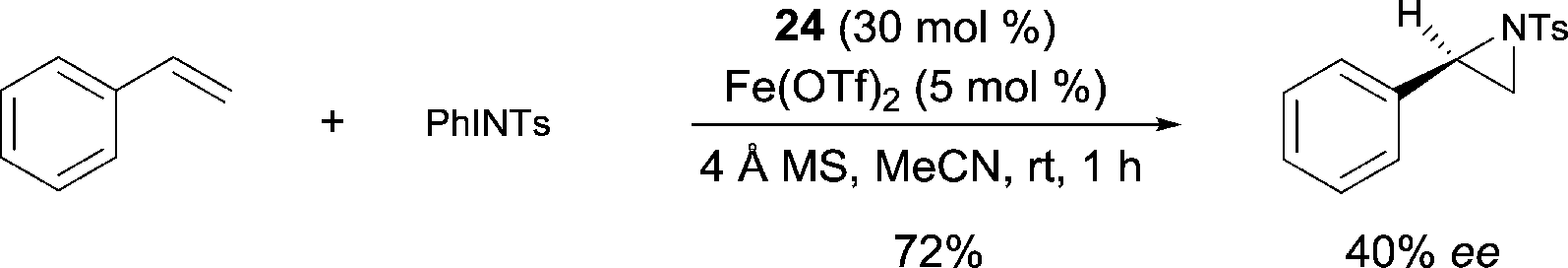 | ||
| Scheme 22 FeII pyridine-bis(oxazoline) catalyzed aziridination. | ||
In 2010, Itoh disclosed an asymmetric Nazarov cyclization of divinyl ketones catalyzed by FeII complexes, which were prepared from Fe(ClO4)2·6H2O and Fe(OTf)2 (Scheme 23).34 Such FeII complexes also catalyzed the tandem Nazarov cyclization–fluorination reaction of divinyl ketones in good yields but no enantiomeric excess could be observed.
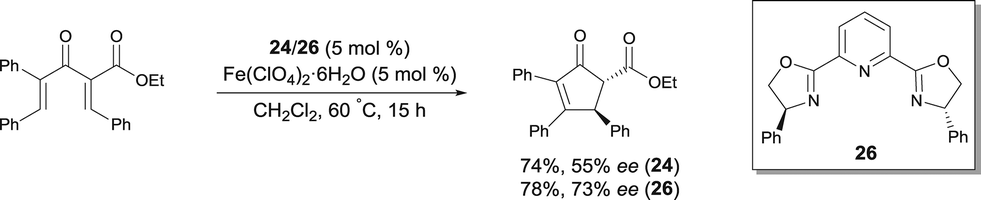 | ||
| Scheme 23 Asymmetric Nazarov cyclization catalyzed by a FeII pyridine-bis(oxazoline) complex. | ||
Bolm reported the first FeIII catalyzed enantioselective sulfimidation reaction using FeIII salts and pyridine-bis(oxazoline) ligand 26, in combination with N-(p-tolylsulfonyl)imino phenyliodinane as the nitrene precursor.35 A variety of optically active sulfimides were afforded in good yields and enantioselectivities (Scheme 24). Interestingly, the reactions could be performed in air, without exclusion of moisture.
 | ||
| Scheme 24 FeIII catalyzed enantioselective sulfimidation. | ||
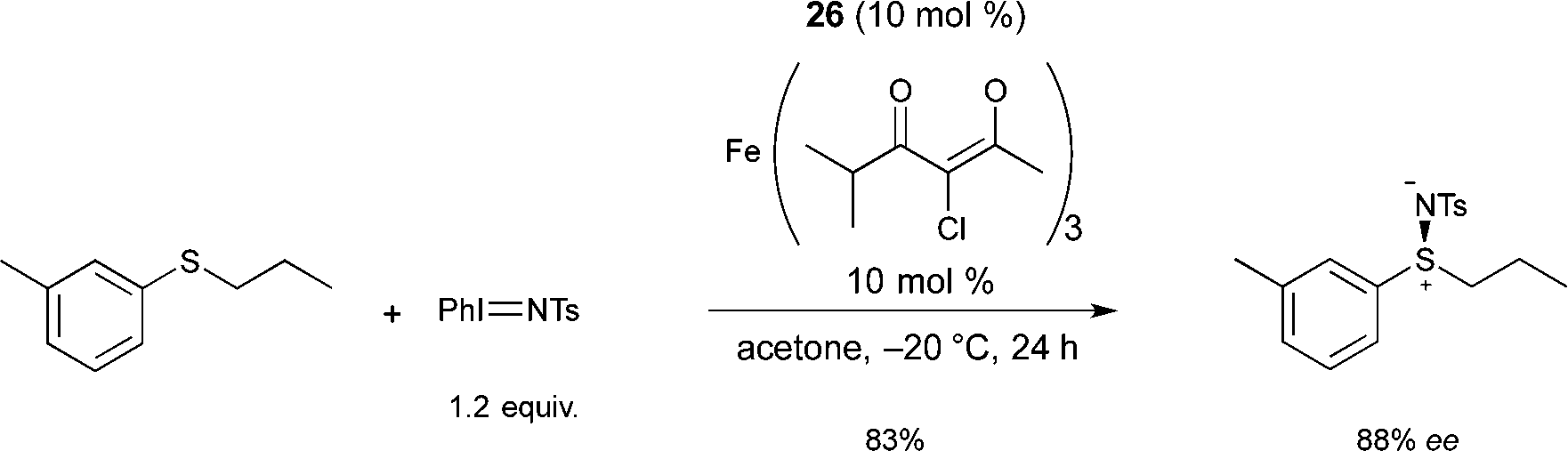
Following on from this, Bolm reported a catalyst, prepared in situ from an FeIII salt and the same ligand 26, allowing the resolution of racemic sulfoxides through catalytic asymmetric nitrene-transfer reactions (Scheme 25).36 This method provides a synthetically useful approach for the synthesis of enantiomerically enriched sulfoximines. In most cases the reaction proceeded smoothly to give the desired product using various sulfides as substrates, Fe+3 4-chloro-2,6-dimethyl-3,5-heptanedionate as the FeIII source, and 26 as chiral ligand. A large variety of enantioenriched sulfimides were prepared in good yields and good enantioselectivities (up to 88% ee). Both iron pre-catalyst and chiral ligand are commercially available. Catalytic loadings of iron source and ligand were used at 10 mol% but a reduction of the catalyst loading from 10 mol% to 5 mol% was possible. Interestingly, the reactions could be performed in air, without exclusion of moisture. Another major advantage of the method is the choice of solvent. Acetone was used as a cheap and environmentally-benign solvent.
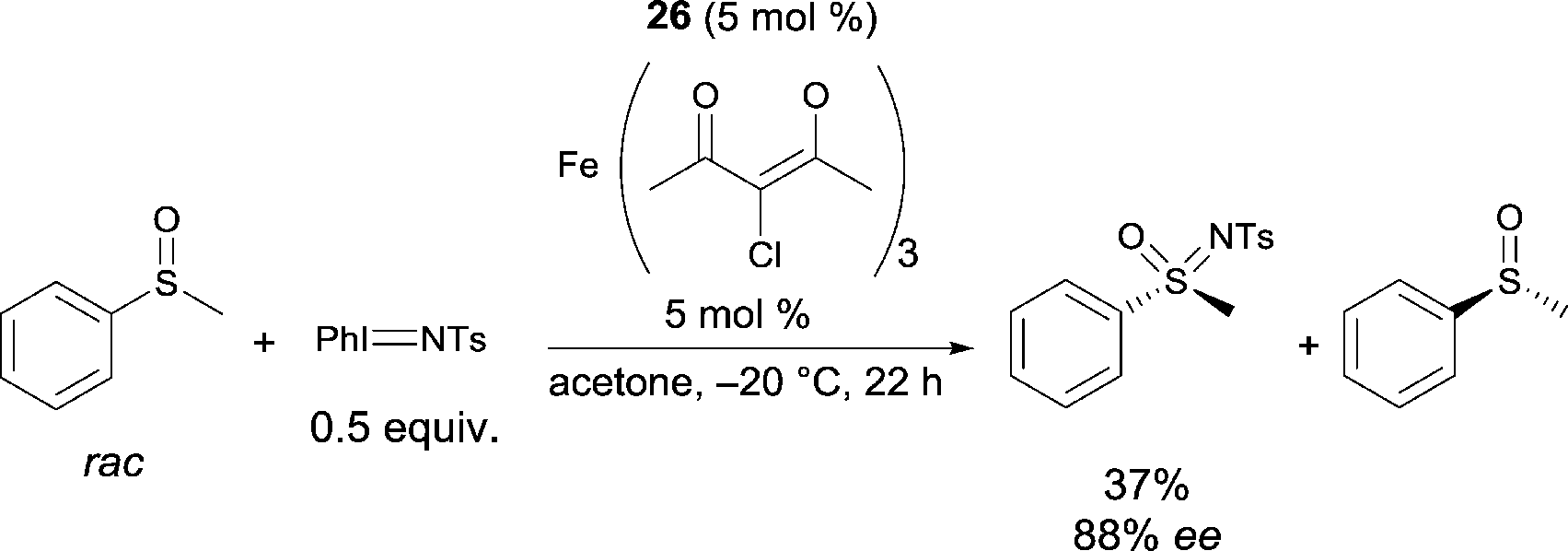 | ||
| Scheme 25 Resolution of racemic sulfoxides through catalytic asymmetric nitrene-transfer reactions. | ||
4. Conclusions
This minireview summarizes many of the important advances in the field of asymmetric catalysis using chiral iron bis(oxazoline) and pyridine-2,6-bis(oxazoline) catalysts. Efficient catalytic asymmetric transformations using iron are definitively high potential processes and demonstrate the future challenges that will need to be addressed in this field. A deeper understanding of iron complex geometries and their influences on enantioselectivity is required for future rational development of new catalysts in enantioselective reactions. Iron catalysts have definitively contributed to the area of environmentally benign catalysts, known as green catalysts. The study of asymmetric catalysis using iron has been garnering increased attention, particularly over the last five years. Given the ongoing need for new economical and green reactions, asymmetric catalysis using iron salts will clearly be a prominent area in the forthcoming years.Acknowledgements
We are grateful to the Natural Sciences and Engineering Research Council of Canada (NSERC) and to the Centre in Green Chemistry and Catalysis (CGCC) for financial support of our program.References
- (a) I. Bauer and H.-J. Knoelker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (b) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS PubMed; (c) Iron Catalysis II, ed. E. Bauer, Springer, Heidelberg, 2015, vol. 50 Search PubMed; (d) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317 CrossRef CAS PubMed; (e) L. C. Misal Castro, H. Li, J.-B. Sortais and C. Darcel, Green Chem., 2015, 17, 2283 RSC.
- (a) M. Darwish and M. Wills, Catal. Sci. Technol., 2012, 2, 243 RSC; (b) K. Gopalaiah, Chem. Rev., 2013, 113, 3248 CrossRef CAS PubMed; (c) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC; (d) P. E. Sues, K. Z. Demmans and R. H. Morris, Dalton Trans., 2014, 43, 7650 RSC; (e) T. Ollevier and H. Keipour, in Iron Catalysis II, ed. E. Bauer, Springer, Heidelberg, 2015, vol. 50, pp. 259–310 Search PubMed; (f) S. Enthaler, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2013, pp. 549–562 Search PubMed; (g) A. Fingerhut, O. V. Serdyuk and S. B. Tsogoeva, Green Chem., 2015, 17, 2042 RSC.
- (a) B. D. Ward and L. H. Gade, Chem. Commun., 2012, 48, 10587 RSC; (b) L. M. Stanley and M. P. Sibi, in Privileged Chiral Ligands and Catalysts, ed. Q. L. Zhou, Wiley-VCH, Weinheim, 2011, pp. 171–219 Search PubMed; (c) G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2011, 111, PR284 CrossRef CAS PubMed; (d) G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2003, 103, 3119 CrossRef CAS PubMed; (e) R. Rasappan, D. Laventine and O. Reiser, Coord. Chem. Rev., 2008, 252, 702 CrossRef CAS.
- (a) E. J. Corey, N. Imai and H. Y. Zhang, J. Am. Chem. Soc., 1991, 113, 728 CrossRef CAS; (b) E. J. Corey and K. Ishihara, Tetrahedron Lett., 1992, 33, 6807 CrossRef CAS.
- M. P. Sibi, S. Manyem and H. Palencia, J. Am. Chem. Soc., 2006, 128, 13660 CrossRef CAS PubMed.
- J. M. Takacs and S. C. Boito, Tetrahedron Lett., 1995, 36, 2941 CrossRef CAS.
- G. Guillemot, M. Neuburger and A. Pfaltz, Chem. – Eur. J., 2007, 13, 8960 CrossRef CAS PubMed.
- S.-F. Zhu, Y. Cai, H.-X. Mao, J.-H. Xie and Q.-L. Zhou, Nat. Chem., 2010, 2, 546 CrossRef CAS PubMed.
- Y. Cai, S.-F. Zhu, G.-P. Wang and Q.-L. Zhou, Adv. Synth. Catal., 2011, 353, 2939 CrossRef CAS.
- J.-J. Shen, S.-F. Zhu, Y. Cai, H. Xu, X.-L. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13188 CrossRef CAS PubMed.
- T. Niwa and M. Nakada, J. Am. Chem. Soc., 2012, 134, 13538 CrossRef CAS PubMed.
- H. Nishiyama and A. Furuta, Chem. Commun., 2007, 760 RSC.
- T. Inagaki, T. Phong Le, A. Furuta, J.-i. Ito and H. Nishiyama, Chem. – Eur. J., 2010, 16, 3090 CrossRef CAS PubMed.
- T. Inagaki, A. Ito, J.-I. Ito and H. Nishiyama, Angew. Chem., Int. Ed., 2010, 49, 9384 CrossRef CAS PubMed.
- S. Kanemasa, Y. Oderaotoshi, H. Yamamoto, J. Tanaka, E. Wada and D. P. Curran, J. Org. Chem., 1997, 62, 6454 CrossRef CAS.
- S. Kanemasa, Y. Oderaotoshi, S.-I. Sakaguchi, H. Yamamoto, J. Tanaka, E. Wada and D. P. Curran, J. Am. Chem. Soc., 1998, 120, 3074 CrossRef CAS.
- K. S. Williamson and T. P. Yoon, J. Am. Chem. Soc., 2012, 134, 12370 CrossRef CAS PubMed.
- K. S. Williamson, J. W. Sawicki and T. P. Yoon, Chem. Sci., 2014, 5, 3524 RSC.
- (a) G.-S. Liu, Y.-Q. Zhang, Y.-A. Yuan and H. Xu, J. Am. Chem. Soc., 2013, 135, 3343 CrossRef CAS PubMed; (b) Y.-Q. Zhang, Y.-A. Yuan, G.-S. Liu and H. Xu, Org. Lett., 2013, 15, 3910 CrossRef CAS PubMed.
- D.-F. Lu, G.-S. Liu, C.-L. Zhu, B. Yuan and H. Xu, Org. Lett., 2014, 16, 2912 CrossRef CAS PubMed.
- C.-L. Zhu, J.-S. Tian, Z.-Y. Gu, G.-W. Xing and H. Xu, Chem. Sci., 2015, 6, 3044 RSC.
- Q.-H. Deng, T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2013, 135, 5356 CrossRef CAS PubMed.
- T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2015, 137, 2456 CrossRef CAS PubMed.
- W. Dai, G. Li, B. Chen, L. Wang and S. Gao, Org. Lett., 2015, 17, 904 CrossRef CAS PubMed.
- H. Usuda, A. Kuramochi, M. Kanai and M. Shibasaki, Org. Lett., 2004, 6, 4387 CrossRef CAS PubMed.
- Y. Shimizu, S.-L. Shi, H. Usuda, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2010, 49, 1103 CrossRef CAS PubMed.
- J. Jankowska, J. Paradowska and J. Mlynarski, Tetrahedron Lett., 2006, 47, 5281 CrossRef CAS.
- J. Jankowska, J. Paradowska, B. Rakiel and J. Mlynarski, J. Org. Chem., 2007, 72, 2228 CrossRef CAS PubMed.
- S. Hosokawa, J.-I. Ito and H. Nishiyama, Organometallics, 2010, 29, 5773 CrossRef CAS.
- (a) M. Kawatsura, Y. Komatsu, M. Yamamoto, S. Hayase and T. Itoh, Tetrahedron Lett., 2007, 48, 6480 CrossRef CAS; (b) M. Kawatsura, Y. Komatsu, M. Yamamoto, S. Hayase and T. Itoh, Tetrahedron, 2008, 64, 3488 CrossRef CAS.
- A. M. Tondreau, J. M. Darmon, B. M. Wile, S. K. Floyd, E. Lobkovsky and P. J. Chirik, Organometallics, 2009, 28, 3928 CrossRef CAS.
- M. Redlich and M. M. Hossain, Tetrahedron Lett., 2004, 45, 8987 CrossRef CAS.
- M. Nakanishi, A.-F. Salit and C. Bolm, Adv. Synth. Catal., 2008, 350, 1835 CrossRef CAS.
- M. Kawatsura, K. Kajita, S. Hayase and T. Itoh, Synlett, 2010, 1243 CrossRef CAS.
- J. Wang, M. Frings and C. Bolm, Angew. Chem., Int. Ed., 2013, 52, 8661 CrossRef CAS PubMed.
- J. Wang, M. Frings and C. Bolm, Chem. – Eur. J., 2014, 20, 966 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |