
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Host–guest chemistry in two-dimensional supramolecular networks
Joan
Teyssandier
,
Steven De
Feyter
and
Kunal S.
Mali
*
Division of Molecular Imaging and Photonics, Department of Chemistry, KU Leuven–University of Leuven, Celestijnenlaan 200F, B3001 Leuven, Belgium. E-mail: Kunal.Mali@kuleuven.be
First published on 30th August 2016
Abstract
Nanoporous supramolecular networks physisorbed on solid surfaces have been extensively used to immobilize a variety of guest molecules. Host–guest chemistry in such two-dimensional (2D) porous networks is a rapidly expanding field due to potential applications in separation technology, catalysis and nanoscale patterning. Diverse structural topologies with high crystallinity have been obtained to capture molecular guests of different sizes and shapes. A range of non-covalent forces such as hydrogen bonds, van der Waals interactions, coordinate bonds have been employed to assemble the host networks. Recent years have witnessed a surge in the activity in this field with the implementation of rational design strategies for realizing controlled and selective guest capture. In this feature article, we review the development in the field of surface-supported host–guest chemistry as studied by scanning tunneling microscopy (STM). Typical host–guest architectures studied on solid surfaces, both under ambient conditions at the solution–solid interface as well as those formed at the ultrahigh vacuum (UHV)–solid interface, are described. We focus on isoreticular host networks, hosts functionalized pores and dynamic host–guest systems that respond to external stimuli.
 Joan Teyssandier | Joan Teyssandier obtained his PhD in chemistry in 2012 from the Paris Diderot University (France) under the supervision of Dr Philippe Lang. During his doctoral work he studied the growth and properties of molecular and metallic guests confined in nanoporous supramolecular networks. Thereafter, he joined the group of Prof. Anna Proust at the University Pierre and Marie Curie for a postdoctoral position to study functionalized polyoxometalates for surface nanostructuration. He has been a postdoctoral fellow in the De Feyter group at KU Leuven since 2014, where his research involves investigating supramolecular self-assembly on surfaces and functionalization of 2D materials. |
 Steven De Feyter | Steven De Feyter is a professor at KU Leuven in Belgium. After completing his PhD with Frans De Schryver at KU Leuven in 1997, he moved for a postdoctoral position to the group of Ahmed Zewail (California Institute of Technology, Pasadena). His research group investigates various aspects of supramolecular chemistry and self-assembly phenomena of surfaces using scanning probe methods with special attention to liquid–solid interfaces. |
 Kunal S. Mali | Kunal S. Mali obtained his PhD in chemistry in 2008 from University of Mumbai (India) under the supervision of Dr G. B. Dutt. His doctoral work focused on investigation of fast dynamic processes in complex media by employing time-resolved fluorescence spectroscopy. Currently, he is a senior postdoctoral fellow at KU Leuven in the De Feyter group where his research involves different aspects of surface-confined supramolecular self-assembly. |
Introduction
Host–guest chemistry is one of the defining concepts of supramolecular chemistry which describes the formation of unique structural complexes between two or more molecules or ions via non-covalent interactions. Although historically developed in organic and aqueous solutions, there is increasing interest in implementing the principles of supramolecular host–guest chemistry to systems assembling on solid surfaces. The presence of a solid surface not only ensures a high degree of crystallinity in the host network thus enabling an efficient capture of guests, but it also provides additional stability to the resultant host–guest complex via molecule–surface interactions. Surface assembled host networks often exhibit specificity in guest binding akin to that found in enzymes, yet have crystalline structures emulating naturally occurring porous materials such as zeolites. Furthermore, such nanostructured host surfaces1,2 can be readily integrated into real-life functional supramolecular systems leading to potential applications in separation technology, molecular sensing and catalysis.Similar to host–guest chemistry in solution, molecular recognition lies at the heart of host–guest chemistry studied on solid surfaces. The mutually specific recognition between the host network and the guest molecules occurs over very small length scales, however scanning tunneling microscopy (STM)1–4 has made it possible to observe such guest binding in real time at submolecular resolution, provided that the self-assembly takes place on an atomically flat conductive surface. In conventional solution phase host–guest chemistry, a molecular recognition event is often inferred from indirect means such as changes in chemical shifts (NMR), measurement of heat change (calorimetry), or changes in the photophysical properties (UV-Vis absorption). The experimental data from such measurements provides information on the strength and selectivity of intermolecular interactions allowing chemists to construct a step-by-step picture of the process. Although such techniques are now highly evolved and are scientifically rigorous, they lack the immediate visual appeal of microscopy based measurements where one can directly ‘see’ the structure of the host–guest complex. In this context, STM not only provides structural information of the host–guest complexes, but if appropriate conditions are met, it also allows to follow dynamic aspects of such systems, thus capturing molecular recognition events in real time.5 STM has evolved as a versatile surface science technique for studying host–guest interactions over the past two decades and it can function in diverse type of environments ranging from the solution–solid interface6 to ultrahigh vacuum (UHV) conditions.7
A surface-confined host network is often obtained via self-assembly of an organic molecule onto a solid surface. The host network contains voids in the form of shallow (single molecule thick) nanowells where the guest molecules can adsorb. The host networks are typically sustained by either hydrogen bonding or van der Waals (vdW) forces, however halogen bonding8 and metal–ligand coordination9 have also been used. The host networks formed at the solution–solid interface are also believed to be stabilized by (dynamic) co-adsorption of solvent molecules.10 If the size and shape of the guest match with that of the voids, it gets immobilized on the surface within the host network. At the solution–solid interface, immobilization of guest molecules occurs at the expense of solvent desorption as the guest species often have higher adsorption energy compared to solvent molecules. The guest stabilization often occurs via attractive dispersion interactions with the host network as well as with the underlying surface. Thus, host–guest chemistry on surfaces is often ‘surface-assisted’. Alternatively, the host as well as the guest species can be brought onto the surface simultaneously.
Solution–solid interface offers a more dynamic and thus relatively complex environment than UHV conditions due to competitive influence of molecule–solvent and solvent–surface interactions in addition to the intermolecular and molecule–surface interactions which are ubiquitous in the self-assembly processes on solid surfaces. The host and the guest molecules may or may not interact in a typical ‘host–guest’ fashion in solution but such interactions unravel only upon adsorption onto the surface. Furthermore, solution–solid interface provides favorable conditions for molecular dynamics such that guest binding takes place at or close to equilibrium conditions.11 The choice of surfaces however, is limited under ambient conditions. Typically, stable surfaces that do not undergo oxidation are chosen. Highly oriented pyrolytic graphite (HOPG) and Au(111) are most commonly used, however MoS2 has also been used for self-assembly experiments under ambient conditions.12,13 Although the solution–solid interface provides a ‘real-life’ view of the assembly process, the UHV environment has unique attributes such as ultra-clean environment and choice of variety of surfaces. Deposition of molecules is typically carried out using organic molecular beam epitaxy (OMBE) technique. This method allows precise control over the layer thickness and the molecular ratios. A much wider variety of surfaces are accessible which include different crystal facets of metals such as Au, Ag, Cu, Pt, Pd etc.7 SiB(111) has also been used for host–guest chemistry under UHV conditions.14 Since the assembly occurs in vacuum, temperature of the surface can be precisely controlled, which permits both controlled annealing and imaging at low temperatures.
The nature of the surface is crucial factor in host–guest chemistry as it essentially governs the mobility of molecules upon adsorption, and thus, the ability to self-repair. Although annealing at higher temperatures can induce the necessary dynamics (often practiced under UHV conditions), the temperature window accessible for experiments carried out at the solution–solid interface is often limited due to evaporation of the solvent. This becomes a serious concern in the case of metals, which tend to interact relatively strongly with aromatic molecules.15 Therefore, controlling organization and achieving long-range order in self-assembled networks of physisorbed molecules is often challenging on metal surfaces compared with HOPG due to higher diffusion barriers. As a consequence, HOPG has been the surface of choice for studying multicomponent self-assembly under ambient conditions and more than two components have rarely been co-crystallized on metals.16,17
A number of similarities exist between the host–guest strategies employed on surfaces and those exercised in solution. Both intrinsically porous (containing permanent covalent cavities) as well as extrinsically porous host networks are studied on surfaces. Intrinsic porosity is inherent to the chemical structure of the molecule when a single molecule is considered in isolation. Examples of intrinsically porous hosts include macrocyclic compounds such as cyclodextrins,18 crown ethers,19 calixarenes20 and other shape-persistent macrocycles.16 On the other hand, extrinsic porosity results from non-covalent (or covalent) assembly of the constituent molecules, which is usually not intrinsic to the isolated building block. A vast majority of studies carried on solid surfaces have focused on extrinsically porous systems made up of relatively smaller, judiciously chosen molecular components that self-assemble upon adsorption to yield a host network. By default, a host–guest system consists of two-components (with the exception of auto host–guest systems21,22 where the host forming molecules themselves act as guests) however, higher order multicomponent systems consisting of up to four different23,24 molecular components have been reported where more than one type of guest molecules are assembled in a parent host network. It must be noted however, that although every host–guest system is a multicomponent system, every multicomponent system may or may not represent a host–guest system.25 While most 2D host networks formed on solid surfaces are made up of periodically arranged building blocks, non-periodic porous networks have also been reported.26,27
In this feature article, we provide a brief account of the progress made in surface-supported host–guest chemistry by highlighting important examples from literature. The article is structured as follows. After briefly introducing the pioneering examples, we describe in detail, well-characterized families of host networks which exhibit isoreticular topologies with scalable cavities. Novel host systems such as supramolecular organic frameworks (SOFs),28 and covalent organic frameworks (COFs)29 are also described in this section. The second half of the manuscript includes the survey of various aspects of host–guest chemistry including dynamic multicomponent systems, selectivity in guest binding and stimulus responsive systems. In the final section we provide a brief summary and outlook.
Emergence of surface-confined host–guest systems
Single molecule thick, 2D porous networks appeared on the scene in early 2000s. One of the first of such examples is the pioneering report on hydrogen-bonded porous hexagonal network formed by benzene-1,3,5-tricarboxylic acid (trimesic acid, TMA Fig. 2a) on HOPG.30 TMA is an archetypal building block that forms a cyclic hexamer via resonance stabilized hydrogen bonding. The basic unit of the hexagonal porous structure is a hydrogen bonded TMA dimer. In this study, which was carried out under UHV conditions, two different polymorphs, namely chicken-wire (also called honeycomb) and flower structures, were observed (Fig. 1a and b). The flower structure consists of relatively denser arrangement of TMA molecules compared to the chicken wire structure. Both networks exhibit a hexagonal lattice where the TMA molecules form a rim around periodically arranged, supramolecular cavities with an internal vdW diameter of ∼1.1 nm. These hydrogen-bonded networks are extremely versatile and can be fabricated on a variety of solid surfaces both under UHV conditions as well as at the solution–solid interface.32,33 TMA network remains one of the most robust self-assembled host network to date and has been utilized to immobilize molecular guests such as coronene,34 heterocirculenes35 and C6036 based on size and shape complementarity. This early work on TMA cemented the foundation of host networks based on strong, highly directional hydrogen bonding interactions between carboxylic groups.37 | ||
| Fig. 1 Early examples of host–guest systems. STM images of the (a) Chicken wire and (b) flower structure of TMA self-assembled network on HOPG. (c) STM image showing entrapment of heptameric C60 clusters in the PTCDI–melamine host network assembled on Ag terminated silicon surface. (d) Schematic of the PTCDI–melamine host–guest system. Reproduced from ref. 30 and 31 with permission from Wiley-VCH and Macmillan Publishers Ltd, respectively. | ||
One of the early examples of surface-confined host–guest chemistry involved a fairly complex bicomponent host network. It was obtained upon co-adsorption of PTCDI with melamine.31 These two molecules have complementary hydrogen bonding sites such that each melamine molecule forms three hydrogen bonds with PTCDI. Co-deposition of the two molecules onto a silver terminated silicon surface resulted in the formation of an open honeycomb network with melamine adsorbing on the vertices and PTCDI forming the edges of the honeycomb lattice. Sublimation of C60 on such preformed host network resulted in entrapment of heptameric C60 clusters inside the hexagonal voids (Fig. 1c and d). The surface coverage of C60 guests could be tuned by increasing its dosage leading to a C60 terminated bilayer which was supported by the underlying PTCDI–melamine host network.31 These early reports provided classic examples where a supramolecular synthon strategy realized in solution and/or in the solid state was directly applied to surface assembly.38
Isoreticular self-assembled host networks
The search for novel materials and functions has remained one of the major driving forces behind supramolecular chemistry research. Given that structure determines function, the study and manipulation of supramolecular structures is the elemental step in the pursuit of that goal. Scalability of voids within porous structures is a challenging aspect in supramolecular chemistry and material science. The basic strategy consists of changing the pore dimensions and/or the chemical functionality of the host network by changing the size of the building block while maintaining the same network topology. Commonly known as isoreticular synthesis – a term coined first in the context of metal–organic frameworks (MOFs), this strategy reflects the high-fidelity of supramolecular synthons. Higher pore size leads to the possibility of trapping either larger guest species or higher number of guests per cavity.While increasing the size of the organic building block can be readily achieved via organic synthesis, translation of the increased size into higher pore dimensions is not always straightforward. This is because, molecular packing, whether in the solid state or on surfaces, is largely governed by considerations of size and shape – the so called principle of ‘close-packing’.39 Such close-packing is enthalpically favored due to intimate intermolecular contact. Thus, creating an open porous network at an interface is often energetically expensive due to the lower adsorption enthalpy per unit area of the resultant network. While smaller molecules such as TMA can sustain open porous networks via strong hydrogen bonds, scaling up the size of the building block alters the balance between long-range anisotropic forces such as hydrogen bonds and medium-range isotropic forces such as van der Waals interactions. This often leads to collapse of the porous networks into denser structures unless the enthalpic loss in the formation of the open structure is compensated via co-adsorption of guest species or solvent molecules. Furthermore, the structures of self-assembled networks critically depend on the type of surface, solvent, and solution concentration (or coverage in UHV). Creating isoreticular host networks thus requires a thorough understanding of intermolecular and interfacial interactions. Research efforts over the past decade have culminated into fabrication of isoreticular host-networks with pore diameters up to 7.5 nm.40 In the following section we highlight a few families of isoreticular host networks.
Based on hydrogen bonding between carboxylic groups
Hydrogen-bonded host architectures are one of the most frequently encountered motifs due to the relatively strong and directional nature of hydrogen bonds. Carboxyl groups are widely exploited synthons for such motifs since they are endowed with unique “self-complementary” hydrogen bonding ability where the oxygen atom of the carbonyl group acts as a hydrogen bond acceptor and the hydroxyl group acts as a hydrogen bond donor. Thus, two carboxylic groups can form a cyclic dimer interconnected by two equivalent hydrogen bonds. However, apart from the cyclic dimers, other binding arrangements such as trimers and catemers are also known to exist both in the solid state as well as in surface assembled networks. The mere presence of a carboxyl group however, is not a sufficient criterion for obtaining a 2D (porous) network. At least three appropriately placed carboxyl groups are required to form an extended network based on hydrogen bonding. Phthalic acid, isophthalic acid (ISA), terephthalic acid (TA) and TMA all contain carboxyl groups, however only TMA forms an extended porous network sustained by hydrogen bonds. Porous networks of terephthalic acid have been reported though such assemblies are stabilized by metal–organic coordinate bonds (vide infra).9Although the first example on nanoporous TMA networks was reported at the UHV–solid interface, a number of interesting results were obtained while studying the self-assembled system at the solution–solid interface (Fig. 2e and f). Typically, fatty acids are used as solvents on HOPG surface. The dimensions of the host cavities could be increased by adding rigid spacers between the central benzene ring and the peripheral carboxylic groups such that the original 3-fold symmetry is preserved. 1,3,5-Tris(4-carboxyphenyl)benzene (BTB, Fig. 2b) is a larger analogue of TMA which consists of an additional phenyl spacer between the central phenyl ring and each carboxyl group. Similar to TMA, BTB self-assembles into a honeycomb porous network sustained by resonance stabilized hydrogen bonds (Fig. 2f and j). The porous BTB network has been obtained both under UHV conditions42 as well as the solution–solid43–46 interface. It offers larger hexagonal cavities with a vdW diameter of ∼2.8 nm, more than two-fold increase than the cavities of the TMA network. BTB shows rich self-assembling properties with three additional structural polymorphs, the relative occurrence of which depends on type of solvent,44,46 temperature,42,47 solution concentration,43 and the polarity of voltage applied to the sample.43,45
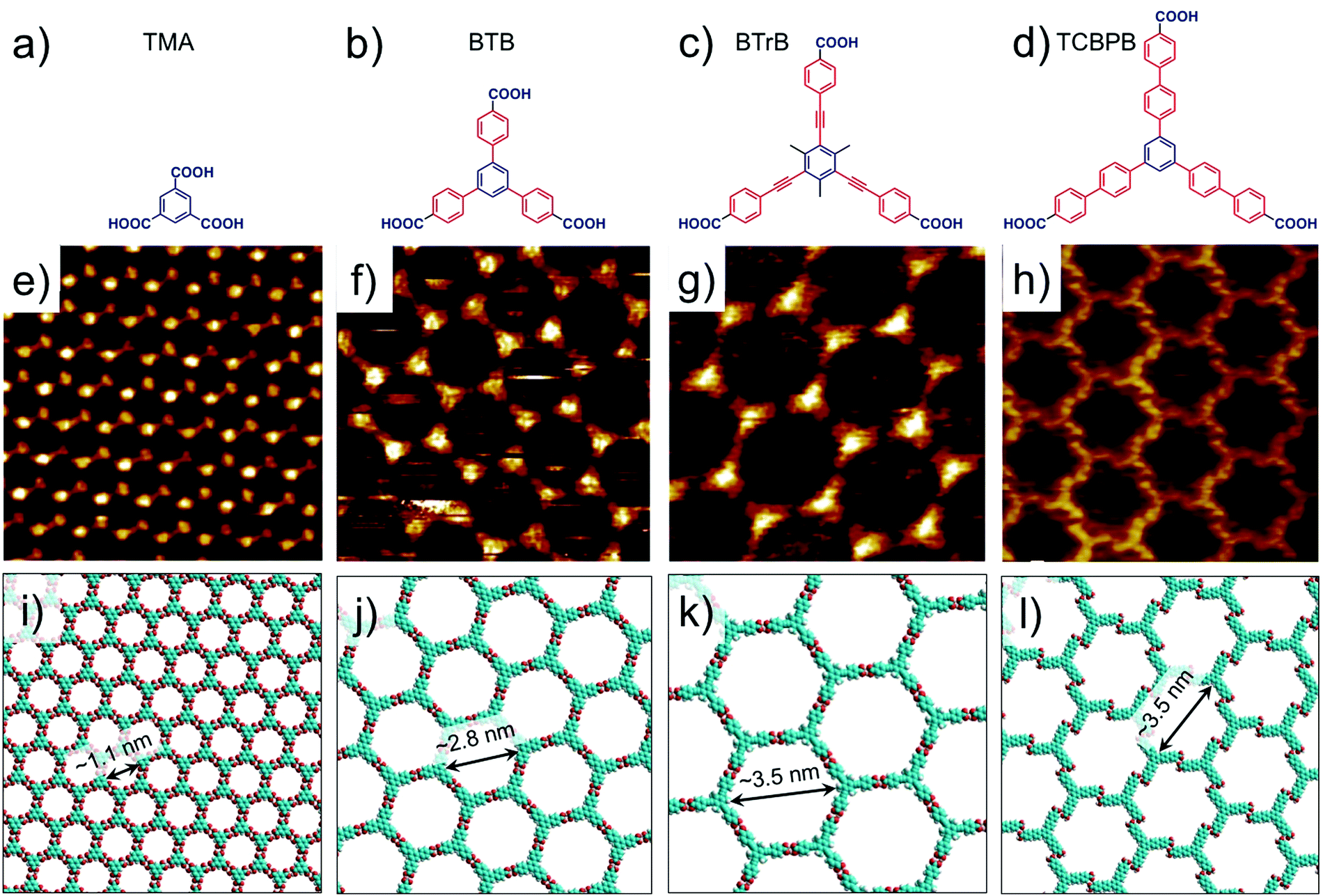 | ||
| Fig. 2 Isoreticular host networks based on hydrogen bonding between carboxyl groups. (a–d) Molecular structures of TMA, BTB, BTrB and TCBPB. (e–h) STM images of porous networks formed by TMA, BTB, BTrB and TCBPB, respectively. TCBPB forms a displaced chicken wire network. Panels (i–l) show molecular models for the corresponding porous networks, respectively. Reproduced from ref. 37 and 41 with permission from the American Chemical Society. | ||
Another homologue of TMA was obtained by insertion of a phenylethyne spacer between the phenyl rings and the carboxyl groups (BTrB, Fig. 2c). This compound also formed honeycomb porous network at the solution–solid interface with cavity diameter of ∼3.5 nm (Fig. 2g and k).48 The next larger homologue, TCBPB (Fig. 2d), however did not yield the expected isotopological honeycomb network. TCBPB has a biphenyl linker between the central phenyl ring and the carboxylic groups, relative to the structure of TMA. Contrary to the aromatic carboxylic acids described above, TCBPB furnished a deformed hexagonal porous network based on energetically inferior Ar–CH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) hydrogen bonding instead of the anticipated one based on carboxyl group dimers. Estimations of Gibbs free energy indicated that the so called ‘displaced chicken-wire’ structure with vdW cavity diameter of ∼3.5 nm is thermodynamically favored compared to the hypothetical ideal honeycomb network (Fig. 2h and i). The large size of the molecules ensures that the molecule–surface interactions dominate the assembly process. The higher packing density of the displaced chicken-wire structure provides significantly large gain in adsorption enthalpy which cannot be compensated by the enthalpic gain obtained upon formation of ideal hydrogen bonds, which would lead to a network with lower packing density.41
hydrogen bonding instead of the anticipated one based on carboxyl group dimers. Estimations of Gibbs free energy indicated that the so called ‘displaced chicken-wire’ structure with vdW cavity diameter of ∼3.5 nm is thermodynamically favored compared to the hypothetical ideal honeycomb network (Fig. 2h and i). The large size of the molecules ensures that the molecule–surface interactions dominate the assembly process. The higher packing density of the displaced chicken-wire structure provides significantly large gain in adsorption enthalpy which cannot be compensated by the enthalpic gain obtained upon formation of ideal hydrogen bonds, which would lead to a network with lower packing density.41
An alternate strategy towards TMA based isoreticular networks consists of introduction of an alkoxy chain in between the phenyl ring and the carboxylic group. A series of such compounds up to 10 carbon atoms in the alkoxy chain have been reported to form nanoporous networks with varying cavity sizes. Only the structural analogue with a carboxymethoxy spacer formed an isotopological network akin to TMA, while other derivatives yielded distorted porous networks upon surface adsorption. The origin of network distortion lies in the competitive influence of van der Waals interactions between the alkoxy chains (which have a tendency to close-pack) and directional hydrogen bonding interactions between the terminal carboxyl groups. While the networks are not necessarily isotopological to TMA, this design strategy is useful in building relatively flexible host networks based on carboxyl hydrogen bonding. The self-assembly and the host–guest chemistry of such ‘telechelic’ TMA derivatives has been summarized recently.49
Besides the strong hydrogen bonds between carboxylic acid groups, relatively weaker hydrogen bonding interactions are also known to stabilize open porous structures. A unique example consists of anthraquinone molecules self-assembled on Cu(111) under UHV conditions.50 This honeycomb network is sustained by hydrogen bonds formed between carbonyl oxygens and aromatic hydrogen atoms and offers pore diameter of ∼5.0 nm. The primary unit is made up of a trimer of anthraquinone molecules. The origin of this unusual host motif lies in the delicate balance between intermolecular attraction and substrate-mediated long-range repulsion. This unusual host network further shows equally unusual guest binding behavior. In contrast to the typical guest immobilization observed in surface-confined networks, where the host network directly interacts with the guest entity, carbon monoxide molecules were found to be immobilized in the center of the honeycomb cavity, away from the walls of the network. This extraordinary capture of CO molecules within the host cavities was ascribed to presence of confined surface states. Gradual increase in the surface coverage of CO molecules revealed a discrete sequence in which CO guests occupy specific locations within the cavity. Detailed calculations revealed that the sequence in which these locations are occupied matches closely with the energetic succession of the corresponding confined state is reminiscent of the filling of electrons into an atomic orbital diagram.51
Based on van der Waals interactions between alkyl chains
Although van der Waal interactions intrinsically lack the strength and directionality of hydrogen bonds, when used in combination with an appropriate surface and molecular design strategy, they are extremely effective in directing surface self-assembly. Possibly the most commonly discussed type of van der Waals interactions are those between close-packed (interdigitated) alkyl chains. The calculated interaction energy for such chains is 7.9 × 10−21 J per methylene group, given that the alkyl chain is flanked by other alkyl chains. On the other hand, the energy of a two-fold O–H⋯O hydrogen bond between two carboxylic acid groups is approximately 60 kJ mol−1. It must be noted however that, although the energy of van der Waals interactions is typically less than that of hydrogen bonds, collectively these interactions can compete with hydrogen bonds. This means that sufficiently long alkyl chains can stabilize the supramolecular network as good or better than a single hydrogen bonding unit. Furthermore, alkanes/alkyl chains are known to interact strongly with graphite surface via attractive van der Waals interactions. Linear alkanes thus form close-packed 2D lamellae on graphite, which are stabilized via molecule–surface and molecule–molecule van der Waals interactions. The strong adsorption of alkanes on the graphite surface is a result of structural similarities between the alkane backbone and the graphite lattice. The basal (0001) plane of graphite has a 3-fold symmetry and the zigzag orientation of carbon atoms along any C3 axis matches with that of an all-trans alkyl chain. Moreover, the in-plane lattice constant of graphite (2.46 Å) matches closely with the distance between every alternate methylene group (2.58 Å) in an alkyl chain. This fortuitous match allows the methylene groups of an all trans alkyl chain to rest over the voids of the hexagons of graphite lattice thereby providing an approximately commensurate packing. The lateral spacing between alkanes is also dictated by the distance between every other carbon row along the (1100) directions of graphite (4.24 Å). Thus, graphite lattice provides epitaxial stabilization to alkanes/alkylated molecules.
A typical example where van der Waals forces have been used as potent directional intermolecular interactions is the porous networks of triangular phenylene-ethynylene macrocycles, commonly known as DBAs. These building blocks consist of a rigid triangular or rhombic dehydrobenzo[12]annulene core substituted with alkoxy or alkyl chains. The peripheral chains stabilize the self-assembled network not only via van der Waals interactions with the surface, but they also function as highly directional intermolecular linkages by forming a characteristic binding pattern commonly known as interdigitation. The basic unit of the honeycomb porous network consist of a dimer of DBA molecules where the two molecules interact with each other via van der Waals forces between their interdigitated alkyl chains. The length of the alkyl chains governs the distance between the DBA cores and thus also the size of the hexagonal voids produced within the self-assembled network.52
While increasing the chain length appears a rather straightforward strategy, fabrication of large porous DBA networks was not accomplished until the concentration dependence of surface self-assembly was discovered. A unique facet of molecular assembly at the solution–solid interface, the influence of solution concentration on structure formation first came to light in the case of DBAs.53 At relatively high concentrations, DBA derivatives form a dense non-porous pattern however, the network morphology changes to honeycomb porous when dilute solutions are used. The two structures coexist at intermediate concentrations. The two networks also differ in the way the DBA molecules are adsorbed on the surface. All six alkyl chains per molecule are adsorbed on the surface in the porous network whereas one or more alkyl chains are desorbed from the surface in the dense packing. The concentration dependence arises from the different stabilities and molecular densities of the two structures formed. At higher concentrations, adsorption energy per unit area governs the network formation and thus the close-packed dense network is favored. At lower concentrations, the number of molecules available to cover the surface is reduced and under such circumstances, porous honeycomb structure is favored in order to maximize the adsorption energy per molecule.
Furthermore, the concentration range over which the dense to porous structural transition occurs depends the alkyl chain length. The surface coverage of the honeycomb network follows a linear relation with concentration for DBA derivatives with smaller alkoxy chains, whereas for DBAs with longer alkoxy chains, this relation is exponential. The adsorption energy per unit area for the dense and the porous patterns are comparable for DBAs with shorter chain lengths. As a consequence, DBAs with shorter chain lengths preferentially form porous networks over a wide concentration range. However, the energy difference increases with increasing chain length thus favoring the close-packed non-porous structure for DBAs with longer chains.53
Understanding of the concentration dependence of molecular self-assembly laid the foundation of isoreticular host–guest networks based on van der Waals interactions between interdigitating alkyl chains. With an increment of 1.25 Å per methylene group in the alkyl chain length, the size of the hexagonal pores increases linearly. Using this design, porous networks with voids ranging in size from ∼2.6 nm up to ∼7.5 nm have been fabricated on the graphite surface (Fig. 3). DBAs represent a classic example where molecular design, organic synthesis and supramolecular surface science strategies have been effectively employed to realize surface networks with different functions. These networks have been used for immobilization of a variety of guest species in the form of (hetero)molecular clusters24,55,56 and large shape-persistent macrocycles (vide infra).16,40
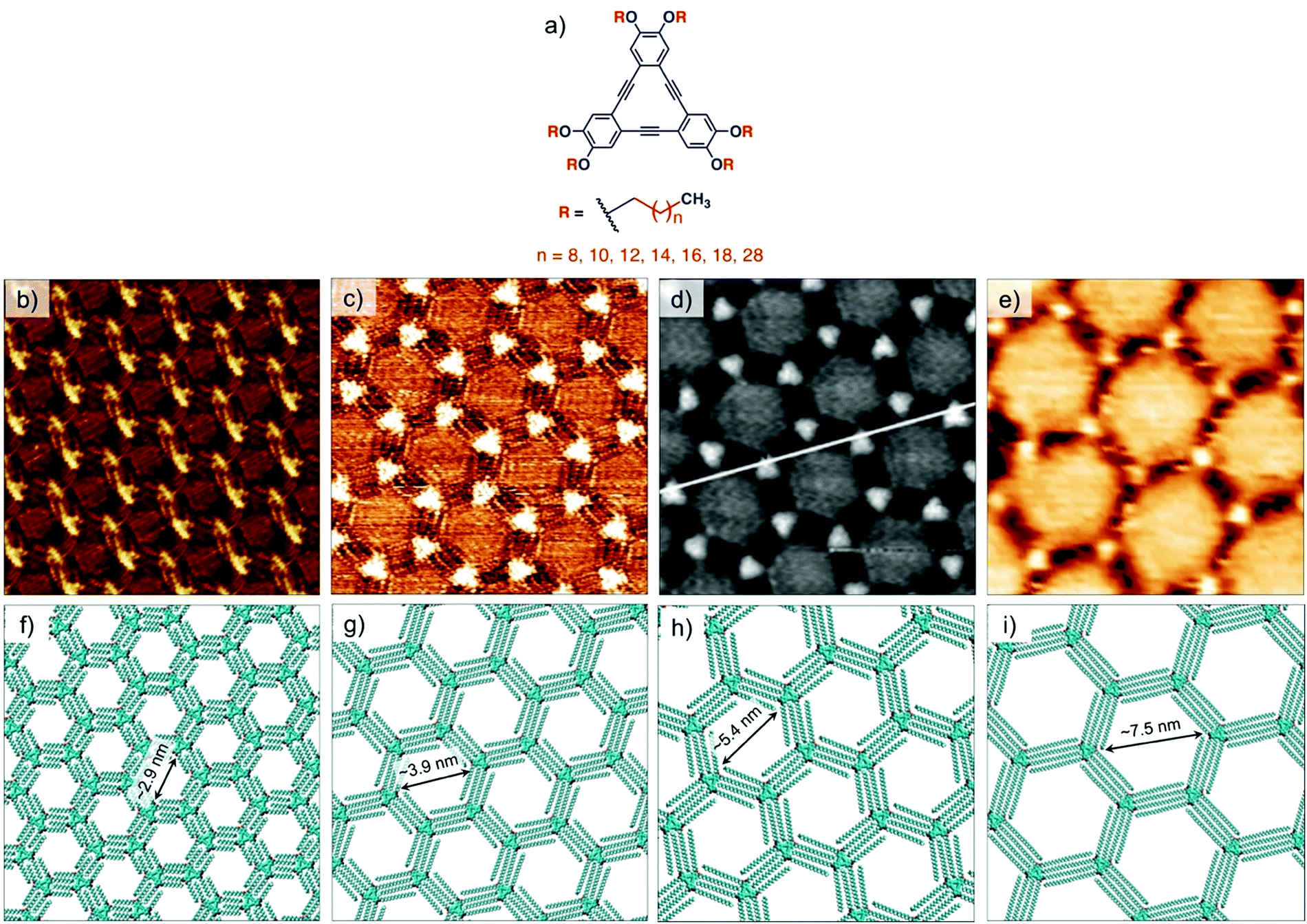 | ||
| Fig. 3 Isoreticular host networks based on van der Waals interactions between alkyl chains. (a) Molecular structure of the DBA derivatives. STM images of honeycomb porous networks formed by (b) DBA-OC10 (c) DBA-OC14, (d) DBA-OC20 and (e) DBA-OC30. Panels (f–i) show molecular models for the corresponding porous networks, respectively. Reproduced from ref. 54, 53 and 40 with permission from the American Chemical Society, Wiley-VCH and the Royal Society of Chemistry, respectively. | ||
Based on metal–ligand coordination
Metal–ligand coordinate bonds provide an alternative pathway for engineering of 2D porous networks. These forces are akin to hydrogen bonds as far as specificity and directionality of interaction are concerned. While stronger than most hydrogen bonding interactions, metal–ligand co-ordinate bonds are relatively more labile than covalent bonds and thus allow error correction during network formation.7 Given that metal–ligand complexes are often chemically labile, a large body of work on metallo-supramolecular networks has been carried out under UHV conditions. A typical strategy consists of combining judiciously chosen organic ligands with metal centers under UHV conditions. The metal centers are either provided by thermal evaporation of high purity materials or they are extracted from the step-edges of the metal surface as metal ad-atoms. Commonly used metals include gold, copper and silver with different crystal facets while organic ligands based on carboxylate, pyridine, pyrrole, hydroxyl and carbonitrile functional groups have been employed.57Honeycomb porous networks formed by dicarbonitrile-polyphenylenes (NC-Phn-CN, where n is the number of phenyl groups) constitutes an excellent example of isoreticular host networks based on metal–ligand co-ordination. The size of these building blocks can be varied by adding an extra phenyl ring to the oligophenylene backbone. Dicarbonitrile-polyphenylenes ranging in size from 1.66 nm (n = 3)58 up to 2.96 nm (n = 6)59 have been studied on Ag(111) surface (Fig. 4a–d).
 | ||
| Fig. 4 Isoreticular host networks based on metal–ligand coordination. (a–d) Molecular structures of dicarbonitrile-polyphenylene (NC-Phn-CN) building blocks with increasing the length of the oligophenylene backbone. Corresponding STM images of the host networks for (e) n = 3 (f) n = 4 (g) n = 5 and (h) n = 6. Panels (i–l) show molecular models for the corresponding porous networks, respectively. Reproduced from ref. 58 and 59 with permission from the American Chemical Society. | ||
Typical experimental protocol involves sublimation of submonolayer amount of organic ligand onto the silver surface followed by exposure to a beam of Co atoms at 300 K. The metal–organic networks are then characterized by STM at ∼10 K. Coordination of the carbonitrile groups with cobalt atoms drives the honeycomb network formation. The NC-Phn-CN ligands form the walls of the hexagonal voids while Co atoms are positioned at the vertices. Each nodal Co atom coordinates with three ligands. These metal–organic networks are commensurate with the underlying silver lattice wherein the orientation of the network is dominated by the interaction of the polyphenylene backbone with the surface. Using this strategy, isoreticular honeycomb porous networks with cavity sizes ranging from ∼4.2 nm to ∼6.7 nm have been fabricated on Ag(111) surface (Fig. 4e–l). By carefully fine tuning the stoichiometry of the metal versus the ligand, the NC-Phn-CN molecules themselves are captured into the host cavities giving rise to an auto host–guest system.60 These host networks were found to be robust and survived annealing at higher temperature. It must be noted that the robustness and the ability assemble into a honeycomb porous network is a result of the strength of the metal–ligand co-ordinate bonds. In absence of cobalt atoms, NC-Phn-CN molecules form a variety of complex open porous structures on Ag(111) surface which are sustained by relatively weak hydrogen bonding between Ar–CH⋯NC– interactions.61
Another notable class of metallo-supramolecular networks is based on metal–carboxylate systems. One of the early examples included Fe–carboxylates of aromatic bis-carboxylic acids namely, terephthalic acid (TPA) and 4,1′,4′,1′′-terphenyl-1,4′′-dicarboxylic acid (TDA).9 TDA is an extended analogue of TPA with an extra phenyl ring in the backbone. Both compounds form metal–organic co-ordination complexes with co-adsorbed Fe atoms on Cu(100). In contrast to the host networks discussed so far, the metal–carboxylate networks of TPA and TDA on Cu(100) are rectangular in shape due to the symmetry of the surface lattice. The Fe–carboxylate networks of TPA and TDA were used to immobilize C60 guests. This strategy was further extended to a rather complex three component system involving linear bis-carboxylic acids, bipyridines and Fe atoms on a Cu(110) surface. The self-assembled metallo-supramolecular motif consists of co-ordination of two carboxylates and two pyridyl ligands to a Fe dimer. The co-deposition of any of the binary combinations with Fe atoms yielded highly ordered, extended coordination networks. The dimensions of the rectangular voids could be varied in a modular way via the length of the molecular ligand leading isoreticular cavities with areas ranging from 1.9 nm2 to 4.2 nm2.62
Novel host systems
Supramolecular organic frameworks (SOFs)
The structure of surface-supported supramolecular networks is often defined by the surface lattice underneath. Thus, most of the host networks and their resultant host–guest properties are often realized only in presence of an appropriate surface. The surface-adsorbed networks therefore do not exist as ‘networks’ in solution. Supramolecular polymers on the other hand, maintain their polymeric properties in solution. They consist of arrays of monomeric units held together via non-covalent interactions. These relatively novel materials were developed by application of supramolecular chemistry principles (reversibility, self-organization, weak interactions etc.) to polymer science. While the initial design concepts focused only on hydrogen bonded units, later strategies employed a variety of other types of interactions.63–65 Most examples of supramolecular polymers are reported in solution and only a handful of macrocycle-based supramolecular polymers, all forming non-porous networks have been analyzed on a solid surface.66–68Similar to surface-supported networks, monomers with two binding sites form linear polymeric architectures whereas those bearing three or more binding sites yield extended porous networks. The 2D self-association of such planar triangular, square or hexagonal monomers leads to formation of extended supramolecular organic frameworks (SOFs). The association can occur between the same type of building blocks or the framework may consist of another rod-like ditopic monomer. Typically the binding of the homomeric or heteromeric units is achieved by encapsulation of the binding sites using a covalent macrocyclic host. Such SOFs offer alternative systems for host–guest chemistry. Structural aspects of a number of different types of SOFs are already being explored.28 These 2D frameworks can be easily prepared in water and recent examples demonstrate formation of long-range ordered, free-standing films extending several square micrometers.69,70
A recent example of extended SOF films used host–guest enhanced donor–acceptor interactions between tris-(methoxynaphthyl) and N-methyl viologenyl units installed on two different monomers (Fig. 5a and b). The two monomers strongly associate together in presence of cucurbit[8]uril (Fig. 5c) to yield a robust, free standing SOF. The supramolecular polymerization was carried out at the liquid–liquid (toluene–water, Fig. 5d–g) interface which prevents out-of-plane polymerization leading to a homogenous monolayer SOF which could cover an area of up to 0.25 cm2 (Fig. 5h–j).69 The emergence of such free standing supramolecular membranes is beneficial for host–guest chemistry as these films can be transferred to arbitrary surfaces thereby widening their applicability.
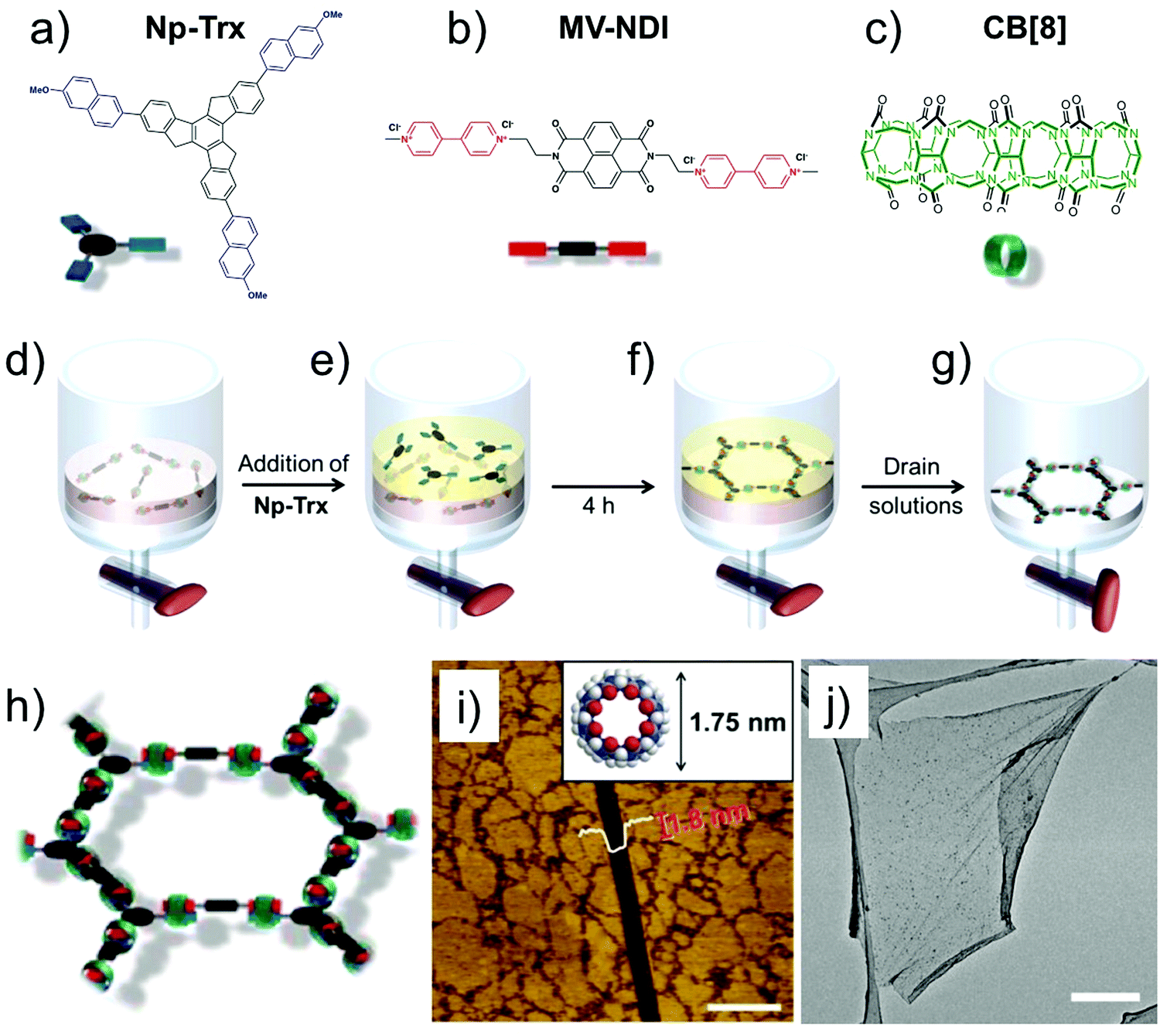 | ||
| Fig. 5 2D-SOF. (a–c) Molecular structures of the building blocks. (d and e) Schematic showing the process of SOF formation at the liquid–liquid interface. (h) A schematic of the honeycomb SOF (i) AFM image of the 2D-SOF with a scratch with a depth of 1.8 nm. The inset shows the molecular structure of CB[8] with an outer diameter of 1.75 nm, providing evidence for monolayer thickness of the film. The scale bar is 1 μm. (j) TEM-image showing slightly wrinkled, free standing 2D-SOF. The scale bar is 2 μm. Reproduced from ref. 69 with permission from the American Chemical Society. | ||
Covalent organic frameworks (COFs)
Covalent organic frameworks represent an emerging class of crystalline porous materials made up of light elements. They represent an all-organic equivalent of zeolites or MOFs. In contrast to SOFs, the building blocks of COFs are linked together via covalent bonds. The solution synthesis of bulk COFs has been extensively studied in the last decade. It is largely driven by potential applications in gas storage, catalysis, optoelectronics and photovoltaics.71 Isolation of single layers of COFs however, remains a major challenge. Surface synthesis of COFs has been explored, which allows in situ characterization of single layered material using scanning probe methods. While limiting the growth of the surface-synthesized material to a single layer is often challenging, a number of examples have already been reported describing the nanoscale characterization of COFs based on boronic acids72,73 and imines74–76 using STM.Bulk isoreticular COFs, typically synthesized using solvothermal methods, have been routinely reported. Surface synthesis of isoreticular monolayer COFs however, was only demonstrated recently.72,73,76 Boronic acid self-condensation and Schiff’s base formation are the two most commonly explored chemistries for surface synthesis of 2D COFs. Defect-free, long-range ordered COF films can be obtained using these reactions as they are reversible and can be carried out under mild conditions. The self-condensation of three diboronic acid molecules leads to formation of covalent sheets with hexagonal arrangement of boroxine (B3O3) rings (Fig. 6a) interconnected by the organic backbone of the diboronic acid monomer. Reversibility is usually ensured by addition of small amount of water in the reactor which evaporates during the course of the reaction thus shifting the equilibrium to the dehydrated product. A series of isoreticular 2D COFs were obtained by self-condensation of para-boronic acids with increasing size of the organic linker from phenyl to quaterphenyl (Fig. 6b–e). These covalent host networks offer cavity sizes ranging from ∼1.0 nm to ∼3.2 nm (Fig. 6f–m).
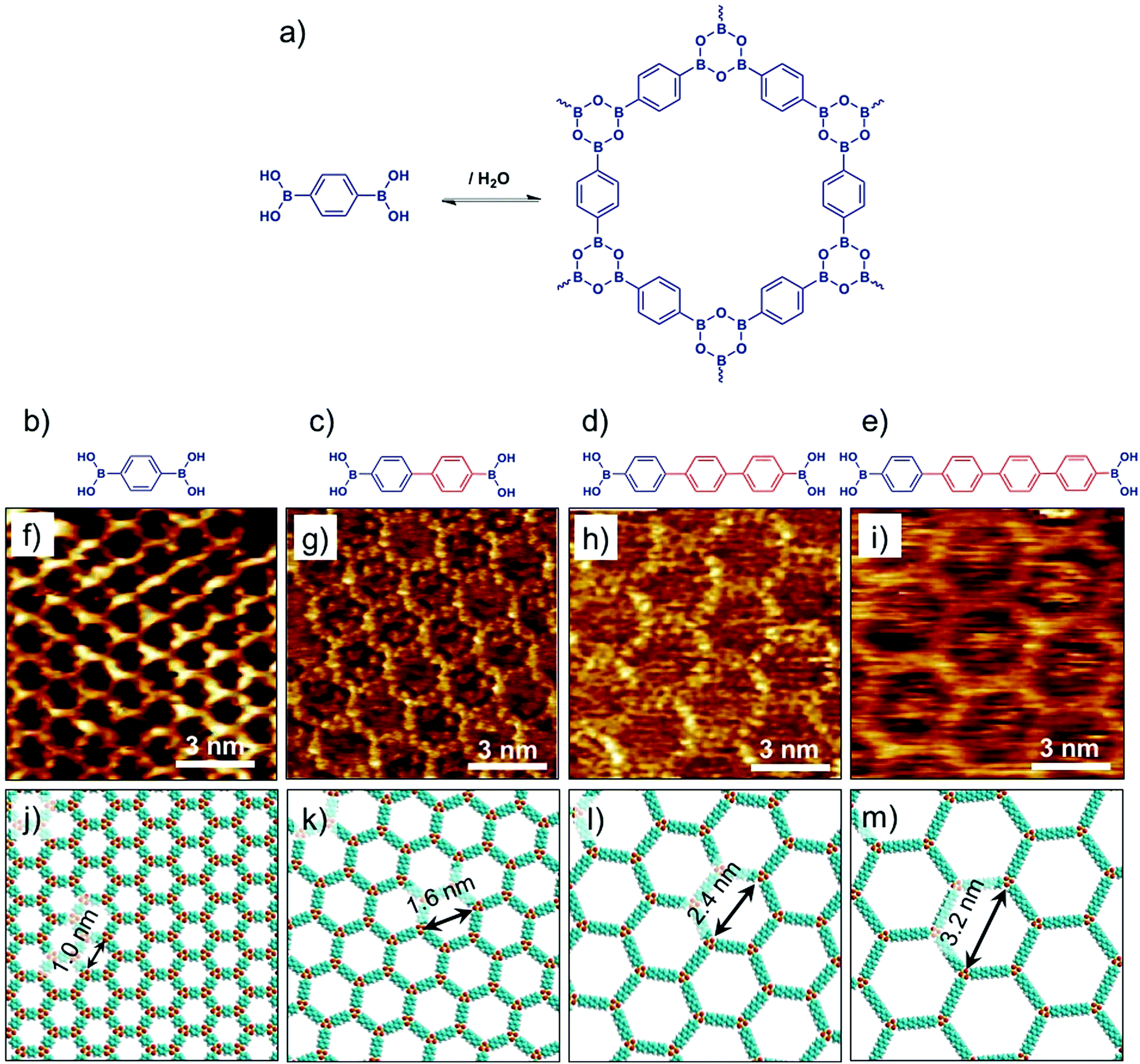 | ||
| Fig. 6 Surface synthesis of isoreticular 2D-COFs. (a) A reaction scheme showing boronic acid self-condensation to yield boroxine based COF. (b–e) Molecular structures of the boronic acid building blocks with increasing length of the oligophenylene backbone. (f–i) Corresponding STM images of 2D-COF films synthesized on graphite surface. (j–m) Molecular models of the isoreticular COF networks. Reproduced from ref. 72, 73 with permission from the American Chemical Society. | ||
Isoreticular synthesis of imine based COFs obtained via Schiff’s base reaction has also been reported recently on graphite and provides access to covalent films with ordered cavities ranging in size from ∼1.7 nm to 3.5 nm.76 A recent exciting development in this field is the synthesis of monolayer films of imine based COFs at the air–liquid interface. A number of challenges associated with the bulk as well as surface synthesis of COFs are alleviated when the synthesis is carried out at the air–liquid interface. This allows the transfer of these so-called 2D polymers to arbitrary surfaces for their detailed characterization. Two notable examples of 2D polymers synthesized at the air–liquid interface include the photopolymerization of an amphiphilic anthraquinone based monomer77 and an imine COF realized using dynamic covalent chemistry.78 The novel strategies for the fabrication of supramolecular as well covalent frameworks described above are beneficial for the field of host–guest chemistry on surfaces in general.
In addition to the covalently and non-covalently assembled host networks, inorganic surfaces such as hexagonal boron nitride (h-BN) nanomesh also serve as weakly interacting hosts. h-BN nanomesh is a single sheet of hexagonal boron nitride formed on Rh(111). It has highly corrugated nanostructured surface which appears like self-assembled hexagonal pores. The periodicity of the pores is ∼3.2 nm whereas the pore diameter is ∼2.0 nm. The porosity of the surface is the result of varying interaction of the h-BN nitride layer with the Rh lattice. The lower regions or the ‘pores’ bind strongly to the metal whereas the walls of the nanowells or the so called ‘wires’ are regions where the interaction with the surface is relatively weak. It has been recently shown that the pores of such nanomesh not only serve as adsorption sites for organic molecules79,80 and water81 but can also be used as nanoscale reactors.75
Host–guest chemistry in 2D supramolecular networks
As mentioned in the introduction, host–guest chemistry on solids has been carried using hosts with intrinsic as well as extrinsic porosity. The host cavity in the case of former is a result of synthesis whereas it is a consequence of supramolecular self-assembly for the later. Both types however involve extended 2D networks. A critical limitation of host networks employing intrinsic cavities is the tedious organic synthesis of the building blocks. For instance, realization of isoreticular host networks using host molecules with intrinsic cavities is a massive task. Furthermore, introducing a desired modification into the structure of intrinsically porous host molecules is often far from simple. Despite these limitations, host–guest chemistry using porous organic molecules has been explored and in the following section we describe a few examples.Using intrinsically porous building blocks
For host networks with extrinsic cavities, the individual building blocks are incapable of capturing guest species on their own. On the contrary, intrinsically porous hosts often interact with an ideal guest both in solution as well as on the surface. The high affinity between the two components results in highly specific guest binding with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry. Typically, small molecules such as C60 and cations serve as the guest species, however larger molecules such as hexa-peri-hexabenzocoronene (HBC)82 and a macrocyclic peptide valinomycin83 have also been immobilized using giant macrocyclic cavities.
:1 stoichiometry. Typically, small molecules such as C60 and cations serve as the guest species, however larger molecules such as hexa-peri-hexabenzocoronene (HBC)82 and a macrocyclic peptide valinomycin83 have also been immobilized using giant macrocyclic cavities.
Host–guest complexes formed between a cyclothiophene macrocycle (Fig. 7a) and C60 have been characterized at the organic solution–solid interface. In addition to the π–π interactions, these host–guest complexes are stabilized by donor–acceptor interactions between the electron rich cyclothiophene macrocycle and the electron accepting C60. These donor–acceptor interactions are highly specific and ensure that the C60 molecules are preferentially complexed to the rim of the macrocycle instead of the covalent cavity (Fig. 7b and c). The stoichiometry at the monolayer surface is always 1:1 due to electrostatic interactions between the two molecules. This is because, binding of the C60 guest to one side of the rim significantly alters the electron density of the macrocycle thus creating an intrinsic dipole. As a consequence, the other end of the rim becomes electron deficient and cannot bind another C60 molecule.84
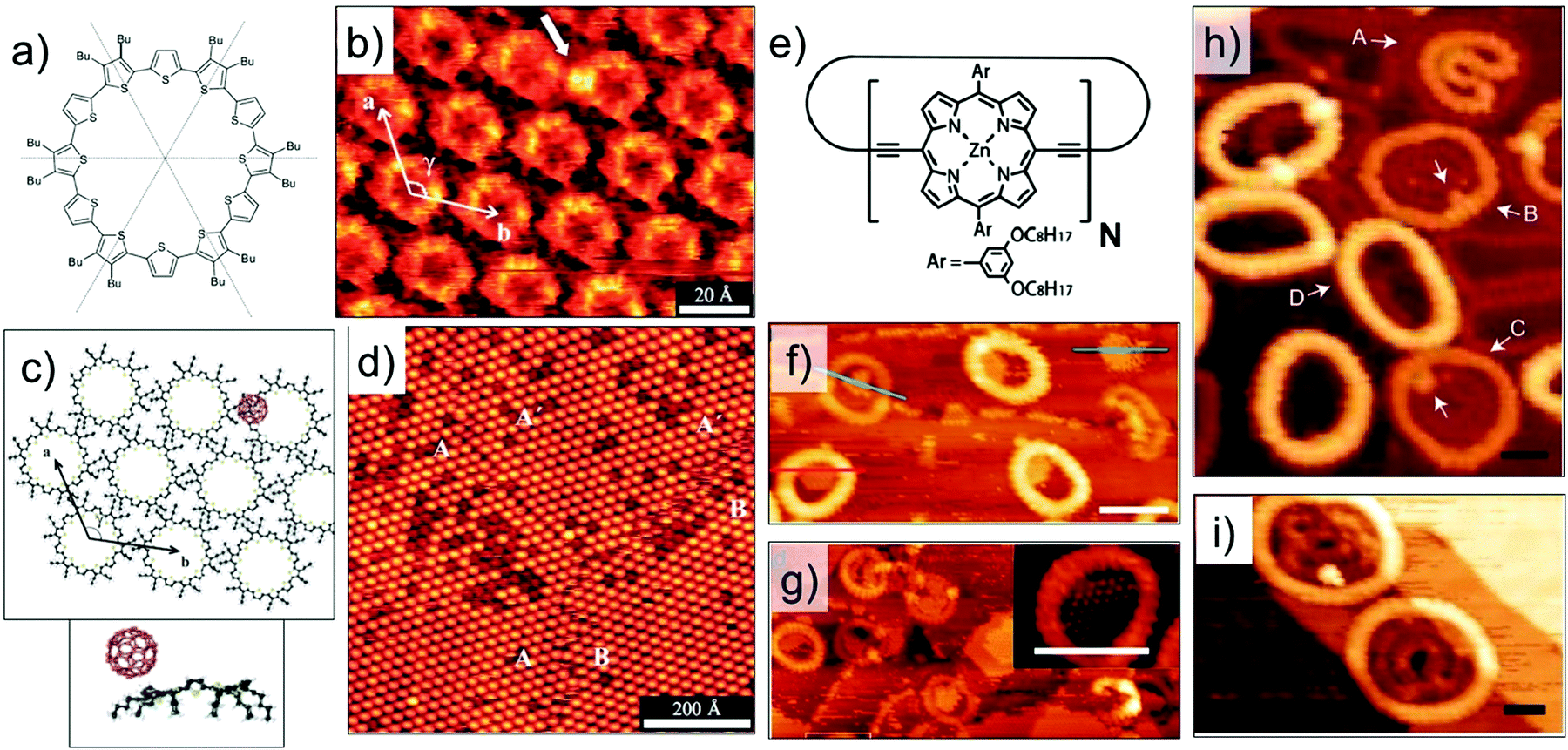 | ||
| Fig. 7 Host–guest chemistry using intrinsically porous building blocks. (a) Molecular structure of the cyclothiophene macrocycle. (b) STM image showing capture of C60 (white arrow) on the rim of the macrocycle. (c) Calculated model of a closely packed monolayer of the cyclothiophene macrocycle with a hexagonal arrangement together with the side view of the calculated energy minimum conformation of the complex. (d) Large scale STM image showing the host–guest complexation. (e) Molecular structure of the cyclic porphyrin polymer. (f) STM image showing stacked nanorings for a cyclic polymer with N = 24. (g) Capture of C60 by the nanorings. Panels (h) and (i) show STM images where the polymers show auto host–guest type encapsulation for polymers with N = 30 and N = 40, respectively. Reproduced from ref. 84, 85 and 86 with permission from Wiley-VCH, Macmillan Publishers Ltd, and the Royal Society of Chemistry, respectively. | ||
Organic macrocyclic hosts popularly used in solution phase host–guest chemistry have also been studied on surfaces. Apart from their typical guest binding ability, peculiar effects due to close packing and surface-confinement have been reported. For example, a crown ether substituted phthalocyanine derivative forms an ordered array on Au(111) surface and is capable of binding calcium ions. However, despite the availability of four binding sites per molecule, inclusion of Ca2+ ions in only two crown ether sites was observed. The unusual binding behavior is the result of electrostatic repulsion. On filling the remaining crown ether moieties, the bound Ca2+ ions would get too close to the crown ethers of neighboring hosts that already contain ionic guests. Furthermore, the ion binding depends on the crystallographic orientation of the Au surface. No complexation of Ca2+ ions was observed when Au(100) was used for the assembly of host array.87 In addition to this, the binding of K+,88 Na+, H+ and Cs+89 ions to dibenzo crown ethers has been studied by STM on different surfaces. STM studies of anion binding on the other hand, are rather scarce.90–92 Recent examples include iodide binding to a tricarbazolo triazolophane macrocycle92 and binding of hexafluorophosphate anions to a class of macrocycles called ‘cyanostars’,91 both studied on the graphite surface. In both these examples anion binding promoted formation of higher order stacks of macrocycles, thus demonstrating the active role played by guest binding.
Host architectures based on large shape-persistent macrocycles have received considerable attention in the recent past. Highly evolved synthetic strategies have provided access to large (metal–) organic macrocycles.16,85,86,93,94 While solution–solid interface is ideally suited for the surface assembly of such compounds, recent progress in experimental protocols for deposition of high molecular weight compounds such as electrospray ionization, has opened new frontiers of host–guest chemistry under clean UHV conditions. A notable example is giant macrocycles synthesized using the so-called Vernier templating approach.94 An elegant method, where molecular recognition is combined with organic synthesis, this strategy has been employed to synthesize large cyclic porphyrin polymers (Fig. 7e) ranging in diameter from 4.7 nm to 21 nm. Electrospray ionization allows deposition of these giant molecules on Au(111) and their subsequent structural characterization using STM under UHV conditions. The surface-adsorbed structures of a nanoring with N = 24, where N is the number of porphyrin units in the polymer, reveal columnar stacks up to 4 layers high. The nanorings capture C60 in their covalent cavities and the capture of C60 has been found to depend on the number layers in the stack (Fig. 7f and g).85 Larger nanorings with 30 or more repeat units exhibit a unique supramolecular ‘nesting’ behavior where one molecule adsorbs as folded ring inside another circular nanoring (Fig. 7h and i). Such auto host–guest behavior was observed under UHV conditions as well as at the solution–solid interface.86
Using self-assembled nanoporous networks
Due to unpredictable entropic factors, bulk self-assembly of molecules into discrete, porous architectures is often elusive. However, 2D confinement against a surface restricts several degrees of translational, rotational and vibrational freedom thus allowing formation of well-defined porous architectures. A vast majority of host–guest systems are studied at the solution–solid interface under ambient conditions, possibly due to ease of experimental procedure. Furthermore, monitoring the dynamic aspects of host–guest interactions becomes possible at the solution–solid interface. Incorporation as well as exchange dynamics of molecular guests within typical host networks has been reported by using time-dependent in situ STM imaging. Such studies shed light not only on the dynamic aspects of host–guest chemistry but they also reveal important details about the mechanistic aspects of the binding process.95,96 Host–guest chemistry under UHV conditions is often limited by the ability to sublime the guest molecules which is linked to its molecular weight. Most studies under UHV conditions have focused on C60 as guest, due to ease of its sublimation. In the following sections, we highlight some interesting aspects of host–guest chemistry on surfaces.The first of such examples consisted of transition of an otherwise non-porous self-assembled network into a porous one in response to addition of a guest. Alkoxy substituted DBAs with n = 14 and n = 16 form a densely packed non-porous network at relatively high concentrations. Addition of ten-fold excess of coronene (COR) to the preformed network led to conversion of the non-porous structure to the honeycomb porous structure wherein the guest cavities are occupied by COR molecules. Comparison with other guest molecules revealed that only planar guest molecules with large π conjugated backbones induce the non-porous to porous transition irrespective of their symmetry whereas non-planar or smaller guests do not affect the dense network. This observation indicates that the open porous network is thermodynamically stabilized via gain in the free energy upon adsorption of guest molecules thereby overcoming the intrinsic energy penalty associated with large empty voids.97 Similar type of phase transitions are reported for van der Waals host networks using HBC as guest.98
Guest induced transitions however, are not limited to van der Waals based host systems. A quaterphenyl tertracarboxylic acid derivative (QPTC, Fig. 8a) forms a relatively close packed parallel structure upon adsorption at the solution–solid interface (Fig. 8b). The energetic preference of the system can be changed by addition of coronene as a guest template which directs the formation of a 2D Kagomé network (Fig. 8c).99 Guest-induced transitions are not unique to planar guest molecules either. Recently, immobilization of C60 in a hydrogen-bonded porous 2D network was found to promote the growth of a second layer of the host network in an orthogonal direction to the graphite surface. A shorter analogue of QPTC, p-terphenyl-3,5,3′′,5′′-tetracarboxylic acid (TPTC) forms an open porous network with hexagonal voids via in-plane hydrogen bonding between carboxyl groups (Fig. 8d). Addition of saturated solution of C60 to the preformed network of TPTC induced the growth of a second layer of TPTC monolayer. The second layer is templated by the adsorbed C60 guests (Fig. 8e and f). The templating of the bilayer is a highly co-operative process as neither adsorption of C60 nor bilayer formation of TPTC was observed in isolation. The upper layer is sustained by host–guest interactions with C60 as well as via π stacking interactions with the lower TPTC layer directly in contact with the graphite surface (Fig. 8g). Thanks to the dynamic nature of the solution–solid interface, the TPTC–C60 bilayer network can be readily converted to TPTC–COR monolayer network by addition of COR which is a preferred guest in view of its higher adsorption energy and better fit into the pores. This system represents an important step towards realization of 3D architectures based on 2D patterns.100
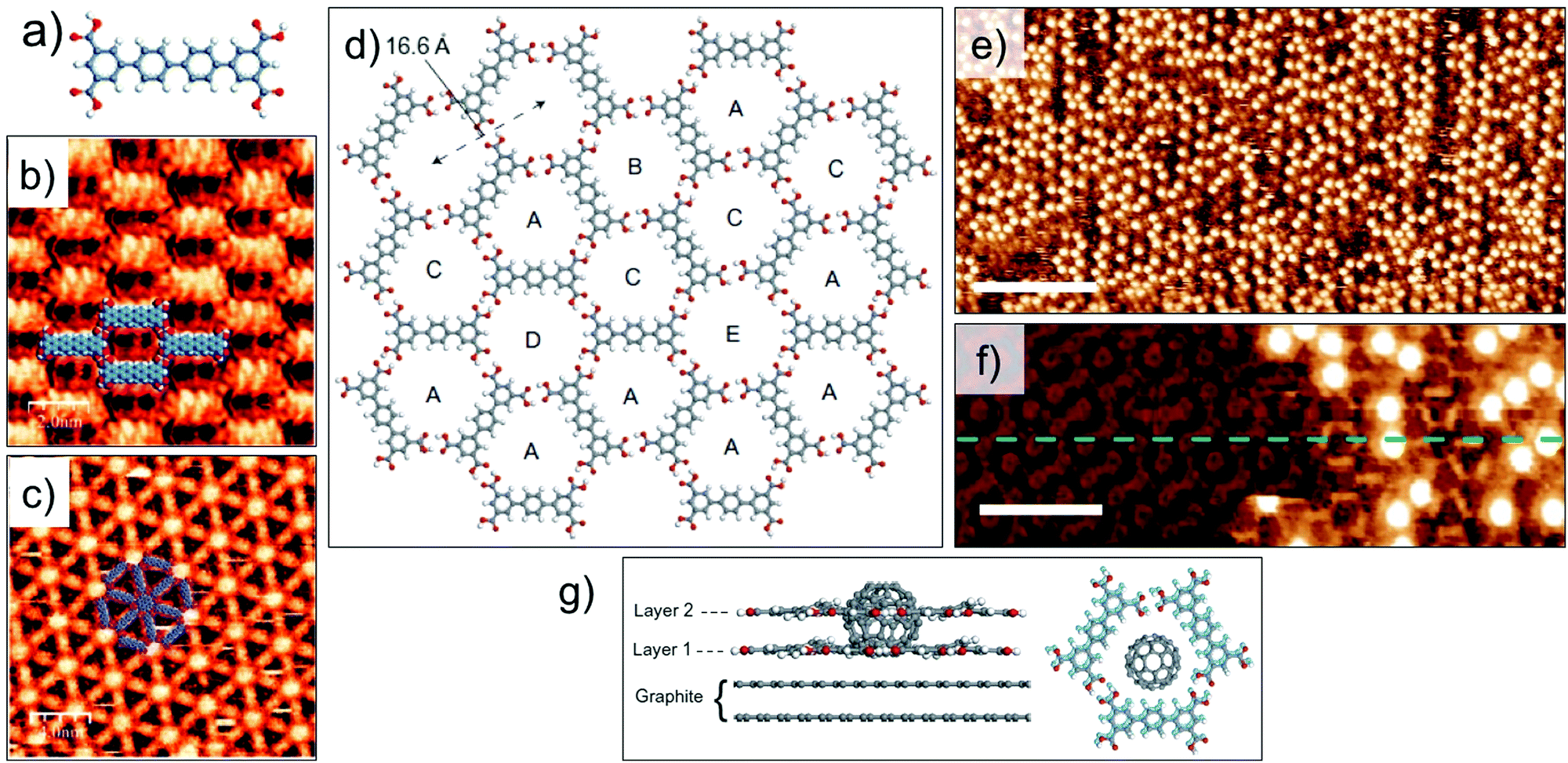 | ||
| Fig. 8 Guest induced structural transitions in host networks. (a) Molecular structure of QPTC. (b) Close packed network of QPTC in absence of guests. (c) Host–guest architecture formed upon addition of COR to the close packed network. (d) Molecular model showing host network of TPTC formed at the nonanoic acid/HOPG interface. (e) STM image showing TPTC-C60 host–guest architecture. (f) STM image of TPTC network immediately after C60 deposition. The initial layer of TPTC network is visible with an altered contrast and the TPTC molecules in the second layer appear as bright, rod-like features surrounding the C60 molecules which appear as bright blobs. Reproduced from ref. 99 and 100 with permission from the Royal Society of Chemistry and Macmillan Publishers Ltd, respectively. | ||
Similar transitions have been reported in systems which are stabilized by a balance of different supramolecular interactions. A recent example of such guest-induced dynamic host–guest chemistry includes supramolecular networks of alkoxy substituted isophthalic acids which are sustained by a balance between van der Waals interactions between interdigitating alkoxy chains (leading to a dense assembly) and hydrogen bonding between carboxyl groups (giving rise to a porous structure). In this case also addition of coronene favored the formation of the porous structure.102
A unique example of metal ion triggered dynamic assembly and re-assembly of supramolecular host networks was reported recently. A N9-alkylguanine derivative (Fig. 9a) self-assembles into a ribbon-like architecture (Fig. 9b) at the 1,2,4-trichlorobenzene (TCB)/HOPG interface. In situ addition of potassium picrate solution in TCB to the ribbon-like network lead to a structural transition where the initial motif was converted into a G4 quartet (Fig. 9c). This structure consists of a hydrogen-bonded cyclic tetramer of guanine molecules. The transition could be reversed by addition of a [2.2.2]cryptand which complexes with potassium ion thus removing it from the quartet and leading to the collapse of the cyclic tetramer back to the ribbon-like network (Fig. 9d). Finally, in situ addition of trifluoromethanesulfonic acid (HTf) lead to the release of K+ ions from the cryptand thus making them available for complexation with alkylguanine derivative which again transitions into the G4 quartet structure due to ion complexation (Fig. 9e). The dynamic supramolecular cycle was completed by addition of cryptand which converted the quartet back to the ribbon-like network (Fig. 9f).101 It must be noted that the common feature of the systems discussed above is that they are all assembled at the solution–graphite interface. The presence of a solvent and a weakly interacting surface such as graphite seems to be a key to such dynamic behavior.
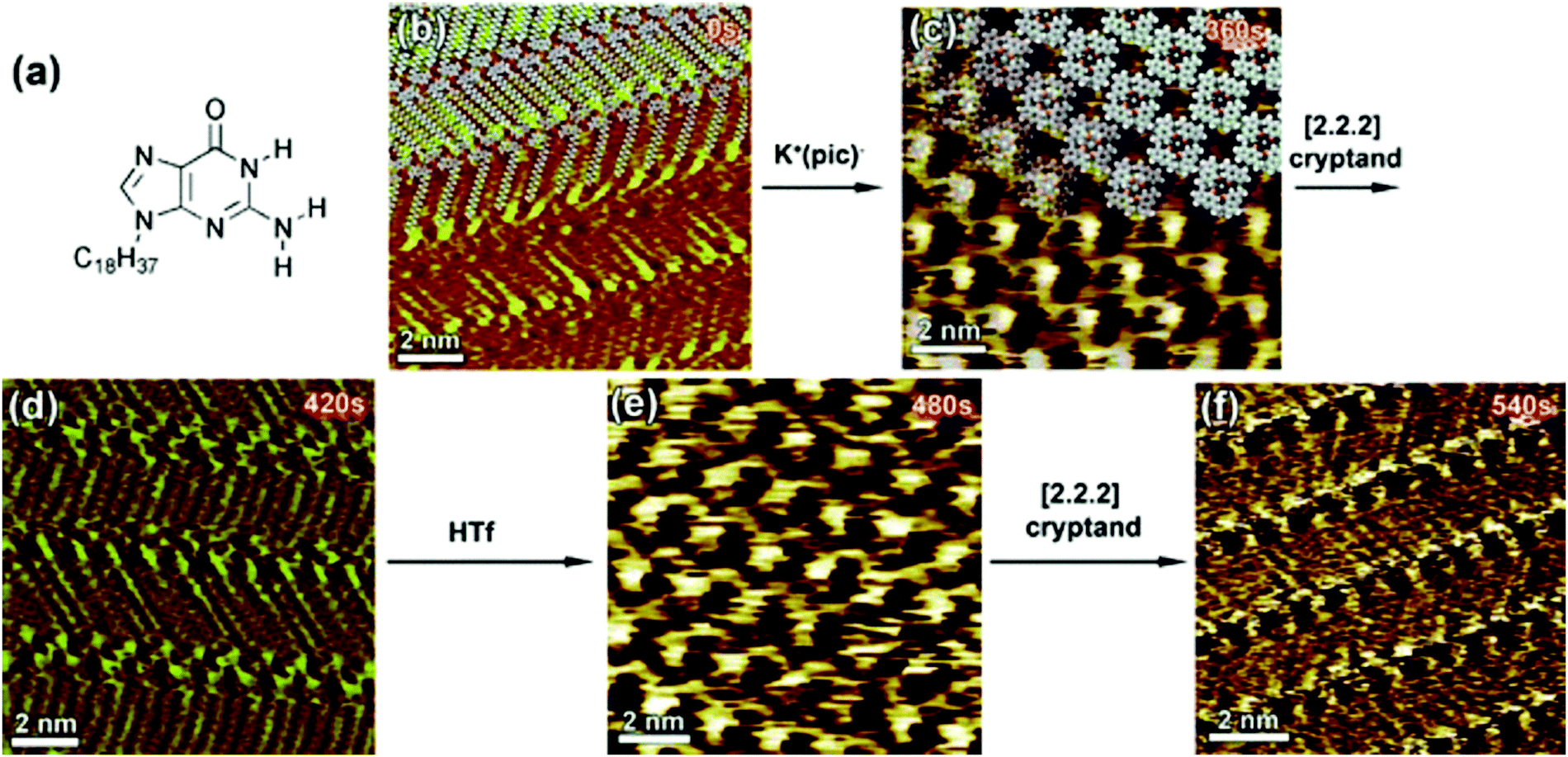 | ||
| Fig. 9 Ion-induced transitions in supramolecular networks. (a) Molecular structure of the N9-alkylguanine derivative. Panels (b–f) present consecutive STM images showing the structural evolution of the N9-alkylguanine monolayer over a 9 min time scale (time range displays in the upper right part of the images correspond to the time that was needed to reach the equilibrium after addition of reacting agents). Images (b), (d), and (f) show ribbon-like structure, whereas (c) and (e) exhibit G4-based architectures. Reproduced from ref. 101 with permission from Wiley-VCH. | ||
The complexity of hierarchical self-assembly was further extended to a three component host–guest architecture assembling at the solution–solid interface. Instead of using a single of type of guest, a heteromolecular guest cluster composed of COR and ISA could be immobilized in host network formed by DBA-OC10 derivative. The trapping of the guest cluster is based on size and shape complementarity. This is a unique system where guest induced transitions occurs at two different levels. At the COR–ISA level: ISA by itself does not form cyclic hexamers at the solution–solid interface but self-assembles into a densely packed zigzag structure dictated by hydrogen bonding interactions between carboxyl groups. However, COR templates the formation of COR–ISA cluster which consist of a COR molecule surrounded by a hydrogen bonded hexamer of ISA (Fig. 10 a–c). In the concentration range employed in this study, DBA-OC10 forms only the densely packed structure at the 1-octanoic acid/HOPG interface. However, addition of a solution containing COR–ISA to the preassembled network of DBA-OC10 resulted in a structural transition from the dense to a honeycomb porous network, the cavities of which are occupied by COR–ISA heteroclusters (Fig. 10d and e). All the clusters have the same composition and symmetry indicating a highly specific recognition with the host cavity. Similar results were obtained upon premixing the three components in solution followed by drop casting the HOPG surface.56
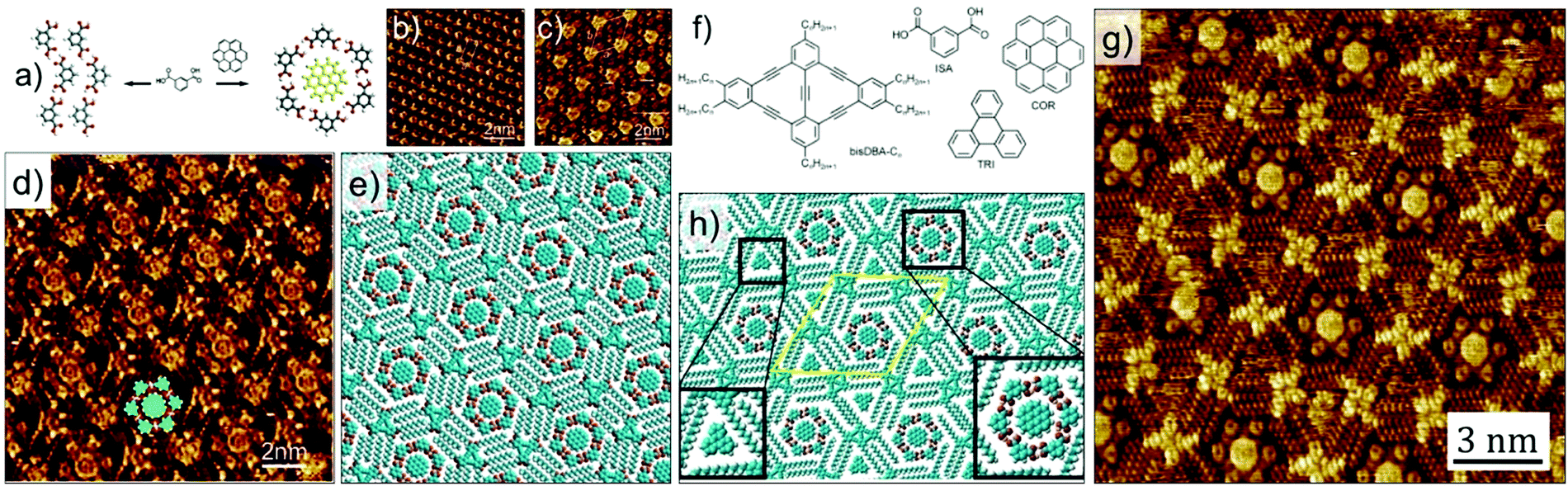 | ||
| Fig. 10 Multicomponent host–guest system obtained using DBA derivatives at the octanoic acid/HOPG interface. (a) Molecular model showing the COR templated assembly of ISA into cyclic hexamers. (b) Zigzag network of ISA obtained in absence of COR. (c) COR–ISA host–guest architecture. (d) A three component host–guest network involving DBA-OC10, COR and ISA. (e) Molecular model for the three component network. (f) Molecular structures of the constituents of the four component host–guest architecture. (g) STM image showing the four component host–guest network. (h) Molecular model for the four component network. Reproduced from ref. 56 and 24 with permission from the American Chemical Society and Wiley-VCH, respectively. | ||
A modified approach in 2D supramolecular engineering of DBAs yielded an even more complex four-component host–guest architecture. In order to accommodate more guests, a geometrically different host offering two different types of cavities was employed. Rhombus shaped bisDBA derivatives (Fig. 10f) readily form a Kagomé network which offers spatially well-ordered hexagonal and triangular voids. However, bisDBA-C12 (chosen due to the similar size of the hexagonal voids) does not form a Kagomé network at the 1-octanoic acid/HOPG interface. Only when COR and ISA are added, a stable three-component network is obtained, with the hexagonal cavity filled with a COR–ISA cluster. By adding a triangular guest molecule such as triphenylene (TRI) a four-component 2D host–guest structure is successfully fabricated, with the triangular cavities filled with TRI (Fig. 10g and h). An important finding is that, upon proper concentration control, this four-component pattern spontaneously emerges at the solvent/graphite interface upon simply depositing a drop of the solution containing the mixture. Control experiments revealed that the four-component host–guest assembly is a highly cooperative process involving the action of all components at the same time. The exact size matching of the guests (clusters) to both types of pores is crucial for attaining a stable four-component self-assembly.24
As mentioned earlier, controlling the organization of more than one component on metal surfaces is often challenging compared with that on graphite due to increased diffusion barriers in the case of the former. The influence of higher diffusion barriers for planar aromatic molecules on the process of nucleation and subsequent growth of a multicomponent networks was revealed when the DBA–COR–ISA system was studied on Au(111). The three component host–guest system forms a fundamentally different supramolecular structure on Au(111) surface. For understanding the differences in the two types structures one needs to understand the expression of chirality in DBA host networks. The rim of each hexagonal cavity in the DBA host network consists of a pair of alkyl chains from one DBA molecule, interdigitated with a pair from an adjacent molecule. When adsorbed on a surface, this interdigitation becomes chiral with two distinct interdigitation motifs, labelled arbitrarily (−) and (+) (Fig. 11a). The combination of interdigitation motifs lining an individual cavity can produce either chiral or achiral host cavity. Chiral host cavities have a combination of six identical interdigitation motifs. Achiral pores, on the other hand, have a combination of three (−)- and three (+)-type interdigitation motifs arranged in an alternating pattern (Fig. 11b). On Au(111), the DBA–COR–ISA multi-component network displays an ordered superlattice arrangement of chiral and achiral pores (Fig. 11c and d). In comparison, similar network on HOPG displays only chiral pores (Fig. 10d and e).56 The unique superlattice structure observed on Au(111) is related to the lower energetic preference for chiral pores than on HOPG and increased diffusion barriers for guest molecules. The increased diffusion barriers for guests allow them to act as nucleation sites for the formation of achiral pores. Following the initial nucleation of an achiral pore, restrictions imposed by the accommodation of guests within the porous network ensure that subsequent growth naturally leads to the formation of the superlattice structure (Fig. 11e).17
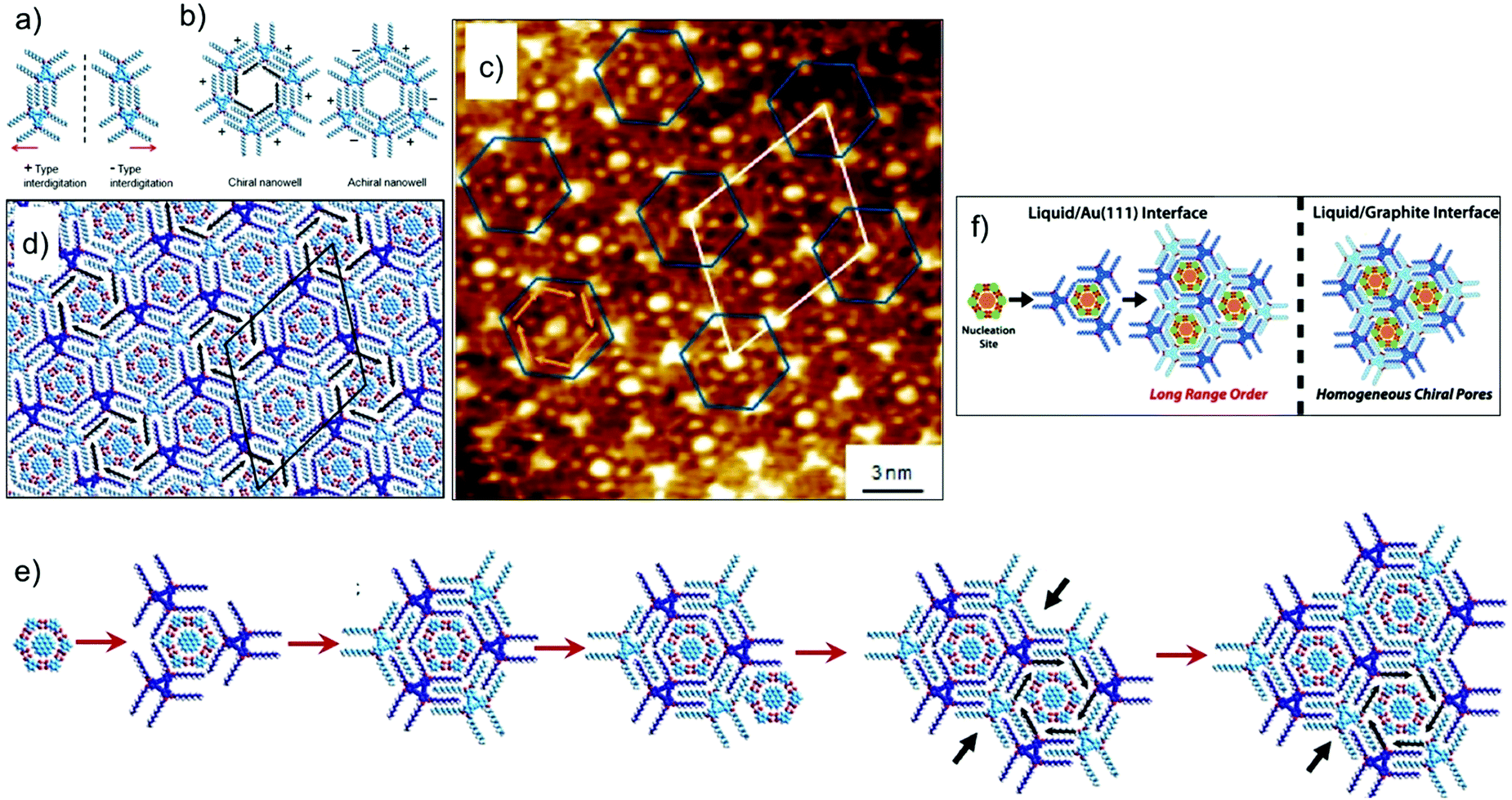 | ||
| Fig. 11 Multicomponent host–guest system obtained using DBA derivatives at the octanoic acid/Au(111) interface. (a and b) Expression of chirality in DBA networks. (c) STM image of the DBA–COR–ISA three component network on Au(111). (d) Molecular model for the DBA–COR–ISA network. (e) Molecular models showing step-by-step growth of the superlattice. The nucleation of the COR–ISA cluster is followed by adsorption of the alkyl chains of three DBA molecules surrounding the cluster. The only way to complete a pore is by the formation of an achiral pore. A COR/ISA cluster is captured in one of the neighboring sites, which leads to formation of either a chiral or an achiral nanowell around it. Assuming formation of a chiral nanowell, the third adjacent nanowell (black arrows) can only adapt an achiral arrangement. The black arrows indicate positions where the structural arrangement of nanowells is predetermined by the combination of structures for the initial two nanowells. (f) A schematic showing the difference in the host–guest patterns obtained on HOPG and Au(111) surface. Reproduced from ref. 17 with permission from the American Chemical Society. | ||
The PTCDI–melamine bicomponent host network described earlier has been used for trapping C60 as well as higher fullerenes.103 This physisorbed host network can also be assembled under ambient conditions and has been used as a template for directing the chemisorption of thiol self-assembled monolayers. The host network remains intact after thiol chemisorption. Such hybrid self-assembled monolayers were stable in the liquid environment and could be processed further by electrochemically depositing copper between the thiols and the Au(111) surface. Cu was only inserted between the thiols and the surface and not between the host network and the surface. Such a combination of physisorbed noncovalent networks with chemisorbed SAMs offers considerable design flexibility, with the network providing a well-defined confinement of structures within the surface plane, and the SAM permitting orthogonal modification of the surface.104
The first case scenario is exemplified by the enantioselective adsorption of chiral DBA (cDBA, Fig. 12a) derivatives in porous host network formed by structurally equivalent achiral DBA.105 When chiral and achiral analogues of DBAs are co-assembled at the solution–solid interface, the cDBA derivative alters the chiral balance of the system by co-adsorbing in the network. This is the well-known ‘sergeant–soldiers’ principle, where the handedness of the supramolecular network is defined by the handedness of the chiral DBA. Thus, a mixture of cDBA-OC12(S) (the sergeant) and DBA-OC12 (the soldier), leads to the formation of a porous network predominantly made up of clockwise (CW) nanowells (Fig. 12b).106 However, besides adsorbing as a part of the network, the chiral DBA molecules also occupy the cavities of the porous network. Fascinatingly, they do so in an enantioselective manner. The cDBAs show a pronounced tendency to adsorb in nanowells with handedness that is opposite to the one they induce on the surface. For example, although cDBA-OC12(S) induces formation of CW nanowells on the surface, it preferentially adsorbs as a guest in the CCW nanowells by adapting a windmill like conformation through in plane bending of its chiral chains (Fig. 12c and d). The chiral DBAs preferentially adsorb as guests compared to achiral DBAs. This is because, the guest conformation allows better van der Waals contact of the cDBA molecule with HOPG by bending the chiral methyl groups away from the solid surface. Calculations revealed that adsorption in the CCW nanowells is favored by 6 kcal mol−1. Molecular models revealed that the van der Waals contact between the alkyl chains of the chiral guest and those of host network is optimal when the handedness of the host nanowell (CW) does not match with the windmill-like conformation of the guest (CCW) (Fig. 12e and f). Similar behavior was observed for the DBA-OC12–cDBA-OC12(R) pair.105
 | ||
| Fig. 12 Selectivity of guest binding based on chirality. Enantioselective adsorption of chiral DBA derivatives. (a) Molecular structures of the chiral DBA derivatives. (b) Expression of chirality in DBA nanowells. (c) STM image showing enantioselective adsorption of cDBA-OC12(S) in CCW nanowells. The white line highlights the domains border between domains containing CW and CCW nanowells. The guest occupancy in the CCW nanowells is notably high compared to that in CW nanowells. (d) STM image showing a domain with CCW nanowells where the immobilized chiral guests are well-resolved. Molecular models in (e) and (f) show the structure of the confined chiral guests in the nanowells. Reproduced from ref. 105 with permission from the Royal Society of Chemistry. | ||
An example of the second type(a) was illustrated by host–guest assembly between an azo-bis-isophthalic acid (NN4A) derivative and fullerenes at the solution solid interface. NN4A forms an open porous Kagomé network via hydrogen bonding between the isophthalic acid units. No selectivity was observed for the adsorption of C60 which was captured in both hexagonal (type A) as well as triangular (type B) cavities. Larger fullerenes such as C80 and Sc3N@C80 exclusively occupied the larger hexagonal cavities displaying site selective binding. Furthermore, the higher electronegativity of Sc3N@C80 due to caged metal atom lead to stronger affinity with the host network affording a stable, well-ordered host–guest network.107
Recently, self-sorting of molecular guests using a sophisticated DBA host network was reported. Built using a tailored DBA derivative, this host network consists of periodically functionalized cavities of different sizes. The design strategy is based on the use of a DBA derivative with one functionalized alkoxy chain and five simple alkoxy chains. Such a building block would possibly lead to formation of two different patterns, A and B. In pattern A, the functional groups are randomly placed in the nanowells. On the contrary, if the functional groups are programmed to cluster together in the same nanowell, the resulting network would consist of periodically functionalized host cavities (Fig. 13a). The working hypothesis and the strategy were verified using a DBA derivative having an isophthalic acid unit at the end of one alkoxy chain connected with an azobenzene linker (iso-DBA, Fig. 13b) and five simple tetradecyloxy chains. This design exploits the ability of isophthalic acid units to form a hydrogen-bonded cyclic hexamer under appropriate conditions. Iso-DBA self-assembles at the solution–graphite interface exclusively in pattern B upon annealing. The network presents hexagonal cavities in which each nanowell containing the cyclic hexamer of isophthalic acid units is surrounded by six non-functionalized nanowells (Fig. 13d and e). Although entropically disfavored, the nanoscale separation of isophthalic acid unit containing cavities is favored due to the enthalpic gain associated with the formation of hydrogen bonds between six isophthalic acid units. The periodically spaced nanowells differ in size and were used for site-selective immobilization of COR and a large aromatic guest HPEPEB (Fig. 13c, f and g).108 Similar site-selective guest sorting behavior was reported for mixed monolayers formed by co-adsorption of two types of butadiyne-bridged planar macrocycles.109
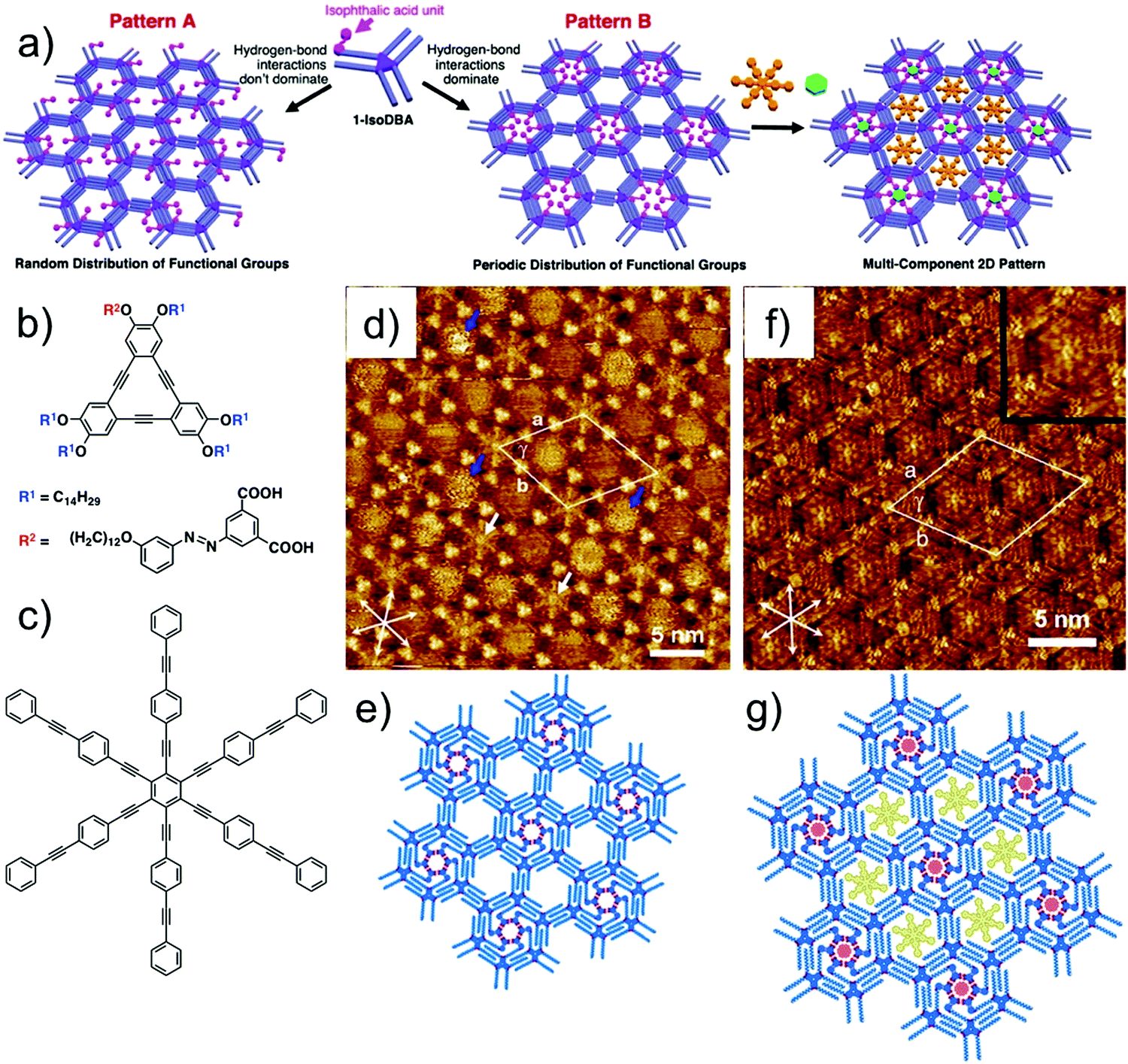 | ||
| Fig. 13 Site-selective immobilization of guest molecules in periodically functionalized host cavities. (a) Design strategy for periodically functionalized pores showing two possible outcomes of the self-assembly. (b) Molecular structure of the azo-DBA derivative. (c) Molecular structure of HPEPEB guest. (d) Self-assembled host network of azo-DBA showing periodically functionalized cavities with ISA units (white arrows) surrounded by unfunctionalized cavities. (e) Molecular model for the azo-DBA network. (f) Site selective immobilization of COR and HPEPEB in the two types of azo-DBA network. The inset shows digital zoom of the larger cavity hosting HPEPEB. (g) Molecular model for the host–guest network. Reproduced from ref. 108 with permission from the American Chemical Society. | ||
The dicarboxyazobenzene unit discussed above has also been employed for realizing photoresponsive host cavities in DBA networks. In contrast to the design used for periodic functionalization of host cavities, this strategy uses a DBA derivative in which the dicarboxyazobenzene units are installed on alternating alkoxy chains (Fig. 14a). Such design allows formation of a honeycomb network with functionalized pores containing the dicarboxyazobenzene units in all the host cavities (Fig. 14b). Given that the azobenzene derivatives are also known to undergo photoisomerization on surfaces,111,112 the host cavities are thus tailored to respond to irradiation of light of appropriate wavelength. The structural difference between the planar trans- configuration and the non-planar cis- configuration is then anticipated to change the pore size and shape upon photoisomerization. STM images revealed that the host network consists of hexagonal nanowells with nearly all the azobenzene units directed towards the center of the nanowell, suggesting the formation of a cyclic hexamer of the dicarboxyazobenzene units (Fig. 14d). This network ideally captures a single COR molecule per cavity (Fig. 14e). The guest binding ability was further explored by in situ irradiation of the honeycomb network adsorbed on the graphite surface. Irradiation of the surface with 320 nm light followed by addition of COR solution to the system revealed that the number of cavities containing more than two COR molecules increased (Fig. 14f). Molecular models show that the trans-to-cis isomerization followed by desorption of one of the azobenzene units generates enough space to accommodate an additional COR molecule. Since the isomerization is a reversible process, the pore size can be reduced by triggering the cis- to trans- isomerization by irradiation with longer wavelength light (λ > 400 nm). The reduction in the pore size forces excess COR guests to desorb from the host cavity (Fig. 14g).110
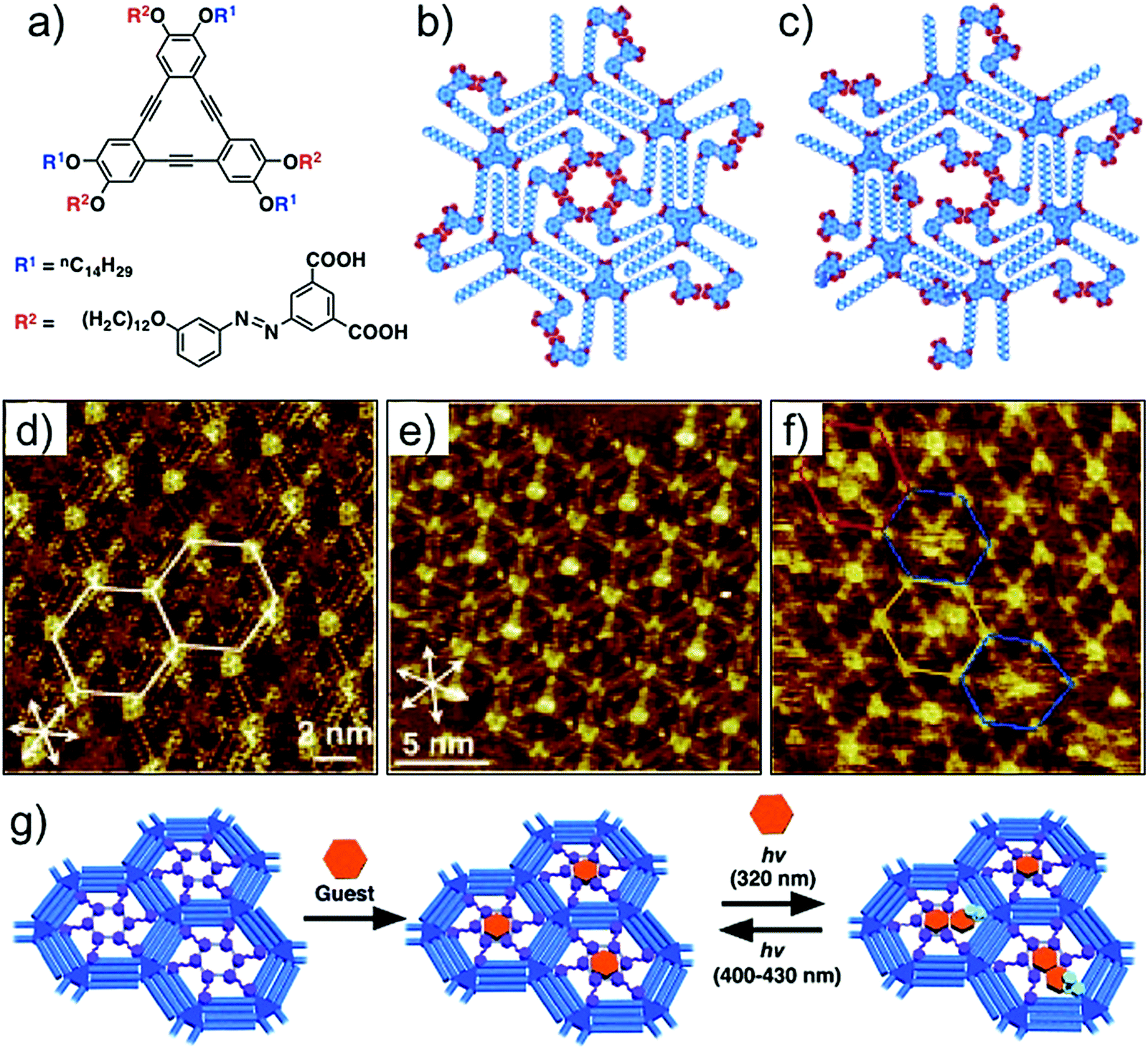 | ||
| Fig. 14 Photoresponsive host–guest system. (a) Molecular structure of dicarboxyazobenzene substituted DBA. (b) Molecular model for the all trans configuration in the nanowell. (c) Molecular model showing a nanowell where one of the dicarboxyazobenzene units has adopted cis configuration. (d) STM image of the porous host network in all trans configuration. (e) Host–guest architecture with COR as a guest. (f) STM image obtained after in situ irradiation of the monolayer with UV light. The colored hexagons indicate the pores containing four CORs (red), two CORs (yellow), and those with fuzzy images (blue). (g) Schematic of the light responsive host–guest system. Reproduced from ref. 110 with permission from Wiley-VCH. | ||
Photoresponsive host systems based on intrinsically porous building blocks have also been reported. The isomerizing azo units are integrated in the backbone of an azobenzenophane type 4-NN macrocycle. Although it does not form an extended 2D network on its own, it can be immobilized into the porous self-assembled network formed by TCDB (Fig. 15a) at the heptanoic acid/HOPG interface. Depending on their relative solution stoichiometry, the TCDB network captures either a monomer or a dimer of 4-NN macrocycle (Fig. 15b and c). STM data revealed that all the azo groups are in the trans- configuration. Irradiation of the monolayer by 366 nm light triggers the isomerization within the rim of the macrocycle leading to formation of different photoisomers which could be identified from the shape of the macrocycle. From the original all trans (t,t,t,t) configuration, the azobenzene units give rise to different isomers including trans–trans–trans–cis (t,t,t,c) and trans–cis–trans–cis (t,c,t,c) isomers after irradiation with UV light (Fig. 15d and e).113 The light induced change in the shape and size of 4-NN macrocycle has been used to capture and release COR molecules. The covalent cavity of the all trans (t,t,t,t) isomer is too small to host COR as a guest. As a consequence, addition of COR to a preformed TCDB/4-NN architecture does not lead to its immobilization. The COR guests rather sit atop the monolayer (Fig. 15f and g). Upon UV irradiation, a new supramolecular arrangement is observed in which the voids of the host network are occupied by immobilized COR molecules. The shape of the macrocycle changes from parallelogram to ellipsoidal after irradiation, and it is attributed to the photoinduced transformation of the all trans (t,t,t,t) configuration to a trans–cis–trans–cis (t,c,t,c) configuration. This transformation increases the effective area of the voids leading to immobilization of COR molecules in the voids (Fig. 15h and i). Irradiation with visible light causes the reverse transition to the all trans configuration where COR molecules are expelled from the surface owing to shrinkage of the covalent cavities.114
 | ||
| Fig. 15 Photoresponsive host–guest system based on intrinsically porous building block. (a) Molecular structures of 4-NN macrocycle (top) and TCDB (bottom). (b) Host–guest system wherein 4-NN macrocycle occupies cavities of the TCDB host network. (c) Molecular model for the TCDB–4-NN macrocycle system. (d) STM image showing formation of different isomers of the 4-NN macrocycle on graphite surface upon irradiation with UV light. (d) Corresponding molecular model. (f) TCDB–4-NN host–guest system after addition of coronene. (g) Molecular model showing that COR guests are not immobilized in the host cavities. (h) STM image of the TCDB–4-NN macrocycle–COR ternary host–guest system upon irradiation with UV light. (i) Molecular model corresponding to the STM image provided in (h) showing immobilization of COR in the photoisomerized cavities. Reproduced from ref. 113 and 114 with permission from the American Chemical Society. | ||
Changes in electric field and temperature have also been recently employed to bring about reversible transformations in supramolecular host networks. A recent example of stimulus responsive host–guest system was demonstrated for hydrogen bonded networks of BTB formed at the octanoic acid/HOPG interface. As already described in detail earlier, BTB forms hydrogen bonded honeycomb porous networks which could be used for immobilization of planar aromatic guests such as COR and nanographene. The BTB network however, can be reversibly switched between porous and non-porous topology by changing the polarity of the voltage applied to the surface.43 Similarly, increasing the temperature of the surface to 55 °C also triggers a transition of the porous network into a much more densely packed structure where the BTB molecules are oriented vertically with respect to the graphite surface.47 Both transitions lead to squeezing of the guest molecules from the host network due to its compression. The reverse transitions can be accomplished seamlessly and capture the guest molecules in controlled manner.45 Such supramolecular nanostructures that can be externally triggered to contract or expand in a controlled fashion are highly desirable in the rapidly developing field of stimuli-responsive materials.
 | ||
| Fig. 16 Host–guest chemistry in covalent organic frameworks. (a) STM image showing the boronic acid COF imaged at the 1-phenyloctane-HOPG interface. (b) Molecular model for the COF. (c) COF–C60 host–guest system. (d) Molecular model for the host–guest system. Reproduced from ref. 115 with permission from the Royal Society of Chemistry. | ||
Summary and outlook
Since its inception in the 1980s, host–guest chemistry – a defining tenet of supramolecular chemistry, has rapidly developed, thanks to several generations of hosts: from crown ethers and cryptands to calixarenes and pillararenes. Exploration of host–guest chemistry on solid surfaces however began relatively late, possibly due to lack of techniques that can access the buried and often experimentally inaccessible solution–solid interface. STM, especially STM at the solution–solid interface, has made it possible to characterize the structures of complex host–guest networks at submolecular resolution. Such crystalline porous networks are highly desirable as their long-range order and scalability allows fine structural control needed in applications such as molecular separations.As surveyed in detail above, host–guest chemistry on solid surfaces has evolved significantly over the last decade. The early systems used simple host networks and the choice of guest molecules is still typically limited to coronene and C60, both at the UHV– and the solution–solid interface. Diverse host architectures have been fabricated on different solid surfaces leading to a better understanding of their stability and guest binding ability. While the emergence of rational design strategies in the recent years has shown great promise towards the development of complex and functional host systems, demonstration of chemical and chiral selectivity of guest binding is only recent. Both UHV–STM as well as solution–solid interfaces have so far provided solid examples of host–guest binding albeit these are mostly qualitative results. The solution–solid interface appears to be an approach that can be scaled up for future applications. However, studies carried out under UHV conditions will continue to have special importance in view of the ultraclean conditions they provide. Such clean conditions together with the absence of solvent are key for unravelling important mechanistic aspects of guest binding processes.
Recent trend in the field indicates combined use of molecular design, supramolecular synthesis and surface science principles to realize host–guest systems designed for specific functions. Recently reported novel design strategies have provided access to sophisticated host networks that exhibit guest-binding behavior that is selective and responsive to external stimuli. The literature surveyed here reveals that enough is already known so that the principles of supramolecular chemistry can be profitably employed in the design functional ‘real-life’ systems. As far as the real-life applications are concerned, host–guest binding studied on solid surfaces is poised to move forward in two different directions: when successfully scaled up to quantitative measurements (for example, using high-surface area powdered materials), host–guest systems realized at the solution–solid interfaces can find applications in molecular separations. On the other hand, qualitative measurements can be valuable for sensing small quantities of chemicals, thus finding application in molecular sensing.
Moving forward, the host–guest chemistry on surfaces will be greatly benefited by more radical design strategies that allow modification of the chemical/chiral environment inside the interior of the 2D cavities. Such modification will permit selective recognition based on the chemical and/or chiral complementarity between the host and the guest. An interesting possibility is to use the confined space inside these nano-sized cavities for carrying out chemical transformations. Such confinement-induced chemistry may allow access to reaction pathways and products that are neither available in solution nor on large terraces of solid surfaces. Given that the dimensions of the host cavities can be precisely tuned, such soft membranes represent an accurately controlled reaction field. Precise control over the open (porous) and closed (dense) network topologies is another desirable attribute for future host systems. This will allow storage of target molecules as long as the network is open and one can release them by closing the system using an external stimulus. While a few recent examples discussed above already possess these desirable properties, there is certainly room for further exploration. Complementary analytical techniques such as optical absorption/emission spectroscopy could be used to track changes in solution concentration upon guest release or capture, provided guest release/capture occurs on a quantitative scale. Furthermore, qualitative measurements on host–guest systems where chemically distinct guests compete for adsorption could greatly benefit from tip enhanced Raman spectroscopy which is sensitive to the chemical nature of guest species.
Acknowledgements
We thank the support of the Fund of Scientific Research – Flanders (FWO), Internal Funds KU Leuven, Belgian Federal Science Policy Office (IAP-7/05) and European Research Council under the European Union’s Seventh Framework Programme (FP7/2007–2013)/ERC grant agreement no. 340324.References
- J. A. A. W. Elemans, S. Lei and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7298–7332 CrossRef CAS PubMed.
- T. Kudernac, S. Lei, J. A. A. W. Elemans and S. De Feyter, Chem. Soc. Rev., 2009, 38, 402–421 RSC.
- X. Zhang, Q. Zeng and C. Wang, RSC Adv., 2013, 3, 11351–11366 RSC.
- A. Ciesielski, C.-A. Palma, M. Bonini and P. Samorì, Adv. Mater., 2010, 22, 3506–3520 CrossRef CAS PubMed.
- S. De Feyter, H. Xu and K. Mali, Chimia, 2012, 66, 38–43 CrossRef CAS PubMed.
- Y. Yang and C. Wang, Chem. Soc. Rev., 2009, 38, 2576–2589 RSC.
- J. V. Barth, Annu. Rev. Phys. Chem., 2007, 58, 375–407 CrossRef CAS PubMed.
- Q.-N. Zheng, X.-H. Liu, T. Chen, H.-J. Yan, T. Cook, D. Wang, P. J. Stang and L.-J. Wan, J. Am. Chem. Soc., 2015, 137, 6128–6131 CrossRef CAS PubMed.
- S. Stepanow, M. Lingenfelder, A. Dmitriev, H. Spillmann, E. Delvigne, N. Lin, X. Deng, C. Cai, J. V. Barth and K. Kern, Nat. Mater., 2004, 3, 229–233 CrossRef CAS PubMed.
- Y. Yang and C. Wang, Curr. Opin. Colloid Interface Sci., 2009, 14, 135–147 CrossRef CAS.
- S. De Feyter and F. C. De Schryver, J. Phys. Chem. B, 2005, 109, 4290–4302 CrossRef CAS PubMed.
- N. Afsharimani, A. Minoia, C. Volcke, M. Surin, R. Lazzaroni, J.-Y. Balandier, C. Niebel, Y. H. Geerts and B. Nysten, J. Phys. Chem. C, 2013, 117, 21743–21751 CAS.
- V. V. Korolkov, S. A. Svatek, S. Allen, C. J. Roberts, S. J. B. Tendler, T. Taniguchi, K. Watanabe, N. R. Champness and P. H. Beton, Chem. Commun., 2014, 50, 8882–8885 RSC.
- B. Baris, V. Luzet, E. Duverger, P. Sonnet, F. Palmino and F. Cherioux, Angew. Chem., Int. Ed., 2011, 50, 4094–4098 CrossRef CAS PubMed.
- A. Bhattarai, U. Mazur and K. W. Hipps, J. Am. Chem. Soc., 2014, 136, 2142–2148 CrossRef CAS PubMed.
- K. Cui, F. Schlütter, O. Ivasenko, M. Kivala, M. G. Schwab, S.-L. Lee, S. F. L. Mertens, K. Tahara, Y. Tobe, K. Müllen, K. S. Mali and S. De Feyter, Chem. – Eur. J., 2015, 21, 1652–1659 CrossRef CAS PubMed.
- T. Balandina, K. Tahara, N. Sändig, M. O. Blunt, J. Adisoejoso, S. Lei, F. Zerbetto, Y. Tobe and S. De Feyter, ACS Nano, 2012, 6, 8381–8389 CrossRef CAS PubMed.
- Y. Liu, H. Wang, P. Liang and H.-Y. Zhang, Angew. Chem., Int. Ed., 2004, 43, 2690–2694 CrossRef CAS.
- S. Yoshimoto, K. Suto, K. Itaya and N. Kobayashi, Chem. Commun., 2003, 2174–2175 RSC.
- G.-B. Pan, J.-M. Liu, H.-M. Zhang, L.-J. Wan, Q.-Y. Zheng and C.-L. Bai, Angew. Chem., Int. Ed., 2003, 42, 2747–2751 CrossRef CAS PubMed.
- H. Bertrand, F. Silly, M.-P. Teulade-Fichou, L. Tortech and D. Fichou, Chem. Commun., 2011, 47, 10091–10093 RSC.
- N. Wintjes, D. Bonifazi, F. Cheng, A. Kiebele, M. Stöhr, T. Jung, H. Spillmann and F. Diederich, Angew. Chem., Int. Ed., 2007, 46, 4089–4092 CrossRef CAS PubMed.
- K. Tahara, K. Kaneko, K. Katayama, S. Itano, C. H. Nguyen, D. D. D. Amorim, S. De Feyter and Y. Tobe, Langmuir, 2015, 31, 7032–7040 CrossRef CAS PubMed.
- J. Adisoejoso, K. Tahara, S. Okuhata, S. Lei, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7353–7357 CrossRef CAS PubMed.
- Y. Xue and M. B. Zimmt, J. Am. Chem. Soc., 2012, 134, 4513–4516 CrossRef CAS PubMed.
- J. I. Urgel, D. Écija, G. Lyu, R. Zhang, C.-A. Palma, W. Auwärter, N. Lin and J. V. Barth, Nat. Chem., 2016, 8, 657–662 CrossRef CAS PubMed.
- L. Cao, L. Xu, D. Zhao, K. Tahara, Y. Tobe, S. De Feyter and S. Lei, Chem. Commun., 2014, 50, 11946–11949 RSC.
- J. Tian, L. Chen, D.-W. Zhang, Y. Liu and Z.-T. Li, Chem. Commun., 2016, 52, 6351–6362 RSC.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- M. L. S. Griessl, M. Edelwirth, M. Hietschold and W. M. Heckl, Single Mol., 2002, 3, 25–31 CrossRef.
- J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029–1031 CrossRef CAS PubMed.
- Y. Ye, W. Sun, Y. Wang, X. Shao, X. Xu, F. Cheng, J. Li and K. Wu, J. Phys. Chem. C, 2007, 111, 10138–10141 CAS.
- Z. Li, B. Han, L. J. Wan and T. Wandlowski, Langmuir, 2005, 21, 6915–6928 CrossRef CAS PubMed.
- S. J. H. Griessl, M. Lackinger, F. Jamitzky, T. Markert, M. Hietschold and W. M. Heckl, Langmuir, 2004, 20, 9403–9407 CrossRef CAS PubMed.
- O. Ivasenko, J. M. MacLeod, K. Y. Chernichenko, E. S. Balenkova, R. V. Shpanchenko, V. G. Nenajdenko, F. Rosei and D. F. Perepichka, Chem. Commun., 2009, 1192–1194 RSC.
- S. J. H. Griessl, M. Lackinger, F. Jamitzky, T. Markert, M. Hietschold and W. M. Heckl, J. Phys. Chem. B, 2004, 108, 11556–11560 CrossRef CAS.
- M. Lackinger and W. M. Heckl, Langmuir, 2009, 25, 11307–11321 CrossRef CAS PubMed.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- A. Kitaigorodskii, Acta Crystallogr., 1965, 18, 585–590 CrossRef CAS.
- K. Tahara, S. Lei, D. Mossinger, H. Kozuma, K. Inukai, M. Van der Auweraer, F. C. De Schryver, S. Hoger, Y. Tobe and S. De Feyter, Chem. Commun., 2008, 3897–3899 RSC.
- J. F. Dienstmaier, K. Mahata, H. Walch, W. M. Heckl, M. Schmittel and M. Lackinger, Langmuir, 2010, 26, 10708–10716 CrossRef CAS PubMed.
- M. Ruben, D. Payer, A. Landa, A. Comisso, C. Gattinoni, N. Lin, J.-P. Collin, J.-P. Sauvage, A. De Vita and K. Kern, J. Am. Chem. Soc., 2006, 128, 15644–15651 CrossRef CAS PubMed.
- F. P. Cometto, K. Kern and M. Lingenfelder, ACS Nano, 2015, 9, 5544–5550 CrossRef CAS PubMed.
- L. Kampschulte, M. Lackinger, A.-K. Maier, R. S. K. Kishore, S. Griessl, M. Schmittel and W. M. Heckl, J. Phys. Chem. B, 2006, 110, 10829–10836 CrossRef CAS PubMed.
- S.-L. Lee, Y. Fang, G. Velpula, F. P. Cometto, M. Lingenfelder, K. Müllen, K. S. Mali and S. De Feyter, ACS Nano, 2015, 9, 11608–11617 CrossRef CAS PubMed.
- F. Silly, J. Phys. Chem. C, 2012, 116, 10029–10032 CAS.
- R. Gutzler, T. Sirtl, J. r. F. Dienstmaier, K. Mahata, W. M. Heckl, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2010, 132, 5084–5090 CrossRef CAS PubMed.
- R. Gutzler, S. Lappe, K. Mahata, M. Schmittel, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 680–682 RSC.
- H.-J. Yan, J. Liu, D. Wang and L.-J. Wan, Philos. Trans. R. Soc., A, 2013, 371 DOI:10.1098/rsta.2012.0302.
- G. Pawin, K. L. Wong, K.-Y. Kwon and L. Bartels, Science, 2006, 313, 961–962 CrossRef CAS PubMed.
- Z. Cheng, J. Wyrick, M. Luo, D. Sun, D. Kim, Y. Zhu, W. Lu, K. Kim, T. L. Einstein and L. Bartels, Phys. Rev. Lett., 2010, 105, 066104 CrossRef PubMed.
- K. Tahara, S. Lei, J. Adisoejoso, S. De Feyter and Y. Tobe, Chem. Commun., 2010, 46, 8507–8525 RSC.
- S. Lei, K. Tahara, F. C. De Schryver, M. Van der Auweraer, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2008, 47, 2964–2968 CrossRef CAS PubMed.
- I. Destoop, E. Ghijsens, K. Katayama, K. Tahara, K. S. Mali, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2012, 134, 19568–19571 CrossRef CAS PubMed.
- S. Lei, K. Tahara, X. Feng, S. Furukawa, F. C. De Schryver, K. Müllen, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2008, 130, 7119–7129 CrossRef CAS PubMed.
- S. Lei, M. Surin, K. Tahara, J. Adisoejoso, R. Lazzaroni, Y. Tobe and S. D. Feyter, Nano Lett., 2008, 8, 2541–2546 CrossRef CAS PubMed.
- N. Lin, S. Stepanow, M. Ruben and J. V. Barth, in Top. Curr. Chem., ed. P. Broekmann, K.-H. Dötz and C. A. Schalley, Springer Berlin Heidelberg, Berlin, Heidelberg, 2009, pp. 1–44, DOI: http://10.1007/128_2008_150 Search PubMed.
- U. Schlickum, R. Decker, F. Klappenberger, G. Zoppellaro, S. Klyatskaya, M. Ruben, I. Silanes, A. Arnau, K. Kern, H. Brune and J. V. Barth, Nano Lett., 2007, 7, 3813–3817 CrossRef CAS PubMed.
- D. Kühne, F. Klappenberger, R. Decker, U. Schlickum, H. Brune, S. Klyatskaya, M. Ruben and J. V. Barth, J. Am. Chem. Soc., 2009, 131, 3881–3883 CrossRef PubMed.
- D. Kühne, F. Klappenberger, W. Krenner, S. Klyatskaya, M. Ruben and J. V. Barth, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21332–21336 CrossRef PubMed.
- D. Kühne, F. Klappenberger, R. Decker, U. Schlickum, H. Brune, S. Klyatskaya, M. Ruben and J. V. Barth, J. Phys. Chem. C, 2009, 113, 17851–17859 Search PubMed.
- A. Langner, S. L. Tait, N. Lin, C. Rajadurai, M. Ruben and K. Kern, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17927–17930 CrossRef CAS PubMed.
- T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171–173 CrossRef CAS PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS PubMed.
- L. Zhu, M. Lu, Q. Zhang, D. Qu and H. Tian, Macromolecules, 2011, 44, 4092–4097 CrossRef CAS.
- M. Fathalla, A. Neuberger, S.-C. Li, R. Schmehl, U. Diebold and J. Jayawickramarajah, J. Am. Chem. Soc., 2010, 132, 9966–9967 CrossRef CAS PubMed.
- H. Isla, E. M. Pérez and N. Martín, Angew. Chem., Int. Ed., 2014, 126, 5735–5739 CrossRef.
- M. Pfeffermann, R. Dong, R. Graf, W. Zajaczkowski, T. Gorelik, W. Pisula, A. Narita, K. Müllen and X. Feng, J. Am. Chem. Soc., 2015, 137, 14525–14532 CrossRef CAS PubMed.
- K.-D. Zhang, J. Tian, D. Hanifi, Y. Zhang, A. C.-H. Sue, T.-Y. Zhou, L. Zhang, X. Zhao, Y. Liu and Z.-T. Li, J. Am. Chem. Soc., 2013, 135, 17913–17918 CrossRef CAS PubMed.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- J. F. Dienstmaier, A. M. Gigler, A. J. Goetz, P. Knochel, T. Bein, A. Lyapin, S. Reichlmaier, W. M. Heckl and M. Lackinger, ACS Nano, 2011, 5, 9737–9745 CrossRef CAS PubMed.
- J. F. Dienstmaier, D. D. Medina, M. Dogru, P. Knochel, T. Bein, W. M. Heckl and M. Lackinger, ACS Nano, 2012, 6, 7234–7242 CrossRef CAS PubMed.
- X.-H. Liu, C.-Z. Guan, S.-Y. Ding, W. Wang, H.-J. Yan, D. Wang and L.-J. Wan, J. Am. Chem. Soc., 2013, 135, 10470–10474 CrossRef CAS PubMed.
- R. Tanoue, R. Higuchi, N. Enoki, Y. Miyasato, S. Uemura, N. Kimizuka, A. Z. Stieg, J. K. Gimzewski and M. Kunitake, ACS Nano, 2011, 5, 3923–3929 CrossRef CAS PubMed.
- L. Xu, X. Zhou, Y. Yu, W. Q. Tian, J. Ma and S. Lei, ACS Nano, 2013, 7, 8066–8073 CrossRef CAS PubMed.
- D. J. Murray, D. D. Patterson, P. Payamyar, R. Bhola, W. Song, M. Lackinger, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2015, 137, 3450–3453 CrossRef CAS PubMed.
- W. Dai, F. Shao, J. Szczerbiński, R. McCaffrey, R. Zenobi, Y. Jin, A. D. Schlüter and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 213–217 CrossRef CAS PubMed.
- M. Iannuzzi, F. Tran, R. Widmer, T. Dienel, K. Radican, Y. Ding, J. Hutter and O. Groning, Phys. Chem. Chem. Phys., 2014, 16, 12374–12384 RSC.
- T. Dienel, J. Gómez-Díaz, A. P. Seitsonen, R. Widmer, M. Iannuzzi, K. Radican, H. Sachdev, K. Müllen, J. Hutter and O. Gröning, ACS Nano, 2014, 8, 6571–6579 CrossRef CAS PubMed.
- Y. Ding, M. Iannuzzi and J. Hutter, J. Phys. Chem. C, 2011, 115, 13685–13692 CAS.
- B. Schmaltz, A. Rouhanipour, H. J. Räder, W. Pisula and K. Müllen, Angew. Chem., Int. Ed., 2009, 48, 720–724 CrossRef CAS PubMed.
- Y. Li, C. Liu, Y. Xie, X. Li, X. Fan, L. Yuan and Q. Zeng, Chem. Commun., 2013, 49, 9021–9023 RSC.
- E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2006, 18, 447–451 CrossRef CAS.
- M. B. Wieland, L. M. A. Perdigao, D. V. Kondratuk, J. N. O'Shea, H. L. Anderson and P. H. Beton, Chem. Commun., 2014, 50, 7332–7335 RSC.
- D. V. Kondratuk, L. M. A. Perdigao, A. M. S. Esmail, J. N. O'Shea, P. H. Beton and H. L. Anderson, Nat. Chem., 2015, 7, 317–322 CrossRef CAS PubMed.
- S. Yoshimoto, K. Suto, A. Tada, N. Kobayashi and K. Itaya, J. Am. Chem. Soc., 2004, 126, 8020–8027 CrossRef CAS PubMed.
- A. Ohira, M. Sakata, C. Hirayama and M. Kunitake, Org. Biomol. Chem., 2003, 1, 251–253 CAS.
- N. Thontasen, G. Levita, N. Malinowski, Z. Deng, S. Rauschenbach and K. Kern, J. Phys. Chem. C, 2010, 114, 17768–17772 CAS.
- B. E. Hirsch, K. P. McDonald, B. Qiao, A. H. Flood and S. L. Tait, ACS Nano, 2014, 8, 10858–10869 CrossRef CAS PubMed.
- B. E. Hirsch, S. Lee, B. Qiao, C.-H. Chen, K. P. McDonald, S. L. Tait and A. H. Flood, Chem. Commun., 2014, 50, 9827–9830 RSC.
- S. Lee, B. E. Hirsch, Y. Liu, J. R. Dobscha, D. W. Burke, S. L. Tait and A. H. Flood, Chem. – Eur. J., 2016, 22, 560–569 CrossRef CAS PubMed.
- T. Chen, G.-B. Pan, H. Wettach, M. Fritzsche, S. Höger, L.-J. Wan, H.-B. Yang, B. H. Northrop and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 1328–1333 CrossRef CAS PubMed.
- M. C. O’Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O’Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 CrossRef PubMed.
- G. Eder, S. Kloft, N. Martsinovich, K. Mahata, M. Schmittel, W. M. Heckl and M. Lackinger, Langmuir, 2011, 27, 13563–13571 CrossRef CAS PubMed.
- G. Schull, L. Douillard, C. Fiorini-Debuisschert, F. Charra, F. Mathevet, D. Kreher and A. J. Attias, Adv. Mater., 2006, 18, 2954–2957 CrossRef CAS.
- S. Furukawa, K. Tahara, F. C. De Schryver, M. Van der Auweraer, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2007, 46, 2831–2834 CrossRef CAS PubMed.
- D. Bléger, D. Kreher, F. Mathevet, A.-J. Attias, G. Schull, A. Huard, L. Douillard, C. Fiorini-Debuischert and F. Charra, Angew. Chem., Int. Ed., 2007, 46, 7404–7407 CrossRef PubMed.
- M. Blunt, X. Lin, M. d. C. Gimenez-Lopez, M. Schroder, N. R. Champness and P. H. Beton, Chem. Commun., 2008, 2304–2306 RSC.
- M. O. Blunt, J. C. Russell, M. d. C. Gimenez-Lopez, N. Taleb, X. Lin, M. Schröder, N. R. Champness and P. H. Beton, Nat. Chem., 2011, 3, 74–78 CrossRef CAS PubMed.
- A. Ciesielski, S. Lena, S. Masiero, G. P. Spada and P. Samorì, Angew. Chem., Int. Ed., 2010, 49, 1963–1966 CrossRef CAS PubMed.
- K.-W. Park, J. Adisoejoso, J. Plas, J. Hong, K. Müllen and S. De Feyter, Langmuir, 2014, 30, 15206–15211 CrossRef CAS PubMed.
- F. Silly, A. Q. Shaw, K. Porfyrakis, G. A. D. Briggs and M. R. Castell, Appl. Phys. Lett., 2007, 91, 253109 CrossRef.
- R. Madueno, M. T. Räisänen, C. Silien and M. Buck, Nature, 2008, 454, 618–621 CrossRef CAS PubMed.
- E. Ghijsens, H. Cao, A. Noguchi, O. Ivasenko, Y. Fang, K. Tahara, Y. Tobe and S. De Feyter, Chem. Commun., 2015, 51, 4766–4769 RSC.
- K. Tahara, H. Yamaga, E. Ghijsens, K. Inukai, J. Adisoejoso, M. O. Blunt, S. De Feyter and Y. Tobe, Nat. Chem., 2011, 3, 714–719 CrossRef CAS PubMed.
- M. Li, K. Deng, S.-B. Lei, Y.-L. Yang, T.-S. Wang, Y.-T. Shen, C.-R. Wang, Q.-D. Zeng and C. Wang, Angew. Chem., Int. Ed., 2008, 47, 6717–6721 CrossRef CAS PubMed.
- K. Tahara, K. Nakatani, K. Iritani, S. De Feyter and Y. Tobe, ACS Nano, 2016, 10, 2113–2120 CrossRef CAS PubMed.
- K. Tahara, S. Lei, W. Mamdouh, Y. Yamaguchi, T. Ichikawa, H. Uji-i, M. Sonoda, K. Hirose, F. C. De Schryver, S. De Feyter and Y. Tobe, J. Am. Chem. Soc., 2008, 130, 6666–6667 CrossRef CAS PubMed.
- K. Tahara, K. Inukai, J. Adisoejoso, H. Yamaga, T. Balandina, M. O. Blunt, S. De Feyter and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 8373–8376 CrossRef CAS PubMed.
- D. Bléger, A. Ciesielski, P. Samorì and S. Hecht, Chem. – Eur. J., 2010, 16, 14256–14260 CrossRef PubMed.
- X. Zhang, S. Wang, Y. Shen, Y. Guo, Q. Zeng and C. Wang, Nanoscale, 2012, 4, 5039–5042 RSC.
- Y.-T. Shen, L. Guan, X.-Y. Zhu, Q.-D. Zeng and C. Wang, J. Am. Chem. Soc., 2009, 131, 6174–6180 CrossRef CAS PubMed.
- Y.-T. Shen, K. Deng, X.-M. Zhang, W. Feng, Q.-D. Zeng, C. Wang and J. R. Gong, Nano Lett., 2011, 11, 3245–3250 CrossRef CAS PubMed.
- J. Plas, O. Ivasenko, N. Martsinovich, M. Lackinger and S. De Feyter, Chem. Commun., 2016, 52, 68–71 RSC.
- D. Cui, J. M. MacLeod, M. Ebrahimi, D. F. Perepichka and F. Rosei, Chem. Commun., 2015, 51, 16510–16513 RSC.
- M. O. Blunt, J. C. Russell, N. R. Champness and P. H. Beton, Chem. Commun., 2010, 46, 7157–7159 RSC.
- C. Liu, W. Zhang, Q. Zeng and S. Lei, Chem. – Eur. J., 2016, 22, 6768–6773 CrossRef CAS PubMed.
- J. Sun, X. Zhou and S. Lei, Chem. Commun., 2016, 52, 8691–8694 RSC.
| This journal is © The Royal Society of Chemistry 2016 |