
Catalytic transformation of esters of 1,2-azido alcohols into α-amido ketones†
Yongjin
Kim
,
Han Kyu
Pak
,
Young Ho
Rhee
* and
Jaiwook
Park
*
Department of Chemistry, POSTECH (Pohang University of Science and Technology), Pohang 790-784, Korea. E-mail: pjw@postech.ac.kr; Web: http://oml.postech.ac.kr
First published on 13th April 2016
Abstract
The esters of 1,2-azido alcohols were transformed into α-amido ketones without external oxidants through the Ru-catalyzed formation of N–H imines with the liberation of N2 followed by intramolecular migration of the acyl moiety. A wide range of α-amido ketones were obtained, and one-pot transformation into the corresponding oxazoles (or a thiazole) was demonstrated.
α-Amido ketones are biologically relevant molecules and useful building blocks for valuable compounds in organic synthesis.1 In addition, they are useful substrates in various organic transformations such as the Robinson–Gabriel reaction to oxazoles2 and thiazoles,2e the Norrish–Yang photocyclization to 2-aminocyclobutanols,3 the epoxy-annulation reaction to epoxide-fused heterocycles4 and the reaction with ammonium acetate (or primary amines) to imidazoles.5
For the versatile transformations, α-amido ketones have been synthesized by various methods, including Pd-catalyzed coupling reaction of methylene aziridines with carboxylic acids,6 Rh-catalyzed denitrogenative hydration of N-sulfonyl-1,2,3-triazoles,7 the Dakin–West reaction of α-amino acids with acid anhydrides,8 the Neber rearrangement of ketoxime sulfonates9 and a radical cascade reaction of alkynes with N-fluoroarylsulfonimides and alcohols.10 However these methods suffer from the difficulty in preparing substrates, harsh reaction conditions, and/or limitations of the substrate scope.
Additional and noticeable methods are compared with our new finding in Scheme 1. The aza-benzoin condensation reaction of aldehydes with N-acyl imines is an interesting method using thiazolium organocatalysts.5c,11 However, the synthesis of tosylamides from tosylsulfinic acid, amides, and aldehydes is required to generate the intermediate N-acyl imines, and is not effective for enolizable aldehydes.12 The asymmetric hydrogenation of α-dehydroamido ketones can provide optically active α-amido ketones,13 but the scope is limited by the intrinsic regioselectivity problem in the condensation reaction of 1,2-diketones and primary amides. An old method employing 1,2-amino alcohols as the starting substrates looks simple but suffers practically from inefficiency in the N-acylation and the subsequent oxidation.5c,14 A carboxyl-activating agent and an oxidant are required in a stoichiometric amount in the acylation and the oxidation, respectively. Meanwhile, 1,2-amino alcohols are frequently prepared from 1,2-azido alcohols by the Staudinger reaction using triphenylphosphine as a reductant. Herein we wish to report an efficient synthesis of α-amido ketones from 1,2-azido alcohols without oxidation and reduction steps through a novel one-step catalytic transformation of 1,2-azido esters under neutral and mild conditions.
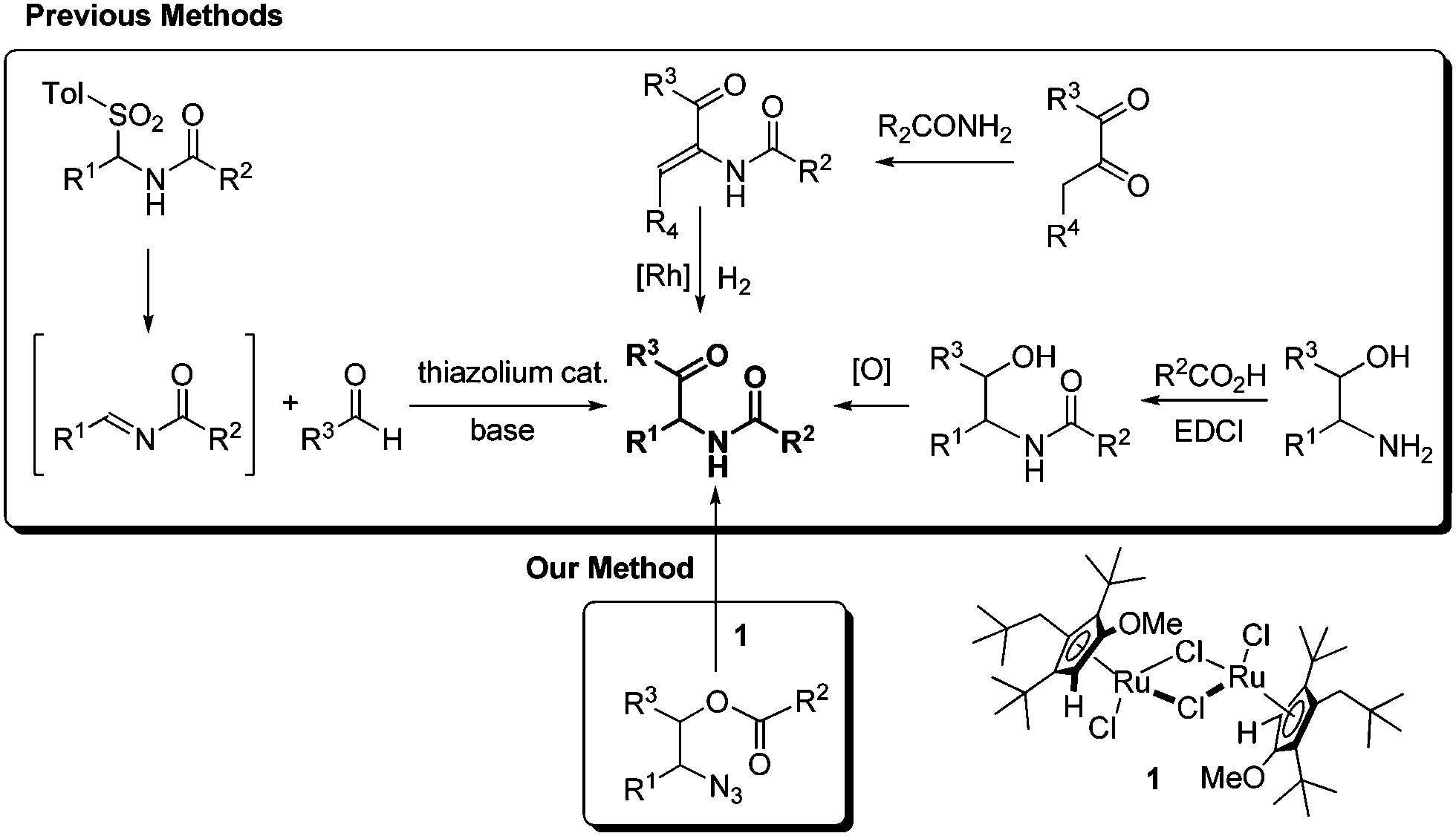 | ||
| Scheme 1 Synthetic methods for α-amido ketones. | ||
Recently we found an interesting Ru-catalyzed transformation of alkyl azides to N–H imines.15 As an application of the catalytic transformation, we have developed an efficient method for the synthesis of enamides from alkyl azides and acyl donors utilizing the N-acylation of intermediate N–H imines.16 In a related study on the N-acylation of N–H imines containing a hydroxyl group, we observed the unexpected formation of α-amido ketones in the catalytic reactions of 1,2-azido alcohols. For example, N-(2-oxo-1,2-diphenylethyl)acetamide (3a) was obtained in 55% yield by the reaction of 2-azido-1,2-diphenylethanol with acetic anhydride in the presence of the ruthenium catalyst 1 (Scheme 2). Then we envisioned that its intramolecular version would improve the efficiency of the transformation. We examined the transformation of 2-azido-1,2-diphenylethyl acetate (2a) under various conditions (Table 1). The transformation was more efficient in polar solvents than in non-polar ones such as THF and toluene (entries 1 and 2). In dimethylformamide (DMF), 3a was formed in 89% yield (entry 3). Noticeably, the transformation was effective in ionic liquids,17 which have some advantages such as being experimentally safe and recycled. In particular 3a was formed in almost quantitative yield in 1-butyl-3-methylimidazolium chloride ([bmim]Cl) (entry 4). A gram-scale reaction was also effective to give 3a in 91% isolated yield (entry 5), and recycling of [bmim]Cl was possible simply by removing water from the aqueous phase by heating after the workup procedure (entry 6).18 Decreasing the reaction temperature to 50 °C significantly lowered the yield of 3a (entry 7), while increasing it to 100 °C was not beneficial (entry 8). As in the synthesis of enamides involving N-acylation of N–H imines,16 a catalytic amount of triethylamine was helpful for the formation of 3a (entry 9).17
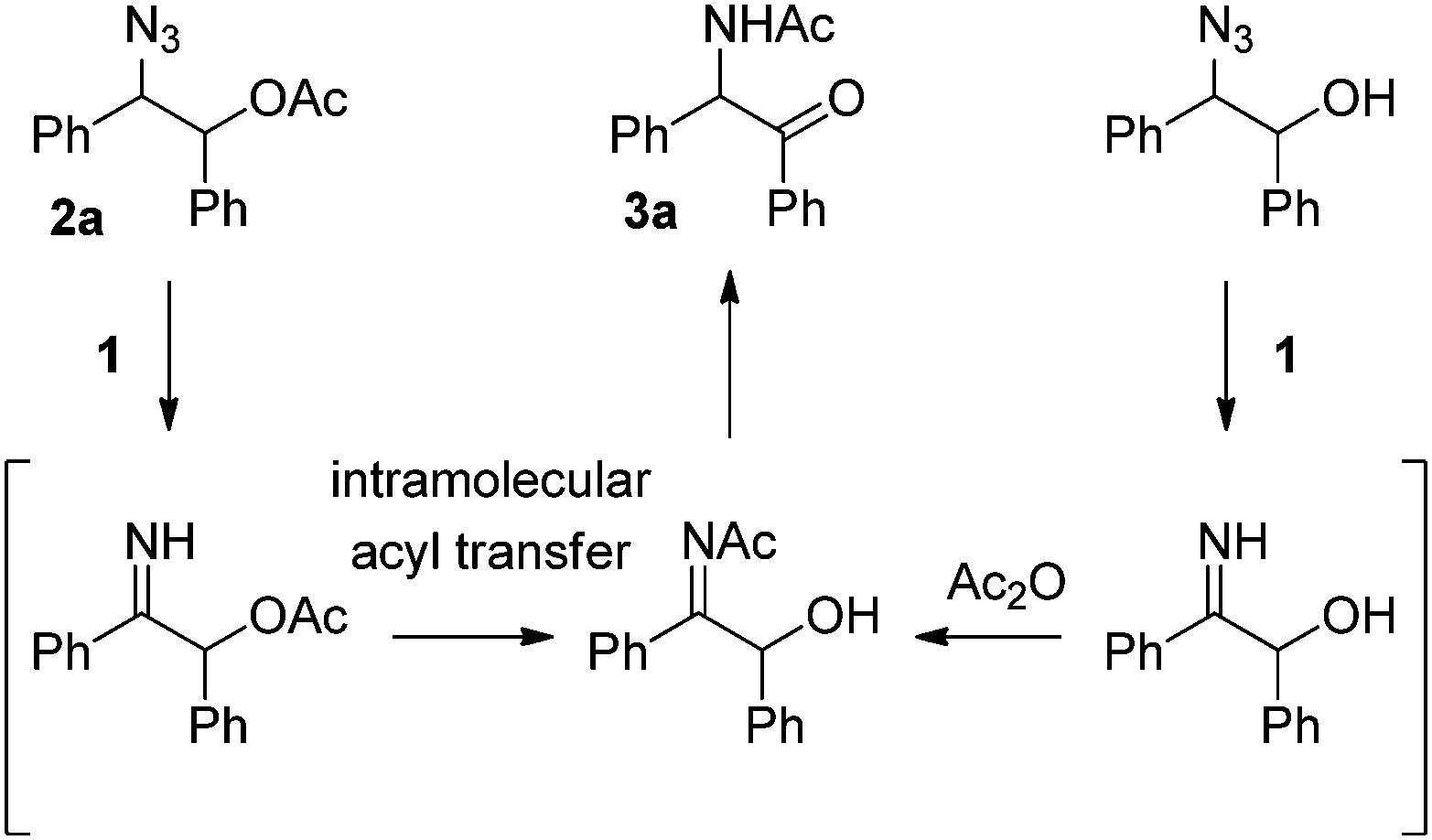 | ||
| Scheme 2 Formation of α-amido ketone 3a from 1,2-azido acetate 2a or from the corresponding 1,2-azido alcohol. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Additive | Temp. (°C) | Yieldb (%) |
| a Typical reaction conditions: a solution of an azide (0.25 mmol), 1 (1.0 mol%) and Et3N (2.0 mol%) in a solvent (1.0 mL) was stirred for 12 h. b Estimated by 1H NMR using nitromethane as an internal standard. c Isolated yield. d A large scale reaction employing 1.06 g (3.6 mmol) of 2a and 15 mg (0.5 mol%) of 1 in 6.0 mL of [bmim]Cl at 70 °C for 36 h. e The yield of the reaction using [bmim]Cl recovered from the 5th recycling reaction. | ||||
| 1 | THF | Et3N | 70 | 15 |
| 2 | Toluene | Et3N | 70 | 28 |
| 3 | DMF | Et3N | 70 | 89 |
| 4 | [bmim]Cl | Et3N | 70 | 96 (94)c |
| 5 | [bmim]Cl | Et3N | 70 | 91c,d |
| 6 | [bmim]Cl | Et3N | 70 | 90e |
| 7 | [bmim]Cl | Et3N | 50 | 15 |
| 8 | [bmim]Cl | Et3N | 100 | 91 |
| 9 | [bmim]Cl | None | 70 | 85 |

The transformation to α-amido ketones was applicable for a broad range of acetates of 1,2-azido alcohols (Table 2). The electronic effect of the substituents of aromatic rings was not so significant (3a–3c and 3g–3h). The yields of α-amido ketones were high in the transformation of the derivatives having alkyl groups (3d–3j). The low yield of 3i was due to the formation of unidentified side-products, and the use of DMF as a solvent gave 3i in 62% yield. The transformation of esters of primary β-hydroxy azides to α-amido ketones (3k–3r) was also successful despite the fact that the intermediates are unstable N–H aldimines. The transformation was effective for various derivatives containing functional groups on aromatic rings such as methyl, methoxy, halides and nitrile substituents. The yield of the α-amido ketone (3s), which has a benzyl moiety, was moderate with the formation of unidentified side products. The transformation of cyclic substrates (3t–3w) was less efficient than that of linear ones, probably due to the rigidity of ring structures. A six-membered cyclic α-amido ketone (3u) was obtained in moderate yield, while a five-membered one (3t) was not formed. However, interestingly, a seven-membered cyclic one (3w) was obtained in high yield, and a benzofused six-membered bicyclic one (3v) was formed in a much higher yield than the monocyclic one (3u).
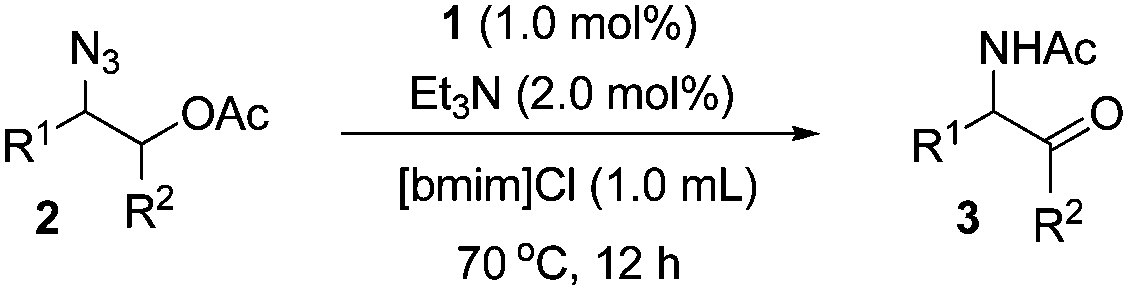

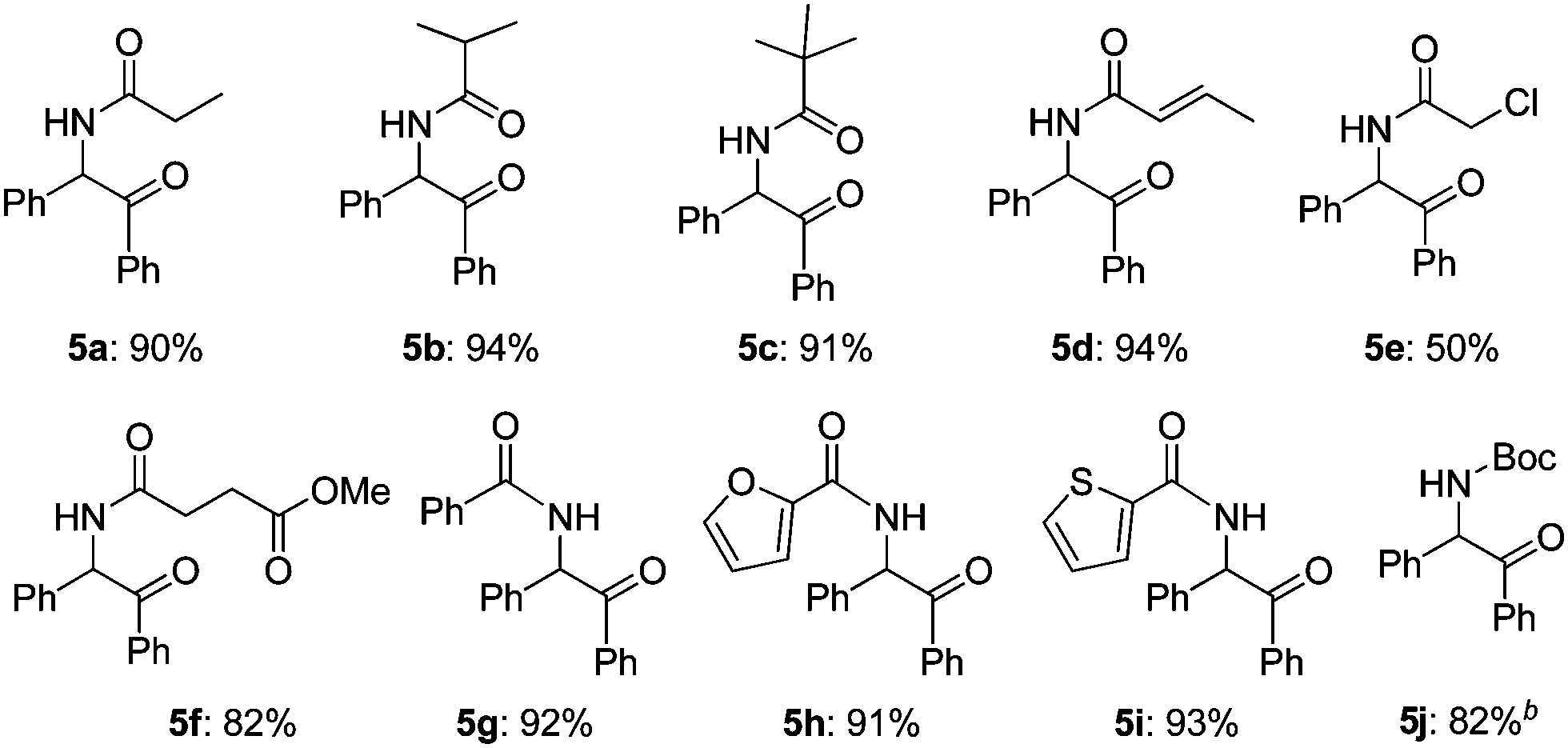
Then, the scope of α-amido ketones was explored for the derivatives having various N-acyl groups (Table 3). R3 in the α-amido ketones 5 could be varied not only to an ethyl (5a), isopropyl (5b), or a tert-butyl (5c) group but also to a conjugated alkenyl (5d), chloromethyl (5e), or an ester (5f) group. The derivatives containing phenyl (5g), furyl (5h), and thiofuryl (5i) groups were also obtained in high yields. The migration of the butyloxycarbonyl (Boc) group was possible, although heating at a higher temperature for a longer reaction time was required to give an N-Boc protected derivative (5j) in good yield.
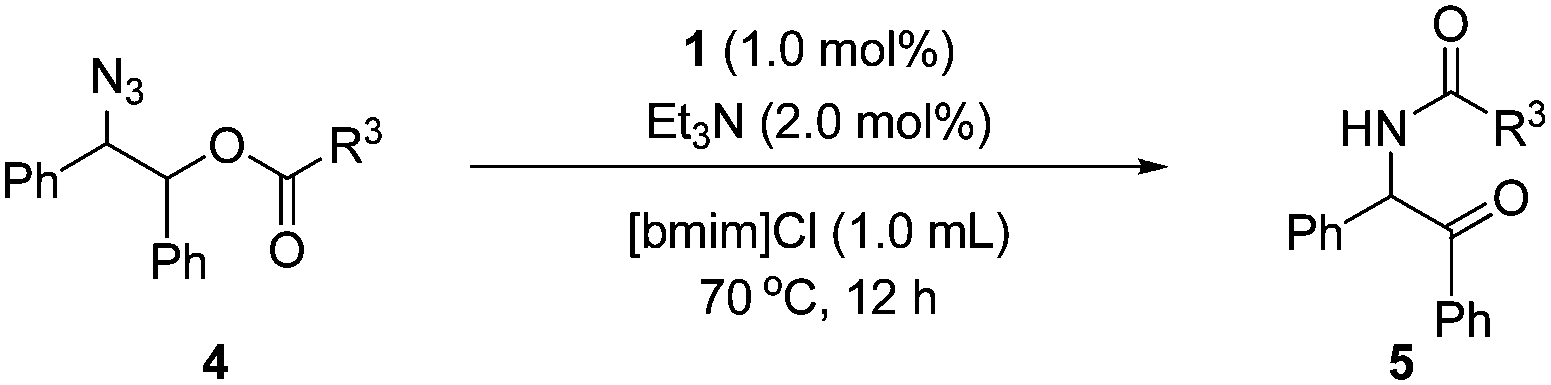
To demonstrate the utility of our synthesis of α-amido ketones, we carried out one-pot transformations to oxazoles (6a–c) and a thiazole (7) (Scheme 3). Treatment of 3ain situ generated from 2a with sulfuric acid afforded oxazole 6a in 94% yield. The corresponding thiazole (7) was obtained by the treatment with Lawesson's reagent in 87% yield. Noticeably, oxaprozin (6b), which is a well-known non-steroidal anti-inflammatory drug,19 was obtained directly from 4f in 89% yield. The stereochemistry of 4k at the α-position was practically maintained during the one-pot transformation to 6c,20 although the intermediate α-amido ketone was formed as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture.
:1 diastereomeric mixture.
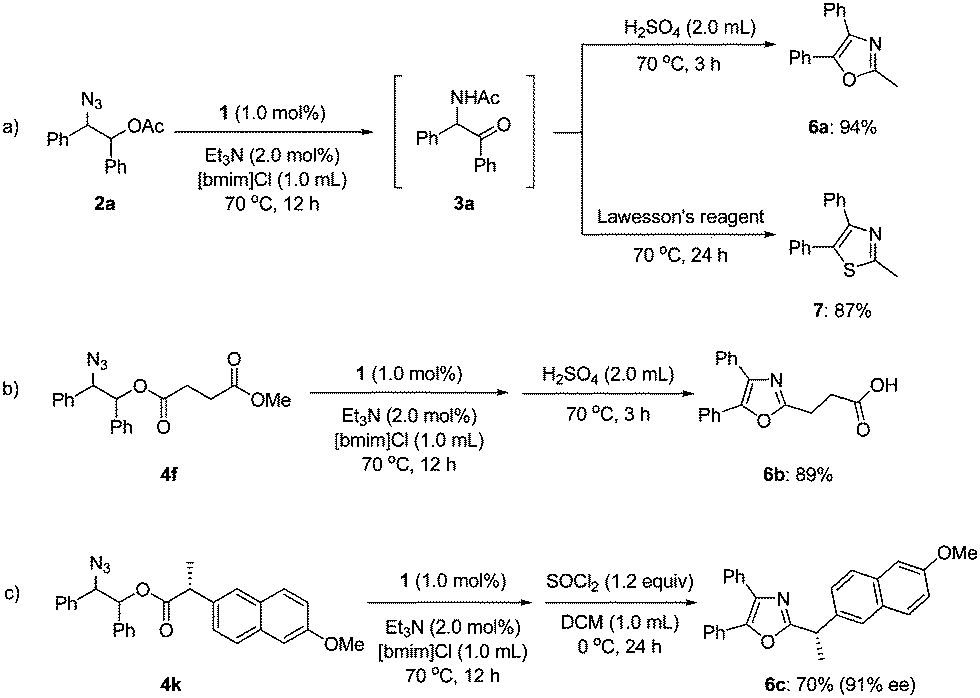 | ||
| Scheme 3 One-pot transformations to oxazoles and a thiazole. | ||
To obtain mechanistic insights into the transformation of 1,2-azido esters to α-amido ketones, a crossover experiment and the generation of an enol amide were examined: only non-crossover products (3a and 9) were formed in high yields in the transformation of a mixture of the 1,2-azido acetate 2a and another azide (8) containing a benzoyl group (Scheme 4a), and the α-amido ketone 3a was obtained in 76% yield in the deprotection reaction of a MOM-protected enol amide (10) (Scheme 4b).21
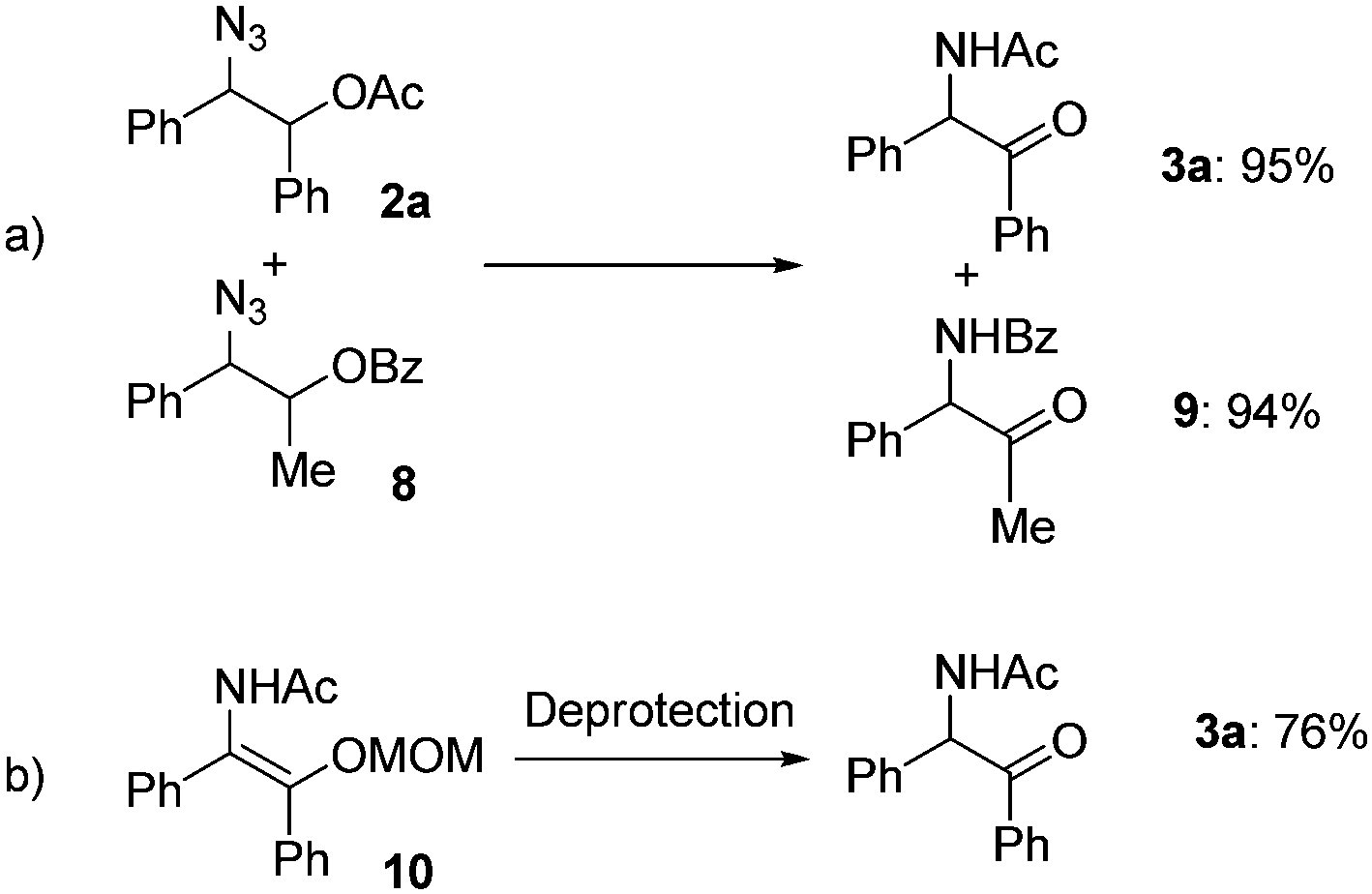 | ||
| Scheme 4 Mechanistic investigation. | ||
Now we can propose a plausible pathway for the transformation of the esters of 1,2-azido alcohols into α-amido ketones (Scheme 5). On the basis of our previous reports on the formation of enamides from N-acyl imines,16 the results of the crossover experiment support intramolecular migration of the acyl group in the intermediate N–H imine A to give the α-hydroxyl N-acylimine B. And the result of the deprotection reaction of 10 is indicative of the intermediacy of the enol amide C, which is tautomerized to the final α-amido ketone product.
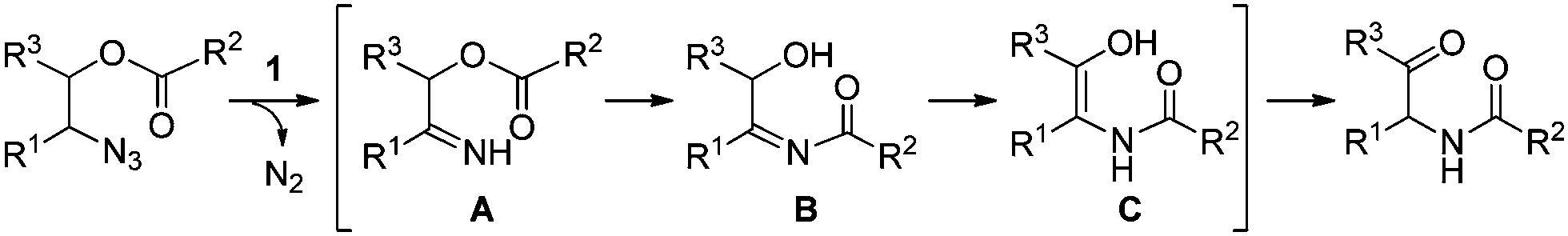 | ||
| Scheme 5 Plausible pathway for the formation of α-amido ketones. | ||
In summary, we developed a new and simple method for the synthesis of α-amido ketones from the esters of 1,2-azido alcohols just by the liberation of molecular nitrogen under mild conditions. Our method is effective for the synthesis of a wide range of multi-substituted α-amido ketones, and efficient for gram scale synthesis in recyclable ionic liquids. In addition, we demonstrated the one-pot synthesis of oxazoles and a thiazole using α-amido ketones as intermediates.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (2015R1A2A2A01008130).
Notes and references
- (a) A. Lee, L. Huang and J. A. Ellman, J. Am. Chem. Soc., 1999, 121, 9907 CrossRef CAS; (b) C. Béguin, S. V. Andurkar, A. Y. Jin, J. P. Stables, D. F. Weaver and H. Kohn, Bioorg. Med. Chem., 2003, 11, 4275 CrossRef; (c) A. Białas, J. Grembecka, D. Krowarsch, J. Otlewski, J. Potempa and A. Mucha, J. Med. Chem., 2006, 49, 1744 CrossRef PubMed; (d) H. Azuma, S. Ijichi, M. Kataoka, A. Masuda, T. Izumi, T. Yoshimoto and T. Tachibana, Bioorg. Med. Chem., 2007, 15, 2860 CrossRef CAS PubMed; (e) A. El-Dahshan, S. I. Al-Gharabli, S. Radetzki, T. H. Al-Tel, P. Kumar and J. Rademann, Bioorg. Med. Chem., 2014, 22, 5506 CrossRef CAS PubMed.
- (a) P. Wipf and C. P. Miller, J. Org. Chem., 1993, 58, 3604 CrossRef CAS; (b) T. Morwick, M. Hrapchak, M. DeTuri and S. Campbell, Org. Lett., 2002, 4, 2665 CrossRef CAS PubMed; (c) K. C. Nicolaou, J. Hao, M. V. Reddy, P. B. Rao, G. Rassias, S. A. Snyder, X. Huang, D. Y. K. Chen, W. E. Brenzovich, N. Giuseppone, P. Giannakakou and A. O'Brate, J. Am. Chem. Soc., 2004, 126, 12897 CrossRef CAS PubMed; (d) M. Keni and J. J. Tepe, J. Org. Chem., 2005, 70, 4211 CrossRef CAS PubMed; (e) E. Biron, J. Chatterjee and H. Kessler, Org. Lett., 2006, 8, 2417 CrossRef CAS PubMed; (f) J. Zhang and M. A. Ciufolini, Org. Lett., 2011, 13, 390 CrossRef CAS PubMed.
- (a) A. G. Griesbeck, H. Heckroth and J. Lex, Chem. Commun., 1999, 1109 RSC; (b) A. G. Griesbeck and H. Heckroth, J. Am. Chem. Soc., 2002, 124, 396 CrossRef CAS PubMed.
- (a) M. G. Unthank, N. Hussain and V. K. Aggarwal, Angew. Chem., Int. Ed., 2006, 45, 7066 CrossRef CAS PubMed; (b) M. G. Unthank, B. Tavassoli and V. K. Aggarwal, Org. Lett., 2008, 10, 1501 CrossRef CAS PubMed.
- (a) T. N. Sorrell and W. E. Allen, J. Org. Chem., 1994, 59, 1589 CrossRef CAS; (b) H. B. Lee and S. Balasubramanian, Org. Lett., 2000, 2, 323 CrossRef CAS PubMed; (c) D. E. Frantz, L. Morency, A. Soheili, J. A. Murry, E. J. J. Grabowski and R. D. Tillyer, Org. Lett., 2004, 6, 843 CrossRef CAS PubMed.
- B. H. Oh, I. Nakamura and Y. Yamamoto, J. Org. Chem., 2004, 69, 2856 CrossRef CAS PubMed.
- T. Miura, T. Biyajima, T. Fujii and M. Murakami, J. Am. Chem. Soc., 2012, 134, 194 CrossRef CAS PubMed.
- (a) N. L. Allinger, G. L. Wang and B. B. Dewhurst, J. Org. Chem., 1974, 39, 1730 CrossRef CAS; (b) G. L. Buchanan, Chem. Soc. Rev., 1988, 17, 91 RSC; (c) A. G. Godfrey, D. A. Brooks, L. A. Hay, M. Peters, J. R. McCarthy and D. Mitchell, J. Org. Chem., 2003, 68, 2623 CrossRef CAS PubMed; (d) R. C. Wende, A. Seitz, D. Niedek, S. M. M. Schuler, C. Hofmann, J. Becker and P. R. Schreiner, Angew. Chem., Int. Ed., 2016, 55, 2719 CrossRef CAS PubMed.
- (a) C. O'Brien, Chem. Rev., 1964, 64, 81 CrossRef; (b) T. Ooi, M. Takahashi, K. Doda and K. Maruoka, J. Am. Chem. Soc., 2002, 124, 7640 CrossRef CAS PubMed.
- G. Zheng, Y. Li, J. Han, T. Xiong and Q. Zhang, Nat. Commun., 2015, 6, 7011 CrossRef CAS PubMed.
- (a) J. A. Murry, D. E. Frantz, A. Soheili, R. Tillyer, E. J. J. Grabowski and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 9696 CrossRef CAS PubMed; (b) A. E. Mattson and K. A. Scheidt, Org. Lett., 2004, 6, 4363 CrossRef CAS PubMed; (c) S. M. Mennen, J. D. Gipson, Y. R. Kim and S. J. Miller, J. Am. Chem. Soc., 2005, 127, 1654 CrossRef CAS PubMed; (d) D. A. DiRocco and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 5904 CrossRef CAS PubMed; (e) M. M. D. Wilde and M. Gravel, Org. Lett., 2014, 16, 5308 CrossRef CAS PubMed.
- T. Mecozzi and M. Petrini, J. Org. Chem., 1999, 64, 8970 CrossRef CAS PubMed.
- T. Sun, G. Hou, M. Ma and X. Zhang, Adv. Synth. Catal., 2011, 353, 253 CrossRef CAS.
- K. H. Bleicher, F. Gerber, Y. Wüthrich, A. Alanine and A. Capretta, Tetrahedron Lett., 2002, 43, 7687 CrossRef CAS.
- J. H. Lee, S. Gupta, W. Jeong, Y. H. Rhee and J. Park, Angew. Chem., Int. Ed., 2012, 51, 10851 CrossRef CAS PubMed.
- (a) J. Han, M. Jeon, H. K. Pak, Y. H. Rhee and J. Park, Adv. Synth. Catal., 2014, 356, 2769 CrossRef CAS; (b) H. K. Pak, J. Han, M. Jeon, Y. Kim, Y. Kwon, J. Y. Park, Y. H. Rhee and J. Park, ChemCatChem, 2015, 7, 4030 CrossRef CAS.
- For screening of ionic liquids and additives, see the ESI†.
- For more detailed results for the recycling of [bmim]Cl, see the ESI†.
- D. J. Greenblatt, R. Matlis, J. M. Scavone, G. T. Blyden, J. S. Harmatz and R. I. Shader, Br. J. Clin. Pharmacol., 1985, 19, 373–378 CrossRef CAS PubMed.
- A. K. Ghosh, N. Kumaragurubaran, L. Hong, H. Lei, K. A. Hussain, C.-F. Liu, T. Devasamudram, V. Weerasena, R. Turner, G. Koelsch, G. Bilcer and J. Tang, J. Am. Chem. Soc., 2006, 128, 5310–5311 CrossRef CAS PubMed.
- H. Han, Y. E. Kwon, J.-H. Sohn and D. H. Ryu, Tetrahedron, 2010, 66, 1673 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc02063a |
| This journal is © The Royal Society of Chemistry 2016 |