
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of mucoadhesive thiol-bearing microgels from 2-(acetylthio)ethylacrylate and 2-hydroxyethylmethacrylate: novel drug delivery systems for chemotherapeutic agents to the bladder†
M. T.
Cook
a,
S. A.
Schmidt
b,
E.
Lee
b,
W.
Samprasit
c,
P.
Opanasopit
c and
V. V.
Khutoryanskiy
*b
aDepartment of Pharmacy, Life and Medical Sciences, University of Hertfordshire, Hatfield, UK
bSchool of Pharmacy, University of Reading, Reading, RG6 6UR, UK. E-mail: V.Khutoryanskiy@Reading.ac.uk
cPharmaceutical Development of Green Innovations Group (PDGIG), Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, 73000, Thailand
First published on 15th July 2015
Abstract
Thiol-bearing microgels have been synthesised from copolymerisation of 2-(acetylthio)ethylacrylate and 2-hydroxyethylmethacrylate, and subsequent deprotection using sodium thiomethoxide. The concentration of thiol groups on these microgels could be tailored by use of different molar ratios of the two monomers. These thiol-bearing microgels were shown to adhere to ex vivo porcine urinary bladder, which was correlated with their level of thiolation. By simply mixing solutions of thiol-bearing microgels and doxorubicin, high levels of drug loading into the microgels could be achieved. Thiol-bearing microgels controlled the release of doxorubicin in a time-dependent manner over several hours. These doxorubicin-loaded thiol-bearing microgels could have application in the treatment of early-stage bladder cancers. The method used represents a new ‘bottom-up’ approach for the synthesis of novel mucoadhesive microgels.
Introduction
Polymeric materials capable of adhering to mucosal surfaces have attracted significant interest of pharmaceutical researchers for the development of novel drug delivery systems. The mucoadhesive properties of these materials achieve better retention of dosage forms on mucosal surfaces, which in turn will result in increased time for absorption and improved drug bioavailability.1,2Many different hydrophilic polymers have been identified as mucoadhesive. The mucoadhesion of these polymers can be significantly enhanced when these materials are modified to bear specific functional groups such as thiols,3 acrylates,4 or catechols.5 Polymers bearing thiol functional groups, termed polymeric thiomers, have been developed and widely investigated for a range of transmucosal drug delivery applications (oral, ocular, vaginal, intravesical, etc.).6 The principal method developed by Bernkop-Schnürch et al.3 to make polymeric thiomers involves the covalent conjugation of water-soluble polymers with thiol-containing functional molecules such as cysteine or thioglycolic acid. These thiomers could then be converted into particulate formulations through various methods, such as chemical cross-linking.7 We also have demonstrated that the mucoadhesive thiolated nanoparticles could be synthesised using a ‘bottom-up’ approach from polyfunctional thiol-bearing small molecules, such as 3-mercaptopropyltrimethoxysilane that undergoes self-condensation to form particulates,8 or by reacting tetrakis(3-mercapto-propionate) with pentaerythritol tetraacrylate using thiol–ene click chemistry.9
The development of thiolated polymers using polymerisation of protected thiol-bearing monomers has recently been demonstrated by Liras and co-workers.10 They have synthesised 2-(acetylthio)ethylmethacrylate as a new monomer with a thioester group and polymerised it using atom transfer radical polymerisation. The thioester groups could subsequently be deprotected to form a series of well-defined thiolated thermosensitive polymers, nanoparticles or hydrogels and multidentate ligands for functionalising CdSe quantum dots, or graphene.10–14 These materials could, however, also have application in mucoadhesion.
One potential application of mucoadhesive dosage forms is in delivery of chemotherapeutics to the bladder.15 The surface of the bladder is known to possess mucins,16 of the MUC1 and MUC3 type,17 and cancers of the bladder also express bladder-cancer-associated mucins.18 These appear to possess cysteine-rich domains, so may be a potential target of thiomers. Zhang et al.19 and Barthelmes et al.15 have demonstrated that thiol-bearing particles may adhere to these membranes. Bladder cancers typically progress from the lining of the bladder, and with time proliferate into the surrounding tissues. Treatment of the early stage tumors is conducted either by cystectomy or chemotherapy,19 if surgery is not suitable for the patient. Chemotherapy to the bladder is typically administered intravesicular, and the catheter may need to remain in the patient, or the patient may need to not urinate for one to two hours in order to maintain the chemotherapeutic agent in the site of administration until absorption.20 The design of a microparticulate delivery system which was able to adhere to the mucosa and deliver chemotherapy locally may offer an alternative to this procedure.
In the present study we have synthesised a new monomer with protected thiol-groups – 2-(acetylthio)ethylacrylate (ATEA) and have used a surfactant-free emulsion free-radical polymerisation with subsequent deprotection to develop a range of microgels with thiol-bearing surfaces, with a tunable thiol content depending on composition. The advantage of this approach is that free thiol groups act as chain transfer agents during polymerisation, and without protection polymerisation could occur from the sulfur atom. These microgels were confirmed to be mucoadhesive and it was found that doxorubicin, an anticancer drug, could easily be incorporated up to doses that are therapeutically relevant for the treatment of bladder cancer, and released from the microgels in a time-dependent manner. This ‘bottom-up’ method of synthesis could be considered as a new paradigm for the development of mucoadhesive thiol-bearing materials for drug delivery.
Experimental
Materials
2-Hydroxyethyl methacrylate (HEMA) was a gift from Vista Optics (UK). 2-Bromoethanol, ethylene glycol dimethacrylate, potassium thioacetate, L-cysteine hydrochloride, doxorubicin hydrochloride, Tris-buffered saline, FITC-dextran (10 kDa) and 5,5′-dithiobis-(2-nitrobenzoic acid) were purchased from Sigma-Aldrich (UK). Sodium thiomethoxide and acryloyl chloride were purchased from Alfa Aesar (UK). Dialysis membranes (12–14 kDa MWCO) were purchased from Visking (UK). Triethylamine was dried over 3 Å molecular sieves before use. 2-Hydroxyethyl methacrylate was purified by vacuum distillation, all other reagents were used without further purification. Artificial urine was composed of sodium chloride (105.5 mM), potassium chloride (63.7 mM), sodium citrate dihydrate (3.2 mM), sodium dihydrogen phosphate (3.2 mM) and sodium sulfate (17 mM).21 Porcine bladder tissues were received from PC Turner abattoir (Farmborough, UK), taken on the day of slaughter, and dissected to yield approximately 2 × 2 cm2 squares of internal mucosa, which were frozen in cling-film and stored at −20 °C.Synthesis of 2-(hydroxyethyl) thioacetate
A procedure was modified from Liras et al.10 2-bromoethanol (5.6 mL, 158 mmol, 1 eq.) and potassium thioacetate (20 g, 176 mmol, 1.1 eq.) were dissolved in acetone (400 mL). The solution was then stirred overnight at room temperature. The solution was then filtered and concentrated in vacuo. The crude product was then re-dissolved in dichloromethane (400 mL) and washed with deionised water (3 × 40 mL). After drying over MgSO4 and filtering, the mixture was concentrated in vacuo once more to yield a yellow-orange liquid (11.14 g). Yield: 59%. 1H NMR (400 MHz, DMSO-d6, δ): 5.16 (s, 1H, OH), 3.45 (t, 2H, SCH2CH2), 2.92 (t, 2H, SCH2CH2), 2.34 (s, 3H, CH3).Synthesis of 2-(acetylthio)ethyl acrylate
2-(Hydroxyethyl) thioacetate (6 g, 34 mmol) and triethylamine (7.7 mL, 54 mmol, 1.6 eq.) were dissolved in dichloromethane (60 mL). Acryloyl chloride (4.5 mL, 54 mmol, 1.6 eq.) was added dropwise over the course of 1 h. The mixture was then heated to reflux and reaction allowed to proceed overnight. After cooling, the solution was filtered, and washed with deionised water (12 mL). The resulting solution was dried over MgSO4 and concentrated in vacuo. The resulting liquid was vacuum distilled to yield a pale yellow liquid (1.9 g). Yield: 22%. 1H NMR (400 MHz, DMSO-d6, δ): 6.24 (d, 1H, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHH), 6.16 (dd, 1H, HCCHH), 5.98 (d, 1H, HCCHH), 4.22 (t, 2H, SCH2CH2), 3.16 (t, 2H, SCH2CH2), 2.36 (s, 3H, CH3).
CHH), 6.16 (dd, 1H, HCCHH), 5.98 (d, 1H, HCCHH), 4.22 (t, 2H, SCH2CH2), 3.16 (t, 2H, SCH2CH2), 2.36 (s, 3H, CH3).
Synthesis of microgels
Microgels were synthesised with 100, 80, 50, 30, 10, and 0 mol% ATEA – the remaining monomer mass being HEMA. The mass of each monomer used in the feed mixture for each batch of microgels is contained in Table S1 (ESI†). The general procedure was as follows: Deionised water (45 mL) was added to a three-necked round-bottom flask, and bubbled with nitrogen for 20 min. Meanwhile, ammonium persulfate (15 mg) was added to water (1 mL), and stirred. Monomers (1 g) and ethylene glycol dimethacrylate (100 mg, 83 μL) were then added to the degassed water, and nitrogen bubbled for a further 5 min. The solution of ammonium persulfate was then added, and the polymerisation mixture heated to 70 °C. After 6 h, the cloudy solution of microgels was allowed to cool and purified by dialysis, before lyophilisation to yield a white solid. All products were stored at 4–8 °C under nitrogen before use.Dynamic light scattering and ζ-potential measurements
The z-average hydrodynamic diameter and ζ potential was determined using a Nano Zetasizer at 25 °C using 100 μg mL−1 samples in water, diluted 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fold.
000 fold.
Deprotection of thiol groups
The thioester moieties found in the ATEA-containing microgels were cleaved to yield free thiol groups. The general method was as follows: microgels (20 mg) were dissolved in methanol (6 mL), and degassed with nitrogen bubbling. A methanolic solution of 0.1 M sodium thiomethoxide (1.5 mL) was then added via syringe and reaction allowed to proceed for 30 minutes with nitrogen bubbling at room temperature. 0.1 M HCl solution (20 mL) was then added to quench the reaction, and the product immediately dialysed with regular changes of the water over 24 h. The resulting cloudy liquid was lyophilised to yield deprotected microgels.Determination of thiol group content
The number of thiol groups per gram of microgels was determined using Ellman's assay.22 Microgels (3 mg) were dissolved in 0.5 M phosphate buffer at pH 8 (10 mL). L-Cysteine hydrochloride was used as a standard solution for the calibration curve, so a range of cysteine solutions (200–3 μg mL−1) were also prepared in phosphate buffer. Ellman's reagent, was then prepared by dissolution of 5,5′-dithiobis-(2-nitrobenzoic acid) (3 mg) in phosphate buffer (10 mL). Ellman's reagent (500 μL) was added to an aliquot (500 μL) of each microgel and standard solution. The solutions were then left to react for 2 h in the dark. After incubation, the microgel samples were centrifuged to get rid of any particulates which may lead to turbidity. Three aliquots (200 μL) of each sample and standard were placed into a 96-well plate, and the absorbance was measured at 420 nm using a plate reader. The concentration of thiol groups was calculated by comparison with the calibration curve of L-cysteine HCl, which was linear over the range measured.Loading of doxorubicin hydrochloride into microgels
30 mol% and 80 mol% ATEA microgels (2 mg) were suspended in Tris buffer adjusted to pH 7 (1 mL). 30 mM aqueous doxorubicin hydrochloride solution (90 μL) was then added to the microgel suspensions. The mixtures were stirred for 16 h in dark conditions to allow doxorubicin to enter the microgels. The resulting suspensions were centrifuged for 8 min at 13000 rpm. The supernatants were removed using a pipette and the remaining pellets were gently washed with 1 mL Tris buffer, leaving microgels with encapsulated doxorubicin.
Confocal microscopy of doxorubicin-loaded microgels
30 mol% ATEA microgels loaded with doxorubicin were dried onto a glass slide. They were then examined using a Nikon A1R confocal microscope with an excitation wavelength of 488 nm used to induce fluorescence.Release of doxorubicin from microgels in artificial urine
Doxorubicin-loaded microgels (1 mg) were suspended in artificial urine (1 mL). The suspended particles were then transferred to individual dialysis membranes and placed into artificial urine (80 mL) at 37 °C. The beakers were placed in an incubator and the temperature was maintained at 37 °C. Aliquots (3 mL) were taken from the dialysate at regular intervals and replaced with fresh artificial urine (3 mL). The concentration of doxorubicin in the dialysate was determined by spectrophotometry (Jasco V-530) at 480 nm.Labelling of microgels with 5-(iodoacetamido)fluorescein
30% and 80% ATEA microgels (10 mg) were suspended in Tris buffer (pH 7, 5 mL). 5-(Iodoacetamido)fluorescein (3.6 mg) was added directly into the microgel suspension and the mixture was stirred for 16 h at 4 °C. The fluorescently labelled microgels were purified by dialysis in 1 L of deionised water, which was replaced hourly until no fluorophore was present in the dialysate, as measured by fluorometry (Jasco FP-6200) at λex: 480 nm and λem: 520 nm.Retention of microgels on ex vivo porcine bladder
The retention of microgels on porcine bladder was assessed using a method developed in-house, modified from a published procedure.23,24 Solutions of microgels or FITC-dextran (1 mg mL−1) were prepared in artificial urine. An aliquot (20 μL) was pipetted onto a 1 × 1 cm2 piece of ex vivo porcine bladder mucosa, which was then placed into a sloped channel, and artificial urine washed over the surface using a syringe pump. At pre-defined intervals, fluorescence images of the whole tissue were acquired using a fluorescence stereomicroscope (Leica MZ10F) with a GFP filter (Fig. S1, ESI†). The fluorescence images were then analysed with ImageJ to measure the pixel intensity of the images, which was linearly correlated with the concentration of microgels present, as determined in preliminary experiments. A ‘blank’ value of pixel intensity was subtracted from each measurement, using an image of the bladder mucosa without fluorescent microgels present. The data is presented as a percent of microgels remaining on the mucosa surface, with volume of artificial urine washed over the surface. In order to assess whether the retention of microgels was specific to mucosa, their retention on a control substrate of PTFE was also measured. The retention data was analysed by two-way analysis of variance (ANOVA) with Bonferroni post hoc test to determine significant differences in the measurements with elution volume. The final retention % was also compared using one-way ANOVA with Bonferroni post hoc test.Results and discussion
Synthesis of protected microgels
Firstly, ATEA was synthesised using a two-step method (Fig. 1a), purified by vacuum distillation, and characterised by NMR. This confirmed that ATEA and its precursor 2-(hydroxyethyl) thioacetate had only small levels of impurity. The yield was 22%, which might be improved by up-scaling, due to the difficulty in distilling small volumes of liquid. A range of microgels were then synthesised using various molar ratios of ATEA to 2-hydroxyethylmethacrylate, a water-soluble monomer which could improve the colloidal stability of the resulting microgels. Polymerisation to form microgels was simply conducted by addition of the two monomers to an aqueous solution of cross-linker (ethylene glycol dimethacrylate), followed by thermal initiation using ammonium persulfate (Fig. 1b), in a surfactant-free emulsion polymerisation-type procedure.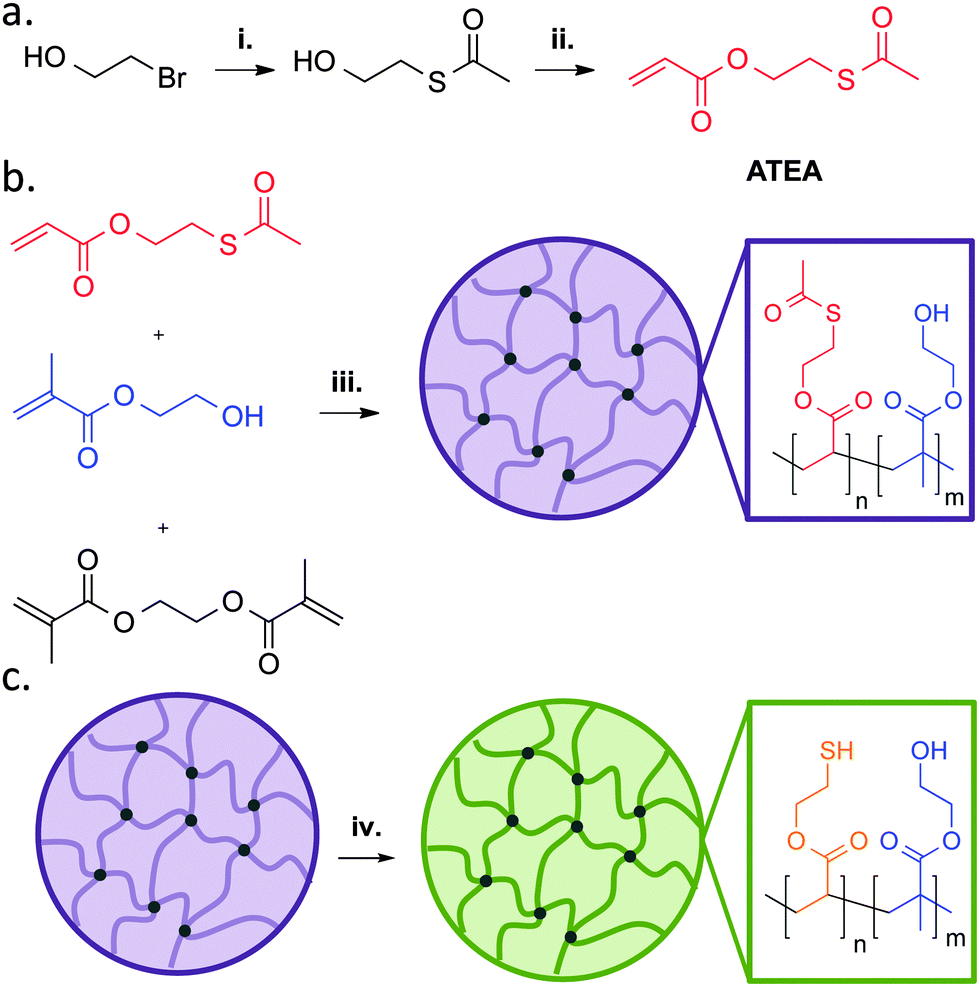 | ||
| Fig. 1 (a) Synthetic route to ATEA, a protected thiomonomer. (b) Polymerisation to form ATEA:HEMA copolymer microgels, displaying the pendant functionalities present. (c) Deprotection of ATEA using sodium thiomethoxide to yield thiol-bearing microgels. (i) Potassium thioacetate, acetone, 24 h. (ii) Acryloyl chloride, triethylamine, DCM, reflux, 24 h. (iii) Ammonium persulfate, ethylene glycol dimethacrylate, water, 70 °C, 6 h. (iv) Sodium thiomethoxide, methanol, 30 min. | ||
This method resulted in microgels of between 635 and 977 nm diameter, with a highly negative ζ-potential in every case, indicating good colloidal stability (Table 1). IR spectroscopy (Fig. S2, ESI†) on the ATEA:HEMA copolymer microgels indicated that they also contained both monomers, characterised by the presence of ATEA's C–S thioester stretch at approximately 1000 cm−1 and HEMA's broad O–H stretch at 3300 cm−1 in spectra of copolymer microgels. The actual ATEA content in the microgels was estimated by 1H NMR, and it was found that the conversion of HEMA was higher than ATEA, possibly due to the higher stability of the methacrylate radical.
| ATEA concentration (mol%) | Size (nm) | PDI | ζ-Potential | Thiol content (μmol g−1) | ATEA content (mol%) |
|---|---|---|---|---|---|
| Protected | |||||
| 10 | 977 | 0.280 | −45 | N/A | N.D. |
| 30 | 648 | 0.319 | −38 | N/A | 17 |
| 50 | 635 | 0.133 | −36 | N/A | 33 |
| 80 | 850 | 0.244 | −40 | N/A | 73 |
| Deprotected | |||||
| 10 | 659 | 0.723 | −29 | 21 ± 23 | |
| 30 | 547 | 0.163 | −30 | 49 ± 24 | |
| 50 | 828 | 0.116 | −23 | 143 ± 53 | |
| 80 | 1348 | 0.328 | −26 | 453 ± 133 | |
Deprotection of microgels
The previously-synthesised ATEA-containing microgels were successfully deprotected using sodium thiomethoxide (Fig. 1c). This deprotection method was adopted because sodium thiomethoxide is chemoselective in its cleavage of thioester groups in the presence of esters.25 The removal of the acetate protecting group from ATEA could be seen by NMR (Fig. S3 and S4, ESI†) and FTIR (Fig. S5, ESI†), and the number of thiol groups accessible to solvent was quantified using Ellman's assay (Table 1). Ellman's assay determined that the thiol content of ATEA-containing microgels was dependent on the concentration of ATEA in the particles, with maximum thiol content reached at 80 mol% ATEA in the feed mixture. 100 mol% ATEA particles were synthesised; however, they were not dispersible in water after deprotection. Swelling of 50 and 80 mol% ATEA particles was apparent after deprotection (Fig. 2), which may be a result of the acidity of the thiol groups (pKa ∼ 8–10),26 relative to thioesters (pKa ∼ 35), leading to a greater uptake of solvent and charged species. Transmission electron microscopy (TEM) confirmed that microgels were formed (Fig. 2), which appeared to form clusters during sample preparation. For further experiments in drug loading and mucoadhesion, 30 mol% and 80 mol% ATEA microgels were taken forward, to represent particles with a low and high level of thiol groups on their surface, respectively.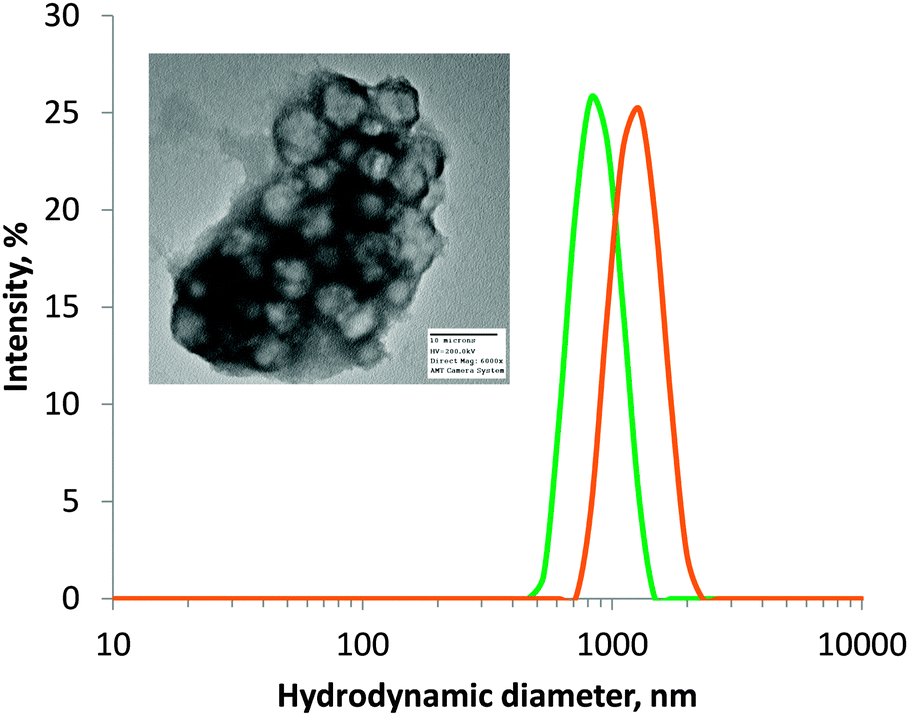 | ||
| Fig. 2 Hydrodynamic diameter of 80 mol% ATEA microgels before (green) and after (orange) deprotection with sodium thiomethoxide, as measured by dynamic light scattering. Deprotection appears to lead to the swelling of microgels. Inset: a TEM image of thiol-bearing microgels. | ||
Retention of microgels on porcine urinary bladder mucosa
In order to assess the mucoadhesive properties of these thiol-bearing microgels, their retention on ex vivo porcine urinary bladder was assessed (Fig. 3). Thiol-bearing 30 mol% and 80 mol% ATEA microgels were labelled with the fluorescein and their retention on urinary bladder mucosa during washing with artificial urine determined using fluorescence stereomicroscopy. In this case, 80 mol% ATEA was chosen as it showed the highest levels of thiol groups on its surface, and 30 mol% ATEA was a control, having very low levels of thiol groups. It was found that the 80 mol% ATEA microgels were retained significantly (p < 0.01, two-way ANOVA) more on the bladder mucosa than the 30 mol% microgels. This is most likely a result of the increased number of thiol groups on the surface of the microgels binding to cysteine-rich domains of mucins present on the surface of the bladder epithelium via disulfide bridges (inset, Fig. 3). Interestingly, the retention profile of the 80 mol% ATEA microgels plateaued after the elution of 20 mL artificial urine. This suggests that there may be saturation of the cysteine binding sites occurring, leading to improved retention of those microgels which are strongly bound by covalent attachment to these sites. The percentage of 80 mol% ATEA microgels bound at the end of the experiment was significantly more than for 30 mol% ATEA microgels, by one-way ANOVA (p < 0.05). FITC-dextran was used as a non-mucoadhesive control, and showed no significant difference to the 30 mol% ATEA microgels (p > 0.05). The retention of 30 mol% and 80 mol% ATEA microgels on an apolar polytetrafluoroethylene surface was also determined. It was found that the microgels were washed from the surface after 3 mL of artificial urine was eluted, indicating that there is some selectivity in the retention of these particles, and that the adhesion process is not simply a result of the rheological properties of the microgels, but likely due to interactions between the microgels and the surface of the mucosa. The ζ-potential of the microgels is comparable and highly-negative for both microgels studied, so the adhesion process is not likely due to poor colloidal-stability leading to deposition. As the ζ-potential is similar for both systems, it is unlikely that the retention is due to surface charge. These results point to disulphide bridge formation as the driving force for mucoadhesion (inset, Fig. 3). Mucoadhesives acting in this manner have been well-reported by Bernkop-Schnürch and colleagues.3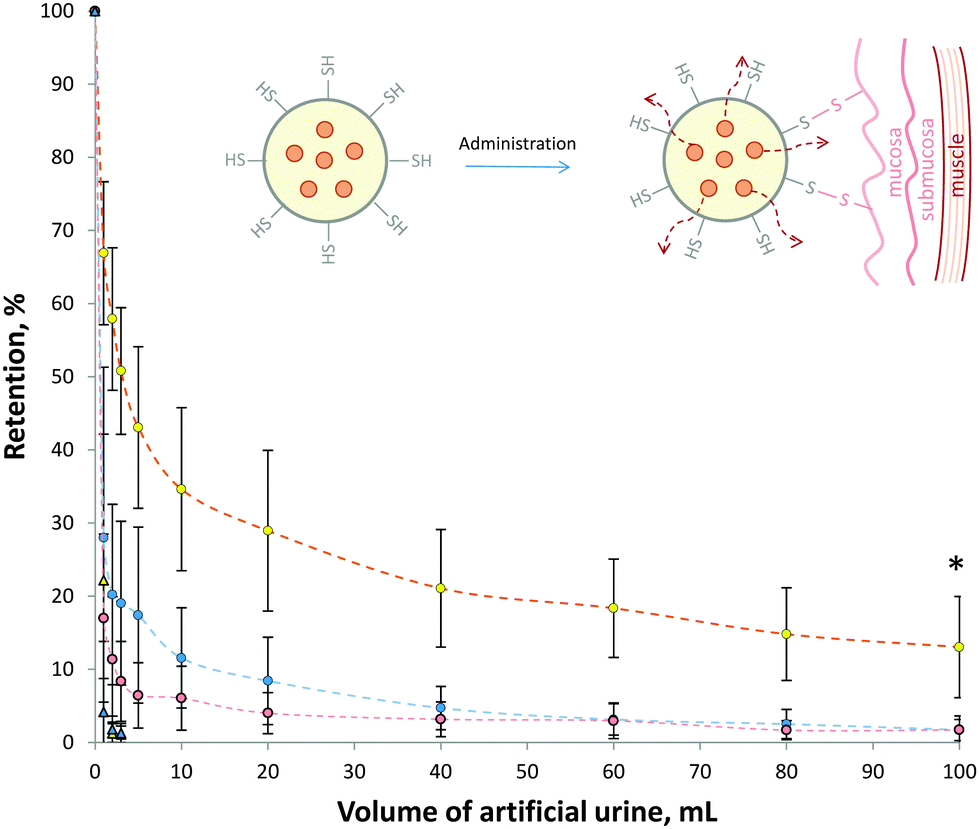 | ||
| Fig. 3 Retention of thiol-bearing 30 mol% (blue circles) and 80 mol% (yellow circles) ATEA microgels on ex vivo porcine urinary bladder. FITC-dextran (red circles) was used as a control. The retention of 30 mol% (blue triangles) and 80 mol% (yellow triangles) ATEA microgels on PTFE was also examined. Data expressed as mean ± standard deviation (n = 3). Inset: schematic illustration demonstrating the mechanism by which we propose these microgels bind to the urinary bladder's mucosal surface, at which point, in vivo, they would deliver doxorubicin. | ||
Loading of doxorubicin hydrochloride into microgels
The highly-negative ζ-potential of the deprotected microgels indicates good colloidal stability, but also gives the opportunity for incorporation of cationic species into the particles. The ATEA:HEMA copolymer microgels were able to take-up the chemotherapeutic agent doxorubicin hydrochloride simply by incubation of the drug with an aqueous dispersion of microgels. The microgels were able to uptake doxorubicin with high encapsulation efficiencies – 75 ± 15%, and 86 ± 8% for 30 mol% and 80 mol% particles, respectively (Table S2, ESI†). The difference in encapsulation efficiency (eqn (S1), ESI†) between the different microgel compositions was found to be statistically insignificant (p > 0.05, t-test). This encapsulation efficiency resulted in a high drug loading (eqn (S2), ESI†) of between 37 and 40% by mass. Doxorubicin hydrochloride is typically administered to bladder cancer patients as 30–100 mL of a 1 mg mL−1 solution via catheter.20 Achieving this dose of doxorubicin is possible even after loading into ATEA:HEMA microgels, requiring 2.5 and 2.7 mg mL−1 of the doxorubicin-loaded 30 mol% and 80 mol% ATEA microgels, respectively.Doxorubicin is a fluorescent drug, and its presence in the thiol-bearing microgels could be seen by confocal microscopy (Fig. 4). Microscopy appeared to indicate that doxorubicin was absorbed into the matrix of the microgel, rather than adsorbed to the periphery. The microgels were ∼0.5 μm in diameter, consistent with dynamic light scattering measurements.
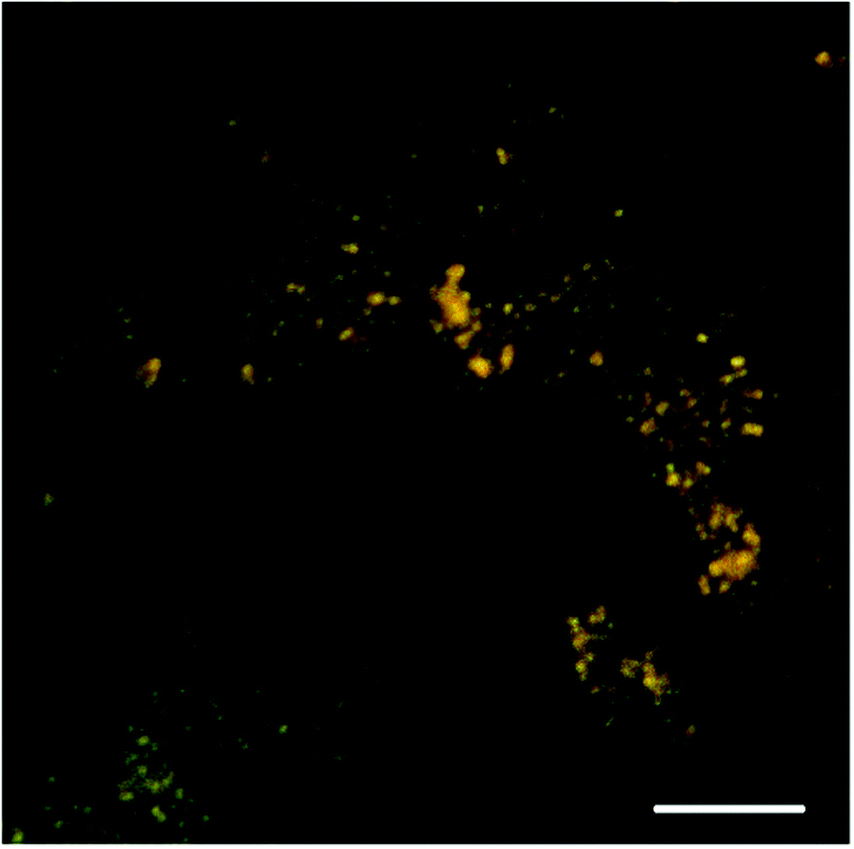 | ||
| Fig. 4 A confocal image of doxorubicin-loaded 30 mol% ATEA microgels. Scale: 10 μm. | ||
Release of doxorubicin from microgels in vitro
In order to act on a patient, doxorubicin would have to be released from the microgels in vivo. The release behavior of the particles was assessed by exposure of the doxorubicin-loaded microgels to an artificial urine solution maintained at 37 °C. The exact volume of urine contained in the bladder is difficult to precisely replicate, due to its dynamic nature. The functional capacity of the bladder is ∼300 mL before micturition,27 so large volumes may be unrealistic in vivo. A set volume of 80 mL was used in order to ensure that sink conditions were met for these experiments, whilst ensuring that the functional capacity of the bladder was not exceeded. These release experiments (Fig. 5) indicated that there was an initial burst release of doxorubicin from the microgels, likely attributable to loosely bound doxorubicin on the surface, followed by a time-dependent release over 300 min. This indicates that if administered into the bladder, the doxorubicin load should be delivered to the patient over several hours. The data is expressed as percentage release in Fig. S6 (ESI†). The difference in drug release from the two different particle compositions was statistically insignificant (p > 0.05, two-way ANOVA). This data fit well to the Higuchi model of drug release,28,29 with an R2 value of 0.99 in both cases (Fig. S7, ESI†). This indicates that the drug release is consistent with Fickian diffusion from an insoluble matrix.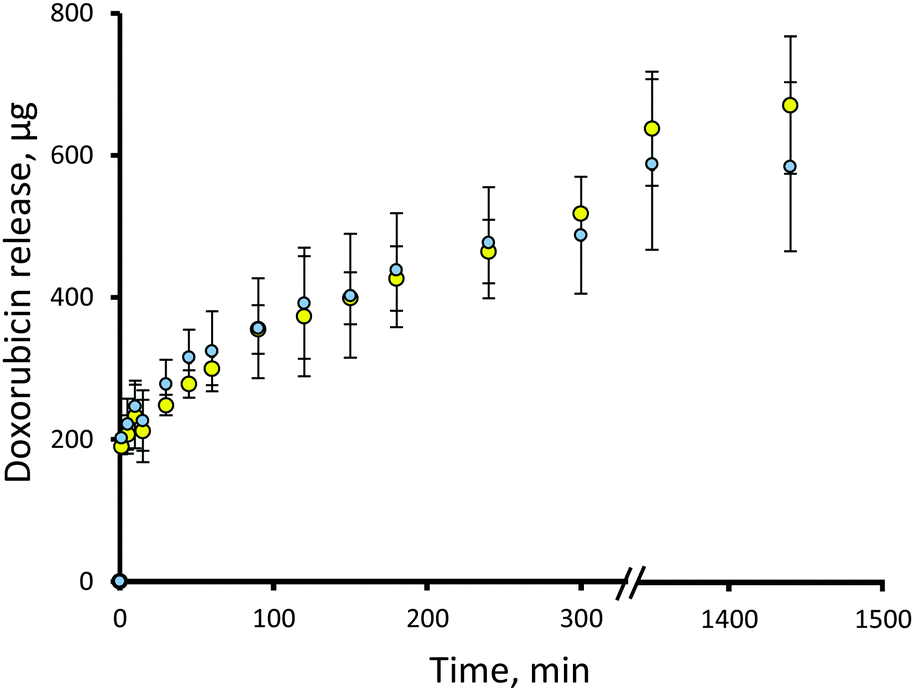 | ||
| Fig. 5 Release of doxorubicin from deprotected 30 mol% (blue) and 80 mol% (yellow) ATEA microgels. Data expressed as mean ± standard deviation (n = 3). | ||
Conclusions
A series of copolymer microgels have been designed from HEMA and ATEA, a protected thiomonomer synthesised for this study. These microgels were between 635 and 977 nm in diameter, and could subsequently be deprotected to yield particles bearing varying levels of thiol groups on their surface. A high level of thiol groups on the surface of the particles improved the mucoadhesion capability of the copolymer microgels on porcine urinary bladder tissue. It was also found that these microgels can be loaded with doxorubicin, with potential for site-specific delivery to the bladder via urethral catheter. This could lead to materials which allow micturition after the admission of chemotherapy, without loss of the drug. This bottom-up approach could be used in future research to yield a new generation of mucoadhesive materials.Acknowledgements
The Leverhulme Trust is thanked for funding (RPG-2013-017). The authors wish to thank the Commission of Higher Education (Thailand), the Thailand Research Funds through the Royal Golden Jubilee PhD Program (Grant No. PHD/0001/2553) for financial support and providing 6 months placement for WS at the University of Reading. Kreisberufsschulzentrum Ellwangen (Germany) is thanked for providing a placement at the University of Reading for SAS. The Chemical Analysis Facility at the University of Reading is thanked for assistance with analysis.References
- A. Sosnik, J. das Neves and B. Sarmento, Prog. Polym. Sci., 2014, 39, 2030 CrossRef CAS PubMed.
- V. V Khutoryanskiy, Macromol. Biosci., 2011, 11, 748 CrossRef PubMed.
- A. Bernkop-Schnürch and A. Greimel, Am. J. Drug Delivery, 2005, 3, 141 CrossRef.
- M. Davidovich-Pinhas and H. Bianco-Peled, Acta Biomater., 2011, 7, 625 CrossRef CAS PubMed.
- J. Xu, G. M. Soliman, J. Barralet and M. Cerruti, Langmuir, 2012, 28, 14010 CrossRef CAS PubMed.
- S. Bonengel and A. Bernkop-Schnürch, J. Controlled Release, 2014, 195, 120 CrossRef CAS PubMed.
- J. Barthelmes, S. Dunnhaupt, J. Hombach and A. Bernkop-Schnürch, Drug Delivery, 2011, 18, 613 CAS.
- G. S. Irmukhametova, G. A. Mun and V. V. Khutoryanskiy, Langmuir, 2011, 27, 9551 CrossRef CAS PubMed.
- A. Štorha, E. A. Mun and V. V. Khutoryanskiy, RSC Adv., 2013, 3, 12275 RSC.
- M. Liras, O. García, N. Guarrotxena, M. Palacios-Cuesta and I. Quijada-Garrido, Polym. Chem., 2013, 4, 5751 RSC.
- M. Liras, E. Peinado, P. Cañamero, I. Quijada-Garrido and O. García, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3087 CrossRef CAS PubMed.
- M. P. Montero-Rama, M. Liras, O. García and I. Quijada-Garrido, Eur. Polym. J., 2015, 63, 37 CrossRef CAS PubMed.
- M. Liras, O. García, I. Quijada-Garrido, G. Ellis and H. J. Salavagione, J. Mater. Chem. C, 2014, 2, 1723 RSC.
- M. Liras, M. González-Béjar, E. Peinado, L. Francés-Soriano, J. Pérez-Prieto, I. Quijada-Garrido and O. García, Chem. Mater., 2014, 26, 4014 CrossRef CAS.
- J. Barthelmes, S. Dünnhaupt, S. Unterhofer, G. Perera, W. Schlocker and A. Bernkop-Schnürch, Nanomedicine, 2012, 8, 65 CrossRef PubMed.
- C. L. Parsons and S. G. Mulholland, Am. J. Pathol., 1978, 93, 423 CAS.
- J. N'Dow, J. P. Pearson, M. K. Bennett, D. E. Neal and C. N. Robson, J. Urol., 2000, 164, 1398 CrossRef.
- A. Bergeron, H. LaRue and Y. Fradet, Biochem. J., 1997, 895, 889 Search PubMed.
- Q. Zhang, K. G. Neoh, L. Xu, S. Lu, E. T. Kang, R. Mahendran and E. Chiong, Langmuir, 2014, 30, 6151 CrossRef CAS PubMed.
- C. L. Amling, Curr. Probl. Cancer, 2001, 25, 219 CrossRef CAS.
- F. J. Opalko, J. H. Adair and S. R. Khan, J. Cryst. Growth, 1997, 181, 410 CrossRef CAS.
- I. Bravo-Osuna, D. Teutonico, S. Arpicco, C. Vauthier and G. Ponchel, Int. J. Pharm., 2007, 340, 173 CrossRef CAS PubMed.
- R. A. Cave, J. P. Cook, C. J. Connon and V. V Khutoryanskiy, Int. J. Pharm., 2012, 428, 96 CrossRef CAS PubMed.
- C. A. Withers, M. T. Cook, L. Methven, G. A. Gosney and V. V. Khutoryanskiy, Food Funct., 2013, 4(11), 1668 CAS.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805 CrossRef CAS PubMed.
- K. S. Jensen, J. T. Pedersen, J. R. Winther and K. Teilum, Biochemistry, 2014, 53, 2533 CrossRef CAS PubMed.
- C. L. Amundsen, M. Parsons, B. Tissot, L. Cardozo, A. Diokno and A. C. Coats, Neurourol. Urodyn., 2007, 26, 341 CrossRef PubMed.
- J. Siepmann and N. A. Peppas, Int. J. Pharm., 2011, 418, 6 CrossRef CAS PubMed.
- T. Higuchi, J. Pharm. Sci., 1961, 50, 874–875 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR and NMR spectra of microgels, % drug release and fitting, exemplar retention images, drug loading information, compositional information, and equations. See DOI: 10.1039/c5tb00834d |
| This journal is © The Royal Society of Chemistry 2015 |