
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA supermolecular building layer approach for gas separation and storage applications: the eea and rtl MOF platforms for CO2 capture and hydrocarbon separation†
Zhijie
Chen
,
Karim
Adil
,
Łukasz J.
Weseliński
,
Youssef
Belmabkhout
and
Mohamed
Eddaoudi
*
Functional Materials Design, Discovery and Development Research Group (FMD3), Advanced Membranes and Porous Materials Center (AMPM), Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: mohamed.eddaoudi@kaust.edu.sa
First published on 11th February 2015
Abstract
The supermolecular building layer (SBL) approach was employed to deliberately synthesize five novel metal–organic frameworks (1–5) with an exposed array of amide or amine functionalities within their pore system. The ability to decorate the pores with nitrogen donor moieties offers potential to evaluate/elucidate the structure–adsorption property relationship. Two MOF platforms, eea-MOF and rtl-MOF, based on pillaring of kgm-a or sql-a layers with heterofunctional 3-connected organic building blocks were targeted and constructed to purposely introduce and expose the desired amide or amine functionalities. Interestingly, gas adsorption properties of eea-MOF-4 (1) and eea-MOF-5 (2) showed that by simply altering the nitrogen donor position within the ligand, it is possible to relatively reduce the pore size of the related eea-MOF material and subsequently increase the associated CO2 uptake. The slightly confined pore space in 2, relative to 1, has enabled an enhancement of the pore local charge density and thus the observed relative increase in the CO2 and H2 isosteric heat of adsorption (Qst). In addition, light hydrocarbon adsorption studies revealed that 2 is more selective toward C2H6 and C3H8 over CH4 than 1, as exemplified for C2H6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH4 (5:95) or C3H8:CH4 (5:95) binary gas mixtures.
:CH4 (5:95) or C3H8:CH4 (5:95) binary gas mixtures.
1. Introduction
In recent years, metal–organic frameworks (MOFs) have emerged as an attractive class of solid-state materials owing to their associated exceptional wide range of properties pertinent to gas storage/separation,1–7 catalysis,8 surface chemistry and sensors9,10 and drug controlled release.11–13 Importantly, advances in MOF crystal chemistry offer potential to ideally practice the building block approach based-assembly for the construction of a given MOF with desired and appropriate chemical and structural features for specific applications.14 Namely, the ability to design and construct a MOF with the appropriate pore size, shape and functionality offers prospects to rationally correlate properties with the structure, and subsequently access appropriate MOF platforms15 that can address some difficult and energy intensive gas separations, e.g. CO2 capture16,17 and hydrocarbon separation.18,19 Nevertheless, the unequivocal prediction of a given MOF structure prior to its assembly remains an ongoing challenge; our group has recently published an elaborate tutorial review highlighting distinct strategies that can assist/aid chemists to rationally assemble desired functional MOFs.20A unique and powerful assembly strategy, detailed in the aforementioned review, is based on the use of pre-targeted 2-periodic MOF layers as supermolecular building layers (SBLs) for the deliberate construction of 3-periodic functional MOFs.20 In the present study, we employed the SBL approach to deliberately synthesize functional MOFs with an exposed array of amide or amine functionalities within their pore system. Specifically, two MOF platforms based on (3,6)-connected nets with eea and rtl underlying topologies were targeted, i.e.eea-MOF and rtl-MOF platforms.21–24 The elected two MOF platforms consist of inter-connected layers and thus can be regarded as 3-periodic MOFs based on pillared 2-periodic SBLs.20 Indeed, our research group has employed successfully the SBL approach to target and construct isoreticular tbo-MOFs,25 exhibiting large surface areas and exceptional CH4 storage working capacity for stationary applications.18 Similarly, other 3-periodic porous MOFs can be potentially targeted via pillaring SBLs based on one of the sole five edge-transitive 2-periodic nets: sql (square lattice), kgm (Kagomé), hcb (honeycomb), kgd (Kagomé dual) and hex (hexagonal lattice).15 Noticeably, augmenting the aforementioned nets, i.e. replacing a vertex with a vertex figure, reveals only two distinct possibilities for the assembly of squares into edge-transitive 2-periodic nets, namely sql-a and kgm-a.
The distinctive kgm-a and sql-a nets can be regarded as ideal blueprints to target 2-periodic MOFs based on the assembly of square building units, derived from well-known metal paddlewheel clusters as molecular building blocks (MBBs).26 Subsequently, the resultant MOF layers can be perceived as SBLs that are amenable to pillaring through 2-, 3-, 4- or 6-connected organic building blocks for the construction of desired 3-periodic MOF platforms.20
Here, the ligand-to-axial (L-A) pillaring strategy, successfully introduced by us21 and demonstrated by others,23,24 has been employed to introduce the desired functionality into the resultant MOF via pillar moieties. Purposely, triangular hetero-functional ligands were conceived to contain concurrently (i) the isophthalic moiety, needed for the formation of the layers in combination with dinuclear cluster paddlewheel MBBs, (ii) nitrogen donor moiety necessary for connecting neighboring layers via its coordination to an apical position in the dinuclear cluster, and (iii) specific functional groups such as acetylamide27–30 or amines31,32 that offer potential to enhance the affinity between CO2 molecules and the pore system in the resultant (3,6)-connected 3-periodic MOF.
Indeed, 3-connected organic building blocks (5-R-isophthalic acid with R = isonicotinamido, nicotinamido, pyridin-4-ylamino, pyridin-3-ylamino, and pyrimidin-5-ylamino) were synthesized and employed in combination with the dinuclear copper paddlewheel to yield the targeted eea-MOF-4 (1), eea-MOF-5 (2), rtl-MOF-2 (3), rtl-MOF-3 (4) and rtl-MOF-4 (5). Interestingly, gas adsorption properties of eea-MOF-4 (1) and eea-MOF-5 (2) showed that by simply altering the nitrogen donor position within the ligand, it is possible to relatively reduce the pore size of the related eea-MOF material and subsequently increase the associated CO2 uptake and relative affinity toward light hydrocarbons.
2. Results and discussion
The solvothermal reaction of copper nitrate and 5-(isonicotinamido)isophthalic acid (H2L1) in N,N′-dimethylacetamide (DMA)/acetic acid (HOAc) at 115 °C for 24 h yielded green block microcrystals. The structure of the as-synthesized compound was determined by single-crystal X-ray diffraction (SCXRD) and formulated as Cu(L1)·(solv)x (1), eea-MOF-4 (Fig. 1). The phase purity of the crystalline material was confirmed by similarities between the experimental and calculated powder X-ray diffraction (PXRD) patterns (ESI, Fig. S9†). | ||
| Fig. 1 (a) Layer segment of a kgm-MOF. (b) Left: 5-(isonicotinamido)isophthalic acid (H2L1) and right: 5-(nicotinamido)isophthalic acid (H2L2). (c) Hourglass-shaped channels with two primary types of cavities. C = gray, O = red, N = blue, Cu = plum; H atoms are omitted for clarity. | ||
The resultant 3-periodic eea-MOF-4 can be regarded as kgm-MOF layers interconnected via an L-A pillaring approach, where L1 serves as a 3-connected node and the paddlewheel MBB as a 6-connected octahedral building unit. Topological analysis reveals that 1 has the anticipated eea underlying net topology (ESI, Fig. S7†). It is worth noting that while writing the present manuscript, a similar compound appeared in the literature.30
The kgm-MOF sheets in 1 are pillared in an arrangement where pairs of three-membered ring windows of neighboring sheets are further interconnected via interfacing six-membered ring windows (i.e., 3, 3, 6, 3, 3) to yield hourglass-shaped channels (Fig. 1c). These hourglass-shaped channels are comprised of two alternating kinds of cavities (7.8 Å and 10.5 Å diameters), one is delimited by neighboring three membered rings and the second is generated from enclosing hexagonal windows by two three-membered ring windows from the two neighboring layers, as shown in Fig. 1. The calculated total accessible volume for the as-synthesized 1, upon removal of guest solvent molecules, was estimated to be 6887 Å3 per unit cell volume (11558 Å3) or 59.6% v/v.
The eea-MOF platform offers potential to control and fine-tune the pore size via shifting the N-donor group of the pyridine rings within the 3-connected organic building block. Accordingly, we synthesized and used 5-(nicotinamido)isophthalic acid (H2L2) under similar reaction conditions to construct the anticipated isoreticular eea-MOF-5 (2). The single-crystal analysis confirms the expected crystal structure formulated as Cu(L2)·(solv)x (Fig. 1). The replacement of isonicotinamide by the nicotinamide group resulted in slight changes in the size and shape of the two cavities (7.6 Å and 9.8 Å diameters).
The experimental PXRD of the bulk material matches perfectly the calculated PXRD pattern based on the crystal structure of 2, confirming the phase purity of the resultant as-synthesized 2 (ESI, Fig. S9†). The thermogravimetric analysis (TGA) of compounds 1 and 2 reveals the stability of compounds 1 and 2 up to 523 K (ESI, Fig. S11†).
In order to confirm the versatility of the SBL approach for the synthesis of isoreticular MOFs, we synthesized three additional 3-connected organic building blocks (5-R-isophthalic acid with R = pyridin-4-ylamino, pyridin-3-ylamino, and pyrimidin-5-ylamino). The reaction between copper nitrate and 5-(pyridin-4-ylamino)isophthalic acid (H2L3) in a mixture of N,N′-dimethylformamide (DMF), ethanol and water resulted in a 3-periodic MOF, Cu(L3)·(solv)x, 3, based on a triangular organic building block interconnecting 2-periodic sql-MOF layers in a ligand-to-axial (L-A) pillaring fashion (Fig. 2). Topological analysis reveals that 3 is consistent with the anticipated rtl topology, where L3 serves as a 3-connected node and the paddlewheel MBB as a 6-connected node (ESI, Fig. S8†).
 | ||
| Fig. 2 (a) Layer segment of a sql-MOF. (b) Left, up: 5-(pyridin-4-ylamino)isophthalic acid (H2L3). Right, up: 5-(pyridin-3-ylamino)isophthalic acid (H2L4). Bottom: 5-(pyrimidin-5-ylamino)isophthalic acid (H2L5). (c) Projection along the a-axis of the rtl-MOF-2 displaying channel. C = gray, O = red, N = blue, and Cu = plum; H atoms are omitted for clarity. | ||
The sql-a MOF layer comprises square windows where a pair of adjacent benzene moieties in any given four-membered ring points up while the other adjacent pair points down, i.e. benzene moieties in the four-membered ring are arranged in a 1,2-alternate fashion. The quadrangular windows of neighboring 2D sql MOF layers are superimposed to generate channels (2.9 × 7.1 Å) running along the c-axis. The calculated total accessible volume for the as-synthesized 3, upon removal of guest solvent molecules, was estimated to be 3216 Å3 per unit cell volume (6203 Å3) or 51.9% v/v.
As expected, the reaction between copper nitrate and 5-(pyridin-3-ylamino)isophthalic acid (H2L4), or 5-(pyrimidin-5-ylamino)isophthalic acid (H2L5) yielded the formation of two isoreticular rtl-MOF materials as a result of L-A pillaring of sql-a MOF layers, Cu(L4)·(solv)x and Cu(L5)·(solv)x, respectively (Fig. 2). Similar to eea-MOF-4 and eea-MOF-5, the use of different isomers/analogs resulted in slight changes in the associated channel size dimensions (3.55 × 5.6 Å).
The experimental PXRD of the as-synthesized bulk materials matches perfectly the respective calculated PXRD pattern based on the associated crystal structure, confirming the phase purity of the resultant as-synthesized 3–5 (ESI, Fig. S10†).
The permanent porosity of 1 and 2 has been confirmed by argon (Ar) adsorption isotherms at 87 K (Fig. 3). Ar adsorption experiments on compound 1 show a fully reversible type-I isotherm, characteristic of a microporous material with permanent microporosity. The apparent Langmuir surface area of 1 was estimated to be 1860 m2 g−1 and the experimental total pore volume derived from the Ar isotherm was found to be 0.65 cm3 g−1, which is in good agreement with the calculated pore volume from the crystal structure (0.66 cm3 g−1). In contrast, 2 displayed an unusual two-step adsorption isotherm with stepwise adsorption behaviour and a noticeable wide-open hysteresis, which can be symptomatic of plausible framework flexibility. The amount of argon adsorbed up to the first plateau was estimated to be ∼320 cm3 g−1, and the total amount of argon adsorbed in the framework was estimated to be ∼430 cm3 g−1, equivalent to an uptake increase of nearly ∼110 cm3 g−1 between the two adsorption plateaus. Accordingly, the apparent Langmuir surface area of compound 2, determined using the adsorption data leading to the first plateau, was estimated to be 1200 m2 g−1. The estimated total pore volume of 2 derived from the Ar isotherm was estimated to be 0.54 cm3 g−1, matching the total uptake at the second plateau, and correlates remarkably with the calculated total pore volume from the crystal structure (0.52 cm3 g−1). Notably, the total pore volume of 2 (0.54 cm3 g−1) is smaller than the total volume of 1 (0.66 cm3 g−1), confirming the contraction of the pore system in 2 as a result of nitrogen position in the pillaring ligand.
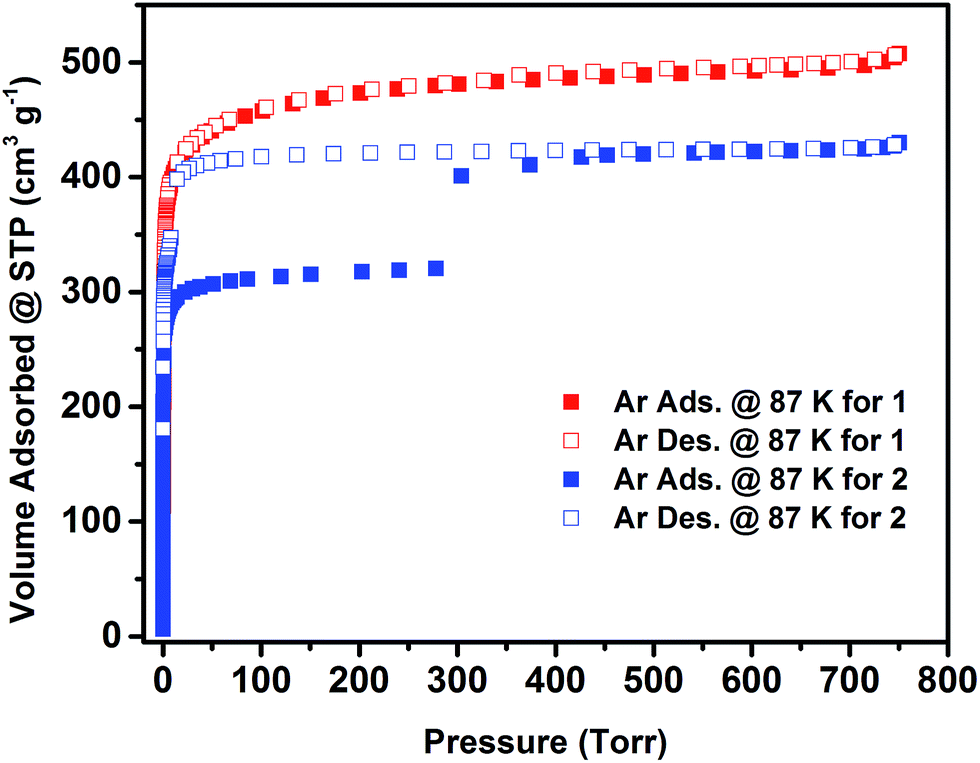 | ||
| Fig. 3 Low pressure argon adsorption (solid squares) and desorption (open squares) isotherms at 87 K for the acetone exchanged 1 and 2. | ||
The shapes of the Ar adsorption/desorption isotherms at cryogenic temperature suggest a plausible gate effect occurring around 14 mmol g−1 of adsorbed Ar at 87 K. In order to verify the occurrence of this adsorption gate effect, we explored the adsorption of different other probe molecules, namely CO2, C3H8 and n-C4H10. Markedly, the aforementioned adsorption gate effect was also observed with these probe molecules at relatively low and high pressures (ESI, Fig. S14–S18†).
Interestingly, the adsorbed amounts corresponding to the first plateau were reduced from CO2 (7 mmol g−1), C3H8 (4.2 mmol g−1) to n-C4H10 (3.75 mmol g−1) in accordance with the increase in the sorbate kinetic diameter and polarizability (ESI, Fig. S19†).
In order to further explore the MOF associated adsorption properties, CO2, H2, N2, and light hydrocarbon adsorption experiments were carried out at various temperatures and pressures. The CO2 adsorption uptake (298 K) of 1 at 0.15 and 1 bar was 0.56 mmol g−1 and 3 mmol g−1, respectively (Fig. 4). By contracting the pore size, the CO2 adsorption uptake (298 K) of 2 at 0.15 and 1 bar increased by 67% (0.94 mmol g−1) and 50% (4.5 mmol g−1), respectively. The CO2 adsorption uptake of 2 at 1 bar and 298 K is higher than most of the acylamide functional MOFs.29 In contrast to the increased uptake of CO2, 2 shows similar uptake of methane and nitrogen at 298 K to 1, which can only adsorb limited amounts of CH4 (0.9 mmol g−1) and N2 (0.18 mmol g−1) at 298 K and 1 bar. Ideal adsorbed solution theory (IAST)33 was used to predict the separation factor of equimolar CO2/CH4 and CO2/N2 (10:90) mixtures using experimentally obtained single gas adsorption isotherms (Fig. 5).
 | ||
| Fig. 4 Single component gas adsorption isotherms for CO2, CH4 and N2 at 298 K and CO2Qst (inset) for 1 and 2. | ||
 | ||
| Fig. 5 IAST selectivities for 50:50 CO2/CH4 (squares) and 10:90 CO2/N2 (circles) binary mixtures predicted at 298 K for 1 (blue) and 2 (red). | ||
IAST selectivity prediction is a valid methodology when the same elaborate model fits matchlessly the experimental data from single gas adsorption isotherms.34 Accordingly, IAST can be confidently used to evaluate gas selectivities in eea-MOFs. Compared with 1, the predicted separation factors of CO2/N2 and CO2/CH4 for 2 are significantly improved from 19.2 to 39.3 and from 4.3 to 7.3, respectively. To better understand the energetics of the interaction between CO2 and associated MOFs, the isosteric heat of adsorption (Qst) over the entire studied range was calculated for 1 and 2 using CO2 isotherms at 258, 273, 288 and 298 K (ESI, Fig. S14 and S15†). The Qst of CO2 adsorption for 2 is slightly higher than 1 over the whole range, with the highest point at ca. 27.5 over ca. 24.6 kJ mol−1 (Fig. 4, inset). The Qst of CO2 adsorption for 2 is relatively high among MOFs without open metal sites.35 Clearly, the confined space promoted relatively favorable interactions (high localized charge density) between the CO2 and the framework and subsequently improved the CO2 adsorption energetics and uptake.36
With regard to hydrogen adsorption, Qst of 2 was found to be higher than 1 over the entire adsorption range (e.g., 7.15 vs. 6.15 kJ mol−1 at low loading); nevertheless, the uptake at 77 K and 1 bar was comparable for 1 and 2 (1.96 vs. 1.98 wt%) (ESI, Fig. S12 and S13†). In this case, the observed increase in the H2 adsorption energetics for 2 can be interpreted as a result of the confined space effect, promoting a higher localized charge density in a more comparatively confined space.37
Adsorption studies of CH4, C2H6 and C3H8 were conducted in order to evaluate 1 and 2 separation potential of light hydrocarbons.
The C2H6 and C3H8 adsorption uptake for 1 at 1 bar and 298 K is 5.21 and 5.99 mmol g−1, which are higher than the C2H6 and C3H8 uptake for 2 (4.31 and 4.77 mmol g−1, respectively) (Fig. 6). This observed enhanced hydrocarbon adsorption uptake for 1 can be attributed to a comparatively larger available total pore volume in 1 relative to 2. Nevertheless, the predicted adsorption separation factors (using IAST) of 1 and 2 for C2H6 and C3H8 over CH4 in 5:95 C2H6/CH4 and 5:95 C3H8/CH4 binary gas mixtures reveal a slightly higher selectivity for 2 (156 and 22, respectively) than 1 (136 and 17, respectively) at 298 K and 1 bar (Fig. 7). These results support, as in the case of the CO2 separation, the impact of pore size reduction on the affinity enhancement for light hydrocarbons versus methane.
 | ||
| Fig. 6 Single component gas adsorption isotherms for C3H8, C2H6 and CH4 at 298 K for 1 and 2. | ||
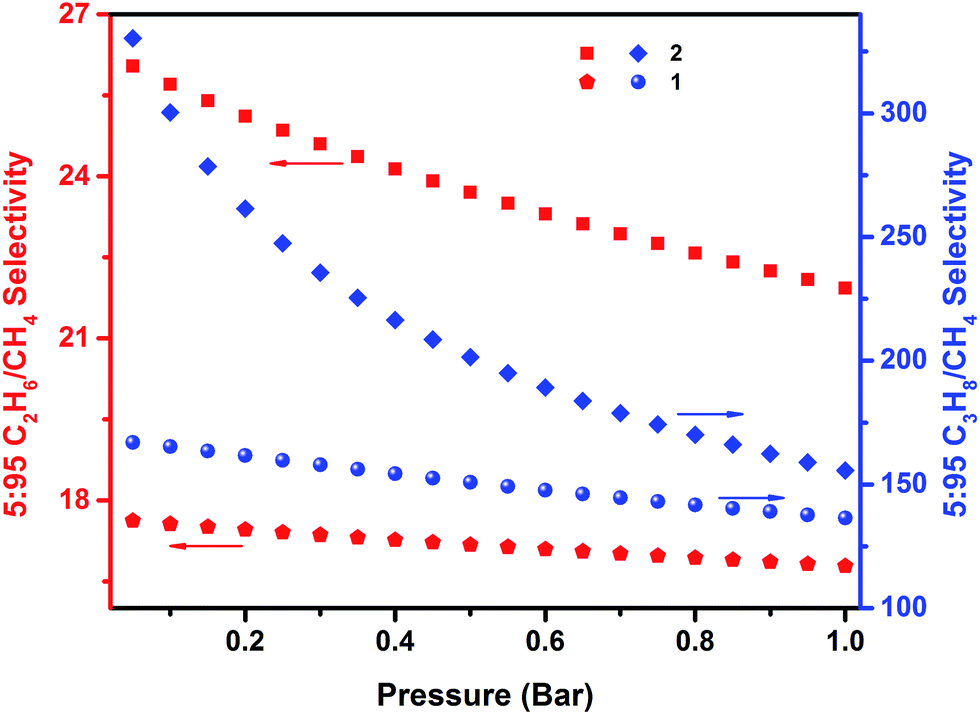 | ||
| Fig. 7 IAST selectivities for 5:95 C2H6/CH4 (red; left ordinate) and 5:95 C3H8/CH4 (blue; right ordinate) binary mixtures predicted at 298 K for 1 and 2. | ||
3. Conclusions
We have successfully utilized the supermolecular building layer (SBL) approach for the design and construction of isoreticular (3,6)-connected eea-MOFs and rtl-MOFs. The acylamide-functionalized eea-MOF showed a relatively good capacity for CO2 storage, as well as good selectivity toward CO2 and C3H8 at 298 K. The deliberate pore size reduction resulted in an enhanced local charge density and subsequently an increased CO2 uptake at both relatively low pressures (0.15 bar) and atmospheric pressures. Light hydrocarbon separation studies suggest that C2H6 and C3H8 could be selectively removed from natural gas to produce a relatively valuable commodity such as CH4. This MOF platform, based on the ligand-to-axial (L-A) pillaring approach, offers opportunities to readily functionalize and fine-tune the MOF pores with moieties that can favorably interact with a given probe molecule and subsequently provide access to functional MOF materials with improved attributes for gas separation and storage.Acknowledgements
Research reported in this publication was supported by the King Abdullah University of Science and Technology (KAUST).Notes and references
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724–781 CrossRef PubMed.
- O. K. Farha, A. O. Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed.
- H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836–868 CrossRef CAS PubMed.
- Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 CAS.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- M. C. So, M. H. Beyzavi, R. Sawhney, O. Shekhah, M. Eddaoudi, S. S. Al-Juaid, J. T. Hupp and O. K. Farha, Chem. Commun., 2015, 51, 85–88 RSC.
- D. Cunha, M. B. Yahia, S. Hall, S. R. Miller, H. Chevreau, E. Elkaïm, G. Maurin, P. Horcajada and C. Serre, Chem. Mater., 2013, 25, 2767–2776 CrossRef CAS.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CAS.
- X. Duan, R. Song, J. Yu, H. Wang, Y. Cui, Y. Yang, B. Chen and G. Qian, RSC Adv., 2014, 4, 36419–36424 RSC.
- Y. Belmabkhout, H. Mouttaki, J. F. Eubank, V. Guillerm and M. Eddaoudi, RSC Adv., 2014, 4, 63855–63859 RSC.
- Z. R. Herm, E. D. Bloch and J. R. Long, Chem. Mater., 2014, 26, 323–338 CrossRef CAS.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- J. F. Eubank, L. Wojtas, M. R. Hight, T. Bousquet, V. C. Kravtsov and M. Eddaoudi, J. Am. Chem. Soc., 2011, 133, 17532–17535 CrossRef CAS PubMed.
- S. Xiang, J. Huang, L. Li, J. Zhang, L. Jiang, X. Kuang and C.-Y. Su, Inorg. Chem., 2011, 50, 1743–1748 CrossRef CAS PubMed.
- X. Liu, M. Oh and M. S. Lah, Inorg. Chem., 2011, 50, 5044–5053 CrossRef CAS PubMed.
- X. Liu, M. Oh and M. S. Lah, Cryst. Growth Des., 2011, 11, 5064–5071 CAS.
- J. F. Eubank, H. Mouttaki, A. J. Cairns, Y. Belmabkhout, L. Wojtas, R. Luebke, M. Alkordi and M. Eddaoudi, J. Am. Chem. Soc., 2011, 133, 14204–14207 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571–8572 CrossRef CAS.
- B. Zheng, H. Liu, Z. Wang, X. Yu, P. Yi and J. Bai, CrystEngComm, 2013, 15, 3517–3520 RSC.
- K. Tang, R. Yun, Z. Lu, L. Du, M. Zhang, Q. Wang and H. Liu, Cryst. Growth Des., 2013, 13, 1382–1385 CAS.
- B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2010, 133, 748–751 CrossRef PubMed.
- Y. Xiong, Y.-Z. Fan, R. Yang, S. Chen, M. Pan, J.-J. Jiang and C.-Y. Su, Chem. Commun., 2014, 50, 14631–14634 RSC.
- R. Vaidhyanathan, S. S. Iremonger, K. W. Dawson and G. K. H. Shimizu, Chem. Commun., 2009, 5230–5232 RSC.
- N. Planas, A. L. Dzubak, R. Poloni, L. C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Long, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402–7405 CrossRef CAS PubMed.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- Y. Belmabkhout and A. Sayari, Chem. Eng. Sci., 2009, 64, 3729–3735 CrossRef CAS PubMed.
- S. D. Burd, S. Ma, J. A. Perman, B. J. Sikora, R. Q. Snurr, P. K. Thallapally, J. Tian, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 3663–3666 CrossRef CAS PubMed.
- L. Du, Z. Lu, K. Zheng, J. Wang, X. Zheng, Y. Pan, X. You and J. Bai, J. Am. Chem. Soc., 2012, 135, 562–565 CrossRef PubMed.
- Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278–3283 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthesis of ligands, NMR spectra, synthesis of MOFs, additional structural figures, PXRD, TGA, low and high pressure gas adsorption isotherms, Qst analysis and IAST calculations, Crystallographic Information Files (CIF). CCDC 1036004–1036007 and 1044642. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ta07115h |
| This journal is © The Royal Society of Chemistry 2015 |