DOI:
10.1039/C4RA17314G
(Paper)
RSC Adv., 2015,
5, 16641-16653
Remarkable visible light-triggered cytotoxicity of mitochondria targeting mixed-ligand cobalt(III) complexes of curcumin and phenanthroline bases binding to human serum albumin†
Received
31st December 2014
, Accepted 27th January 2015
First published on 28th January 2015
Abstract
Six new mixed-ligand cobalt(III) complexes of formulation [Co(N–N)2(O–O)](ClO4)2 (1–6), where N–N is a N,N-donor phenanthroline base, namely, 1,10-phenanthroline (phen in 1, 2), dipyrido[3,2-d:2′,3′-f]quinoxaline (dpq in 3, 4), and dipyrido[3,2-a:2′,3′-c]phenazine (dppz in 5, 6), O–O is acetylacetonate (acac in 1, 3, 5) or curcumin (bis(4-hydroxy-3-methoxyphenyl)-1,6-diene-3,5-dione, cur in 2, 4, 6), have been synthesized and characterized. The X-ray crystal structures of complex 1 (as PF6− salt, 1a) and 3 show distorted octahedral geometries formed by the CoN4O2 core. The complexes 1, 3 and 5 having the simple acac ligand are prepared as control species to understand the role of curcumin. The optimized geometries and the frontier orbitals of the curcumin complexes 2, 4, and 6 are obtained from the DFT calculations. The complexes 2, 4, and 6 having the photoactive curcumin moiety display an absorption band in the visible region near 420 nm and show remarkable photocytotoxicity in HeLa cancer cells with respective IC50 values of 7.4 μM, 5.1 μM and 1.6 μM while being much less toxic in dark. MTT assay using complex 6 shows that it is not significantly photocytotoxic to MCF-10A normal cells. The control complexes having the acac ligand are non-toxic both in the presence and absence of light. The cell death is apoptotic in nature and triggered by the photogeneration of reactive oxygen species. Fluorescence imaging experiments on HeLa cells reveals that complex 6 accumulated primarily inside the mitochondria. Human serum albumin (HSA) binding experiments show that the complexes bind HSA with good affinity, but 6 binds with the highest affinity, with a Kb value of 9.8 × 105 M−1. Thus, complex 6 with its negligible toxicity in the dark and in normal cells but remarkable toxicity in visible light holds significant photochemotherapeutic potential.
Introduction
Photodynamic therapy (PDT) is an emerging treatment modality of cancer that essentially involves administering a photosensitizer drug either systemically or topically followed by exposure of the target tissue to light specifically absorbed by the photosensitizer.1,2 PDT offers a means to overcome the limitations of conventional cancer therapies because of its low systemic toxicity, localized drug action to photoexposed regions, and remarkably low level of invasiveness.3 PDT is now considered as a substitute to conventional cancer therapies in dermatology and for endoscopically approachable tumors.1–5 The organic PDT agents have been extensively investigated and Photofrin®, which is an oligomer of a porphyrin derivative, is now the FDA approved clinical PDT drug.6,7 But the drawbacks of Photofrin® have provided strong impetus to look for alternatives.7 The photoactivatable metal complexes of non-porphyrinic ligands have emerged as potential alternatives to Photofrin® in the PDT of cancer.8–11
Anticancer drugs that target cell organelles other than nucleus are much sought after because of the drawbacks associated with the nucleus targeting drugs such as cisplatin and its analogs.12,13 The resistance of cisplatin can be partially attributed to the extensive repair of the cisplatin–DNA adducts by nucleotide excision repair (NER) pathway.14 For example, drugs that target mitochondria can be potential anticancer agents because mitochondria is a vital organelle for cellular function, and interference with its function is likely to cause cell death by the intrinsic pathway of apoptosis.12,13,15 Further, mitochondria lack a functional nucleotide excision repair mechanism to repair DNA adducts. Using this approach, Neamati and coworkers have reported a selective mitochondria targeted chlorambucil with remarkable cytotoxicity in breast and pancreatic cancer cells.16 Gasser and coworkers have reported mitochondria targeting ruthenium complexes of 2-pyridyl-2-pyrimidine-4-carboxylic acid that show anti-proliferative activity in a number of cell lines.17 Dhar and coworkers have recently shown that a cisplatin analogue, Platin-M, targets the mitochondria and disrupts mitochondrial DNA (mtDNA) instead of the conventional target – the nuclear DNA.18 Similarly, Chakravarty and coworkers have recently reported mitochondria and endoplasmic reticulum (ER) targeted oxidovanadium(IV) complexes that are capable of inducing apoptosis in cancer cells.19,20 Thus, the mitochondrial localization of a drug is a desirable aspect in PDT. The FDA approved PDT drug Photofrin® is known to localize in the mitochondria of cancer cells.21
Curcumin (bis(4-hydroxy-3-methoxyphenyl)-1,6-diene-3,5-dione, Hcur) has received considerable attention in recent years because it shows a plethora of medicinal properties.22,23 Curcumin is a naturally occurring β-diketone pigment isolated from the herb Curcuma longa L. Most notably, it shows anticancer activity against a wide range of cancers.23,24 The anticancer activity of curcumin stems from its ability to induce apoptosis in cancer cells while being innocuous to the normal cells.24 Curcumin is reported to interfere with the function of the transcription factor NF-κB which is a protein complex involved in the transcription of DNA.25 However, the poor bioavailability and pharmacokinetic profile have limited curcumin's applications in clinical settings despite its impressively low level of intrinsic toxicity.23–25 While several modifications of curcumin have been studied in attempts to overcome the drawbacks, one strategy takes advantage of curcumin's metal coordinating ability.26–29 Interestingly, curcumin possesses rich photophysical properties and there are reports of curcumin showing PDT effect.30,31 Although, there are reports on the anticancer activity of curcumin and its metal complexes, the studies on the photodynamic potential of its metal complexes are rare despite its rich photophysical and photochemical properties.26,27 Despite cobalt being a bioessential metal, there is no report of its complexes with curcumin showing photoactivated anticancer activity. Very recently, a mixed-ligand Co(III) complex of curcumin was reported to deliver the coordinated curcumin ligand to cancer cells resulting from a hypoxia-promoted intracellular reduction of Co(III) to Co(II).32 The very low dark toxicity (IC50 = 39 ± 4 μM in DLD-1 colon cancer cells) of the reported complex prompted us to explore the photo-induced cytotoxicity of a series of newly designed Co(III) curcumin complex by exploiting the rich photophysical properties of curcumin.32 Further, the intrinsic green fluorescence of curcumin offers a convenient way to study the localization of the curcumin-bound complex in cancer cells.28–30
Human serum albumin (HSA) is the most abundant protein in blood serum, found at concentrations of 35–55 g L−1.33 HSA is known to facilitate the transport of hydrophilic molecules from blood to target tissues and the overwhelming majority of drugs that are administered intravenously inescapably interact with it.33 For instance, the interaction of the FDA approved anticancer drug Abraxane with HSA has been exploited to deliver paclitaxel.34 Similarly, the interaction of platinum anticancer agents with HSA has been extensively researched.35,36 The success of HSA as a carrier of anticancer drugs stems from its nonimmunotoxicity, long circulation time in blood, and high accumulation in tumor.37
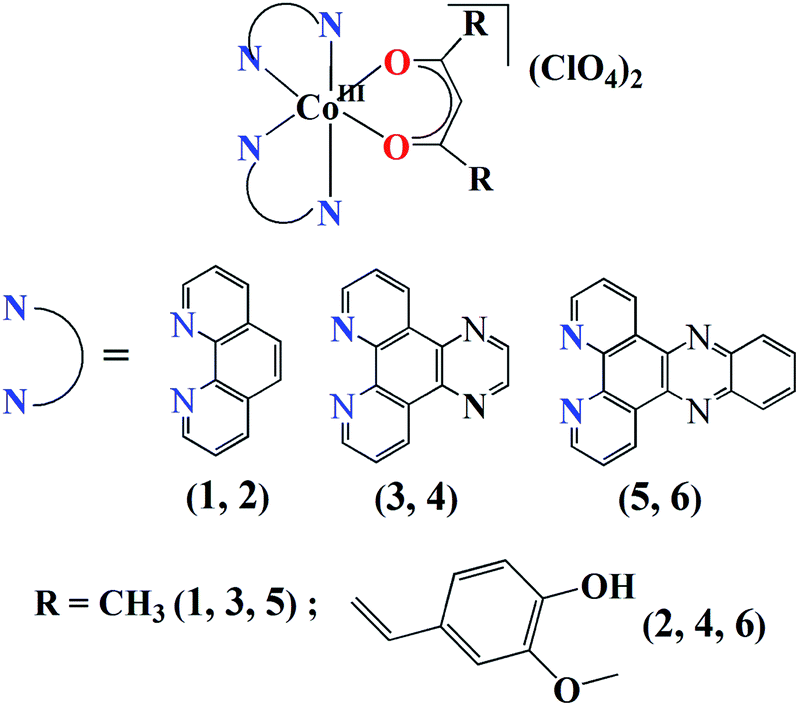
In this paper, we present the first systematic study on the photodynamic potential of a series of cobalt(III) curcumin complexes that are capable of binding to HSA. The strongly chelating phenanthroline bases, namely, phen, dpq and dppz were chosen to achieve stability and additional photoactivity of the resulting complexes. The use of phenanthroline bases, particularly dppz, as chelating photosensitizer ligands for the design of medicinally important metal complexes is fairly numerous in the literature.38–40 The Co(III) complexes of acetylacetone (Hacac) have been prepared as controls to understand the role of curcumin (Fig. 1). We have synthesized and characterized six new complexes of formulation [Co(N–N)2(O–O)](ClO4)2 (1–6), where N–N is a N,N-donor phenanthroline base, namely, 1,10-phenanthroline (phen in 1, 2), dipyrido[3,2-d:2′,3′-f]quinoxaline (dpq in 3, 4), dipyrido[3,2-a:2′,3′-c]phenazine (dppz in 5, 6), O–O is acetylacetonate (acac in 1, 3, 5) or monoanion of curcumin (bis(4-hydroxy-3-methoxyphenyl)-1,6-diene-3,5-dione, cur in 2, 4, 6), and evaluated their photo-triggered cytotoxicity in HeLa cells and HSA binding affinity. Significant results of this study include remarkable photocytotoxicity and HSA binding affinity of the curcumin complexes 2, 4, and 6 and mitochondrial accumulation of complex 6. The control complexes having acac instead of cur as ligand were found to be non-toxic to HeLa cells. Specifically, the dppz complex 6 displayed the greatest photocytotoxic potential and was found to bind to HSA with the highest affinity.
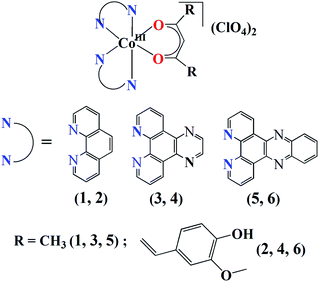 |
| | Fig. 1 The schematic representation of the complexes 1–6. | |
Results and discussion
Synthesis and general aspects
The complexes [Co(N–N)2(O–O)](ClO4)2 (1–6) were synthesized in ∼70% yield by reacting [Co(N–N)2Cl2]Cl with either Hacac or Hcur followed by treatment with NaClO4 (Fig. 1, S1 and S2, ESI†). All the complexes gave satisfactory elemental data. The solid state FT-IR spectra of the complexes displayed IR bands at ∼1080 cm−1 due to the perchlorate (ClO4−) anions and at ∼1510 cm−1 due to the olefinic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the β diketone moiety (Table 1).64 In addition, the complexes 2, 4 and 6 having the cur ligand showed an additional band at ∼3470 cm−1 due to the phenolic hydroxyl groups of cur ligand (Fig. S3–S8, ESI†). The molar conductivity (ΛM) measurements suggested that all the complexes were 1
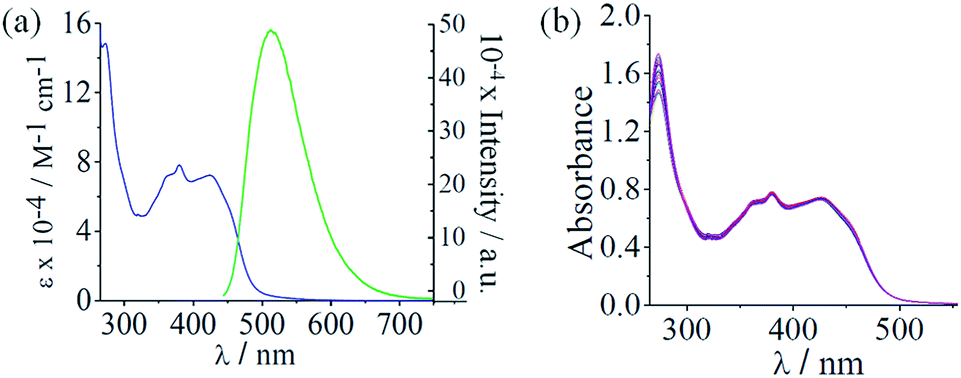
C of the β diketone moiety (Table 1).64 In addition, the complexes 2, 4 and 6 having the cur ligand showed an additional band at ∼3470 cm−1 due to the phenolic hydroxyl groups of cur ligand (Fig. S3–S8, ESI†). The molar conductivity (ΛM) measurements suggested that all the complexes were 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 electrolytes in dimethylformamide (DMF) corresponding the presence of two perchlorate (ClO4−) anions outside the coordination sphere (Table 1).65 The ESI-mass spectra of the complexes recorded in aqueous acetonitrile displayed essentially a single peak corresponding to [M-2ClO4−]2+ species which was supported by the isotopic distribution patterns of the peaks (Fig. S9–S18, ESI†). Complexes 2, 4, and 6 showed a curcumin-based π → π* visible absorption band within 400–500 nm in 5% aqueous DMF. All the complexes displayed a common absorption band around 270 nm assignable to π–π* transition. The dpq complexes 3 and 4 exhibited a band around 340 nm that could be due to n → π* transition involving the quinoxaline moiety.43,44 The dppz complexes 5 and 6 showed two additional bands at 361 nm and 380 nm assignable to the n → π* transitions of the phenazine moiety.39c,44 (Fig. 2(a), S19 and S20, ESI†). All the complexes showed curcumin-based green emission centered at 525 nm in DMF with quantum yield (ϕ) value of ∼0.01 when excited at 420 nm (Fig. 2(a)). 1H-NMR spectra of the complexes measured in DMSO-d6 showed peaks characteristic of the Co(III) bound N–N (phen, dpq or dppz) and β diketonate (acac or cur) ligands.30a,45b The two N–N ligands bound to the complex appeared different in the 1H-NMR spectrum.45b The dppz complexes displayed poor cyclic voltammetric responses in DMF when compared with the dpq and phen analogues (Fig. S21–S26 and Table S1, ESI†). For complex 6, two poor cathodic responses near −0.65 V (Epc1) and 1.10 V (Epc2) and two anodic responses near −0.93 V (Epa1) and 0.00 V (Epa2) were observed. The phen complex 2 and the dpq complex 4, both having the curcumin ligand, showed voltammetric behavior similar to complex 6 with two cathodic and two anodic responses. The complex 5, having the acac ligand, displayed three cathodic responses near −1.70 V (Epc1), −1.06 V (Epc2) and 0.51 V (Epc3) and three anodic responses near −1.18 V (Epa1), −0.90 V (Epa2) and 0.24 V (Epa3). The dpq complex 3 showed voltammetric behavior similar to complex 5 but the complex 1, having phen and acac ligands, showed three cathodic peaks and a number of anodic responses. The curcumin ligand alone displayed a cathodic peak near 0.10 V and an anodic response near −1.11 V in DMF–0.1 M tetrabutylammoniumperchlorate (Fig. S27 and Table S1, ESI†). The dppz ligand alone is known to exhibit a quasi-reversible voltammogram at −1.15 V with ipa/ipc ratio of 0.9.39c The redox data suggest that both Co(III) and the ligands in the ternary structure are involved in the overall redox behavior of the complexes. The poor voltammetric response of complex 6 having dppz and curcumin as ligands indicates the stability of the complex in +3 oxidation state in the absence of reducing agents such as ascorbic acid or glutathione.32
:2 electrolytes in dimethylformamide (DMF) corresponding the presence of two perchlorate (ClO4−) anions outside the coordination sphere (Table 1).65 The ESI-mass spectra of the complexes recorded in aqueous acetonitrile displayed essentially a single peak corresponding to [M-2ClO4−]2+ species which was supported by the isotopic distribution patterns of the peaks (Fig. S9–S18, ESI†). Complexes 2, 4, and 6 showed a curcumin-based π → π* visible absorption band within 400–500 nm in 5% aqueous DMF. All the complexes displayed a common absorption band around 270 nm assignable to π–π* transition. The dpq complexes 3 and 4 exhibited a band around 340 nm that could be due to n → π* transition involving the quinoxaline moiety.43,44 The dppz complexes 5 and 6 showed two additional bands at 361 nm and 380 nm assignable to the n → π* transitions of the phenazine moiety.39c,44 (Fig. 2(a), S19 and S20, ESI†). All the complexes showed curcumin-based green emission centered at 525 nm in DMF with quantum yield (ϕ) value of ∼0.01 when excited at 420 nm (Fig. 2(a)). 1H-NMR spectra of the complexes measured in DMSO-d6 showed peaks characteristic of the Co(III) bound N–N (phen, dpq or dppz) and β diketonate (acac or cur) ligands.30a,45b The two N–N ligands bound to the complex appeared different in the 1H-NMR spectrum.45b The dppz complexes displayed poor cyclic voltammetric responses in DMF when compared with the dpq and phen analogues (Fig. S21–S26 and Table S1, ESI†). For complex 6, two poor cathodic responses near −0.65 V (Epc1) and 1.10 V (Epc2) and two anodic responses near −0.93 V (Epa1) and 0.00 V (Epa2) were observed. The phen complex 2 and the dpq complex 4, both having the curcumin ligand, showed voltammetric behavior similar to complex 6 with two cathodic and two anodic responses. The complex 5, having the acac ligand, displayed three cathodic responses near −1.70 V (Epc1), −1.06 V (Epc2) and 0.51 V (Epc3) and three anodic responses near −1.18 V (Epa1), −0.90 V (Epa2) and 0.24 V (Epa3). The dpq complex 3 showed voltammetric behavior similar to complex 5 but the complex 1, having phen and acac ligands, showed three cathodic peaks and a number of anodic responses. The curcumin ligand alone displayed a cathodic peak near 0.10 V and an anodic response near −1.11 V in DMF–0.1 M tetrabutylammoniumperchlorate (Fig. S27 and Table S1, ESI†). The dppz ligand alone is known to exhibit a quasi-reversible voltammogram at −1.15 V with ipa/ipc ratio of 0.9.39c The redox data suggest that both Co(III) and the ligands in the ternary structure are involved in the overall redox behavior of the complexes. The poor voltammetric response of complex 6 having dppz and curcumin as ligands indicates the stability of the complex in +3 oxidation state in the absence of reducing agents such as ascorbic acid or glutathione.32
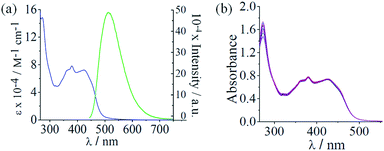 |
| | Fig. 2 (a) The electronic absorption (blue) spectrum of 6 in 5% aqueous DMF and the emission (green) spectrum of complex 6 in DMF (excitation wavelength, 420 nm). (b) Absorption spectral traces of complex 6 in DMSO-10% DMEM medium (1:99 v/v, pH = 7.4, 37 °C). The spectra were recorded after keeping the solution at 37 °C and recorded at different time intervals. The spectral data indicate the stability of the complex in the cellular medium up to 48 hours. | |
Table 1 Selected physicochemical data for complexes 1–6
| Complex |
ESI-MSa (m/z) |
IRb/cm−1 |
ΛMc/S cm2 mol−1 |
Φd/M−1 |
Kbe/M−1 |
| ClO4− |
Olefinic CC |
| The molecular ion peak corresponding to [M-(2ClO4−)]2+ in aqueous acetonitrile. Recorded in solid phase. Molar conductivity values in DMF at 25 °C. Fluorescence quantum yield in DMF solvent, excitation wavelength = 420 nm. The HSA binding constant. |
| 1 |
259.06 |
1080 |
1515 |
155 |
— |
0.5 × 105 |
| 2 |
393.10 |
1085 |
1512 |
135 |
0.007 |
2.5 × 105 |
| 3 |
311.06 |
1080 |
1512 |
150 |
— |
1.1 × 105 |
| 4 |
445.10 |
1080 |
1505 |
138 |
0.01 |
3.6 × 105 |
| 5 |
361.08 |
1085 |
1512 |
142 |
— |
2.9 × 105 |
| 6 |
495.12 |
1082 |
1508 |
132 |
0.01 |
9.8 × 105 |
X-ray crystallography
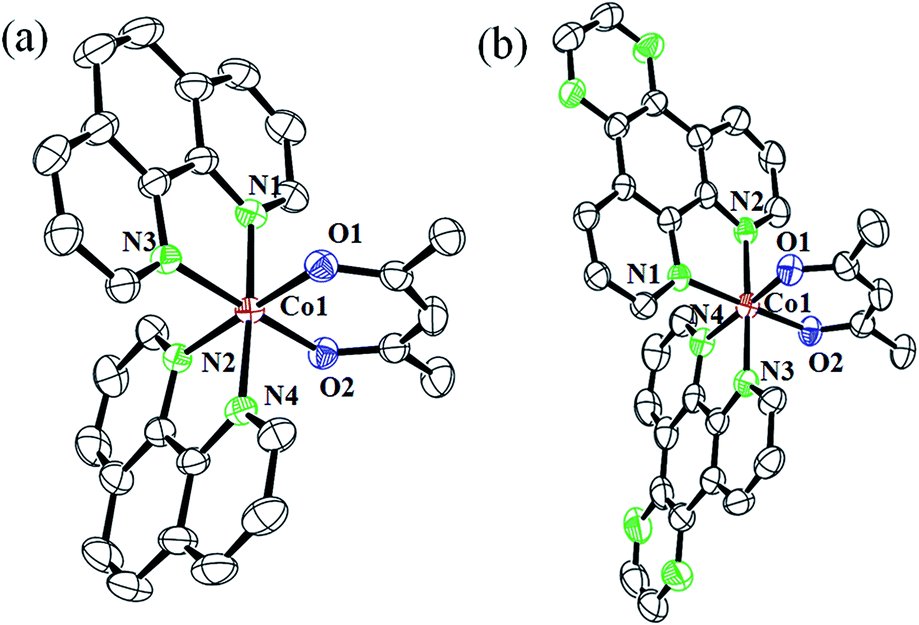
The complexes 1a·CH3COCH3 and 3·H2O were structurally characterized by single crystal X-ray diffraction method (Fig. 3). While 1a·CH3COCH3 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules in the asymmetric unit, 3·H2O crystallized in the monoclinic space group P21/n with four molecules in the asymmetric unit. The complexes are discrete mononuclear species showing a nearly octahedral CoN4O2 core formed by the two bidentate N-donor phen or dpq and one bidentate O-donor acac ligands. The ORTEP views of the complexes are shown in Fig. 2 and the unit cell packing diagrams are provided as ESI (Fig. S28 and S29, ESI†). The Co–N distances are in the range of 1.933(4) Å to 1.958 (3) Å while the Co–O distances are in the range of 1.881(2) Å to 1.893(3) Å. Selected crystallography parameters are given in Table 2 and the bond lengths and bond angles have been enlisted in Table 3.
with two molecules in the asymmetric unit, 3·H2O crystallized in the monoclinic space group P21/n with four molecules in the asymmetric unit. The complexes are discrete mononuclear species showing a nearly octahedral CoN4O2 core formed by the two bidentate N-donor phen or dpq and one bidentate O-donor acac ligands. The ORTEP views of the complexes are shown in Fig. 2 and the unit cell packing diagrams are provided as ESI (Fig. S28 and S29, ESI†). The Co–N distances are in the range of 1.933(4) Å to 1.958 (3) Å while the Co–O distances are in the range of 1.881(2) Å to 1.893(3) Å. Selected crystallography parameters are given in Table 2 and the bond lengths and bond angles have been enlisted in Table 3.
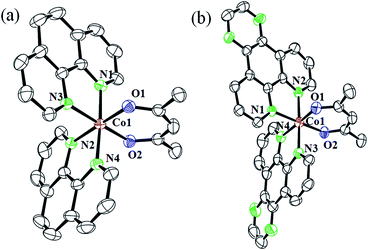 |
| | Fig. 3 ORTEP diagrams of the dications of (a) complex 1a·CH3COCH3, and (b) complex 3·H2O showing the thermal ellipsoids drawn at 50% probability. The hydrogen atoms and the counter anions have been omitted for clarity. The color codes are: metal (red), nitrogen (green), oxygen (blue) and carbon (black). | |
Table 2 Selected crystallographic data and structure refinement parameters for the complexes 1a·acetone and 3·H2O
| Empirical formula |
C32H29N4O3F12P2Co (1a·acetone) |
C33H23N8O10Cl2Co (3·H2O) |
| R = Σ‖Fo| − |Fc‖/Σ|Fo|. wR = {Σ[w(Fo2 − Fo2)2]/Σ[w(Fo)2]}1/2; w = [σ2(Fo)2 + (AP)2 + BP]−1, where P = (Fo2 + 2Fc2)/3, A = 0.1440, B = 1.8460 for complex 1a·acetone; A = 0.1535, B = 0.0000 for complex 3·H2O. |
| Formula weight (g mol−1) |
857.34 |
821.42 |
| Crystal system |
Triclinic |
Monoclinic |
| Space group |
P |
P21/n |
| a (Å) |
10.138(5) |
12.736(5) |
| b (Å) |
10.138(5) |
15.757(5) |
| c (Å) |
15.0670(9) |
17.209(5) |
| α (°) |
95.571(5) |
90.000 |
| β (°) |
104.715(5) |
95.701(5) |
| γ (°) |
105.326(5) |
90.000 |
| V (Å3) |
1502.7(11) |
3436(2) |
| Z |
2 |
4 |
| T (K) |
293(2) |
293(2) |
| ρcalc (g cm−3) |
1.592 |
1.588 |
| λ/Å (Mo-Kα) |
0.71073 |
0.71073 |
| μ (mm−1) |
0.676 |
0.727 |
| Data/restraints/parameters |
5182/0/424 |
8288/0/496 |
| F(000) |
728 |
1672 |
| Goodness-of-fit on F2 |
1.061 |
1.064 |
| R(Fo)a, I > 2σ(I)/wR(Fo)b |
0.0623/0.1967 |
0.0746/0.2238 |
| R (all data)/wR (all data) |
0.0717/0.2151 |
0.1336/0.2537 |
| Largest diff. peak and hole (e Å−3) |
0.944, −0.870 |
0.958, −0.612 |
Table 3 Selected bond distances (Å) and angles (°) with estimated standard deviations (esd) in the parentheses
| [Co(phen)2(acac)](PF6)2·CH3COCH3 (1a·CH3COCH3) |
| Co(1)–O(1) |
1.881(2) |
N(2)–Co(1)–N(3) |
91.88(12) |
| Co(1)–O(2) |
1.881(2) |
N(2)–Co(1)–N(4) |
93.96(12) |
| Co(1)–N(1) |
1.941(3) |
N(3)–Co(1)–N(4) |
84.46(12) |
| Co(1)–N(2) |
1.958(3) |
O(1)–Co(1)–N(2) |
176.24(11) |
| Co(1)–N(3) |
1.953(3) |
O(1)–Co(1)–N(3) |
86.80(11) |
| Co(1)–N(4) |
1.943(3) |
O(1)–Co(1)–N(4) |
89.42(11) |
| N(1)–Co(1)–N(2) |
84.09(12) |
O(1)–Co(1)–O(2) |
95.65(10) |
| N(1)–Co(1)–N(3) |
92.73(11) |
O(2)–Co(1)–N(2) |
85.89(11) |
| N(1)–Co(1)–N(4) |
176.54(12) |
O(2)–Co(1)–N(3) |
175.74(10) |
| N(1)–Co(1)–O(1) |
92.45(11) |
O(2)–Co(1)–N(4) |
92.06(11) |
| N(1)–Co(1)–O(2) |
90.65(11) |
|
|
| [Co(dpq)2(acac)](ClO4)2 (3·H2O) |
| Co(1)–O(1) |
1.893(3) |
N(2)–Co(1)–N(3) |
177.68(14) |
| Co(1)–O(2) |
1.887(3) |
N(2)–Co(1)–N(4) |
94.43(14) |
| Co(1)–N(1) |
1.944(3) |
N(3)–Co(1)–N(4) |
83.79(14) |
| Co(1)–N(2) |
1.933(4) |
O(1)–Co(1)–N(2) |
87.77(13) |
| Co(1)–N(3) |
1.938(4) |
O(1)–Co(1)–N(3) |
93.95(13) |
| Co(1)–N(4) |
1.945(3) |
O(1)–Co(1)–N(4) |
177.04(13) |
| N(1)–Co(1)–N(2) |
83.99(15) |
O(1)–Co(1)–O(2) |
96.53(13) |
| N(1)–Co(1)–N(3) |
94.54(15) |
O(2)–Co(1)–N(2) |
93.98(14) |
| N(1)–Co(1)–N(4) |
91.75(14) |
O(2)–Co(1)–N(3) |
87.38(14) |
| N(1)–Co(1)–O(1) |
86.50(14) |
O(2)–Co(1)–N(4) |
85.31(13) |
| N(1)–Co(1)–O(2) |
176.30(14) |
|
|
Theoretical studies
Density Functional Theory (DFT) calculations were carried out by using b3LYP level theory with LAN2DZ basis set with Gaussian 03 programs to obtain the optimized geometries and insights into the excited state properties of the cobalt(III)–curcumin complexes, namely, complexes 2, 4, and 6. The initial coordinates were obtained from the crystal structure of 3·H2O and were used for optimization. The frontier orbital calculations reveal that the highest occupied molecular orbitals (HOMOs) of all the three complexes are invariably localized on the curcumin ligand and the lowest unoccupied molecular orbitals (LUMOs) of the complexes are spread over the Co(III) centre and the N–N ligands (Fig. 4 and Tables S2–S4, ESI†). This observation suggests that the photo-excitation of the complexes could result in facile electronic transitions from the HOMO centered on the curcumin moiety to the LUMO spread over the Co(III) and N–N ligands. This observation could also explain the remarkable photocytotoxicity of the curcumin complexes.
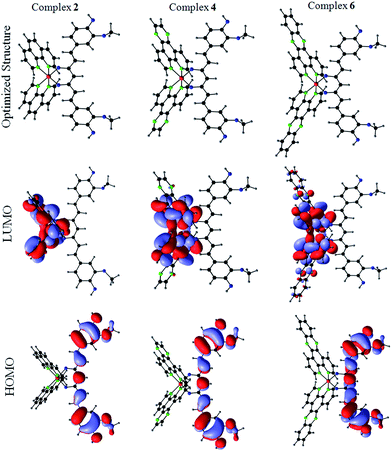 |
| | Fig. 4 The DFT optimized structures (top row), the HOMOs (bottom row) and the LUMOs (middle row) of the complexes 2, 4, and 6. The structures and the frontiers orbitals were obtained by using B3LYP level theory and LanLD2Z basis set. | |
Solubility and stability
The complexes were soluble in acetone, acetonitrile, DMF and DMSO. They were partially soluble in water, methanol, ethanol, tetrahydrofuran, dichloromethane and chloroform but insoluble in hydrocarbon solvents such as hexane, xylene and toluene. To examine the biological activity of the complexes, it is a pre-requisite that they are stable in physiological conditions. The stability of the complexes was thus monitored by UV-visible spectroscopy. The complexes were stable in 10% DMEM (Dulbecco's Modified Eagle's medium) as evidenced from the absorption spectral study on complex 6 showing no apparent spectral change even after 48 hours (Fig. 2(b)). It is to be noted that the free curcumin ligand is susceptible to hydrolytic degradation in aqueous buffer.19 Thus, the observation of no significant spectral changes over 48 hours indicates that binding of curcumin to Co(III) ion arrests its hydrolytic degradation. This finding has been corroborated by earlier studies on metal complexes of curcumin.19,26–28 The emission spectrum of complex 6 monitored over a period of 4 hours both in the absence and presence of added ascorbic acid showed no significant change in intensity. However, in the absence of oxygen, the emission intensity of complex 6 increased gradually upon adding ascorbic acid (Fig. S30 and S31, ESI†). This observation is further corroborated by a recent study on a Co(III)–curcumin complex that releases the coordinated curcumin ligand upon reduction to Co(II) in the absence of oxygen (Fig. S32, ESI†).32
Light-triggered anticancer activity
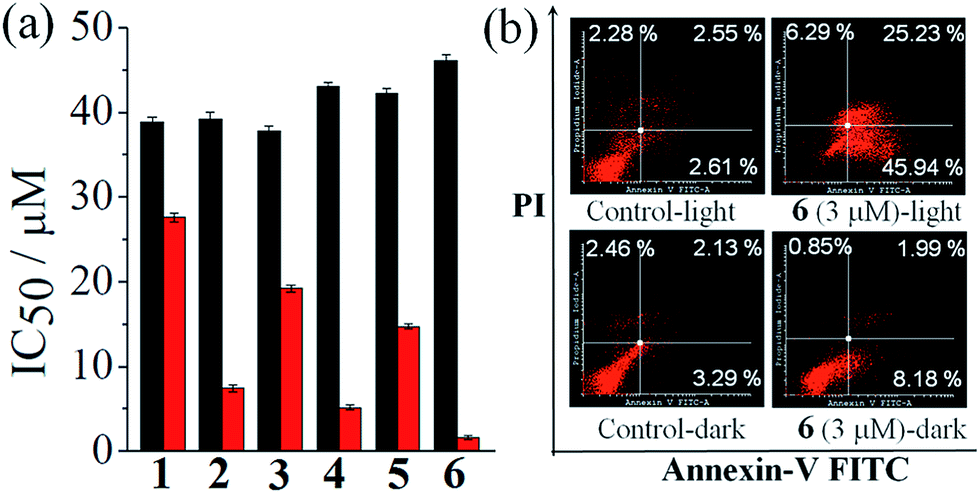
One way to assess the in vitro anticancer activity of a compound is to evaluate its cytotoxicity in terms of its IC50 value in cancer cells by using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Therefore, the anticancer activity of the complexes 1–6 was assessed by MTT assay in human cervical carcinoma (HeLa) cell line in the presence of visible light (Fig. 5(a), Table 4 and Fig. S33–S38, ESI†). The complex 6, having cur and dppz as the photoactive ligand moieties, gave an IC50 value of 1.6 ± 0.2 μM in visible light (400–700 nm, 10 J cm−2) while being non-toxic in dark with an of 46.1 ± 0.7 μM. The other two curcumin complexes, 2 and 4, gave IC50 values of 7.4 ± 0.4 μM and 5.1 ± 0.3 μM, respectively, in light and 39.2 ± 0.8 μM and 43.1 ± 0.4 μM, respectively, in dark. The control complexes, namely, complexes 1, 3, and 5 gave IC50 values of 27.6 ± 0.5 μM, 19.2 ± 0.4 μM and 14.7 ± 0.3 μM, respectively, in light and 37.8 ± 0.6 μM, 37.8 ± 0.6 μM and 42.3 ± 0.5 μM, respectively, in dark. Thus, the control complexes, having simple acac ligand but lacking the photoactive curcumin moiety, are much less photocytotoxic compared to the curcumin complexes. The curcumin ligand alone is known to give an IC50 value of 8.2 ± 0.2 μM in visible light and 85.4 ± 0.6 μM in dark in HeLa cells under similar experimental conditions (Table 4).29a However, the photocytotoxicity observed for complexes 3 and 5 (IC50 = 19.2 ± 0.4 μM and14.7 ± 0.3 μM, respectively) could be due to the presence of photoactive dpq and dppz ligands with their absorption spectral tails entering into the visible region.39c,44 The remarkable photocytotoxicity but insignificant dark toxicity of the curcumin complexes, particularly complex 6, is unprecedented in the chemistry of cobalt based PDT agents. The photocytotoxicity of complex 6 is comparable to the clinically used PDT drug Photofrin® (IC50 = 4.3 ± 0.2 μM in red light of 633 nm in HeLa cells and >41 μM in dark).21a Although, oxovanadium(IV) and Cu(II) complexes of curcumin are known to show photocytotoxicity in visible light, the corresponding Co(III) complexes are virtually unknown in the literature.29,30 To study the selectivity of these curcumin complexes in cancer cells over the normal cells, the MTT assay was carried in breast epithelial MCF-10A cell lines treated with the most photoactive complex 6. Interestingly, complex 6 was found to be significantly less photocytotoxic to MCF-10A cells yielding an IC50 value of 12.8 μM in light and >50 μM in dark. This result shows that there is a distinct selectivity of complex 6 towards cancer cells over the normal cells. This selectivity could be due to preferential internalization of the complexes in cancer cells compared to normal ones since the cancer cells are metabolically more active than the normal cells.
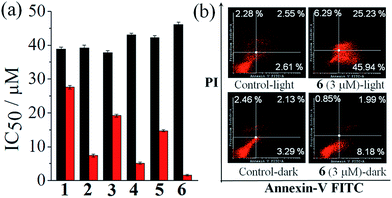 |
| | Fig. 5 (a) Bar diagrams showing the cytotoxicity of the complexes 1–6 in HeLa cells upon photo-irradiation with visible light (400–700 nm, 10 J cm−2, red bars) and in dark (black bars) as determined from MTT assay. (b) Annexin V-FITC/PI coupled flow cytometric analysis showing complex 6 (3 μM) induced apoptosis in the presence of visible light (400–700 nm). | |
Table 4 IC50 values of complexes 1–6 in HeLa cells
| Compound |
Light/μM |
Dark/μM |
| Light of 400–700 nm (10 J cm−2) wavelength. IC50 values of 6 in MCF-10A cell line. Ref. 19. Light source reported: red light of 633 nm. Ref. 21a. |
| 1 |
27.6 ± 0.5 |
37.8 ± 0.6 |
| 2 |
7.4 ± 0.4 |
39.2 ± 0.8 |
| 3 |
19.2 ± 0.4a |
37.8 ± 0.6 |
| 4 |
5.1 ± 0.3 |
43.1 ± 0.4 |
| 5 |
14.7 ± 0.3a |
42.3 ± 0.5 |
| 6 |
1.6 ± 0.2a (12.8 ± 0.3)a,b |
46.1 ± 0.7 (>50)b |
| Curcumin (Hcur)c |
8.2 ± 0.2a |
85.4 ± 0.6 |
| dppzc |
60.1 ± 2.3a |
>100 |
| Photofrind |
4.3 ± 0.2e |
>41e |
Annexin-V-FITC/PI assay
Annexins are a group of Ca(II) and phospholipid binding proteins. Annexin-V-FITC is a fluorescent probe that binds to phosphatidylserine in the presence of calcium(II) ion. In the early apoptotic stage, the cells undergo significant morphological changes including the loss of phospholipid asymmetry. The phosphatidylserine in healthy cells remains in the interior part of the lipid bilayer which is transferred to the external portion of the cell membrane at the onset of apoptosis. This change in cell membrane can be detected by a fluorophore conjugated Annexin-V protein which is known to bind phosphatidylserine. This assay utilizes dual staining of cells by Annexin V-FITC (fluorescein isothiocyanate) and DNA binding dye propidium iodide (PI). Cells undergoing early apoptosis are identified as single positive population for FITC, while necrotic cell population is stained only by PI. The double positive population comprises cells in late apoptosis having compromised cell membrane. To study whether the present complexes are capable of inducing any apoptotic cell death triggered by visible light, we carried out Annexin-V-FITC/PI assay using HeLa cancer cells treated with complex 6 (Fig. 5(b)). The results were analyzed by fluorescence assisted cell sorting (FACS) analysis. PI emits in the red region, while the Annexin V-FITC dye shows green fluorescence. This assay gave an estimate of the apoptotic cells in different stages, viz., early apoptosis: high Annexin V-FITC (Quadrant 4(Q4)), low PI; late apoptosis: high Annexin V-FITC and PI (Q2); viable cells (unstained, only showing auto-fluorescence (Q3)), and necrotic cell populations (stained with PI only (Q1)). The cells were treated with complex 6 (3 μM) followed by 1 h irradiation with visible light (400–700 nm) and the assay was performed 18 h post photo-irradiation. The results show features of early apoptosis in ∼46% of HeLa cells upon light irradiation while no such apoptotic feature was seen in the absence of light (Fig. 5(b)).
Cell cycle analysis
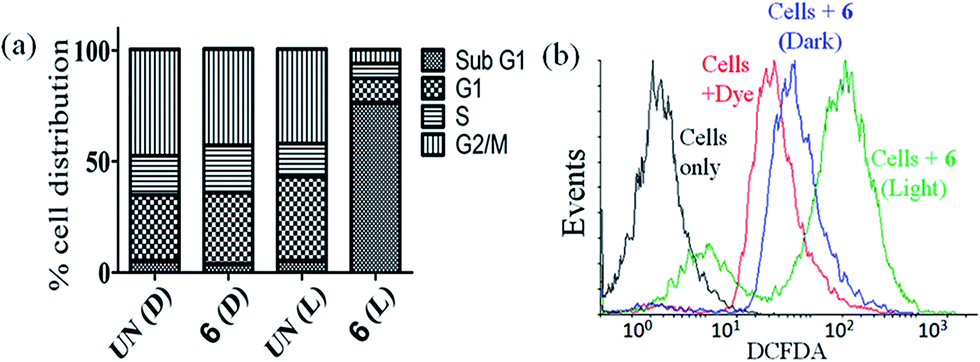
The DNA content of cells was measured with the help of a fluorescent dye, viz., propidium iodide (PI), which provided information about the cell cycle and, consequently, the effect of photo-irradiated metal complexes on the cell cycle. Complex 6 was used for flow cytometric analysis under light and dark conditions at a concentration of 3 μM. The complex did not induce any significant change in the sub-G1 phase (indicative of cell death) in the absence of light, but it did elicit significant cell death (∼74%) on photo-irradiation when compared with the DMSO control (∼3%) indicating the PDT effect of the complex (Fig. 6(a) and S39, ESI†). The sub G1 population of the cells corresponds to those whose membrane has been compromised and have undergone cell death.
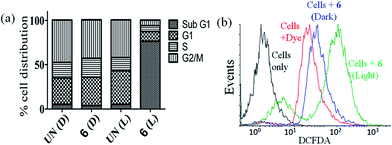 |
| | Fig. 6 (a) Bar diagrammatic representation of flow cytometric analysis of HeLa cells treated with complex 6 showing the percentage cell distribution in different phases of cell cycle. UN(D): untreated (in dark); 6 (D): 6 (in dark); UN(L): untreated (in light); 6 (L): 6 (in light). Complex 6 in the presence of visible light induced apoptosis as seen from the large distribution of the cells in the sub G1 phase. Light source: visible light (400–700 nm, 10 J cm−2). (b) DCFDA/DCF assay performed for the detection of ROS generation by complex 6 in HeLa cells (light source: visible light of 400–700 nm). | |
DCFDA assay
Reactive oxygen species (ROS) plays an important role in the induction of apoptosis by transition metal complexes.66,67 2′,7′-Dichlorofluoresceindiacetate (DCFDA) assay was carried out to study generation of any intracellular ROS by FACS analysis (Fig. 6(b)). The cell-permeable DCFDA upon passive diffusion into the cells is retained in the intracellular level on getting cleaved by the intracellular esterases. Upon oxidation by ROS, the non-fluorescent DCFDA gets converted to the highly fluorescent 2′,7′-dichlorofluorescein (DCF) with an emission band centered at 529 nm. Measurements were done for the fluorescence of the cells (HeLa) alone, cells treated with only DCFDA and cells with DCFDA and complex 6 after photo-irradiation with visible light of 400–700 nm for 1 h or in dark (as controls). Substantial shift in the fluorescence bands was observed for the cells treated with photoexposed complex as compared to the cells treated with the dye alone or the dark controls indicating generation of ROS only on exposure to light. Thus, the ROS generated by the complex could lead to induction of cellular apoptosis via ROS mediated cell stress.
Cellular uptake and localization studies
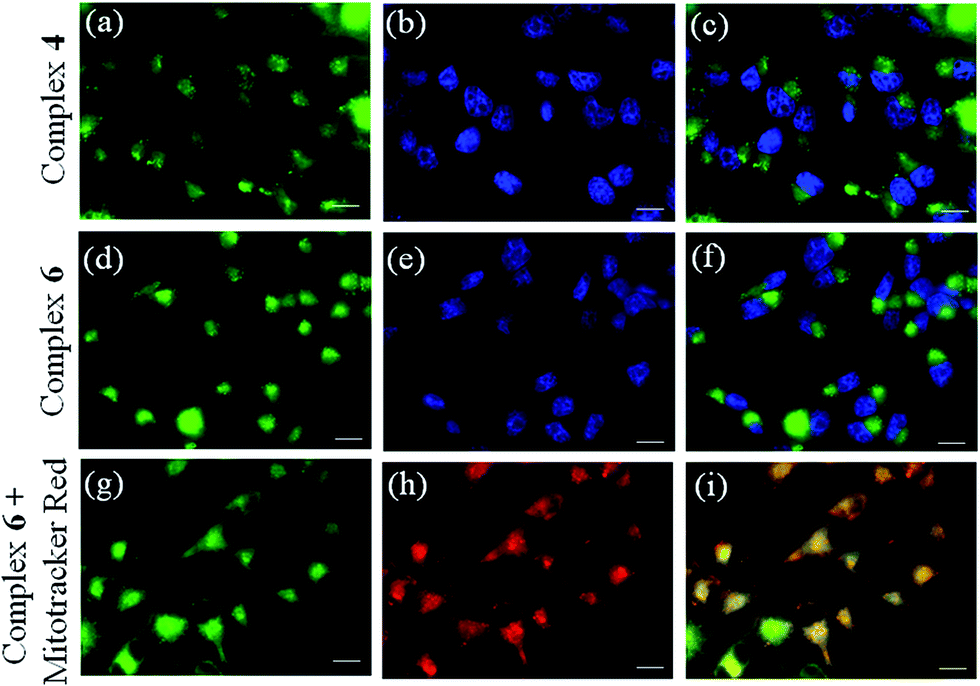
Curcumin-based green emission of complexes 4 and 6 was exploited to study the cellular localization and uptake using fluorescence microscopy (Fig. 7 and S40, ESI†). A significant cellular uptake of complex 6 was observed within 2 h which increased during 4 h of incubation suggesting its accretion inside the cells. A similar uptake was observed for the dpq complex 4 within 4 h of incubation (Fig. 7). Dual staining with the nucleus staining dye Hoechst 33342 (blue emission) showed primarily cytosolic localization of complex 6 within the cells as seen from the merged images (panels (c) and (f) of Fig. 7).
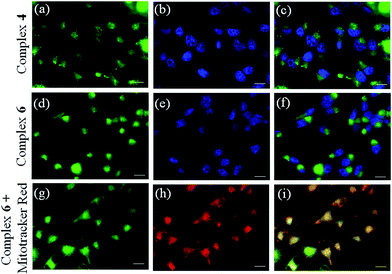 |
| | Fig. 7 Fluorescence microscopic images of the HeLa cancer cells treated with 10 μM of complex 4 or 6 on 4 h incubation and Hoechst 33342 dye (5 μg mL−1): panel (a): emission from complex 4; panel (b): blue emission from the nucleus staining Hoechst dye; panel (c): merged images showing cytosolic localization of complex 4; panel (d): fluorescence emission from complex 6; panel (e): blue emission from the nucleus staining Hoechst dye; panel (f): merged images showing cytosolic localization of complex 6; panel (g) fluorescence emission from complex 6 (10 μM); panel (h): red emission from Mito-tracker red (0.5 μM) showing its mitochondrial localization; panel (i): merged images showing mitochondrial localization of complex 6. The scale bar corresponds to 20 μm. | |
Mitochondrial selectivity
With the knowledge of the complex 6 accumulating in the cytoplasm, we delved further to probe its selectivity of localization to specific cellular organelle. For this specific purpose, we probed the specificity of localization of complex 6 in the mitochondria (panels (g), (h), (i), Fig. 7). Dual staining of complex 6 with the mitochondria staining dye Mitotracker red showed that complex 6 specifically localized in the mitochondria as evidenced from the merged image (panel (h), Fig. 7). The mitochondrial localization of complex 6 is indeed a striking result considering the key role played by mitochondria in mediating the intrinsic pathway of apoptosis.12,13,15–20 There is currently a surge of interest in designing and developing anticancer drugs that target cell organelles other than the nucleus for their chemotherapeutic action.15–21 This is the first demonstration of a mitochondria-targeted Co(III) complex having the photoactive curcumin and dipyridophenazine ligand that shows remarkable photocytotoxicity in visible light. The curcumin ligand is exploited to achieve enhanced photocytotoxicity and for imaging the cells by fluorescence microscopy.
HSA binding studies
Since HSA interacts with most of the drugs that are administered intravenously, we were interested to study the binding propensity of the complexes (1–6) towards HSA by using fluorescence spectroscopy.33–37 The lipophilic nature curcumin and the phenanthroline base could help the complex bind to HSA for transport in the blood.26,30,31 Specifically, the gradual decrease in the fluorescence intensity of HSA due to quenching of emission of the Trp214 residue upon addition of complex was monitored (Fig. 8, S41 and S42, ESI†). Analyses of Stern–Volmer plots (Fig. 8(a)) and Scatchard plots (Fig. 8(b)) showed that complex 6 displayed the highest binding affinity, with a binding constant (Kb) of 9.8 × 105 M−1. The control complexes 1, 3 and 5, lacking the lipophilic curcumin moiety, showed weaker binding affinity with respective Kb values of 0.5 × 105 M−1, 1.1 × 105 M−1 and 2.9 × 105 M−1. The regular increase in the Kb values could be attributed to the gradual increase in planarity and lipophilicity of the phenanthroline bases from phen to dppz, yielding trends 5 > 3 > 1 and 6 > 4 > 2. Further, analysis of a Job plot for 6 revealed a 1:1 binding ratio between the complex and HSA (Fig. S43, ESI†). Thus, the Co(III)–curcumin complexes, particularly complex 6, could be transported by blood for its delivery.
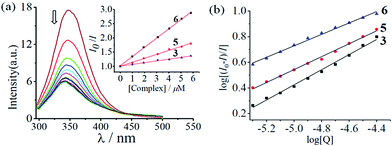 |
| | Fig. 8 Emission spectral traces of human serum albumin (2 μM) in the presence of complex 6. The inset shows the plots of (I0/I) vs. [complex] for complexes 3 ( ), 5 ( ), 5 ( ) and 6 ( ) and 6 ( ). (b) Scatchard plots of log[(I0 − I)/I] vs. log[Q] for 3 ( ). (b) Scatchard plots of log[(I0 − I)/I] vs. log[Q] for 3 ( ), 5 ( ), 5 ( ) and 6 ( ) and 6 ( ) to determine the binding constant (Kb) for HSA–complex interaction. ) to determine the binding constant (Kb) for HSA–complex interaction. | |
Conclusions
In conclusion, we presented the first development of a series of mixed-ligand cobalt(III) complexes of curcumin having phenanthroline bases (phen, dpq, and dppz) that were photocytotoxic in visible light while being non-toxic in the dark. The curcumin complexes 2, 4, and 6 were photocytotoxic to HeLa cells but non-toxic in the absence of light. In particular, the dipyridophenazine complex 6 showed the highest photocytotoxic activity but it was significantly less photocytotoxic to MCF-10A normal cells. The photocytotoxicity of the complexes increased with the increase in planarity of the phenanthroline bases (dppz > dpq > phen). The photocytotoxicity of complex 6 compares well to that of the FDA approved PDT drug Photofrin®. The complex 6 targets the mitochondria of HeLa cells and induces apoptosis which is triggered by the photo-induced formation of ROS. The complexes have the ability to bind HSA with good affinity but complex 6 binds with the strongest affinity and hence it could be transported by HSA in the blood. Thus, the curcumin complexes, particularly complex 6, hold significant chemotherapeutic potential and open up new possibilities to further develop cobalt–curcumin complexes for photochemotherapeutic applications. Further experimental and theoretical efforts are underway for a series of structurally similar cobalt(III) complexes of curcumin with an objective to establish their structure–activity relationship.
Experimental
Materials
Unless otherwise specified, all the reagents and chemicals used in this work were procured from commercial sources (Sigma-Aldrich, U.S.A.; E-Merck; Alfa Aesar, UK; and Rankem, India) and used without further purification. Solvents were purified and distilled prior to use by standard methods.41 L-Ascorbic acid was obtained from E-Merck. Human Serum Albumin (HSA), lyophilized powder (essentially fatty acid free) was procured from Sigma, U.S.A. (A1887) and used as received. Curcumin (95% curcuminoid content, ca. 80% curcumin) was purchased from Sigma-Aldrich, U.S.A. and purified into three individual components by following a literature method.42 Tris-(hydroxymethyl)aminomethane–HCl (Tris–HCl) buffer solution (pH = 7.2) was prepared using deionized and sonicated double distilled water. DMEM, propidium iodide, Hoechst 33342, MTT and 2′,7′-dichlorofluorescein diacetate (DCFDA) were procured from Sigma-Aldrich (U.S.A) and used as received. Mito-Tracker Red was procured from Invitrogen, U.S.A. The N,N-donor heterocyclic bases, namely, dipyrido-[3,2-d:2′,3′-f]-quinoxaline (dpq) and dipyrido[3,2-a:2′,3′-c]phenazine (dppz) were prepared by literature methods using 1,10-phenanthroline-5,6-dione as a precursor reacted with ethylenediamine (for dpq) and 1,2-phenylenediamine (for dppz).43,44 Anhydrous cobalt(II) chloride was prepared by heating the pink red hexahydrate CoCl2·6H2O until it turned sky blue. The complete absence of H2O in the anhydrous salt was confirmed by the disappearance of the broad –OH stretching band of H2O in the FT-IR spectrum. The precursor compounds [Co(N–N)2Cl2]Cl were prepared by a chlorine oxidation method by following reported procedures.45 Dry chlorine gas was prepared in situ by carefully reacting solid KMnO4 with hydrochloric acid followed by bubbling the chlorine gas so produced through concentrated sulfuric acid prior to its use in reaction.
Caution! Chlorine gas, being harmful and oxidizing, its preparation and handling was done strictly inside a well ventilated fume hood with all necessary precautions.
Measurements
Elemental analyses were done using a Thermo Finnigan Flash EA 1112 CHNS analyzer. The infrared (IR) spectra were recorded on a Bruker ATR FT-IR spectrometer. Electronic absorption spectra were recorded on a Perkin Elmer Lambda 650 spectrophotometer. Molar conductivity measurements were performed using a properly calibrated digital conductivity meter (Labtronics, India). 1H-NMR spectra of the cobalt(III) compounds were recorded at room temperature on a Bruker 400 MHz NMR spectrometer. Room temperature electrochemical measurements were carried out on a Biologic SP-50 Potentiostat/Galvanostatic (Biologic Instruments, France) with a three electrode setup consisting of a platinum working electrode, platinum wire auxiliary electrode, and calomel reference electrode (SCE) at a scan rate of 100 mV s−1. The electrochemical measurements were done using solutions of the metal complexes prepared in HPLC grade dimethylformamide (DMF). Tetrabutylammonium perchlorate (TBAP, 0.1 M) was used as a supporting electrolyte in DMF. Electrospray ionization mass spectra (ESI-MS) were recorded using Agilent Technologies 6538 UHD Accurate-mass Q-TOF LC/MS mass spectrometer. Fluorescence quantum yields were determined using a PerkinElmer LS 55 fluorescence spectrometer using coumarin-153 laser dye as a reference with a known quantum yield (Φ) value of 0.56 in acetonitrile.46 The sample for quantum yield determination was deoxygenated prior to spectral measurements. The sample and the reference were excited at 430 nm, maintaining nearly equal absorbance. The integrated emission intensity was calculated using Origin Pro 8.1 software and the quantum yield was calculated using the equation ΦS/ΦR = (AS/AR) × [(OD)R/(OD)S] × [(nS)2/(nR)2], where, ΦS and ΦR are the fluorescence quantum yields of the sample and reference respectively, AS and AR are the area under the fluorescence spectra of the sample and the reference respectively, (OD)S and (OD)R are the respective optical densities of the sample and the reference solution at the wavelength of excitation, and nS and nR are the respective refractive indices of the solvents used for the sample and the ref. 47 and 48 Fluorescence microscopic investigations were carried out using ApoTome.2 fluorescence microscope. Flow cytometric analysis was performed using FACS Calibur (Becton Dickinson (BD) cell analyzer) at FL1 channel (595 nm).
Syntheses of the complexes 1–6
The complexes were synthesized by a general synthetic route. To a methanol suspension (15 mL) of the precursor complex [Co(N–N)2Cl2]Cl (0.524 g phen; 0.628 g dpq; 0.728 g dppz; 1.0 mmol) was added a methanol solution (20 mL) of the corresponding β-diketone ligand (Hacac, 0.101 g; Hcur, 0.368 g, 1.0 mmol) followed by triethylamine (0.10 g, 1.0 mmol). The resulting suspension was stirred at room temperature for 3 hours. The reaction mixture was filtered to remove any insoluble impurities and a reddish brown solution was obtained. Addition of a methanol solution (5 mL) of NaClO4·H2O (0.280 g, 2.0 mmol) afforded a brick red precipitate which was filtered, washed with cold methanol followed by diethyl ether and finally dried in vacuum over phosphorous pentoxide (Fig. 1).
Caution! Metal perchlorates are oxidizing and treacherously explosive in nature. Special precautions were taken while handling them.
[Co(phen)2(acac)](ClO4)2 (1). Yield: ∼78%. Anal. calcd for C29H23N4O10Cl2Co: C, 48.55; H, 3.23; N, 7.81. Found: C, 48.75; H, 3.35; N, 7.73. FT-IR data/cm−1: 3071 w, 1565 m, 1515 s, 1430 m, 1080 m, 845 s, 714 s, 664 w, 621 s (vs, very strong; s, strong; m, medium; w, weak; sbr, strong broad). UV-visible in 5% aqueous DMF [λmax/nm (ε/M−1 cm−1)]: 277 (61200). 1H-NMR spectrum in DMSO-d6 (δ, ppm): 10.18 (d, 1H of phen, J = 5.9 Hz), 9.49 (d, 1H of phen, J = 5.5 Hz), 9.20 (dd, 2H of phen, J = 8.1, 7.2 Hz), 8.75 (d, 1H of phen, J = 8.4 Hz), 8.62 (d, 1H of phen, J = 8.3 Hz), 8.49 (dd, 2H of phen, J = 5.7, 8.1 Hz), 8.44 (d, 1H of phen, J = 8.1 Hz), 8.40 (d, 1H of phen, J = 7.3 Hz), 8.28 (d, 1H of phen, J = 8.1 Hz), 8.21 (d, 1H of phen, J = 8.0 Hz), 7.6 (dd, 1H of phen, J = 5.4, 8.1 Hz), 7.46 (m, 2H of phen), 7.24 (d, 1H of phen, J = 5.4 Hz), 6.09 (s, 1H of acac), 2.15 (s, 6H from 2CH3 of acac) (s, singlet; t, triplet; m, multiplet; J, coupling constant). Molar conductivity in DMF at 298 K [ΛM/S cm2 mol−1]: 155.
[Co(phen)2(cur)](ClO4)2 (2). Yield: ∼71%. Anal. calcd for C45H35N4O14Cl2Co: C, 54.84; H, 3.58; N, 5.68. Found: C, 54.66; H, 3.49; N, 5.60. ESI-MS in 5% aqueous acetonitrile: m/z 393.09 [M-(2ClO4−)]2+. FT-IR data/cm−1: 3370 sbr (phenolic OH of curcumin), 3080 w, 1583 m, 1512 vs, 1421 w, 1381 m, 1280 s, 1160 m, 1085 vs, 1030 m, 1000 m, 973 m, 840 s, 773 w, 716 w, 660 w, 621 m. UV-visible in 5% aqueous DMF [λmax/nm (ε/M−1 cm−1)]: 410 (16900), 268 (75400). 1H-NMR in DMSO-d6 (δ, ppm): 10.12 (d, 1H of phen, J = 5.5 Hz), 9.51 (d, 1H of phen, J = 5.8 Hz), 9.22 (dd, 2H of phen, J = 8.0, 7.4 Hz), 8.75 (d, 1H of phen, J = 8.2 Hz), 8.60 (d, 1H of phen, J = 8.6 Hz), 8.49 (dd, 2H of phen, J = 5.5, 8.4 Hz), 8.45 (d, 1H of phen, J = 8.3 Hz), 8.40 (d, 1H of phen, J = 7.1 Hz), 8.20 (d, 1H of phen, J = 8.1 Hz), 7.46–7.24 (m, 7H), 7.15 (d, 2H of cur, J = 8.3 Hz), 6.95 (d, 2H of cur CC, J = 5.4 Hz), 6.82 (d, 2H of cur J = 8.1 Hz), 6.15 (s, 1H of cur), 3.98 (s, 6H from 2CH3 of cur). Molar conductivity in DMF at 298 K [ΛM/S cm2 mol−1]: 135.
[Co(dpq)2(acac)](ClO4)2 (3). Yield: ∼74%. Anal. calcd for C33H23N8O10Cl2Co: C, 48.25; H, 2.82; N, 13.64. Found: C, 48.20; H, 2.85; N, 13.58. ESI-MS in 5% aqueous acetonitrile: m/z 311.06 [M-(2ClO4−)]2+. FT-IR data/cm−1: 3074 w, 1550 m, 1512 s, 1490 m, 1409 m, 1387 m, 1355 m, 1282 w, 1213 w, 1080 vs, 849 w, 811 m, 716 s, 614 s. UV-visible in 5% aqueous DMF [λmax/nm (ε/M−1 cm−1)]: 299 sh (68100), 346 sh (16500). 1H-NMR in DMSO-d6 (δ, ppm): 10.06 (d, 2H of dpq, J = 7.7 Hz), 9.75–9.64 (m, 2H of dpq), 9.51 (d, 2H, J = 7.2 Hz), 9.48 (d, 2H of dpq, J = 7.0 Hz), 9.03 (d, 2H of dpq, J = 7.9 Hz), 8.63 (d, 2H of dpq, J = 8.3 Hz), 8.25 (d, 2H of dpq, J = 7.7 Hz), 7.98 (d, 2H of dpq, J = 7.6 Hz), 6.08 (s, 1H of acac), 2.16 (s, 6H from 2CH3 of acac). Molar conductivity in DMF at 298 K [ΛM/S cm2 mol−1]: 150.
[Co(dpq)2(cur)](ClO4)2 (4). Yield: ∼72%. Anal. calcd for C49H35N8O14Cl2Co: C, 54.01; H, 3.24; N, 10.28. Found: C, 54.17; H, 3.32; N, 10.35. ESI-MS in 5% aqueous MeCN: m/z 445.10 [M-(2ClO4−)]2+. FT-IR data/cm−1: 3374 sbr (phenolic OH of curcumin), 3085 w, 1586 m, 1505 s, 1408 w, 1276 m, 1210 w, 1161 w, 1080 vs, 852 w, 808 m, 727 s, 626 m. UV-visible in 5% aqueous DMF [λmax/nm (ε/M−1 cm−1)]: 410 (16700), 342 (35800), 297 sh (68000), 270 (99300). 1H-NMR in DMSO-d6 (δ, ppm): 10.08 (d, 2H of dpq, J = 7.8 Hz), 9.76–9.65 (m, 2H of dpq), 9.50 (d, 2H of dpq, J = 7.0 Hz), 8.46 (d, 2H of dpq, J = 7.2 Hz), 9.03 (d, 2H of dpq, J = 7.8 Hz), 8.63 (d, 2H of dppz, J = 8.3 Hz), 8.25 (d, 2H of dpq, J = 7.6 Hz), 7.98 (d, 2H, J = 7.5 Hz), 7.43 (d, 2H of cur, J = 8.9 Hz), 7.29 (d, 2H of cur, J = 2.5 Hz), 7.18 (d, 2H of cur CC, J = 8.5 Hz), 6.98 (d, 2H of cur, J = 9.0 Hz), 6.18 (s, 1H of cur), 3.98 (s, 6H of 2CH3 of cur). Molar conductivity in DMF at 298 K [ΛM/S cm2 mol−1]: 138.
[Co(dppz)2(acac)](ClO4)2 (5). Yield: ∼70%. Anal. calcd for C41H27N8O10Cl2Co: C, 53.44; H, 2.95; N, 12.16. Found: C, 53.60; H, 2.84; N, 12.25. ESI-MS in 5% aqueous acetonitrile: m/z 361.08 [M-(2ClO4−)]2+. FT-IR data/cm−1: 3084 w, 1555 m, 1512 s, 1423 m, 1360 s, 1282 w, 1085 vs, 945 w, 818 m, 766 m, 729 s, 614 s. UV-visible in 5% aqueous DMF [λmax/nm (ε/M−1 cm−1)]: 380 (41600), 361 (41200), 275 (186300). 1H-NMR in DMSO-d6 (δ, ppm): 10.17–10.12 (m, 2H of dppz, J = 8.2 Hz), 9.95 (d, 2H of dppz, J = 4.6 Hz), 9.01 (d, 2H of dppz, J = 8.1 Hz), 8.93 (d, 2H of dppz, J = 8.3 Hz), 8.84 (d, 2H of dppz, J = 7.8 Hz), 8.76 (d, 2H of dppz, J = 8.0 Hz), 8.65–8.62 (m, 2H of dppz), 8.45 (d, 2H, J = 8.3 Hz), 8.40–8.32 (m, 2H of dppz), 8.01 (d, 2H, J = 7.2 Hz), 6.13 (s, 1H of acac), 2.18 (s, 6H from 2CH3 of acac). Molar conductivity in DMF at 298 K [ΛM/S cm2 mol−1]: 142.
[Co(dppz)2(cur)](ClO4)2 (6). Yield: ∼67%. Anal. calcd for C57H39N8O14Cl2Co: C, 57.54; H, 3.30; N, 9.42. Found: C, 57.67; H, 3.42; N, 9.50. ESI-MS in 5% aqueous acetonitrile: m/z 495.12 [M-(2ClO4−)]2+. FT-IR data/cm−1: 3365 sbr (phenolic OH of curcumin), 2939 w, 1620 m, 1594 m, 1556 w, 1508 vs, 1429 m, 1388 m, 1270 s, 1213 m, 1162 m, 1082 s, 1032 m, 947 m, 964 w, 846 w, 811 w, 766 w, 716 w, 614 w. UV-visible in 5% aqueous DMF [λmax/nm (ε/M−1 cm−1)]: 427 (73000), 380 (44600), 360 (71200), 273 (148800). 1H-NMR in DMSO-d6 (δ, ppm): 10.22–10.13 (m, 2H of dppz, J = 8.3 Hz), 9.98 (d, 2H of dppz, J = 4.8 Hz), 9.09 (d, 2H of dppz, J = 8.3 Hz), 8.95 (d, 2H of dppz, J = 8.5 Hz), 8.87 (d, 2H of dppz, J = 7.7 Hz), 8.72 (d, 2H of dppz, J = 8.2 Hz), 8.66–8.60 (m, 2H of dppz), 8.48 (d, 2H, J = 8.0 Hz), 8.42–8.34 (m, 2H of dppz), 8.04 (d, 2H, J = 7.5 Hz), 7.47 (d, 2H of cur CC, J = 9.1 Hz), 7.28 (d, 2H of cur Ph, J = 2.3 Hz), 7.15 (d, 2H of cur Ph, J = 8.3 Hz), 6.95 (d, 2H of cur CC, J = 9.1 Hz), 6.82 (d, 2H of cur Ph, J = 8.2 Hz), 6.15 (s, 1H of cur), 3.98 (s, 6H of 2CH3 of cur). Molar conductivity in DMF at 298 K [ΛM/S cm2 mol−1]: 132.
X-ray crystallographic procedure
The crystal structures of complexes 1·acetone as its PF6− salt (1a) and 3·H2O were obtained by single crystal X-ray diffraction technique. Single crystals were grown by slow evaporation of a solution of the complex in methanol–acetone (1:1 v/v) mixture. Crystal mounting was done on glass fiber with epoxy cement. All geometric and intensity data were collected at room temperature using an automated Bruker SMART APEX CCD diffractometer equipped with a fine focus 1.75 kW sealed tube Mo-Kα X-ray source (λ = 0.71073 Å) with increasing ω (width of 0.3° per frame) at a scan speed of 6 seconds (for 1a·acetone) and 8 seconds (for 3·H2O) per frame. Intensity data, collected using ω–2θ scan mode, were corrected for Lorentz-polarization effects and for absorption.49 Patterson and Fourier techniques were used to solve the structures and refinement was done by full matrix least squares method using SHELX system of programs.50 The hydrogen atoms belonging to the complexes were in their calculated positions and refined using a riding model. All non-hydrogen atoms were refined anisotropically. The perspective views of the molecules were obtained by ORTEP.51 Selected crystallographic data are given in Table 2. The chemically significant bond distances and bond angles are shown in Table 3.†
Computational studies
To rationalize the photophysical properties of the Co(III) curcumin complexes 2, 4, and 6, computational studies were performed using B3LYP/LANL2DZ level of DFT.52,53 The hybrid B3LYP functional and LANL2DZ basis set were used in all calculations as incorporated in Gaussian 09 package. Visualizations of the DFT optimized structures and the frontier molecular orbitals (HOMOs and LUMOs) of the complexes were performed using Gaussview5.0. To ascertain stationary points, further frequency test was performed.
Cell culture
HeLa (human cervical carcinoma) cells were maintained in DMEM supplemented with 10% FBS, 100 IU mL−1 of penicillin, 100 μg mL−1 of streptomycin and 2 mM of Glutamax at 37 °C in a humidified incubator at 5% CO2. The adherent cultures were grown as monolayer and were passaged once in 4–5 days by trypsinizing with 0.25% trypsin–EDTA.
Cytotoxicity
HeLa cells were analyzed for viability post treatment using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. The assay is based on the ability of mitochondrial dehydrogenases in the viable cells to cleave the tetrazolium rings of MTT to form dark blue membrane impermeable crystals of formazan, which upon solubilization can be measured spectrophotometrically.54 Approximately 15 × 103 HeLa cells were plated in a 96-well culture plate in DMEM supplemented with 10% fetal bovine serum (10% DMEM) and cultured overnight. The stock solutions of the compounds were prepared in DMSO and were first diluted in the culture medium to the desired concentration and then added to the cells in 96 well plates. The final concentration of DMSO was kept constant at 1% for all tests. Then cells were incubated for 4 hours in dark. The medium was then replaced with 50 mmol phosphate buffer (pH 7.4) containing 150 mmol NaCl (PBS) and cells were photo-irradiated for 1 h in visible light of 400–700 nm using Luzchem Photoreactor (Model LZC-1, Ontario, Canada) fitted with Sylvania make 8 fluorescent white tubes with a fluence rate of 2.4 mW cm−2 to provide a total dose of 10 J cm−2. PBS was replaced with 10% DMEM after irradiation and cells were incubated for a further period of 20 h in dark. Post incubation, 25 μL of MTT (4 mg mL−1 in PBS) was added to each well and incubated for 3 h. The culture medium was discarded and 200 μL of dimethyl sulfoxide (DMSO) was added to dissolve the formazan crystals. The intensity of the dark blue colour formed by the formazan complex was estimated at an absorbance of 540 nm using an ELISA microplate reader (BioRad, Hercules, CA, USA). The cytotoxicity of the test compounds was measured as the percentage ratio of the absorbance of the treated cells over the untreated controls. The IC50 values were determined by nonlinear regression analysis (GraphPad Prism).
Annexin-V FITC and propidium iodide (PI) assay
To confirm the apoptotic pathway of cell death, HeLa cells (4 × 105 cells per mL) were treated with complex 6 (3 μM) in 10% DMEM for 4 hours, followed by exposure to visible light for 1 hour. The cells were then cultured for 18 hours in complete medium, harvested and washed twice with chilled PBS at 4 °C. The cells were re-suspended in 100 mL Annexin-V binding buffer (100 mmol HEPES/NaOH, pH 7.4 containing 140 mmol NaCl and 2.5 mmol CaCl2), stained with Annexin-V FITC and PI, and incubated for 15 min at room temperature in the dark. After incubation, 400 μL of binding buffer was added to the cells and analyzed immediately using flow cytometry.55
FACS for cell cycle profiling
To study the effect of complex 6 on the cell cycle, ∼3 × 105 HeLa cells were plated per well of a 6-well tissue culture plate in DMEM containing 10% FBS. After 24 h of incubation at 37 °C in a CO2 incubator, 3 μM of complex 6 in 1% DMSO was added to the cells and incubation was continued for 4 hours in dark. The medium was subsequently replaced with PBS and photo-irradiation was done using visible light (400–700 nm, Luzchem Photoreactor) for 1 hour. After photo-irradiation, PBS was removed and replaced with DMEM-10% FBS and incubation was continued for another period of 18 hours in dark. After incubation, cells were trypsinized and collected into 1.5 mL centrifuge tubes. The cells were washed once with chilled PBS of pH 7.4 and fixed by adding 800 μL of chilled 70% ethanol drop-wise with constant and gentle vortexing to prevent aggregation of cells. The cell suspension was incubated at −20 °C for 6 h. The fixed cells were then washed twice with 1.0 mL of chilled PBS by centrifuging at 4000 rpm for 5 min at 4 °C. The supernatant was discarded by gently inverting the tube and the cell pellet was suspended in 200 μL of PBS containing 10 μg mL−1 DNAse free RNAse for a period of 12 h at 37 °C for digesting the cellular RNA. After digestion, a 20 μL volume of 1.0 mg mL−1 propidium iodide solution was added into the mixture and incubated further for 20 min at 25 °C in dark. Flow cytometric analysis was performed using FACS Calibur (Becton Dickinson “Cell Quest Pro” software) cell analyzer at FL2 channel (595 nm). Data analysis was performed for the percentage of cells in each cell cycle phase by using WinMDI version 2.8.56
DCFDA assay for measurement of ROS generation
The generation of any reactive oxygen species (ROS) was probed using 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) assay.57 Cell permeable DCFDA can be oxidized by cellular ROS to generate fluorescent DCF with an emission maxima at 525 nm.58 The percentage of cells generating ROS can be determined by flow cytometry analysis. To detect ROS generation, HeLa cells were incubated with complex 6 (3 μM) for 4 hours followed by photo-irradiation (400–700 nm) for 1 hour in 50 mmol PBS. The cells were harvested by trypsinization and single cell suspension of 1 × 106 cells per mL was prepared. The cells were then treated with 10 μM DCFDA solution in DMSO in dark for 15 min at room temperature. The distribution of DCFDA stained HeLa cells was determined by flow cytometry.
Fluorescence microscopy
HeLa cells (4 × 104 cells per mm2), plated on cover slips, were incubated with 10 μM of complex 4 or 6 for 2 and 4 hours in the dark, fixed with 4% paraformaldehyde for 10 min at room temperature and washed with PBS. This was followed by staining with Hoechst 33342 for 10 min at 25 °C. The cells were washed, mounted in 90% glycerol solution containing Mowiol, an anti-fade reagent, and sealed. Images were acquired using Apotome.2 fluorescence microscope (Carl Zeiss, Germany) using an oil immersion lens at 63× magnification. The images were analyzed using the AxioVision Rel 4.8.2 (Carl Zeiss, Germany) software.59 To understand the sub-cellular localization of the complex, HeLa cells (4 × 104 cells per mm2) were incubated with 10 μM of complex for 2 and 4 hours in the dark, following which, the cells were treated with 0.5 μmol of mitrotracker Red in serum-free medium for 30 min at 37 °C. The cells were then washed with PBS, mounted on slides and sealed with nail-paint. The images were acquired using ApoTome.2 fluorescence microscope at 63× magnification and analyzed using AxioVision Rel 4.8.2 for sub-cellular localization of the complex.
HSA binding experiment
HSA (essentially free from fatty acids) was used for the binding studies and its concentration in the commercial sample was determined by noting the absorbance of its band at 280 nm with a known molar extinction coefficient of 36600 M−1 cm−1.60 The protein interaction study was performed by tryptophan fluorescence quenching experiments using HSA (2 μM) in phosphate buffer (pH 6.8) containing 15% DMF. Quenching of the emission intensity of tryptophan residue (Trp214) of HSA at 345 nm (excitation wavelength at 295 nm) was monitored using complexes 1–6 as quenchers (Q) by gradually increasing their concentrations.61 Stern–Volmer plots of I0/I vs. [complex] were made using the corrected fluorescence data taking into account the effect of dilution. Linear fit of the data using the equation: I0/I = 1 + KHSA[Q], where I0 and I are the respective emission intensities of HSA in the absence and presence of quencher of concentration [Q], gave the interaction constant (KHSA) values using Origin Pro 8.0 software. Scatchard plots were constructed using the equation log[(I0 − I)/I] = logKb + nlog[Q], where I0 and I are the respective emission intensities of HSA in the absence and presence of quencher of concentration [Q], n is the number of binding sites, and Kb is the binding constant.62 The values of Kb were obtained using a GraphPad Prism software. For the construction of Job plots, a series of solutions of the complex in DMF and that of HSA in phosphate buffer were prepared by keeping the sum of their concentrations constant at 2 μM. The molar fractions were varied from 0.1 to 1.0, and a plot of (I0 − I)[HSA]/[total] against the molar fraction of complex, where I0 and I are the emission intensities of HSA in the absence and presence of complex, respectively, was constructed on a Origin 8.0 software.63
Acknowledgements
This work was supported by a Department of Biotechnology (DBT), Government of India, U-Excel grant (BT/401/NE/U-Excel/2013) to A.H. T.S. and S. B. thank the DBT and the University Grant Commission (UGC) respectively for research fellowships. We acknowledge the X-ray single crystal diffractometer facility of Gauhati University. We are thankful to Prof. Akhil R. Chakravarty and Prof. Anjali A. Karande of Indian Institute of Science (IISc), Bangalore for their generous help in this work. We thank Mr Sanjoy Mukherjee of IISc, Bangalore for his help in computational studies.
References
- A. Juarranz, P. Jaén, F. Sanz-Rodríguez, J. Cuevas and S. González, Clin. Transl. Oncol., 2008, 10, 148–154 CrossRef CAS.
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Photochem. Photobiol., 2009, 85, 1053–1074 CrossRef PubMed.
- H. J. Nyst, I. B. Tan, F. A. Stewart and A. J. M. Balm, Photodiagn. Photodyn. Ther., 2009, 6, 3–11 CrossRef CAS PubMed.
- J. Gray and G. Fullarton, Photodiagn. Photodyn. Ther., 2007, 4, 151–159 CrossRef CAS PubMed.
-
(a) R. R. Allison, R. Cuenca, G. H. Downie, M. E. Randall, V. S. Bagnato and C. H. Sibata, Photodiagn. Photodyn. Ther., 2005, 2, 51–63 CrossRef CAS;
(b) M.-F. Zuluaga and N. Lange, Curr. Med. Chem., 2008, 15, 1655–1673 CrossRef CAS.
-
(a) M. Kar and A. Basak, Chem. Rev., 2007, 107, 2861–2890 CrossRef CAS PubMed;
(b) J. Mao, Y. Zhang, J. Zhu, C. Zhang and Z. Guo, Chem. Commun., 2009, 908–910 RSC;
(c) W.-H. Wei, Z. Wang, T. Mizuno, C. Cortez, L. Fu, M. Sirisawad, L. Naumovski, D. Magda and J. L. Sessler, Dalton Trans., 2006, 1934–1942 RSC.
-
(a) J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed;
(b) I. Kinzler, E. Haseroth, C. Hauser and A. Rűck, Photochem. Photobiol. Sci., 2007, 6, 1332–1340 RSC;
(c) L. B. Li and R. C. Luo, Laser. Med. Sci., 2009, 24, 597–603 CrossRef PubMed;
(d) M. Chakrabarti, N. L. Banik and S. K. Ray, Plos One, 2013, 8, e55652 CAS.
-
(a) R. Bonnett, Metal Complexes for Photodynamic Therapy, in Comprehensive Coordination Chemistry, ed. J. A. McCleverty and T. J. Meyer, Elsevier Pergamon, Oxford, 2004, vol. 2, p. 945 Search PubMed;
(b) N. L. Fry and P. K. Mascharak, Acc. Chem. Res., 2011, 44, 289–298 CrossRef CAS PubMed.
-
(a) N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690–10701 RSC;
(b) N. J. Farrer and P. J. Sadler, Aust. J. Chem., 2008, 61, 669–674 CrossRef CAS.
-
(a) U. Schatzschneider, Eur. J. Inorg. Chem., 2010, 1451–1467 CrossRef CAS;
(b) A. Bergamo and G. Sava, Dalton Trans., 2011, 7817–7823 RSC.
-
(a) H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2005, 38, 146–156 CrossRef CAS PubMed;
(b) A. M. Angeles-Boza, H. T. Chifotides, J. D. Aguirre, A. Chouai, P. K.-L. Fu, K. R. Dunbar and C. Turro, J. Med. Chem., 2006, 49, 6841–6847 CrossRef CAS PubMed.
-
(a) C. E. Wenner, J. Cell. Physiol., 2012, 227, 450–456 CrossRef CAS PubMed;
(b) S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed.
-
(a) J. S. Armstrong, Br. J. Pharmacol., 2006, 147, 239–248 CrossRef CAS PubMed;
(b) G. Damia, L. Imperatori, M. Stefanini and M. D'Incalci, Int. J. Cancer, 1996, 66, 779–783 CrossRef CAS.
- A. Basu and S. Krishnamurthy, J. Nucleic Acids, 2010, 201367 Search PubMed.
- S. Marrachea, S. Tundup, D. A. Harn and S. Dhar, ACS Nano, 2013, 7, 7392–7402 CrossRef PubMed.
- M. Millard, J. D. Gallagher, B. Z. Olenyuk and N. Neamati, J. Med. Chem., 2013, 56, 9170–9179 CrossRef CAS PubMed.
- V. Pierozz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376–20387 CrossRef PubMed.
-
(a) S. Marrache, R. K. Pathak and S. Dhar, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10444–10449 CrossRef CAS PubMed;
(b) S. Marrache and S. Dhar, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16288–16293 CrossRef CAS PubMed.
- S. Banerjee, P. Prasad, A. Hussain, I. Khan, P. Kondaiah and A. R. Chakravarty, Chem. Commun., 2012, 48, 7702–7704 RSC.
- P. Prasad, I. Khan, P. Kondaiah and A. R. Chakravarty, Chem.–Eur. J., 2013, 19, 17445–17455 CrossRef CAS PubMed.
-
(a) E. Delaey, F. V. Laar, D. D. Vos, A. Kamuhabwa, P. Jacobs and P. D. Witte, J. Photochem. Photobiol., B, 2000, 55, 27–36 CrossRef CAS;
(b) J. Saczko, M. Mazurkiewicz, A. Chwilkowska, J. Kulbacka, G. Kramer, M. Ługowski, M. Śnietura and T. Banas, Folia Biol., 2007, 53, 7–12 CAS.
-
(a) F. Yang, G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, S. S. Ambegaokar, P. P. Chen, R. Kayed, C. G. Glabe, S. A. Frautschy and G. M. Cole, J. Biol. Chem., 2005, 280, 5892–5901 CrossRef CAS PubMed;
(b) M. Garcia-Alloza, L. A. Borrelli, A. Rozkalne, B. T. Hyman and B. J. Bacskai, J. Neurochem., 2007, 102, 1095–1104 CrossRef CAS PubMed.
-
(a) B. B. Aggarwal and K. B. Harikumar, Int. J. Biochem. Cell Biol., 2009, 41, 40–59 CrossRef CAS PubMed;
(b) M.-H. Teiten, S. Eifes, M. Dicato and M. Diederich, Toxins, 2010, 2, 128–162 CrossRef CAS PubMed;
(c) B. B. Aggarwal, A. Kumar and A. C. Bharti, Anticancer Res., 2003, 23, 363–398 CAS;
(d) R. A. Sharma, W. P. Steward and A. J. Gescher, Adv. Exp. Med. Biol., 2007, 595, 453–470 CrossRef.
-
(a) N. Dhillon, B. B. Aggarwal, R. A. Newman, R. A. Wolff, A. B. Kunnumakkara, J. L. Abbruzzese, C. S. Ng, V. Badmaev and R. Kurzrock, Clin. Cancer Res., 2008, 14, 4491–4499 CrossRef CAS PubMed;
(b) C. H. Hsu and A. L. Cheng, Adv. Exp. Med. Biol., 2007, 595, 471–480 CrossRef.
-
(a) B. B. Aggarwal, C. Sundaram, N. Malani and H. Ichikawa, Adv. Exp. Med. Biol., 2007, 595, 1–75 CrossRef;
(b) P. Anand, S. G. Thomas, A. B. Kunnumakkara, C. Sundaram, K. B. Harikumar, B. Sung, S. T. Tharakan, K. Misra, I. K. Priyadarsini, K. N. Rajasekharan and B. B. Aggarwal, Biochem. Pharmacol., 2008, 76, 1590–1611 CrossRef CAS PubMed.
- D. Pucci, T. Bellini, A. Crispini, I. D′Agnano, P. F. Liguori, P. Garcia-Orduña, S. Pirillo, A. Valentini and G. Zanchetta, MedChemComm, 2012, 3, 462–468 RSC.
-
(a) S. Banerjee, P. Prasad, I. Khan, A. Hussain, P. Kondaiah and A. R. Chakravarty, Z. Anorg. Allg. Chem., 2014, 640, 1195–1204 CAS;
(b) P. Prasad, I. Pant, I. Khan, P. Kondaiah and A. R. Chakravarty, Eur. J. Inorg. Chem., 2014, 2420–2431 CrossRef CAS.
-
(a) A. Hussain, K. Somyajit, B. Banik, S. Banerjee, G. Nagaraju and A. R. Chakravarty, Dalton Trans., 2013, 182–195 RSC;
(b) T. K. Goswami, S. Gadadhar, B. Gole, A. A. Karande and A. R. Chakravarty, Eur. J. Med. Chem., 2013, 63, 800–810 CrossRef CAS PubMed;
(c) B. Banik, K. Somyajit, G. Nagaraju and A. R. Chakravarty, Dalton Trans., 2014, 13358–13369 RSC.
-
(a) M. Sagnou, D. Benaki, C. Triantis, T. Tsotakos, V. Psycharis, C. P. Raptopoulou, I. Pirmettis, M. Papadopoulos and M. Pelecanou, Inorg. Chem., 2011, 50, 1295–1303 CrossRef CAS PubMed;
(b) A. Valentini, F. Conforti, A. Crispini, A. De Martino, R. Condello, C. Stellitano, G. Rotilio, M. Ghedini, G. Federici, S. Bernardini and D. Pucci, J. Med. Chem., 2009, 52, 484–491 CrossRef CAS PubMed.
-
(a) I. K. Priyadarsini, J. Photochem. Photobiol., C, 2009, 10, 81–95 CrossRef PubMed;
(b) T. A. Dahl, P. Bilski, K. J. Reszka and C. F. Chignell, Photochem. Photobiol., 1994, 59, 290–294 CrossRef CAS PubMed.
-
(a) A. Bernd, Phytochem. Rev., 2014, 13, 183–189 CrossRef CAS PubMed;
(b) K. Park and J.-H. Lee, Oncol. Rep., 2007, 17, 537–540 CAS.
- A. K. Renfrew, N. S. Bryce and T. W. Hambley, Chem. Sci., 2013, 4, 3731–3739 RSC.
- A. D. A. M., Medical Encyclopaedia, A. D. A. M., Inc., Atlanta, GA, 2005, vol. 2014 Search PubMed.
- M. J. Hawkins, P. Soon-Shiong and N. Desai, Adv. Drug Delivery Rev., 2008, 60, 876–885 CrossRef CAS PubMed.
-
(a) A. R. Timerbaev, S. S. Aleksenko, K. Polec-Pawlak, R. Ruzik, O. Semenova, C. G. Hartinger, S. Oszwaldowski, M. Galanski, M. Jarosz and B. K. Keppler, Electrophoresis, 2004, 25, 1988–1995 CrossRef CAS PubMed;
(b) A. I. Ivanov, J. Christodoulou, J. A. Parkinson, K. J. Barnham, A. Tucker, J. Woodrow and P. J. Sadler, J. Biol. Chem., 1998, 273, 14721–14730 CrossRef CAS PubMed.
- B. P. Espósito and R. Najjar, Coord. Chem. Rev., 2002, 232, 137–149 CrossRef.
-
(a) B. Elsadek and F. Kratz, J. Controlled Release, 2012, 157, 4–28 CrossRef CAS PubMed;
(b) F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed;
(c) K. Park, J. Controlled Release, 2012, 157, 3 CrossRef CAS PubMed.
- A. M. Angeles-Boza, P. M. Bradley, P. K.-L. Fu, S. E. Wicke, J. Bacsa, K. R. Dunbar and C. Turro, Inorg. Chem., 2004, 43, 8510–8519 CrossRef CAS PubMed.
-
(a) A. K. Patra, S. Dhar, M. Nethaji and A. R. Chakravarty, Chem. Commun., 2003, 1562–1563 RSC;
(b) S. Dhar, D. Senapati, P. K. Das, P. Chattopadhyay, M. Nethaji and A. R. Chakravarty, J. Am. Chem. Soc., 2003, 125, 12118–12124 CrossRef CAS PubMed;
(c) A. Hussain, D. Lahiri, M. S. A. Begum, S. Saha, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Inorg. Chem., 2010, 49, 4036–4045 CrossRef CAS PubMed.
-
(a) P. K. Sasmal, S. Saha, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Chem. Commun., 2009, 1703–1705 RSC;
(b) S. Saha, R. Majumdar, M. Roy, R. R. Dighe and A. R. Chakravarty, Inorg. Chem., 2009, 48, 2652–2663 CrossRef CAS PubMed.
- D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 1980 Search PubMed.
- O. Vajragupta, P. Boonchoon and L. J. Berliner, Free Radical Res., 2004, 38, 303–314 CrossRef CAS.
-
(a) J. E. Dickeson and L. A. Summers, Aust. J. Chem., 1970, 23, 1023–1027 CrossRef CAS;
(b) J. G. Collins, A. D. Sleeman, J. R. Aldrich-Wright, I. Greguric and T. W. Hambley, Inorg. Chem., 1998, 37, 3133–3141 CrossRef CAS.
- E. Amouyal, A. Homsi, J.-C. Chambron and J.-P. Sauvage, J. Chem. Soc., Dalton Trans., 1990, 1841–1845 RSC.
-
(a) A. A. Vlcek, Inorg. Chem., 1967, 6, 1425–1427 CrossRef CAS;
(b) S. Ghosh, A. C. Barve, A. A. Kumbhar, A. S. Kumbhar, V. G. Puranik, P. A. Datar, U. B. Sonawane and R. R. Joshi, J. Inorg. Biochem., 2006, 100, 331–343 CrossRef CAS PubMed.
- J. Guilford II, R. Jackson, C. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294–300 CrossRef.
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067–1071 RSC.
- N. Walker and D. Stuart, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1983, 39, 158–166 CrossRef.
-
(a) G. M. Sheldrick, SHELX-97, Programs for Crystal Structure Solution and Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed;
(b) L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- C. K. Johnson, ORTEP, Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, TN, 1976.
-
(a) A. D. Becke, Phys. Rev., 1998, A38, 3098–3100 Search PubMed;
(b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed;
(c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS;
(d) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284–298 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, H. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, revision B.4, Gaussian Inc., Pittsburgh, PA, 2003 Search PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- C. Soni and A. A. Karande, Mol. Immunol., 2010, 47, 2458–2466 CrossRef CAS PubMed.
- K. Million, É. Horvilleur, D. Goldschneider, M. Agina, G. Raguénez, F. Tournier, J. Bénard and S. Douc-Rasy, Int. J. Oncol., 2006, 29, 147–154 CAS.
- A. S. Keston and R. Brandt, Anal. Biochem., 1965, 11, 1–5 CrossRef CAS.
- T. Takanashi, Y. Ogura, H. Taguchi, M. Hashizoe and Y. Honda, Invest. Ophthalmol. Visual Sci., 1997, 38, 2721–2728 CAS.
- J. L. McClintock and B. P. Ceresa, Invest. Ophthalmol. Visual Sci., 2010, 51, 3455–3461 Search PubMed.
- A. Singh and M. Darshi, New J. Chem., 2004, 28, 120–126 RSC.
- N. S. Quiming, R. B. Vergel, M. G. Nicolas and J. A. Villanueva, J. Health Sci., 2005, 51, 8–15 CrossRef CAS.
-
(a) X. Z. Feng, Z. Yang, L. J. Wang and C. Bai, Talanta, 1998, 47, 1223–1229 CrossRef CAS;
(b) D. Voet, Biochemistry, John Wiley & Sons, Inc., 3rd edn, 1995 Search PubMed.
- C. Y. Huang, Methods Enzymol., 1982, 87, 509–525 CAS.
- K. Nakamoto, in Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, New York, 3rd edn, 1978 Search PubMed.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- S. Banerjee, A. Dixit, R. N. Shridharan, A. A. Karande and A. R. Chakravarty, Chem. Commun., 2014, 50, 5590–5592 RSC.
- C. Qian, J. Q. Wang, C. L. Song, L. L. Wang, L. N. Ji and H. Chao, Metallomics, 2013, 5, 844–854 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Schemes, FT-IR, mass, UV-visible spectra, cyclic voltammograms, packing diagrams, DFT tables, stability plots, MTT assay, FACS analysis, fluorescence images and HSA binding plots. CCDC 1008427 and 1008428. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra17314g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 


 ), 5 (
), 5 ( ) and 6 (
) and 6 ( ). (b) Scatchard plots of log[(I0 − I)/I] vs. log[Q] for 3 (
). (b) Scatchard plots of log[(I0 − I)/I] vs. log[Q] for 3 ( ), 5 (
), 5 ( ) and 6 (
) and 6 ( ) to determine the binding constant (Kb) for HSA–complex interaction.
) to determine the binding constant (Kb) for HSA–complex interaction.