
Conducting polymers and their inorganic composites for advanced Li-ion batteries: a review
Prakash Sengodu
*a and
Abhay D. Deshmukh
 b
b
aDepartment of Chemistry, National Taiwan Normal University, Taipei, Taiwan. E-mail: prakas.chemist@gmail.com
bEnergy Materials and Devices Laboratory, Department of Physics, RTM Nagpur University, Nagpur, India. E-mail: abhay.d07@gmail.com
First published on 10th April 2015
Abstract
Conducting polymers are promising materials for organic–inorganic hybrid composites in lithium-ion batteries due to their electrical conductivity and high coulombic efficiency and are able to be cycled hundreds or thousands of times with only slight degradation. Inorganic compounds generally possess good lithium storage properties but lack the conductivity and cyclability required for commercial application. Therefore, these problems need to be overcome before they can be used as effective electrodes. Over the past few years, conductive polymers in combination with inorganic compounds have attracted great interest as promising matrices for the construction of lithium-ion batteries. Because conducting polymers can interact synergistically with inorganic compounds, noteworthy improvements in electrode lifetime, rate capabilities, and voltage, as well as mechanical and thermal stability, have been achieved. This review covers recent advances in synthetic methods and functions of conducting polymers in hybrid composites and their application in lithium-ion batteries. We then present a comparison of other synthetic methods for new research and development in batteries. A brief appraisal of avenues for future developments in this area is also presented.
 Prakash Sengodu | Prakash Sengodu did his M.Sc in Chemistry at Sri Ramakrishna Mission Vidyalaya, Tamil Nadu, India in 2006 and he completed his PhD at CSIR-Central Salt and Marine Chemicals Research Institute, Bhavnagar, India. He was awarded a Senior Research Fellowship and Research Internship from CSIR India. He has also worked as a Research Fellow at Central Electrochemical Research Institute, India, on conducting polymers. In 2013, he was awarded a National Science Council (NSC) Postdoctoral Fellowship and worked in National Taiwan Normal University (NTNU), Taiwan with Prof. C. J. Chen. His research interests include conducting polymers for electrochemical applications. He may be reached at E-mail: prakas.chemist@gmail.com. |
 Abhay Deshmukh | Abhay D. Deshmukh obtained his M.Sc. and PhD in Physics from Nagpur University, and CSIR-National Environmental Engineering Research Institute, India. He has worked as a Research Associate at National Physical Laboratory, New Delhi, India. Then he was awarded a National Science Council (NSC) Postdoctoral Fellowship and worked in National Taiwan Normal University (NTNU), Taiwan with Prof. C. J. Chen. He is now working as an assistant professor in the Department of Physics, R.T.M. Nagpur University, India. His research interest is nanomaterials and their optical properties as well as advanced carbon materials and composites for batteries and supercapacitors. He may be reached at E-mail: abhay.d07@gmail.com. |
1. Introduction
The urgent need for clean and secure energy has stimulated a global revival in the search for advanced electrical energy storage systems. The predicted future lithium ion batteries (LIBs) are one of the most promising sources of energy storage. So far, lithium metal electrodes have been troublesome due to their unsatisfactory cycling life, which results eventually in shorting between the two electrodes, less safety and low rate capability.1–3 Commercial LIBs with LiCoO2, graphite and a non-aqueous Li salt as cathode, anode and electrolyte, respectively, based on an intercalation and deintercalation mechanism were developed by Sony Co., Japan in 1991. They are a good sign of solid-state chemistry in action, whose details may be found in the literature.4 Fig. 1 shows promising present and future energy storage systems plotted by price vs. energy. From this plot, the energy density of a Li-ion battery is higher than that of a supercapacitor and a Na-ion battery due to its low-radius ions and numerous atoms, which are easy to insert into electrode materials. However, the energy density of a Li-ion battery is lower than that of the corresponding air and sulfur batteries. The price of a Li-ion battery is higher than that of supercapacitors, Na-ion batteries and Li–S/air batteries. Current LIBs are widely used in hybrid electric vehicles (HEVs), cellular phones, laptops, camcorders, power tools, and medical fields. Sales of LIBs have increased by 1.5 per cent annually to $10.6 billion in 2012 and are expected to rapidly increase to a market value of US $31.4 billion in 2015 and US $53.7 billion in 2020, due to their use in hybrid electric vehicles (HEVs) and plug-in electric vehicles (PEVs).5 However, cost and safety issues are the main problems with the use of LIBs in electrical vehicles and smart grids.5–7 Recently developed high-energy-density systems (Li–S and Li–O2 batteries) would be more suitable for electric vehicles. However, some issues are major concerns for Li–S and Li–O2 batteries due to low levels of dissolution of the electrode materials, which results in capacity decay.5–7 The high rate capability and high energy density of Li-ion supercapacitors make them good systems for replacing present lead–acid batteries for car starter batteries. Research into sodium ion batteries (NIBs) is a newly growing field compared to that into other battery types, due to their abundance and low price. Trona (sodium ore) is 30 times cheaper than lithium carbonate.6 Many efforts are also being made to develop sodium batteries for electric vehicles and grid storage applications, which are the most promising in the near future.7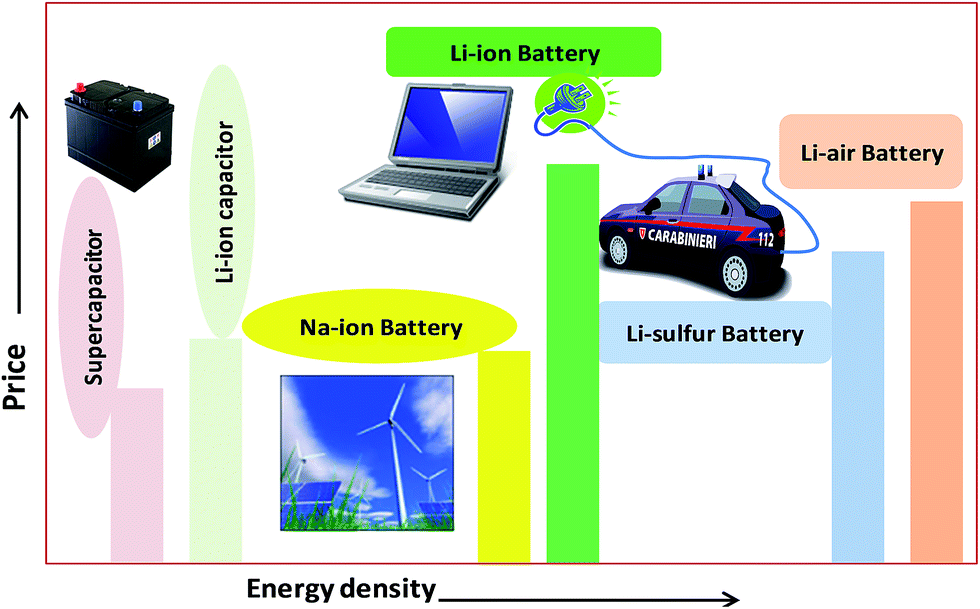 | ||
| Fig. 1 Plot of price vs. energy density for different energy storage systems. | ||
In recent years, the design and fabrication of electrodes has gained much attention in applications of rechargeable batteries. The subject of tuning electrodes has also remained an important area of research activity. With respect to batteries, much emphasis has been placed on the fabrication and characterization of hybrid composites.8,9 These are also of great research interest as emerging electrodes in academia and industry, due to their structural diversity, flexibility and durability as well as high porosity, and therefore a wide spectrum of applications in sensors, solar energy, batteries and supercapacitors have been investigated.10 Interest in inorganic compounds is motivated by their potential applications in lithium-ion batteries,11 due to their easy transport of Li+ ions and electrons allowing better accommodation of ions. Inorganic materials also play diverse roles in different battery systems.12 In general, their good crystallinity, excellent conductivity and redox properties make them suitable as “electrodes” (anodes and cathodes) and photocatalysts (due to their nanometer size and structure).13,14
Conducting polymers (CPs) are not just intended for use in energy storage but are also used to do so independently of their form. Their mechanical properties are like those of a plastic, allowing them to be twisted, contracted, and stiff.15–17 CPs electrodes can be incorporated seamlessly into electronic paper, textiles, data tattoos, structural panels, etc., without including traditional prismatic or cylindrical cells.17–19 They are also used to directly combine the properties of a plastic with polymer solar cells and light-emitting diodes in a streamlined manufacturing process to produce cheap flexible devices.20,21 Furthermore, they have many advantages of synthetic tailorability, cost, and processability, which make them ideal candidates for electrochemical energy storage.22,23 Their properties may be changed by synthetic techniques.24 Also, the raw materials and methods for making these polymers are often cost-effective. New synthetic methods have enhanced their processability via screen printing, doctor blading, or inkjet printing.25,26
Mainly, conducting polymers are ideal materials for hybrid composites in lithium-ion batteries due to their conducting backbone, electrical conductivity and high coulombic efficiency; thus, they can be cycled hundreds or thousands of times with little degradation.20,27–29 Their conductive nature arises from alternating single and double carbon–carbon bonds in polymeric chains, which can be assigned to reversible chemical, electrochemical and physical properties controlled by a doping process. Inorganic materials exhibit high capacity based on a lithium insertion and extraction process but lack the conductivity and cyclability required for commercial application. Overcoming these problems, conducting polymers can interact synergistically with inorganic materials by a mixture of synthetic tuning, processing options, and mechanical flexibility, which can result in polymer power sources that can be designed to suit the device rather than vice versa.30–32 Also, CPs can be used alone as anode/cathode electrodes due to their doping nature and exchange of anions. Therefore, high specific capacity can be achieved by concentrating the electrolyte; however, system batteries with high specific energies at mean cell voltages of up to 3.5 V are possible. CPs not only have advantages with their potential response in a state of charge or discharge, but also disadvantages such as the inclination in their characteristic discharge curve. Of particular note is polyaniline, which was once a cathode in a PANI lithium battery commercially available from Bridgestone–Seiko in the 1980s. Table 1 presents a comparison between commercial electrodes and conducting polymers.33–41
| Electrode | Voltage νs. Li/Li+ | Mw of repeating unit | Capacity (mA h g−1) | Energy density (mW h g−1) | Power density (mW h g−1) | |
|---|---|---|---|---|---|---|
| Theor. | Expt. | |||||
| Graphite33 | ∼0.1 | 12 | 372 | 360 | 372–744 | — |
| LiCoO2 (ref. 34) | ∼3.8 | 97.9 | 274 | 135 | 30–144 | 100–1000 |
| LiFePO4 (ref. 35) | ∼3.2 | 157.8 | 170 | 109 | 90–100 | 23–70 |
| Polythiophene36,37 | 3.1–4.0 | 82.1 | 326 | 82 | 93 | 89 |
| Polypyrrole38,39 | 3.0–4.0 | 65.1 | 412 | 82 | 60 | 10 |
| PEDOT40 | 2.7–4.2 | 140.2 | 191 | 30–70 | 1–4 | 35–2500 |
| Polyaniline41 | 3.0–4.0 | 91.1 | 294 | 100–147 | 300 | 100 |
Popular research into conducting polymer electrodes for lithium-ion batteries has only focused on polyaniline (PANI), polypyrrole, and polythiophene (PT) and their derivatives.42,43 The focus has been on these particular polymers due to the presence of wide background research, the commercial accessibility of both monomers and polymers, and their simplicity of synthesis. Inorganic compounds display low capacity utilization, inferior rate capability, poor cycling stability, or even complete electrochemical inactivity in LIBs. Therefore, numerous articles have been devoted to polymers embedded with inorganic compounds in electrodes as an additive to improve the performance of cathode and anode materials in batteries. Earlier reviews have focused on Si/SiO, Sn/SnO, TiO2, carbon, graphene, and intermetallic compounds as cathode/anode materials,4,44–46 with some attention to binders.47 To the best of our knowledge, there is still no review on conducting polymer–inorganic compound hybrid composite electrodes for battery applications. The fundamental electrochemical aspects of conducting polymers have been discussed in review papers and books covering the literature up to 1997.48 The aim of the present review is to provide information on hybrid composite electrodes in the light of applications of rechargeable batteries in the past decades. Here, we discuss the preparative methods of rechargeable battery electrodes and issues affecting their electrochemical properties. These include low energy, low capacity and power density, oxidative stability, and diffusion limitations on dopant ions. Numerous recent advances in the field have made remarkable progress in addressing these issues and will be discussed in this review.
1.1. Mechanism of conducting polymers in rechargeable batteries
In brief, in rechargeable batteries inorganic ions (M+) are moved freely by the electrolyte from the anode to the cathode as electrons flow through an external circuit from the anode to the cathode, producing power during discharge. During recharge, M+ and electrons are driven in the reverse direction by an external voltage, storing electrical energy as chemical energy in the battery (Fig. 2). Several parameters, including gravimetric energy density (mW h g−1), gravimetric capacity (mA h g−1), volumetric capacity (mA h cm−3), rate capability, cycling ability and self-discharge characteristics, are influenced by the choice of battery materials and battery design. Anode materials function by three different mechanisms, an intercalation–deintercalation mechanism, alloying–dealloying reactions, and redox or “conversion” reactions, whereas cathode materials follow a two-phase mechanism of the classic shrinking core model, anisotropic shrinking core model, and domino-cascade model.48–50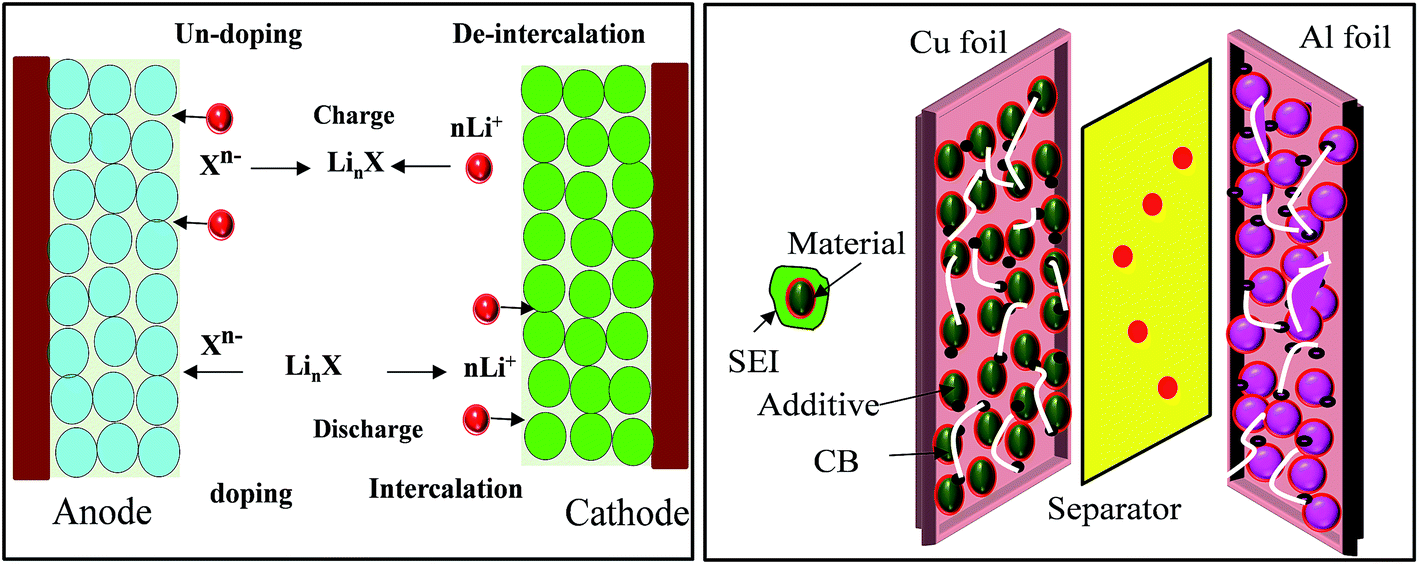 | ||
| Fig. 2 Principle of LIBs based on doping/dedoping (negative) and deintercalation/intercalation (positive) electrodes and a model of the electrodes with their SEI system. | ||
During the charge/discharge process, anode materials undergo a large volume change (>300%), which causes great strain in the material and thus leads to cracking and crumbling of particles. It results in poor reversible capacity and irreversible capacity loss in the first cycle. For example, in Si-based electrodes during transformation from Si to Li4.4Si the volume expansion is about 420%. These electrodes are typically inferior in capacity retention. Early studies on Si anodes confirmed rapid capacity fade during cycling.51–54 During cycling of silicon (average particle size 10 μm), high capacity is reached in the first lithiation, but after that capacity rapidly fades. Even the reversible capacity of a Si electrode drops by 70% after only five cycles. Moreover, the irreversible capacity loss of this Si anode during the first cycle is excessively high (2650 mA h g−1) for practical application, with a coulombic efficiency of 25%. The overall capacity loss and huge initial irreversible capacity loss with Si anodes are caused by large volume changes during the lithium insertion/extraction processes. The solution is a tailored polymer that conducts electricity and binds closely to Li/Na-storing particles with the advantages of low cost and compatibility. In addition, anodes expand to more than three times their volume during charging and then shrink again during discharge. Also, very few studies have claimed a polymer alone as an anode for batteries (usually single experimental cells with a few milligrams of polymer and excess electrolyte) based on a doped (oxidized or reduced) state.55 Conducting polymers have also been cathodes satisfactorily combined with inorganic redox oxides, giving systems suitable for electrochemical Li intercalation with improved performance.48 Polymers can decrease polarization between the cathode particles and the electrolyte and also promote permeation of the electrolyte into the surface of the active particles, hence enhancing M+ insertion–extraction during the cycling process.56 There have been many reports on the role of PANI in cathode materials, such as improving the electrical conductivity, decreasing polarization, and promoting electrolyte permeation.56–58 Conducting polymers alone are used as electrodes in LIBs applications, but their poor capacity limits their practical application.59,60
1.2. Synthetic methods of conducting polymers
Conductive polymers are organic polymers that conduct electricity and may exhibit metallic conductivity or can be semiconductors. They are organic materials and similar to insulating polymers, but are not thermoplastics or thermoformable.15–17 Table 2 shows the conductivity of different conductive polymers.61,62 They offer high electrical conductivity but do not have comparable mechanical properties to other commercially available polymers. Their electrical properties can be fine-tuned using methods of organic synthesis and advanced dispersion techniques.63,64 Heeger, MacDiarmid and Shirakawa were Nobel Laureates for their discovery of metallic conductivity in a polymer in polyacetylene (PA).65 It has been clear that the great potential in the development of organic semiconductors was related to the ability of macromolecular chemists to synthesize new structures having a conjugated backbone. The main aims of synthetic procedures were to overcome the drawbacks of their insolubility and unprocessability.66 Several synthetic methods have been developed in the past three decades. Some of these represented innovative strategies in macromolecular synthesis and were responsible for the evolution of chemistry in the polymer field. The Ziegler–Natta catalyst is foremost for chemical polymerization of acetylene among the various catalyst systems (radiation, cationic, transition/rare-earth metal, etc.) that can be used to initiate polymerization.67 Ti(OBu)4/AlEt3 and NaBH4/Co(NO3)2·6H2O catalysts have also been well studied.68,69 Stable conducting polymers have been synthesized by physically blending conjugated polymers with certain non-conjugated macromolecules70 and chemically synthesizing conducting polymer colloids71 or conjugated oligomers.72 Recent synthetic routes have now been devised for preparing stable conducting polymers, as described below.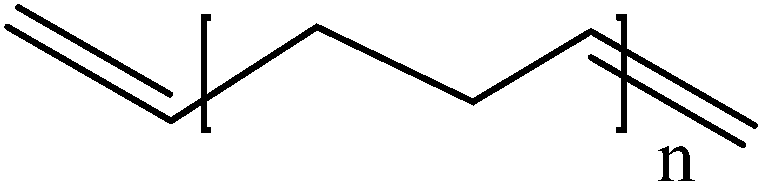
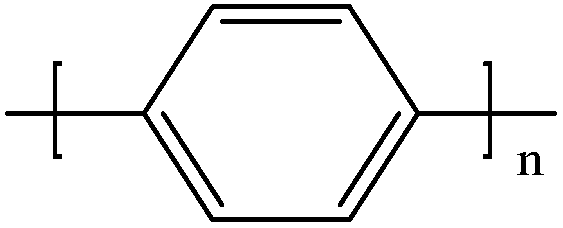

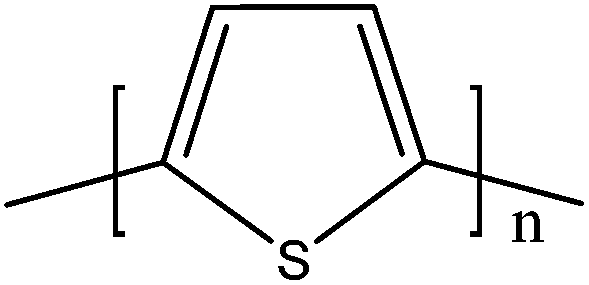

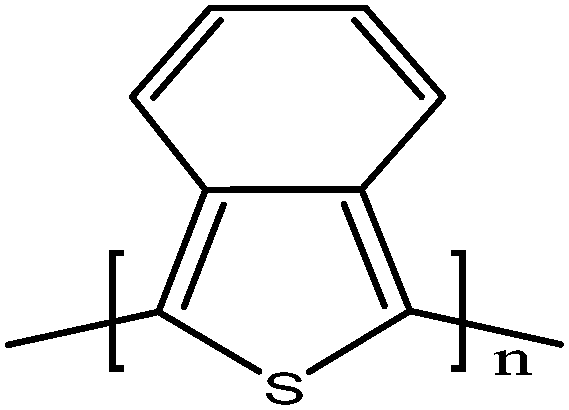
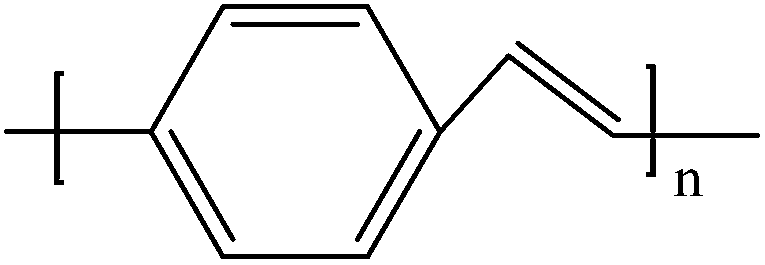
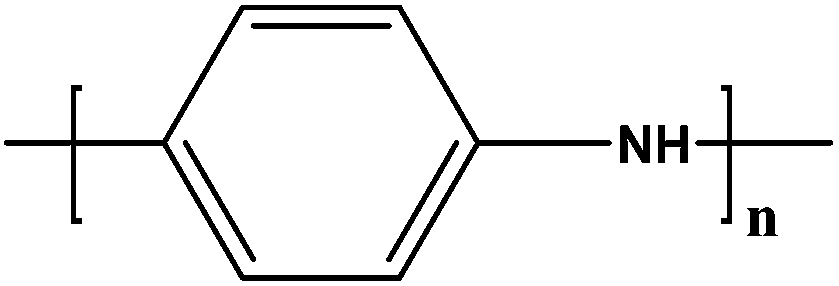
Soluble polymers have been synthesized by different methods such as substitution and copolymerization (random and alternating copolymerization, block and graft copolymerization). Generally, the chemical and physical properties of a polymer material can change with substitution; therefore, soluble forms of various conducting polymers have been prepared by grafting suitable side groups or side chains onto polymer backbones. Yoshino et al. reported polyacetylene grafted with methyl or phenyl side groups.73 Tamao et al. prepared poly(3-alkylthiophenes), which were meltable and soluble in most common organic solvents, from 3-bromothiophene (Scheme 1).74 Soluble polypyrrole and polyaniline have also been synthesized by introducing alkyl side groups,75 and grafting with hydrophilic side groups/chains (–SO3H, –COOH, –OH) even allows the formation of water-soluble conducting polymers.76 Copolymerization of conjugated polymers with various soluble sections provides an alternative way to overcome the uncontrollability of conjugated polymers. The combination of optoelectronic properties characteristic of conjugated structures and the solubility of soluble polymeric segments into a single copolymer chain should, in principle, lead to a material that exhibits properties of both constituent components. Meier et al. have synthesized some highly soluble stilbenoid dendrimers with alkoxy chains substituted on the peripheral benzene rings (Scheme 2).77
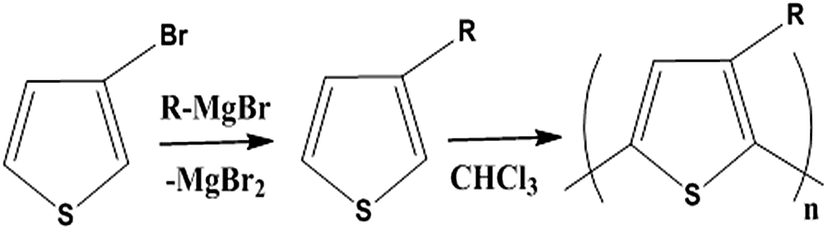 | ||
| Scheme 1 The most common route to poly(3-alkylthiophenes). | ||
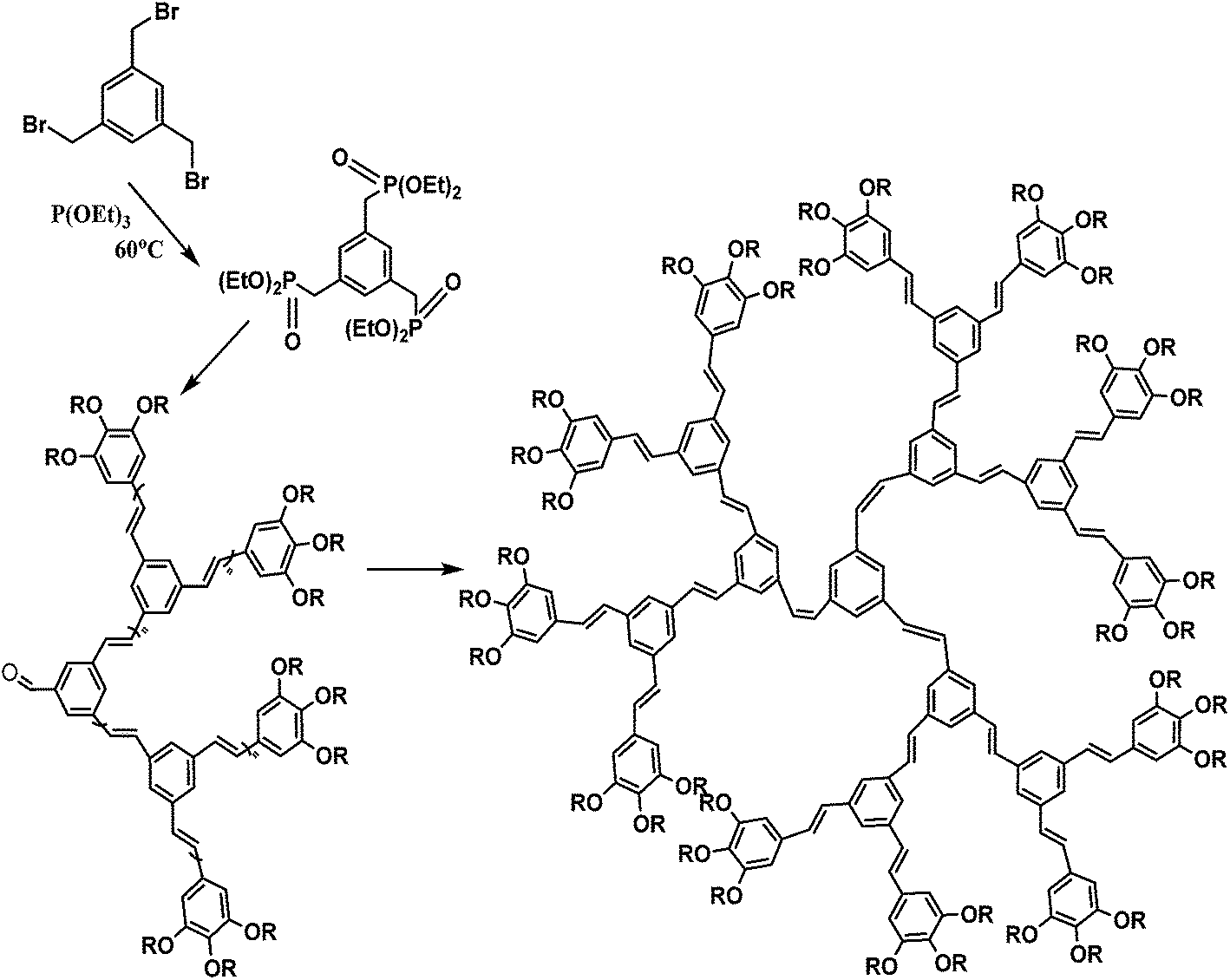 | ||
| Scheme 2 Synthesis of highly soluble stilbenoid dendrimers with alkoxy chains substituted on the peripheral benzene rings. | ||
For the preparation of conjugated polymer films, they developed solution process techniques (spin coating), precursor routes and electropolymerisation methods. The solution process method offers excellent possibilities for the fabrication of advanced devices, but it may cause problems in the quality of polymer films thus formed, such as possible deterioration in the mechanical properties of polymer films by chemical modification of their structures, reduction of the stability of devices by trapped impurities and complications in choosing a suitable solvent for preparing pinhole-free multi-layer polymer thin films. Thermal conversion of soluble non-conjugated precursor polymers has tried to overcome the uncontrollability of the formation of conjugated polymers. Edwards and Feast et al. reported the ring-opening metathesis polymerization (ROMP) of cyclobutene derivatives, followed by thermal conversion into polyacetylene with the evolution of bis(trifluoromethyl)benzene (Scheme 3). Both the potentiostatic method with a constant potential and the potentiodynamic technique by scanning the potential within a certain range of voltages have been used for electrochemical polymerization of electrically conducting polymers. Pyrrole can be polymerized on a suitable anode using a two-electrode electrochemical cell. The mechanism of the electropolymerization of pyrrole is suitable for many other conjugated conducting polymers (Scheme 4).78
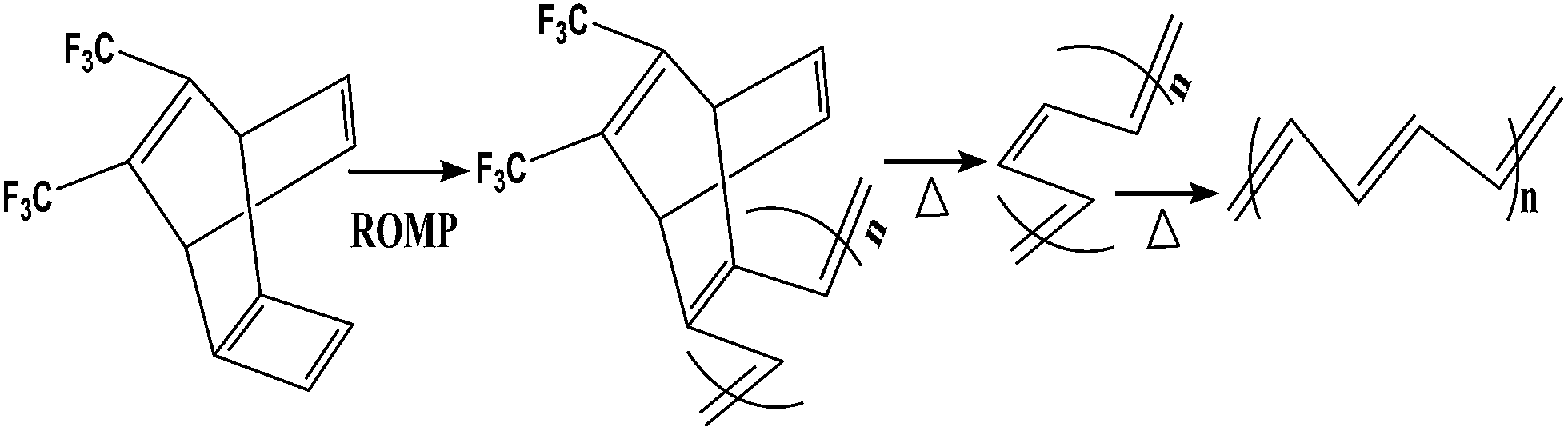 | ||
| Scheme 3 Durham route to polyacetylene (PA). ROMP = ring-opening metathesis polymerization. | ||
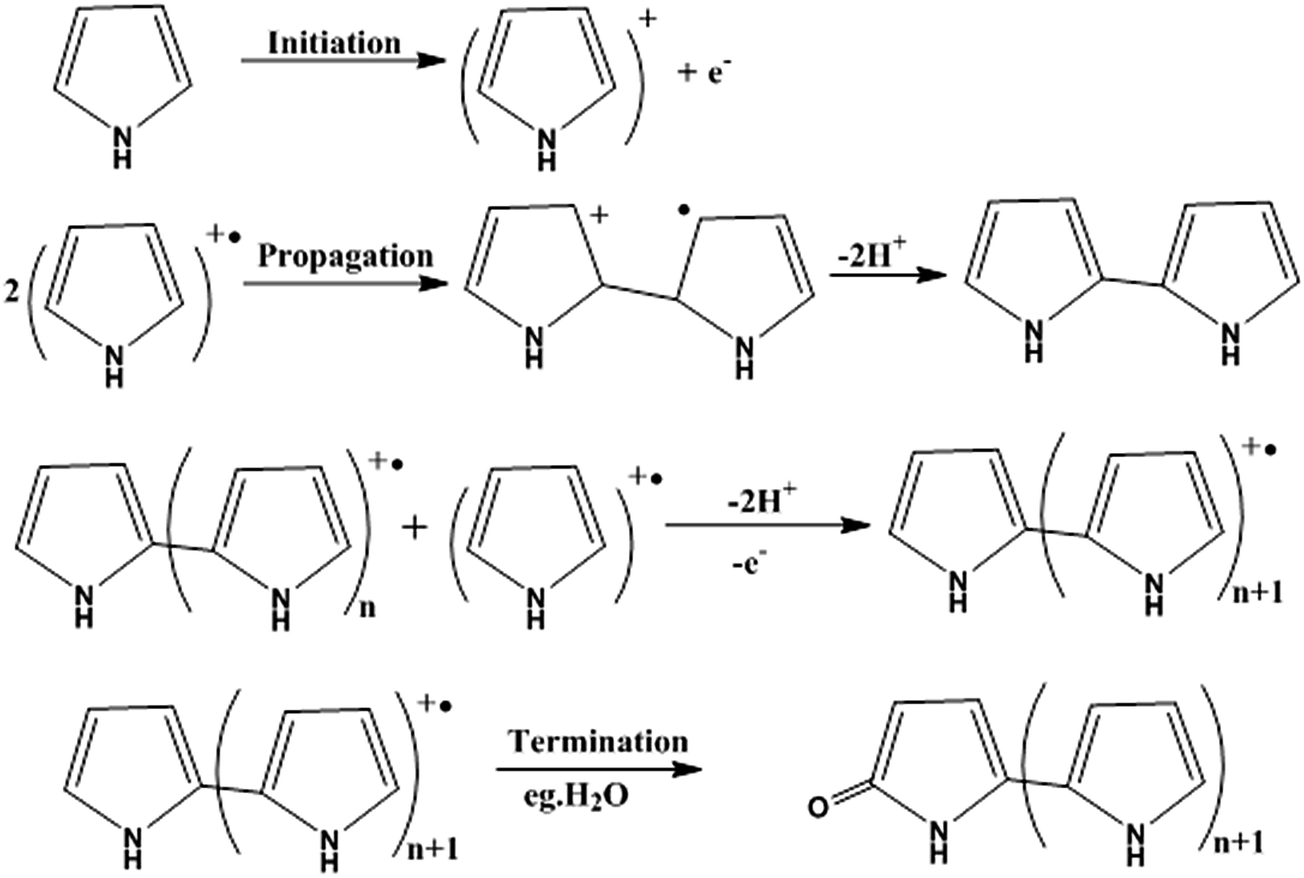 | ||
| Scheme 4 Mechanism for the electropolymerization of pyrrole, which is also valid for many other conjugated conducting polymers. | ||
In general, oxidation is used to provide the driving force for polymer synthesis, whether this is via the addition of a chemical oxidant (ammonium persulfate) or by the application of an oxidizing electrochemical potential to the surface of a submerged electrode.79 As this process is carried out in an oxidizing environment, the ensuing polymers are formed in an already doped state. For example, PANI is synthesized in the emeraldine salt form in oxidative conditions, whereas reductive polymerization produces polythiophene as an n-type polymer.
Recently, nanostructured conducting polymers have been synthesized by versatile methods which are suitable for their applications. Long et al. discussed the synthetic methods in detail.80 The hard-template method provides aligned arrays of tubes and wires with controllable length and diameter, with the disadvantage that a post-synthetic process is needed to remove the template. For example, MnO2/poly(3,4-ethylenedioxythiophene) (PEDOT) coaxial nanowires were synthesized by co-electrodeposition in a porous alumina template. The soft-template method is another powerful and popular technique for producing CPs, but it does not provide uniform morphology in samples, for example, PANI micro/nanostructures. By now, structure-directing molecules, surface micelles, surfactants, liquid crystal or colloidal particles or phases, and aniline oligomers used as soft templates, have been developed, as have rapidly mixed reactions, reverse emulsion polymerization, template-free methods, ultrasonic irradiation, interfacial polymerization, dilute polymerization, and radiolytic synthesis.81–89 Kaner et al. proposed the interfacial polymerization method, which involves step polymerization of two monomers respectively dissolved in two immiscible phases; therefore, the reaction takes place at the interface between the two liquids. It is usually based on self-assembly mechanisms caused by hydrogen bonding, van der Waals forces, double-bond stacking, and electrostatic interactions as driving forces.81,82 Wan proposed the template-free method as simple self-assembly without an external template. By controlling synthesis conditions, including temperature and the molar ratio of monomer to dopant, polyaniline and polypyrrole nanostructures can be synthesized by in situ doping polymerization in the presence of dopants. In the self-assembly formation mechanism the micelles formed by dopants and/or monomers act as soft templates in the process of forming tubes/wires.84 Up to now, a variety of polyaniline micro/nanostructures have been established.
Electrospinning is an efficient approach for constructing lengthy polymer fibers with diameters from microns to the nanoscale using strong electrostatic forces90,91 (Fig. 3). In the electrospinning process, a polymer solution is extruded from an orifice to form a small droplet in the electric field which is present, and jets of the charged solution are extruded from a cone. Usually, extension of the fluid occurs uniformly first, and then the straight lines of flow undergo a vigorous whipping or splitting motion due to fluid instability and electrically driven bending instability. Then, the spun fibers are set down in the form of a non-woven web on a collector. Using an improved or modified electrospinning device, nanofibers with partial or even good orientation could be fabricated.90,91 For example, micro- and nanoscale fibers of polyaniline/polyethylene oxide (PEO) have been prepared.91 Soft lithography is a low-cost approach for shaping an initially flat polymer film using a micro-mold at high temperature or in the presence of solvent vapours. Recently, Huang et al. proposed a technique based on nanoimprint lithography and a lift-off process for patterning CPs.92 Directed by an electrochemical technique for nanowire assembly, CP wire is electrochemically polymerized and assembled onto two biased anode and cathode electrodes immersed in aqueous monomer solutions.93 Nanolithography is used for the fabrication of luminescent nanostructures and conductive polymer nanowires of controlled size using a dip pen.94 Recently, the whisker method has been used for the production of self-assembled conducting polymer nanofibers by anisotropic crystallization in a nematic liquid crystal and achieved large-scale alignment of nanofibers.95
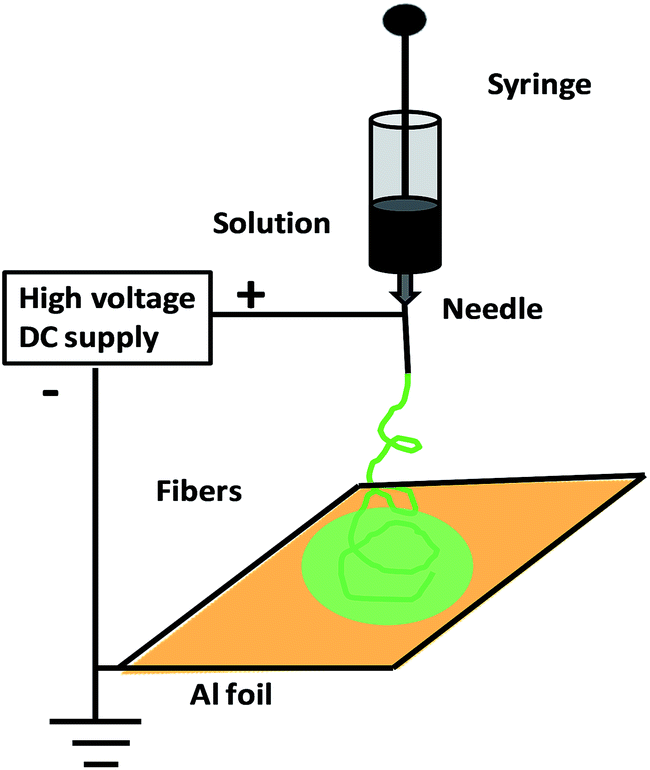 | ||
| Fig. 3 Schematic of electrospinning method for the synthesis of conductive polymer nanofibers. | ||
1.3. Functions of conducting polymers
To realize the functions of conducting polymers within the structure of a battery electrode, firstly we have to assess the basic properties of conducting polymers themselves. Their conductivity and charge storage ability depend on electronic properties not possessed by the aliphatic group of polymers, which arise from the overlap of adjacent p-orbitals. The backbone of a conjugated polymer and its molecular orbitals provide a band structure similar to that observed in inorganic semiconductors.96 The effective conjugation length is the point at which no extra monomer units added to the chain affect the electronic properties of the polymer.97 The energy levels of the HOMO and LUMO conduction band of a polymer help determine its stability properties, its optical absorption wavelengths, and whether a polymer is more likely to be n-type or p-type.98 The HOMO increases in energy and LUMO decreases in energy with increasing conjugation length. A polymer is more susceptible to electrophiles, i.e. more reactive, with a higher HOMO.Polymers are classified as reduced (n-type) and oxidized (p-type) according to their dopant counterions. Due to their ability to stabilize positive charge, electron-rich polymers that have elevated HOMO levels comprise p-type materials. Due to their ability to stabilize negative charge, electron-poor polymers that have low-lying LUMO levels comprise n-type materials.99 The p-doped polymers act as cathodes and there has been much work on them, but little focus on n-doped polymers acting as anodes.100 Both electrodes must be used in flexible polymer batteries and therefore there is a need to research new n-type electrode materials as well. Polymers can be doped via either chemical or electrochemical processes.101 A model of the doping process is presented in Fig. 4 for p-doping and n-doping. Doping generates radical cations or anions (positive/negative polarons), which are delocalized in the polymer backbone and provide a proposed mechanism of conductivity.102 Charge delocalization is coupled with structural reorganization into a quinoidal form and a change in fundamental molecular vibrations.102 UV/vis and IR/Raman spectra can be used to confirm the appearance of new sub-gap energy states and the formation of a quinoidal structure.102 Generally, polymers in their native state have very low conductivities, of the order of 10−4–10−9 S cm−1, but can attain high conductivities when doped.103 Redox reactions coupled with polymer doping coexist with the movement of ions into and out of the polymer matrix to provide charge neutrality.79 Although oxidation generally results in movement of anions within the film, movement of cations occurs in certain conditions as well.104 Movement can also occur for anions during polymer reduction. Growing dense polymer films tend to react more slowly due to ion transport limitations, which cause lower capacities and lower energy and power densities.105 Nanostructured polymers such as nanowires, nanotubes, and nanoparticles can help alleviate trouble associated with ion diffusion by increasing the surface area and porosity.105 By promoting oxidative stability, ion diffusion also influences electrode performance. The specific capacity of an electrode is determined by the number of monomer units over which charge is delocalized. This amounts to one unit of charge for every two to three monomer units, although higher levels of charge are likely for most polymers.79 Higher levels of oxidation or reduction lead to a higher specific capacity in polymers, but high levels of charge can lead to deterioration of the polymer matrix via useless inter-chain interactions or the collapse of polymer chains.106 Highly charged species are much more likely to react with common electrolytes and oxygen in the air as well.102 Low capacities can be alleviated to a limited extent by the selection of dopant ion, electrolyte, or polymer structure.106 There is evidence that strong interactions between highly charged or doped polymers and a complementary polyion can serve to stabilize well-oxidized polymers.107 Deficiencies associated with stability over many charge cycles have seen enhancements via selected composite materials of carbon nanotubes or graphene.108
 | ||
| Fig. 4 Design of polyphenylene electronic and chemical structural changes during (a) oxidation (p-doping) and (b) reduction (n-doping). A radical cation (positive polaron) is formed via oxidation, while a radical anion (negative polaron) is formed during reduction. High states of charge can lead to the formation of positive and negative bipolarons. Cations and anions counterbalance charges in the polymer chain. | ||
1.4. Merits and demerits of conducting polymers
Most research interest in the conducting polymers, such as polyaniline (PANI), polypyrrole, and polythiophene (PT), continues to grow due to their oxidative stability, relatively high theoretical capacity, simple synthesis, and electrochemical reversibility.34 PANI electrodes make use of reversible reactions between fully reduced and partially oxidized salt states. Their cycling stability can break down as the emeraldine salt is irreversibly oxidized to pernigraniline base, due to the reaction pathway to the pernigraniline salt not being favoured at higher potentials.109 There have been some efforts to stabilize the pernigraniline salt and allow an increase in PANI's capacity along with its energy density.41 However, although it has many merits, difficulties with the utilization of PANI stem from its insolubility, diffusion issues, cycling, and stability. Insolubility and uncontrollability are usually considered the main issues obstructing its usage in energy storage.110 These problems had traditionally been addressed by dispersing the polymer in solutions using a series of steps intended to remove aggregates and ensure a stable suspension.111 Polyanions with PANI nanofibers using a template have allowed easy suspension in solution for PANI112 and assembled with a carbon composite have helped maintain conductivity when PANI has been reduced to a non-conductive state, and so offer promising energy storage.110–112 However, they can suffer from inferior capacity vs. redox-active materials due to the non-Faradaic nature of charge storage.113,114 PANI nanostructures can also be produced economically and quickly in solution in the absence of templates, additives, or difficult techniques,115 but they have uncontrollability issues because they are easily suspended in solution and stay so for extended periods of time.112 PANI is a promising material for composites with high-capacity inorganic and carbon-based materials, which allows a facile solution. Our work has demonstrated good conductivity with even lower loadings of PANI/polyelectrolyte (poly(diallyldimethylammonium chloride)), such as 4.5 to 42 μS cm−1.16 Overall, investigation of PANI composites demonstrated significant effects on energy devices because the two components work together synergistically to improve coulombic efficiency and charge transport. There are methods for boosting charge capacity, without compromising the properties of a polymer, by including redox-active components in the polymer structure. Goodenough et al. reported developments in both the capacity and long-term cyclability of polypyrrole by covalently anchoring ferrocene groups to the polymer.116 Ferrocene-functionalized triphenylamine polymer and 2,2,6,6-tetramethylpiperidine-N-oxide-functionalized PT have also been synthesized.117,118 Some enhancement was also observed for PEDOT and PT,115 because, as the Fermi level of the cyanide anion was close to that of the polymers used in the study, the anion served as a mediator for charge transfer between them119. Morphological changes in poly(3-alkylthiophenes) have been linked to enhanced capacitance, conductivity, and stability.120 A major concern is the lack of reliable synthetic methods for making large quantities of n-type polymer. In n-type polyfluorene stabilizing silicon nanoparticles in a composite anode,121 the increased cycle life is attributed to reversible lithium doping in the polymer and the formation of an intimate composite. Furthermore, side-chain functionalization can be used to stabilize polythiophenes for n-doping.122 Another concern is that diffusion limitations cause poor response times at high current densities with low power efficiencies. Self-doped polymers with ionic side chains help to provide fast response times and improved processability, but can suffer from low conductivities.123,124 This can be alleviated to an extent by nanostructuring.124–126 Overcoming diffusion limitations suggested that block copolymers of conducting PT and poly(ethylene oxide) were used to form films; the resulting material was successfully employed in a solid-state battery.127,1282. Battery electrodes based on inorganic nanoparticle-conducting polymer composite
Inorganic nanoparticle–polymer composite electrodes are promising systems for secondary battery applications, due to their unusual combination of properties derived from the different building blocks. The flexibility and stability of composite electrodes arise from their polymer components and functionality. The general concept of designing a composite starts from a nanosized combination of polymers, metal particles and functional molecules. Composite materials are classified based on their interaction: (a) a functionalised polymer or metal precursor undergoes strong interaction via covalent and ionic bonds with amino-functionalised silicon nanoparticles and is polymerised129 and (b) polymers embedded with inorganic nanoparticles, e.g. physical or chemical attachment of an iron complex to the backbone of a conducting polymer leads to stabilization of its charge/discharge characteristics and higher electrode capacities in LiFePO4.1162.1. Materials synthesis
Nanostructured polymer–inorganic hybrid composites can often produce synergistic benefits based on their chemical composition and physical structure for applications as particular functional materials.116 As the fastest-growing battery system, lithium ions (lithium-ion batteries) are used where high energy density and light weight are of prime importance. The device is fragile and a protection circuit is necessary to ensure safety.130 Conducting polymers embedded with inorganic nanoparticles are suitable materials for high-performance electrodes in batteries due to their synergistic effects.131,132 They provide good electrical conductivity, the polymer acts as a stress buffer and they promote transport of both electrons and ions owing to the availability of short diffusion paths and good connectivity. In addition, good catalytic, magnetic, semiconducting and sensing properties, depending on the kind of nanoparticles and polymers and their characteristics, and the effects of the electrolyte concentration as well as the other reaction parameters on the morphology/properties of the final composite should be determined, controlled, improved and characterized in a very precise and careful manner during synthesis reactions.10,80 Recently, several methods have also been employed for the synthesis of hybrid composites for different applications (see Table 3). Using facile one-step seeding-template-assisted oxidative polymerization reactions with different oxidizing agents, these are likely to generate core–shell or network-like composite structures that are composed of conducting polymers with different morphologies decorated with the abovementioned inorganic nanoparticles.133–137 Within 0-D to 3-D matrices of these composites, unique “size-dependent” sensing properties and high catalytic ability were achieved. Furthermore, in hybrid composite electrodes, a considerable change in the shape, size, distribution, or connectivity of each phase during cycling may result in undesirable redistribution or segregation of phases. This may cause electrical isolation of active electrode materials (and so capacity fading), deteriorating connectivity between carbon particles (increased resistance to current collection and charge transfer), and a decline in transport of electroactive reactants to (or products from) active sites (increased resistance to mass transfer and reduced rate of charge and discharge). For example, in a change in morphology during charging, formation of lithium dendrites may cause partial shorting of the two electrodes and, eventually, complete failure of battery operation.138 Several groups have proposed the synthesis of conducting polymers with silicon, tin, vanadium oxide nanoparticles, carbon, etc. Nazar et al.139 and Buttry et al.140 polymerized PANI in the presence of V2O5 to make inorganic/organic hybrid electrodes. The Li+ ion diffusion coefficient for the PANI/V2O5 composite was 10 times that of V2O5, and the capacity was also superior to that of V2O5 xerogel. Cui et al. carried out infusion of a PANI hydrogel into Si-based anodes: the hydrogel was polymerized in situ, resulting in a well-connected three-dimensional network structure consisting of SiNPs conformally coated by PANI. With this anode, a cycle life of 5000 cycles was demonstrated with over 90% capacity retention at 6.0 A g−1 current density.13 A SnNPs/PPy composite was prepared by chemically reducing and coating SnNPs onto a PPy surface. The composite has a much higher surface area than a pure nano-Sn reference sample, due to the higher surface area of porous PPy and the much smaller particle size of Sn in the nano-Sn/PPy composite than in the pure tin nanoparticle sample.141 A polyaniline (PANI)/multi-walled carbon nanotube (CNT) composite cathode was prepared by in situ chemical polymerization of aniline in a well-dispersed CNT solution. The cell delivered a maximum discharge capacity of 86 mA h g−1 at the 80th cycle with an average coulombic efficiency of 98%.142 A secondary lithium metal battery modified by a PANI–CNT nanoporous composite buffer layer was fabricated to decrease the safety risk of the secondary Li metal battery in cycles of recharging processes and improve its cycle life in the future.143| Methods | Advantages | Disadvantages |
|---|---|---|
| Ex situ methods145 | More suitable for large-scale industrial applications than the in situ method | The preparation of particles that possess higher dispersibility in the polymer and exhibit long-term stability against aggregation; in this process the dispersion of nanoparticles in the polymer matrix is difficult |
| In situ methods146 | Prevents the agglomeration of inorganic particles while retaining good spatial distribution in the polymer matrix | The un-reacted products of the in situ reaction might influence the properties of the final materials |
| Mechanical methods154–159 | The cost of power and a grinding medium is low; it is suitable for both batch and continuous operation and open- and closed-circuit grinding and applicable to materials of all degrees of hardness | Contamination, long processing time, no control of particle morphology, agglomerates, and residual strain in the crystallized phase |
| Blending inorganic nanoparticles into polymer matrix160–166 | Reproducible fabrication with controlled levels of electrical conductivity, retaining the desired mechanical properties of the matrix, low-cost synthesis, avoiding oxidants or reductant chemicals | Difficulty in desired structure and properties, uniform low distribution of particles |
| Nanostructured polymer with inorganic particles167–173 | Well-connected inorganic nanoparticles in polymers via chemical or physical bonding, soluble systems if they maintain their solubility after nanoparticle formation, potential use as thin films or free-standing films | Requires choice of the functional group to be introduced to polymers for interaction with inorganic particles |
| Sol–gel process175–184 | This method provides good crystallinity, homogeneous mixing at the atomic or molecular level, low synthesis temperature, short heating time, good stoichiometric control, uniform particle size and small diameter down to the nanometer level | High shrinkage, low yield, high precursor costs, acidic gases evolved during pyrolysis, limited shelf life of sols |
| Surfactant-free solution processes199 | Simple physical attraction process without external surfactants | Relatively poor control of uniformity of shape and diameter |
| Layer-by-layer200 | Simple, cheap, and thin-film deposition method. Its important feature is the high degree of control over thickness, which arises due to linear growth of films with a number of bilayers | Slow deposition, waste of materials, and small surface areas |
| Self-assembly201 | Large variety of shapes and functions on many length scales can be obtained | Synthesis more problematic due to the many free parameters that require control |
| Microemulsion polymerization method215 | Enables fast polymerization rates without loss of temperature control, the viscosity of the reaction medium remains close to that of water, and the final product can be used as is and does not generally need to be altered or processed | An unwanted product is difficult to remove from the final composite and removal of water is an energy-intensive process for isolating composites |
| Template method222 | Controllable length and diameter of nanotubes or wires | A post-synthetic process is needed to remove the template |
| Ion exchange process225 | Reversible process which can be regenerated or loaded with desirable ions by washing with an excess of these ions | The final product formed can be dependent on the size of the ions, their charge, or their structure |
2.2. Ex situ and in situ approaches
Most convenient and attractive routes to the fabrication of polymer composites embedded with inorganic nanoparticles (INPs) involve the following methods. In the ex situ approach, inorganic nanoparticles are first produced by soft-chemistry routes and then dispersed into polymeric matrices, whereas in the in situ approach metals are generated inside a polymer matrix by decomposition (e.g., thermolysis, photolysis, radiolysis, etc.) or chemical reduction of a metallic precursor dissolved in the polymer. These types of polymer implanted with INPs have been extensively studied for energy and electrical devices. The regulated porosity of polymers greatly affects the stability of electrodes in batteries. A porous polymer matrix has vacant space to allow a large expansion in volume and even assist cracked INPs trapped inside CPs with interconnected pores, and a highly conductive coating helps provide good electrical connectivity for INPs during discharge/charge. To enhance the lithium storage abilities of inorganic oxide-based anodes for LIBs, nanopainting with a thin layer of conducting polymer has been investigated.144 A typical layered transition metal sulfide (MoS2) has a S–Mo–S layer with a structure similar to graphite together with van der Waals forces, which can facilitate Li+ insertion/extraction. Poor conductivity between two adjacent S–Mo–S sheets can be improved by PANI via an in situ method to provide anode materials exhibiting high capacity and good cyclability for Li-ion batteries.145 LiFePO4 particles generally have problems associated with slow diffusion of Li ions across the two-phase boundary and/or low conductivity, which were overcome by preparing a composite by an in situ polymerization restriction method, in which Fe3+ acts as a precipitating agent for PO43− and an oxidant for aniline polymerization, and then coating it on graphite (1–2 nm). This material offered a good rate performance of 80 mA h g−1 at 60 C rate (Fig. 5).146 Ding et al. proposed a new anode material CoCO3 using two-step conversion reactions with a total theoretical value of 7 Li per CoCO3. The first reaction involves reduction of CoCO3 to metallic Co and formation of Li2CO3 and the second reaction involves further reduction of Li2CO3 to LixC2 (x = 0, 1, 2), along with formation of Li2O. The low stability of the electrode was increased by mixing it with PPy to form a CoCO3–PPy composite, which exhibited remarkable cycling stability and reversibility, appearing to be a very promising anode material for LIBs (Fig. 5).147 High-capacity Si and Sn anodes underwent huge volume expansion during cycling, which was controlled by PEDOT:PSS. This was embedded by in situ polymerization of EDOT in an aqueous solution of PSS with dispersed nanosized Si particles and subsequent carbonization of Si/PEDOT:PSS, which retained a specific capacity of 768 mA h g−1 and 99.2% CE after 80 cycles.148 A nano-Sn–polypyrrole composite with CMC as binder displayed both superior capacity retention and good rate capability. It demonstrated that both CMC and polypyrrole (PPy) can act as composite binders and prevent the formation of cracks in electrodes during the charge–discharge process, resulting in good cycling stability, despite the large volume changes.141 Generally, poor electronic conductivity of pristine anode and cathode materials hampers commercial applications. To improve electrode performance, conducting polymers are introduced by an in situ/ex situ preparation method. Arbizzani et al. introduced PEDOT onto Li1.03Mn1.97O4 by oxidation and subsequent polymerization of EDOT on Li1.03Mn1.97O4 particles promoted by itself, due to its oxidative property.149 To improve the cycling process of a LiMn2O4/LixV2O5 lithium-ion cell, coating with PPy on the surface of the anode was performed via an in situ polymerization method.150 PANI was intercalated into MnO2, which has a swollen layered structure, with uniform mesoporous structure, typical nano-size, and high surface area, resulting in high electrochemical performance for Li storage.151 Enhanced electrochemical performance of β-AgVO3/PANI triaxial nanowires was achieved via an in situ method.152 A V2O5/PPy hybrid was prepared for improving performance by 20%.153 The basic routes for preparation of hybrid composites are in situ synthesis of INPs, polymers or both components simultaneously. Mixture of final components or precursors can be obtained.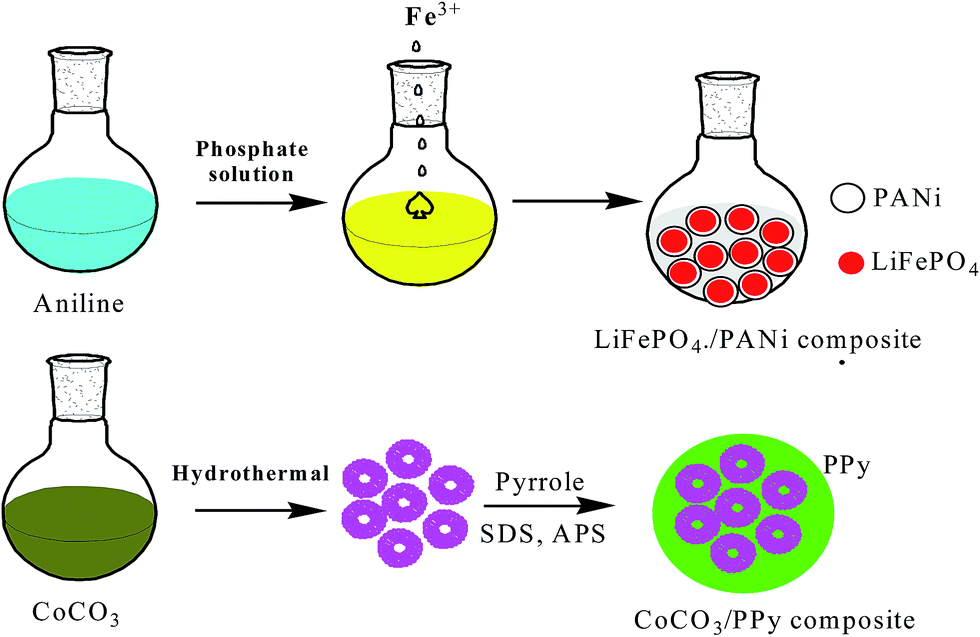 | ||
| Fig. 5 Design of LiFePO4/carbon and CoCO3/PPy composites including an in situ polymerization reaction and two typical restriction processes. | ||
2.3. Mechanical methods
A grinder with high energy is used to crush material into extremely fine powder, which is known as the ball milling method. It is suitable for batch as well as continuous operation, materials of all degrees of hardness, and open- as well as closed-circuit grinding. The vast network of a PPy matrix is a suitable environment to buffer the volume change associated with LixSi alloying and dealloying reactions. The capacities of the composites were dependent on the amount of silicon added. A series of novel high-capacity Si/PPy composites was prepared by high-energy mechanical milling techniques.154 Some 10 wt% PPy nanowires were introduced to Si particles prepared by high-energy mechanical milling to provide better reversibility and cycle life than for silicon, whereas a silicon/PPy microparticles composite displayed no improvement, because PPy NWs and PPy microparticles act as a matrix to hold active silicon grains as they repeatedly alloy with lithium during the operation of LIBs.155 Metal oxides are semiconductors, which cannot provide higher capacity. However, mixing with polymers can deliver a reversible capacity more than ten times that of plain MnO2-based devices. Reversible capacities were in the range 400–1000 mA h g−1 at 120 mA g−1 in the 20th cycle.156 Furthermore, to enhance the long-term cyclability of a Si anode, SiNPs were embedded into a Li+-conducting polymer (polyparaphenylene) (PPP) simply by ball-milling SiNPs with PPP as a core–shell structure, in which nano-Si cores act as an active Li storage phase and the polymeric matrix as a strong buffer to accommodate the volume change, as well as a protective barrier to prevent direct contact of the Si surface with the electrolyte. A Si/PPP composite exhibits a high capacity of 3184 mA h g−1 with 78% initial CE and 1670 mA h g−1 at 16 A g−1, along with long-term cyclability, with 60% capacity retention over 400 cycles (Fig. 6).157 More recently, high-capacity VO4 was synthesized from stoichiometric amounts of In2O3 and V2O5 in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixed using a ball mill for 18 h, which has a surface area of 0.49 to 9.28 m2 g−1 and a capacity about 1200 mA h g−1.158 A substantial change in shape during cycling, resulting in rapid capacity fading, has so far limited the success of this material. Composites that somehow remedy this detrimental effect have been studied, among these composites of silicon with PPy by Guo et al.159
:1 mixed using a ball mill for 18 h, which has a surface area of 0.49 to 9.28 m2 g−1 and a capacity about 1200 mA h g−1.158 A substantial change in shape during cycling, resulting in rapid capacity fading, has so far limited the success of this material. Composites that somehow remedy this detrimental effect have been studied, among these composites of silicon with PPy by Guo et al.159
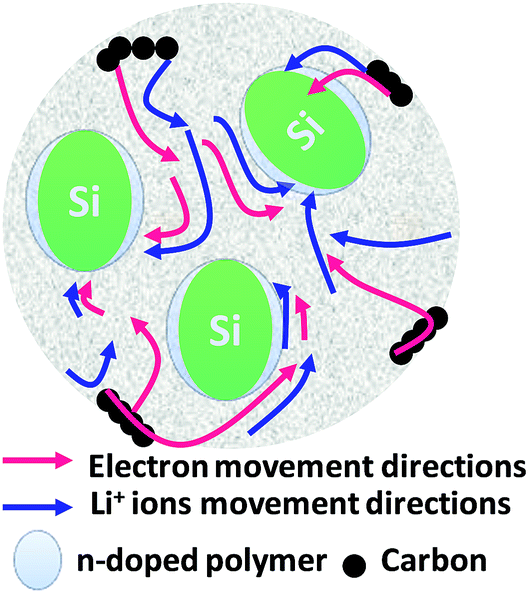 | ||
| Fig. 6 Silicon nanoparticles with n-doped polymer for volume change buffering in nano-Si particles. | ||
2.4. Blending inorganic nanoparticles into polymer matrix
By simply pressing LiMn2O4 and PPy powder without using any binder to give a pellet anode material at potentials ranging between 4.3 and 3.2 V vs. Li/Li+, PPy works well as a conducting matrix for the redox reaction LiMn2O4 ⇌ Li1−xMn2O4 + xLi+ + xe−. PPy also behaved like a capacitor and contributed to the capacity density of the LiMn2O4/PPy composite.160 A thin flexible polypyrrole–lithium iron phosphate (PPy–LiFePO4) composite has been developed to enhance the electrical conductivity of the cathode (Fig. 7).161 Deposition of PPy in hollow spherical crystalline V2O5 yielded a composite with slightly reduced capacity but increased cycling stability.162 PPy coated on small particles of V2O5 evaluated as the positive mass of a lithium-ion battery by Zhao et al. displayed improved cyclability.163 Using pyridine sulfonic acid as an additive during preparation of a V2O5/PPy hybrid material resulted in a capacity increase of 20%.164 Differences between core/shell and host/guest nanocomposites of V2O5/PPy have been studied by Posudievsky et al.165 Therefore, addition of a chemical oxidant and associated problems caused by an excess of this oxidant in the reaction system and possibly the final product could be avoided. The observed capacity of this positive mass was 163 mA h g−1, which was close to the theoretical capacity of 170 mA h g−1 for LiFePO4, with superior high-rate performance attributed to the improved lithium ion diffusivity of the composite material. The influence of the morphology of LiFePO4 particles (donut-shape, dumbbell-shape, nanocrystalline) coated with PEDOT was studied by Dinh et al.166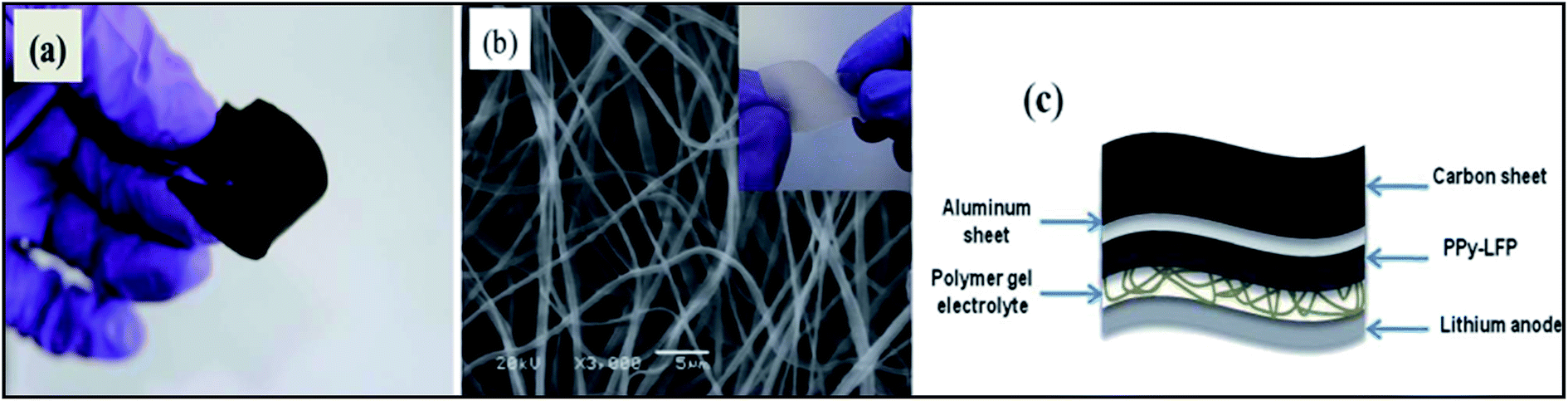 | ||
| Fig. 7 Photograph (a) of a flexible PPy–LFP electrode, (b) SEM of electrospun P(VdF-HFP) matrix and (c) scheme of the flexible battery. Inset of (b) Photograph of electrospun P(VDF-HFP). (Reprinted with permission from ref. 161, copyright 2012, Royal Society and Chemical Journal). | ||
2.5. Nanostructured polymer with inorganic particles
Functional groups of polymers interact with inorganic salts and subsequent reduction or thermal (or other) treatment results in formation of nanoparticles within a functional nanophase. Capping, reducing, stabilising or protective agents (polymers, surfactants) play important roles during the formation of nanoparticles. Incorporation of a PANI–phytic acid hydrogel into Si-based anodes via hydrogen bonding followed by crosslinking resulted in a well-connected three-dimensional network structure consisting of SiNPs conformally coated by PANI. This anode demonstrated a cycle life of 5000 cycles with over 90% capacity retention at a current density of 6.0 A g−1.11 A LiMn2O4–PPy composite was studied for its charge–discharge properties at 3 V and 4 V (Scheme 5).167,168 In order to improve electrode performance, pyrrole dispersed in V2O5 forms a hybrid composite by a chemical polymerisation method, that can influence an acidic dopant169. LiFePO4 that was exposed to hot air and regenerated with PPy as a reductant was studied.170 Excellent performance, in particular at high rates, was observed. This approach was further developed and optimized using EDOT as a monomer and its chemical oxidation by a slightly lithium-poor form of LiFePO4.171 Fabricated high-performance Li-ion battery anodes made by encapsulating SiNPs in a nanostructured 3D porous conductive polymer framework were developed.172 A hierarchical conductive hydrogel framework with carbon nanotubes as an electronic promoter with high porosity was prepared to accommodate the Si particles of a SiNPs/PPy–CNT composite by in situ polymerization for improved cycling performance in LIBs.173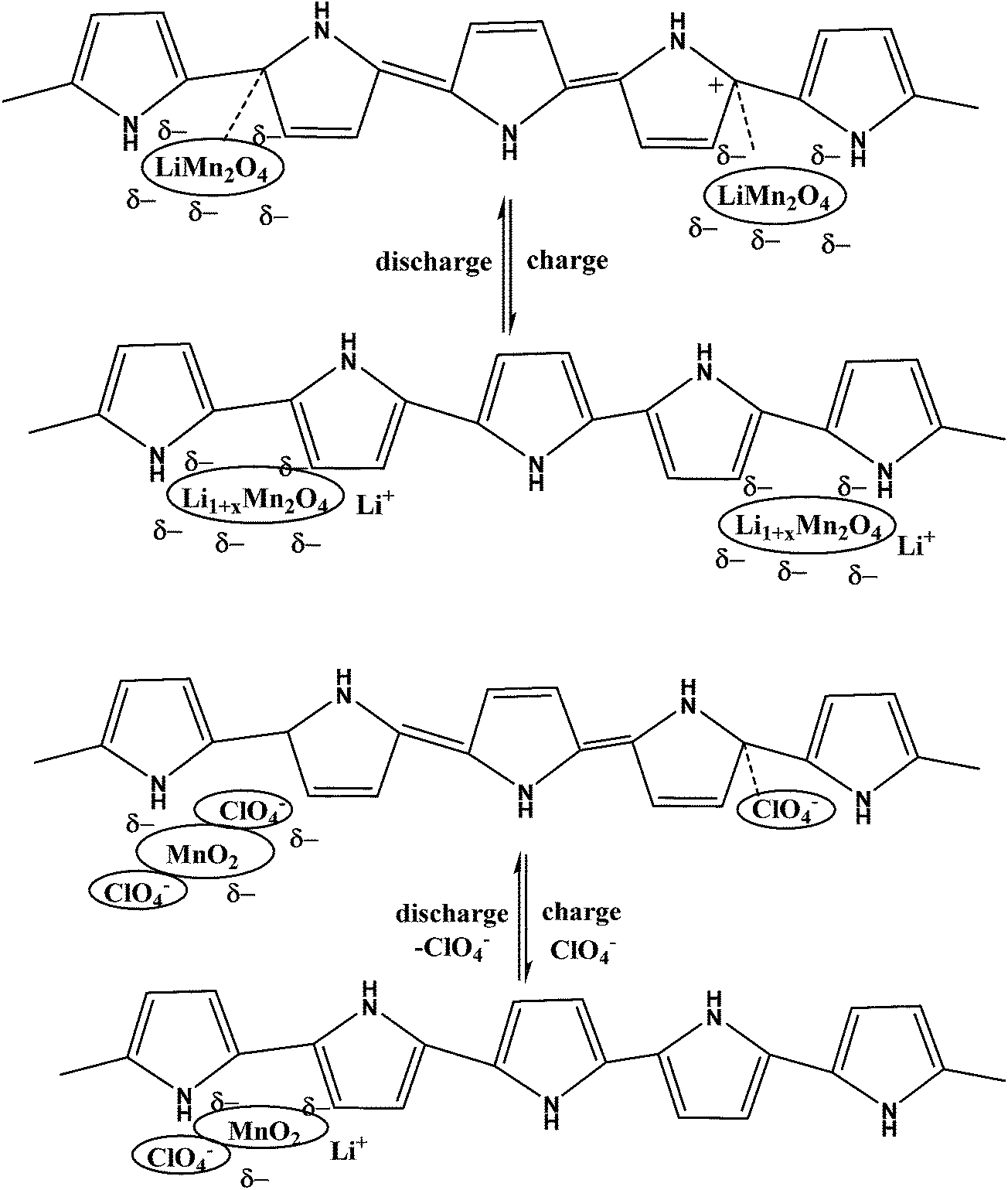 | ||
| Scheme 5 Schematic of changes in β-MnO2/PPy (a) and LiMn2O4/PPy (b) in charge–discharge reactions. | ||
2.6. Sol–gel method
Sol–gel methods are very attractive and instructive processes. They exhibit intense development accompanied by important practical applications in energy conversion and storage systems and possess significant advantages, for example, good crystallinity, homogeneous mixing at the atomic or molecular level, low synthesis temperature, short heating time, good stoichiometric control, uniform particle size and small diameter, down to the nanometer level.174 The basic processing steps of this method are precursor, hydrolysis, reactive monomer, condensation, sol gelation, gel, and treatment. Controlled preparation of a V2O5/PPy composite led to favorable reversible electrochemical properties as suggested by Ren et al.175 Furthermore, V2O5/PPy nanocomposites for Li+ ion transport were proposed.176,177 A V2O5/PPy hydrogel composite was proposed for systematic studies of spectral and structural characterisation.178 The electrochemical lithium intercalation properties of V2O5/PANI were studied.179,180 Park et al. deposited V2O5 on a layer of PANI.181 Li4Ti5O12 was prepared by the sol–gel method, followed by coating with PANI, and exhibited better rate capability and cyclability.182 Tethering a carbon-coated LiFePO4/PPy composite demonstrated enhancement of the rate capability of cathode composites.183 Core/shell structures prepared from small crystals of V2O5 coated with PPy were inferior to host/guest structures synthesized from an aerogel of V2O5 with mechanochemical polymerization of pyrrole. Nanocomposites of PEDOT and V2O5 prepared with microwave assistance displayed an increased capacity of 370 mA h g−1.1842.7. Electrochemical and chemical deposition methods
A thin film is a layer of material ranging from fractions of a nanometer (monolayer) to several micrometers in thickness. Electronic semiconductor devices and optical coatings are the main applications benefiting from thin-film construction. Polymer composite electrodes are mostly synthesized by deposition methods such as chemical or electrochemical processes. A LiFePO4 electrode is modified with PPy via monomer penetration and coating in inner pores, which would reduce surface activity and further decrease undesirable reactions with the electrolyte, while lithium intercalation would not be inhibited. This strategy is used to synthesize PPy films.185 Si/PPy core–shell nanofibers obtained by electropolymerization of a PPy nanofibers electrode followed by CVD of SiNPs are favorable for facile charge delivery and gathering, whereas the porosity of the electrode can efficiently buffer the volume expansion of Si (Fig. 8).186 By electrochemical deposition, PPy-doped LiFePO4 cathodes were prepared, and displayed poor electrochemical properties.187–190 Huang et al. reported a porous NiO/polyaniline (PANI) film obtained by depositing a PANI layer on the surface of a NiO film, which exhibited a capacity of 520 mA h g−1 at 1 C.191 Porous NiO/PEDOT films are prepared by chemical bath and electrodeposition techniques and exhibit weaker polarization and better cycling performance.192 Free-standing PPy/LiFePO4 film electrodes were prepared using the same solution, but with the addition of LiFePO4 powder. A cell with a composite film displayed a higher discharge capacity beyond 50 cycles (80 mA h g−1) than that of a cell with pure PPy (60 mA h g−1).193 Wang et al. prepared by an electrochemical method highly flexible, paper-like, free-standing PPy and PPy–LiFePO4 composite film electrodes and observed that a cell with PPy–LiFePO4 composite film exhibited a higher discharge capacity beyond 50 cycles (80 mA h g−1) than that of a cell with pure PPy (60 mA h g−1).194 LiFePO4, as a typical compound affected by conductance problems, was coated with carbon and embedded in PPy during electropolymerization of the latter compound. Improved specific capacity and rate behavior were reported, with the capacitance attributed to substitution of the inactive carbon matrix (and binder) by redox-active PPy and the rate improvement attributed to the vastly increased electronic conductance of the matrix. Carbon-coated particles of LiFePO4 were tethered to PPy by Huang et al.195 A substantial change in shape during cycling, which resulted in rapid capacity fading, has so far limited the success of this material. Composites that somehow remedy this detrimental effect have been studied, such as silicon with PPy.196 A high-capacity LIB anode SiNWs/PEDOT material was formulated by an electropolymerisation method, which maintained the mechanical integrity of the cycled Si material, along with preserving electrical connections between NWs that would otherwise have become electrically isolated during volume changes.197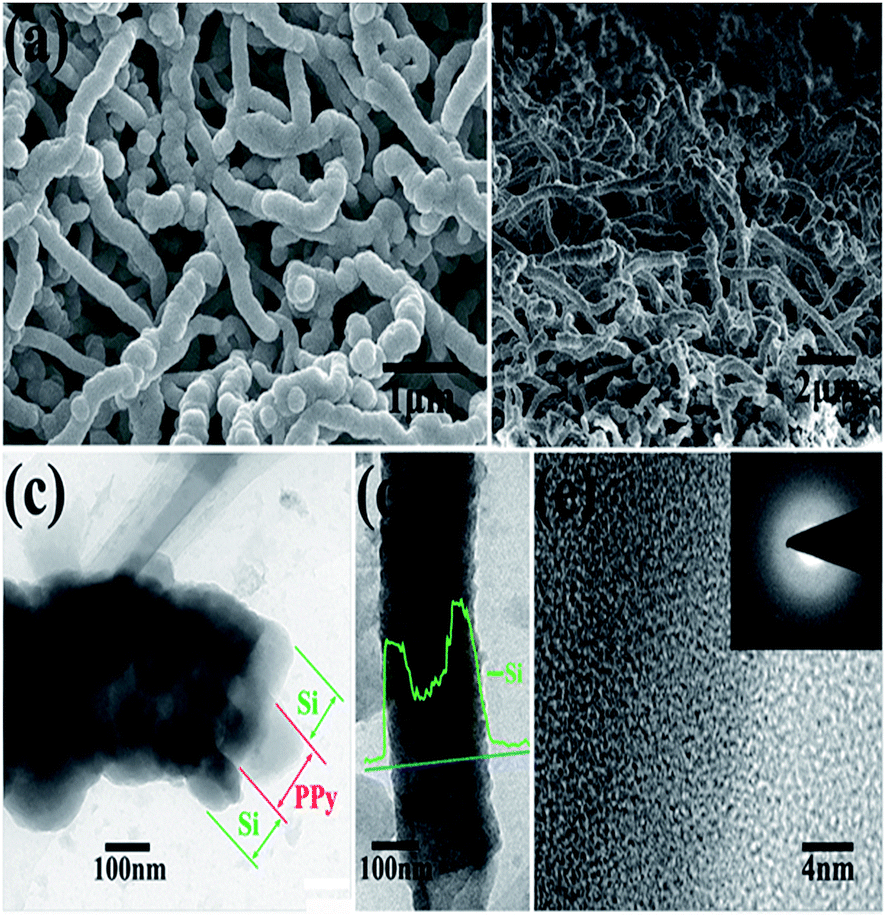 | ||
| Fig. 8 (a) and (b) top and cross-sectional SEM images of PPy–Si core–shell nanofiber network after Si deposition. (c) and (d) TEM images of the core–shell structure of Si@PPy nanocables. (e) High-resolution TEM image of Si coatings. Inset of (e) SAED image of Si coatings (reprinted with permission from ref. 186, copyright 2012, Royal Society and Chemical Journal). | ||
2.8. Miscellaneous methods
A Fe2O3-decorated polyaniline (PANI/Fe2O3) multi-channeled nanotube structure as an anode for an aqueous rechargeable lithium-ion battery (ARLIB) was made using polymerized aniline–Mo3O10 nanowires as a template. The removal of MoOx from intercalated layered MoOx/PANI structures results in a multi-channeled nanotube structure. Subsequent hydrothermal growth of Fe2O3 nanoparticles on the surface of PANI can simultaneously re-dope PANI to provide a highly conductive form, while the surfaces of PANI become relatively smooth, and growth of Fe2O3 is observed on the surface of the PANI/Fe2O3 nanocomposites, where the number of channels in each NT ranges from 2 to 5 (Fig. 9a and b). Study of CVs indicates deintercalation of Li+ at the available tetrahedral sites, and the cathodic curve shows the reverse process. The initial discharge and charge capacities of the PANI/Fe2O3 multi-channeled nanotube anode are 60.5 and 54.2 mA h g−1 at 150 mA g−1, respectively. When fabricated as an ARLIB full cell with the PANI/Fe2O3 multi-channeled nanotube anode and a LiMn2O4 cathode, an initial discharge capacity of 50.5 mA h g−1 is obtained at a current rate of 150 mA g−1, with superior capacity retention of 73.3% after over 1000 charge/discharge cycles (Fig. 9c–f).198 Iron species attached physically or chemically to the backbone of PPy comprising LiFePO4 lead to stabilization of its charge/discharge characteristics and higher electrode capacities.116 Se/PPy/graphene composites were synthesized by surfactant-free solution processes and exhibit a large discharge capacity of 678 Ah kg−1 at a current rate of C/120 when used as a Li–Se battery.199 Layer-by-layer assembly was used to improve the characteristic properties of V2O5 and PANI with a volumetric charge density of 264 mA h cm−3.200 When PANI was deposited by a self-assembly process onto carbon particles with LiFePO4, it was found that capacity increased, in particular at high rates.201 PPy-covered NiO was used to remedy the problems inherent in volume change.202 PPy can also be used to reduce charge transfer resistance associated with the Li+ ion intercalation/deintercalation reaction such as in PPy/MoO3, PPy/V2O5, PPy/LiCoO2 and PPy/LiV3O8.203–207 Javier et al. found that the theoretical capacity value of LiFePO4 was maintained using a block copolymer of poly(3-hexylthiophene) and poly(ethylene oxide).208 An α-LiFeO2–PPy nanocomposite was prepared by a chemical polymerization method as a cathode material to improve reversible capacity and cycling stability (104 mA h g−1 at 0.1 C after 100 cycles).209 To prevent contact of the nano-Si surface with the electrolyte, SiNPs were covered by a Li+-conducting polymer matrix, thus suppressing the continual rupturing and re-formation of SEI films on the Si surface.210 Han et al. discovered a multifunctional PPy/Fe2O3/C nanostructure as a stable and highly efficient anode.211 Controlled Li+ diffusion in anode materials was obtained from SnO2/PPy nanocomposites.212,213 The conductivity of SiNPs was significantly improved by PPy to prevent cracking of the electrode during cycling.214 A microemulsion polymerization method was used for a SnO2/PANI composite in anode material applications.215 SnO2-based composite coaxial nanocables with carbon-related particles (MWCNT, graphene, SWNT) anchored on conducting polymers exhibited excellent stability and high reversible capacity.216–220 TiO2-based conducting polymer composites synthesized by hydrothermal and chemical polymerization methods were developed to enable fast discharge and charge with enhanced electrochemical performance.219–221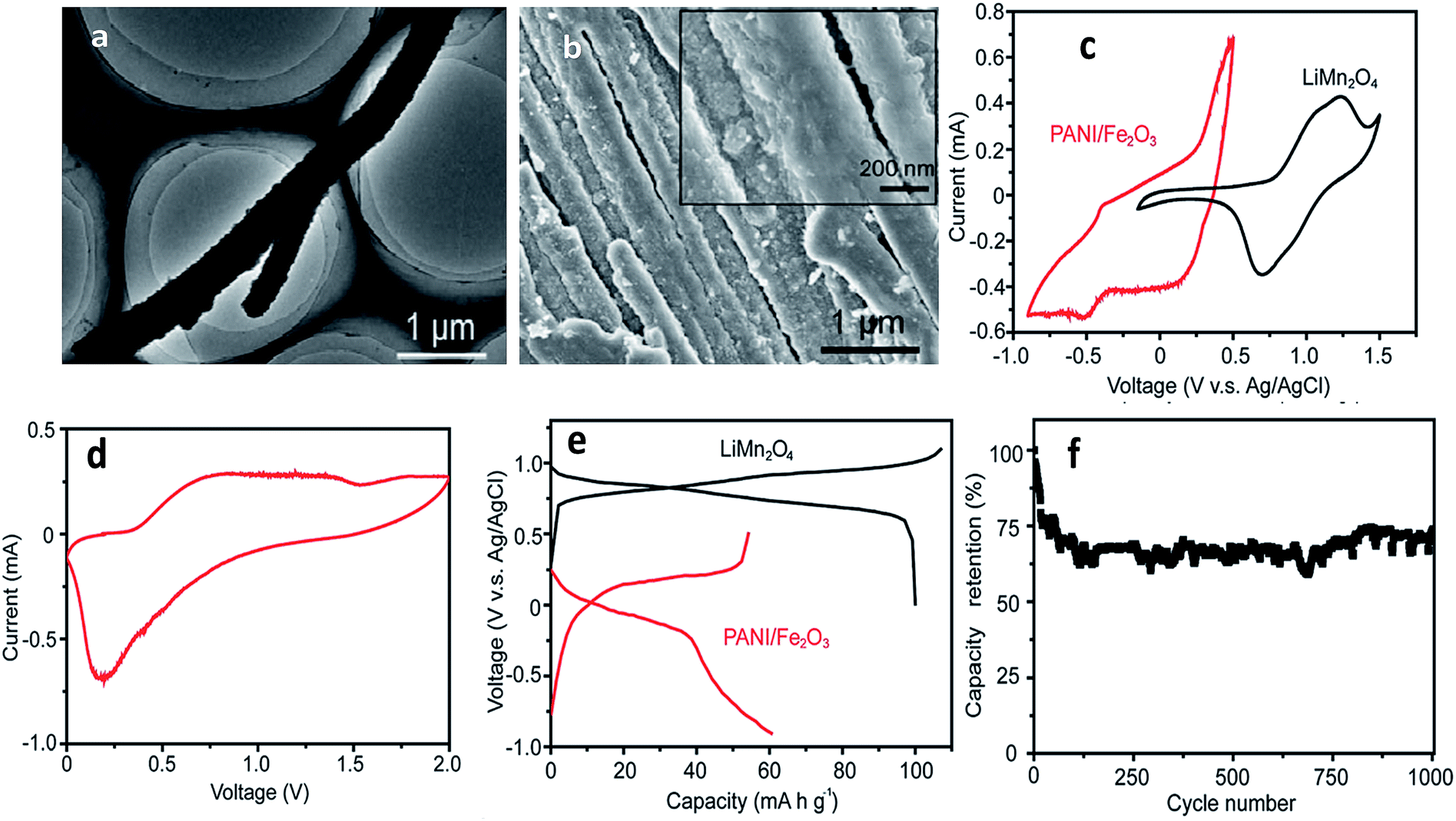 | ||
| Fig. 9 (a) TEM image of a representative re-doped PANI NT, (b) SEM image of PANI/Fe2O3 NTs. Inset: NT surface at a higher magnification, (c) cyclic voltammograms of PANI/Fe2O3 NTs and commercial LiMn2O4 in the first cycle at a scanning rate of 1 mV s−1, (d) cyclic voltammogram of ARLIB, which is assembled using PANI/Fe2O3 and commercial LiMn2O4 as anode and cathode, respectively, in the first cycle at a scanning rate of 1 mV s−1, (e) initial charge and discharge curves of PANI/Fe2O3 NTs and commercial LiMn2O4 at a current rate of 100 mA g−1 and (f) capacity retention of the (PANI/Fe2O3)/LiMn2O4 ARLIB full cell at a current rate of 150 mA g−1 (reprinted with permission from ref. 198, copyright 2014, Royal Society and Chemical Journal). | ||
Utilization of a template in the synthesis of core–shell CuO/PPy composites led to improved stability, electrical conductivity and electrochemical performance.222 Surface adsorption of PDDA on LiMn2O4 particles extended the lifetime of a LIB by retarding dissolution of Mn+, thus increasing the stability of the battery.223 The improved electrochemical performance of a NiO/PPy composite was also studied.224 A cation exchange technique was used for a high-performance cathode material (PPy/VOx–NTs).225 Dissolution of a metal oxide on a LiNi1/3Mn1/3Co1/3O2/LixV2O5/PANI composite was studied.226 Surface coating of PPy on LiFePO4 particles was carried out to improve their electronic conductivity.227 A mixed ion/electron conductor comprising PPy with PAAMPSA was used as an additive in sulfur cathodes in Li–S batteries.228 Nest-like PANI-coated SiNPs were also proposed.229 When the reverse micelle approach to V2O5/PANI was analyzed, the hybrid material seemed to stabilize the capacity due to a probable homogeneous distribution of the stress induced during cycling.230 A simple mixture of a conductive binder (SPAn) and ramsdellite MnO2 was used as a cathodic material for a lithium polymer battery.231 A concomitant ion exchange and polymerization method was used in MoO3/PANI, which provided a moderate increase in cell capacity and improved the reversibility of the Li insertion reaction.232 An organic–inorganic hybrid material was formed from PANI/hexacyanoferrate (HCF) anion; anchoring of the electroactive anion within the polymer caused its application as a functional material that harnessed the activity of the molecular species.233 A pressurized method was used for the synthesis of SnNPs/PANI which was examined for anode properties.234
2.9. Influencing the capacity, rate and life cycle
To operate a real battery, its capacity, charging rate and cycle life are very significant factors. They are associated with volume expansion, surface degradation and dissolution of active material. High-capacity lithium-ion anode materials always lead to problems with volume change and swelling after the electrodes absorb lithium because the structural integrity of the electrode has changed. Many anode materials swell to more than three times their volume when fully charged. Many methods have been suggested to solve this volume change issue. Also, metal nanoparticles need high volumes of a conductive additive (carbon) to ensure conductivity within the particle, which may reduce capacity and provide no mechanical binding force. Flexible conducting polymers can withstand large strain and maintain their conductivity as well as mechanical integrity. Recently, an n-type CP was synthesized and applied as a conductive binder in LIBs. This tailored the energy levels of polymer conduction, which helped provide adequate conductivity, and easily mechanically adhered to a Si particle surface. Electrodes based on polyaniline and PVDF exhibited poor performance due to the insulating nature of the polymer matrix, whereas Si/AB/PVDF and Si/PFFO electrodes display high initial capacity but rapid loss. PFFOMB achieved intimate electrical contact for electron conduction and mechanical integrity with the desired electronic and mechanical properties and provided good specific capacity and stable cycling performance of 2100 mA h g−1 for Si and 1400 mA h g−1 for the electrode after 650 cycles. An isosurface study indicates that electron doping from lithium to PF-type conductive polymers (PFFO) is almost complete, with different LUMO and LUMO + 1. However, for polyaniline, there is strong lithium s- and polyaniline p-hybridization with limited charge transfer. Therefore, an electron is almost completely donated to PFFO once lithium is bound to the carbonyl site of the polymer.235 SnNPs can be cycled with stable high gravimetric capacity (>500 mA h g−1) with a polyfluorene-type conductive polymer binder in composite electrodes due to them offering electrical conductivity and strong adhesion during volume change.236 The enhanced cycling stability of a conductive polymer–Si composite is related to the mesoscale synergistic functions of SiNPs and the conductive polymer. In situ TEM shows volume changes controlled by CPs which helped increase Li ion diffusion.237 A multifunctional conductive polymer binder helped maintain high electronic conductivity, mechanical adhesion, ductility, and electrolyte uptake.238To reduce the charging time, high-rate and high-voltage materials have been used. A core–shell structure of PEDOT and V2O5 with graphite foam was formulated for ultra-fast stable Li-ion storage performance, with higher capacities and improved rate (168 mA h g−1 at 60 C) and cycling capabilities.239 Iron oxide capped with PANI demonstrates a high rate capability and a high capacity of 778 mA h g−1 was retained at a higher current rate of 1.0 A g−1 after 100 cycles.240 PPy/PEG-modified sulfur/CNTs were synthesized using an in situ polymerization method and, remarkably, the battery could work at a very high current density of 8 A g−1 and retained a capacity of 480 mA h g−1 after 100 cycles.241 High rates of charge and discharge of a battery were achieved by substitution of a conductive electroactive polymer on LiFePO4.242 High rate capability of SiNPs was achieved using a PEDOT/PSS/CNT matrix, which possessed a three-dimensionally interconnected hybrid hydrogel system and was able to retain a capacity of 1200 mA h g−1 at a rate of 4.2 A g−1.243 Thin-walled hollow spherical-structured PPy with sulfur which can buffer the volume expansion during charge/discharge have been proposed for Li–S batteries (Fig. 10).244
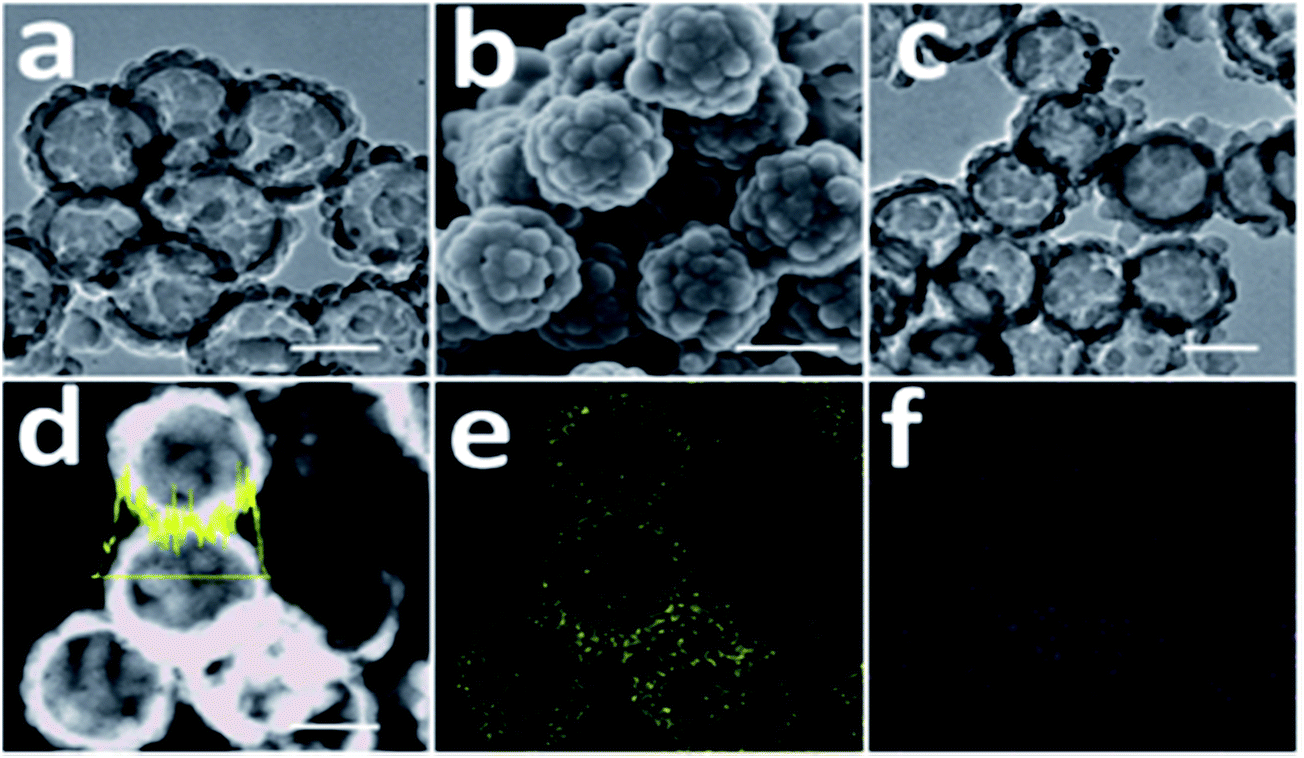 | ||
| Fig. 10 (a) TEM image of T-HSSP, (b) SEM image of T-HSSP, (c) TEM image of S@PPy-300 composite, (d) STEM image and line scan analysis (sulfur) of S@PPy-300 composite, (e) elemental mapping of sulfur in (d) and (f) elemental mapping of carbon in (d). All scale bars: 200 nm (reprinted with permission from ref. 244, copyright 2014, Royal Society and Chemical Journal). | ||
Long-term cycling of an electrode in a real battery is highly dependent on volume expansion and surface degradation of materials. If these factors are well controlled, the battery will function successfully for a long time. A CuHCF cathode and C/PPy anode combined into a full cell displayed good long-term cycling with 100% CE at a 50 C rate after 1000 cycles.245 Cui et al. reported incorporation of a PANI hydrogel into Si-based anodes via an in situ method, resulting in a well-connected 3D network structure which consisted of SiNPs conformally coated by PANI. This exhibited a cycle life of 5000 cycles with over 90% capacity retention at 6.0 A g−1.13 Strong binding of PPy onto the surface of Li2S helps limit intermediate Li2Sn species during cycling and provided a long cycle life (400 cycles). Fig. 11 shows scanning electron microscopy (SEM) images of Li2S particles coated with PPy, the formation of multiple PPy nanocolloids (700 nm) over the surface of the Li2S particles, and the appearance of a raspberry-like Li2S–PPy composite structure.246 A membrane-assisted precipitation technique was used for S/PEDOT nanocomposites and controls the volume expansion for Li–S batteries.247
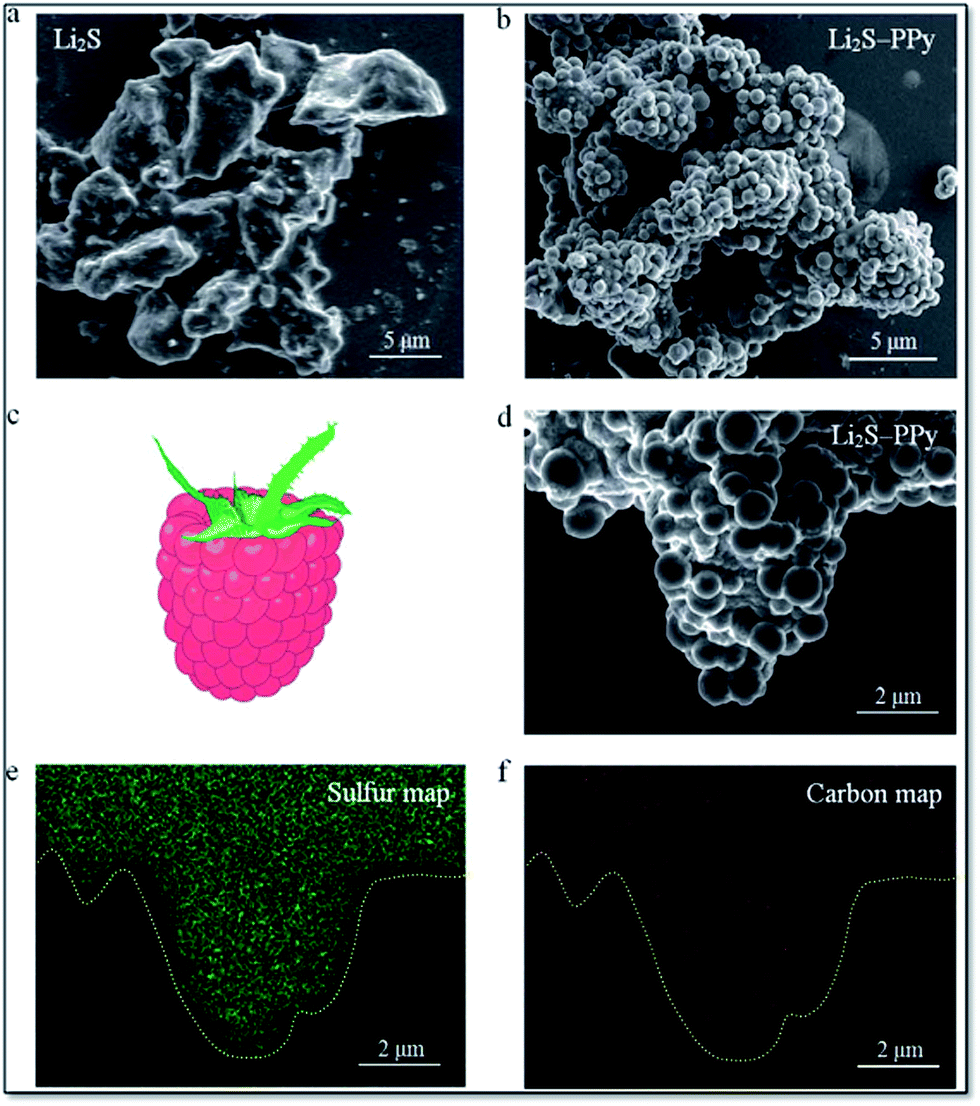 | ||
| Fig. 11 SEM images of (a) pristine Li2S particles and (b) as-synthesized Li2S–PPy composite structures. (c) Schematic of a raspberry and (d) SEM image of a representative Li2S–PPy raspberry-like composite together with the corresponding elemental maps for (e) sulfur and (f) carbon (reprinted with permission from ref. 246, copyright 2014, Royal Society and Chemical Journal). | ||
3. Summary and outlook
LIBs are in demand for powering technologies for today's portable electronics. However, these batteries have lacked commercial success because of their low stability, high cost, and relatively low energy and power density. These problems had traditionally been addressed via characterization of electrodes, electrolytes, and conductive or binder materials. Inorganic particles with polymers are a much-needed vibrant area of chemical research that has offered the opportunity to design innovative energy storage materials for higher capacity and longer cycle life of LIBs. Inorganic–polymer nanocomposites possess a high surface area, which can provide high Li+ ion flux across the electrode interface, short diffusion pathways for Li ions and electrons, abundant active sites for Li storage, and high tolerance for volume change during charge/discharge to enhance the structural stability of the electrodes. Many kinds of chemical reactions have been reported on nanocomposites that include both organic conducting polymers and metal particles using addition and exchange methods. The resulting materials have exhibited new electrochemical properties relevant to storage and catalysis applications. The growth in interest and improved success no doubt coincides with the development of nanocomposites, which provide higher surface areas, improved oxidation/reduction kinetics, and better charge storage and continue to play an important role in many clinical, environmental, industrial, pharmaceutical, defense, and security applications due to their superior properties.Battery electrodes based on inorganic–polymer nanocomposites offer high capacity, the possibility of long-term stability, and a low-cost, simple and reproducible fabrication method, and as such they require a renewed approach to their design and development. Furthermore, this will promote the development of useful and reliable battery devices. However, certain limitations and uncertainty regarding poor safety, reduced capacity, poor rate capability and conductivity are the biggest difficulty with batteries. Also, there is a need to understand the Li storage mechanisms of materials, kinetic transport at the electrode/electrolyte interface, and the development of extrapolating theoretical tools for a better fundamental understanding of the relationships between nanocomposites and their electrochemical properties. Future directions in electrode materials should focus on the search for new electrodes and the design of structures and morphology for improved power density, rate capability, service life, and safety and reduced cost. More sustainable and greener methods should be adopted for producing high-power batteries, which will limit pollution and secure a future for living systems.
Acknowledgements
We thank CRK Rao and M. Vijayan at CSIR labs of IICT and CECRI, respectively, for helpful guidance. One author (Prakash Sengodu) expresses his hearty thanks to Ms Mamali Das for supportive discussion. The authors also thank reviewers for providing valuable suggestions and making a quality paper.Notes and references
- G. A. Nazri and G. Pistoia, Lithium Batteries Science and Technology, Springer, 1st edn, 2003 Search PubMed.
- W. A. Schalkwijk and B. Scrosati, Advances in Lithium-Ion Batteries, Kluwer Academic/Plenum Publishers, 1st edn, 2002 Search PubMed.
- R. A. Huggins, Advanced Batteries Materials Science Aspects, Springer, 2008 Search PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- B. Nicolas, Focus on the US Batteries Market, US Reuters, 5 February 2009 Search PubMed.
- M. S. Islam and C. A. J. Fisher, Chem. Soc. Rev., 2014, 43, 185 RSC.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- X. Gao, W. Luo, C. Zhong, D. Wexler, S. L. Chou, H. K. Liu, Z. Shi, G. Chen, K. Ozawa and J. Z. Wang, Sci. Rep., 2014, 4, 6095 CrossRef CAS PubMed.
- K. C. White, US patent no US20100124702 A1, US 12/272,157, 2010.
- S. Prakash, T. Chakrabarty, A. K. Singh and V. K. Shahi, Biosens. Bioelectron., 2013, 41, 43 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijc, Nat. Mater., 2005, 4, 366 CrossRef PubMed.
- N. Liu, W. Li, M. Pasta and Y. Cui, Front. Phys., 2013, 9, 323 CrossRef.
- B. Liu, P. Soares, C. Checkles, Y. Zhao and G. Yu, Nano Lett., 2013, 13, 3414 CrossRef CAS PubMed.
- S. Prakash, C. R. K. Rao and M. Vijayan, Electrochim. Acta, 2009, 54, 5704 CrossRef.
- S. Prakash, C. R. K. Rao and M. Vijayan, Electrochim. Acta, 2008, 53, 5919 CrossRef.
- D. Wei, D. Cotton and T. Ryhanen, Nanomaterials, 2012, 2, 268 CrossRef CAS.
- G. Wallace, T. Campbell and P. Innis, Fibers Polym., 2007, 8, 135 CrossRef CAS.
- S. Trohalaki, MRS Bull., 2012, 37, 883 CrossRef.
- H. Wu, G. Zheng, N. Liu, T. J. Carney, Y. Yang and Y. Cui, Nano Lett., 2012, 12, 904 CrossRef CAS PubMed.
- K. Bock, Proc. IEEE, 2005, 93, 1400 CrossRef CAS.
- H. E. Katz, P. C. Searson and T. O. Poehler, J. Mater. Res., 2010, 25, 1561 CrossRef CAS.
- M. E. Abdelhamid, A. P. OMullane and G. A. Snook, RSC Adv., 2015, 5, 11611–11626 RSC.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1 CrossRef CAS.
- S. R. Forrest, Nature, 2004, 428, 911 CrossRef CAS PubMed.
- T. Tsutsui and K. Fujita, Adv. Mater., 2002, 14, 949 CrossRef CAS.
- A. Ueda, M. Nagao, A. Inoue, A. Hayashi, Y. Seino, T. Ota and M. Tatsumisago, J. Power Sources, 2013, 244, 597 CrossRef CAS.
- Q. Sun, X. Q. Zhang, F. Han, W. C. Li and A. H. Lu, J. Mater. Chem., 2012, 22, 17049 RSC.
- J. Yin, H. Cao, Z. Zhou, J. Zhang and M. Qu, J. Mater. Chem., 2012, 22, 23963 RSC.
- L. Shao, J. W. Jeon and J. L. Lutkenhaus, Chem. Mater., 2011, 24, 181 CrossRef.
- Y. H. Huang and J. B. Goodenough, Chem. Mater., 2008, 20, 7237 CrossRef CAS.
- G. Liu, S. Xun, N. Vukmirovic, X. Song, O. P. Velasco, H. Zheng, V. S. Battaglia, L. Wang and W. Yang, Adv. Mater., 2011, 23, 4679 CrossRef CAS PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419 CrossRef CAS.
- D. A. Pasquier, I. Plitz, S. Menocal and G. Amatucci, J. Power Sources, 2003, 115, 171 CrossRef.
- M. Dubarry and B. Y. Liaw, J. Power Sources, 2009, 194, 541 CrossRef CAS.
- R. Bittihn, G. Ely, F. Woeffler, H. Munstedt, H. Naarmann and D. Naegele, Makromol. Chem., Macromol. Symp., 1987, 8, 51 CrossRef CAS.
- J. Chen, J. Wang, C. Wang, C. O. Too and G. G. Wallace, J. Power Sources, 2006, 159, 708 CrossRef CAS.
- K. Kaneto, K. Yoshino and Y. Inuishi, Jpn. J. Appl. Phys., 1983, 22, L567 CrossRef.
- C. Arbizzani, M. Mastragostino and M. Rossi, Electrochem. Commun., 2002, 4, 545 CrossRef CAS.
- J. C. Carlberg and O. Inganas, J. Electrochem. Soc., 1997, 144, L61 CrossRef CAS.
- L. Shao, J. W. Jeon and J. L. Lutkenhaus, Chem. Mater., 2011, 24, 181 CrossRef.
- T. F. Otero and I. Cantero, J. Power Sources, 1999, 81, 838 CrossRef.
- J. M. Kim, H. S. Park, J. H. Park, T. H. Kim, H. K. Song and S. Y. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 12789 CAS.
- G. Kucinskis, G. Bajars and J. Kleperis, J. Power Sources, 2013, 240, 66 CrossRef CAS.
- D. Deng, M. G. Kim, J. Y. Lee and J. Cho, Energy Environ. Sci., 2009, 2, 818 CAS.
- M. R. Zamfir, H. T. Nguyen, E. Moyen, Y. H. Lee and D. Pribat, J. Mater. Chem. A, 2013, 1, 9566 CAS.
- S. L. Chou, Y. Pan, J. Z. Wang, H. K. Liu and S. X. Dou, Phys. Chem. Chem. Phys., 2014, 16, 20347 RSC.
- P. Novak, O. Haas, K. S. V. Santhanam and K. Muller, Chem. Rev., 1997, 97, 207 CrossRef CAS PubMed.
- S. B. Chikkannanavar, D. M. Bernardi and L. Liu, J. Power Sources, 2014, 248, 91 CrossRef CAS.
- V. Aravindan, J. Gnanaraj, Y. S. Lee and S. Madhavi, J. Mater. Chem. A, 2013, 1, 3518 CAS.
- J. O. Besenhard, J. Yang and M. Winter, J. Power Sources, 1997, 68, 87 CrossRef CAS.
- C. K. Chan, H. L. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31 CrossRef CAS PubMed.
- M. T. McDowell, S. W. Lee, C. Wang and Y. Cui, Nano Energy, 2012, 1, 401 CrossRef CAS.
- H. Wu and Y. Cu, Nano Today, 2012, 7, 414 CrossRef CAS.
- L. Qie, L. X. Yuan, W. X. Zhang, W. M. Chen and Y. H. Huang, J. Electrochem. Soc., 2012, 159, A1624 CrossRef CAS.
- W. M. Chen, L. Qie, L. X. Yuan, S. A. Xia, X. L. Hu, W. X. Zhang and Y. H. Huang, Electrochim. Acta, 2011, 56, 2689 CrossRef CAS.
- G. Zhang, X. Li, H. Jia, X. Pang, H. Yang, Y. Wang and K. Ding, Int. J. Electrochem. Sci., 2012, 7, 830 CAS.
- K. Park, H. Song, Y. Kim, Y. Kim, S. Mho, W. Cho and I. H. Yeo, Electrochim. Acta, 2010, 55, 8023 CrossRef CAS.
- G. M. Suppes, B. A. Deore and M. S. Freund, Langmuir, 2008, 24, 1064 CrossRef CAS PubMed.
- V. Egan, R. Bernstein, L. Hohmann, T. Tran and R. B. Kaner, Chem. Commun., 2001, 801 RSC.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309 CrossRef CAS.
- S. O. Vilela, M. A. Soto-Oviedo, A. P. F. Albers and R. Faez, Mater. Res., 2007, 10, 297 CrossRef CAS.
- H. Naarmann, Polymers, Electrically Conducting Ullmann's, Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- H. S. Nalwa, Handbook of Nanostructured Materials and Nanotechnology, ed. Academic Press, New York, NY, USA, 2000, vol. 5, p. 501 Search PubMed.
- H. Shirakawa, E. J. Lewis, A. G. MacDiarmid, C. K. Chiang and A. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578 RSC.
- H. J. Boweley, D. L. Gerrard and W. F. Maddams, Makromol. Chem., 1985, 186, 715 CrossRef.
- H. Shirakawa, Angew. Chem., Int. Ed., 2001, 40, 2574 CrossRef.
- T. Ito, H. Shirakawa and I. J. Ikeda, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 11 CrossRef CAS.
- L. Dai, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1999, 39, 273 CrossRef.
- G. L. Baker, in Electronic and Photonic Applications of Polymers, ed. M. J. Bowden and S. R. Turner, Adv. Chem. Ser. 218, ACS, Washington DC, 1988 Search PubMed.
- S. P. Armes, Curr. Opin. Colloid Interface Sci., 1996, 1, 214 CrossRef CAS.
- Carbon Rich Compounds II, Macrocyclic Oligoacetylenes and Other Linearly Conjugated Systems, ed. A. de Meijere, Springer-Verlag, Berlin, 1999 Search PubMed.
- K. Yoshino, M. Hirohata, R. Hidayat, K. Tada, T. Sada, M. Teraguchi, V. M. Frolov, S. M. Shkunov, Z. V. Vardeny and M. Hamaguchi, Synth. Met., 1997, 91, 283 CrossRef CAS.
- K. Tamao, S. Kodama, I. Nakajima, M. Kumada, A. Minato and S. Suzuki, Tetrahedron, 1982, 38, 3347 CrossRef CAS.
- J. M. Tour, Adv. Mater., 1994, 6, 190 CrossRef CAS.
- Conjugated Polymers, ed. J. L. Bredas and R. Silbey, Kluwer Academic, Dordrecht, 1991 Search PubMed.
- H. Meier, M. Lehmann and U. Kolb, Chem.–Eur. J., 2000, 6, 2462 CrossRef CAS.
- Handbook of Conducting Polymers, ed. T. A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, Marcel Dekker, New York, 1998 Search PubMed.
- J. Heinze, B. Frontana, A. Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724 CrossRef CAS PubMed.
- Y. Z. Long, M. M. Li, C. Gu, M. Wan, J. L. Duvail, Z. Liue and Z. Fan, Prog. Polym. Sci., 2011, 36, 1415 CrossRef CAS.
- J. X. Huang, S. Virji, B. H. Weiller and R. B. Kaner, J. Am. Chem. Soc., 2003, 125, 314 CrossRef CAS PubMed.
- J. X. Huang and R. B. Kaner, J. Am. Chem. Soc., 2004, 126, 851 CrossRef CAS PubMed.
- N. R. Chiou and A. J. Epstein, Adv. Mater., 2005, 17, 1679 CrossRef CAS.
- M. X. Wan, Adv. Mater., 2008, 20, 2926 CrossRef CAS.
- M. X. Wan, J. Huang and Y. Q. Shen, Synth. Met., 1999, 101, 708 CrossRef CAS.
- J. X. Huang and R. B. Kaner, Angew. Chem., Int. Ed., 2004, 43, 5817 CrossRef CAS PubMed.
- J. Jang and H. Yoon, Chem. Commun., 2003, 720 RSC.
- H. Liu, X. B. Hu, J. Y. Wang and R. I. Boughton, Macromolecules, 2002, 35, 9414 CrossRef CAS.
- S. K. Pillalamarri, F. D. Blum, A. T. Tokuhiro, J. G. Story and M. F. Bertino, Chem. Mater., 2005, 17, 227 CrossRef CAS.
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223 CrossRef CAS.
- I. D. Norris, M. M. Shaker, F. K. Ko and A. G. MacDiarmid, Synth. Met., 2000, 114, 109 CrossRef CAS.
- C. Y. Huang, B. Dong, N. Lu, N. J. Yang, L. G. Gao, L. Tian, D. P. Qi, Q. Wu and L. F. Chi, Small, 2009, 5, 583 CrossRef CAS PubMed.
- P. S. Thapa, D. J. Yu, J. P. Wicksted, J. A. Hadwiger, J. N. Barisci, R. H. Baughman and B. N. Flanders, Appl. Phys. Lett., 2009, 94, 033104 CrossRef.
- S. Samitsu, T. Shimomura, K. Ito, M. Fujimori, S. Heike and T. Hashizume, Appl. Phys. Lett., 2005, 86, 233103 CrossRef.
- S. Samitsu, Y. Takanishi and J. Yamamota, Macromolecules, 2009, 42, 4366 CrossRef CAS.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 16, 578–580 RSC.
- J. Rissler, Chem. Phys. Lett., 2004, 395, 92 CrossRef CAS.
- A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591 CrossRef CAS.
- T. Michinobu, Chem. Soc. Rev., 2011, 40, 2306 RSC.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797 RSC.
- N. Basescu, Z. X. Liu, D. Moses, A. J. Heeger, H. Naarmann and N. Theophilou, Nature, 1987, 327, 403 CrossRef CAS.
- J. L. Bredas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971 CrossRef CAS PubMed.
- M. Mueller, M. Fabretto, D. Evans, P. Hojati-Talemi, C. Gruber and P. Murphy, Polymer, 2012, 53, 2146 CrossRef CAS.
- S. Bruckenstein, K. Brzezinska and A. R. Hillman, Electrochim. Acta, 2000, 45, 3801 CrossRef CAS.
- S. I. Cho and S. B. Lee, Acc. Chem. Res., 2008, 41, 699 CrossRef CAS PubMed.
- M. D. Levi and D. Aurbach, J. Power Sources, 2008, 180, 902 CrossRef CAS.
- J. W. Jeon, J. O'Neal, L. Shao and J. L. Lutkenhaus, ACS Appl. Mater. Interfaces, 2013, 5, 10127 CAS.
- S. W. Lee, B. M. Gallant, H. R. Byon, P. T. Hammond and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 1972 CAS.
- D. A. Pasquier, A. Laforgue, P. Simon, G. G. Amatucci and J. F. Fauvarque, J. Electrochem. Soc., 2002, 149, A302 CrossRef.
- S. Bhadra, D. Khastgir, N. K. Singha and J. H. Lee, Prog. Polym. Sci., 2009, 34, 783 CrossRef CAS.
- J. H. Cheung, W. B. Stockton and M. F. Rubner, Macromolecules, 1997, 30, 2712 CrossRef CAS.
- D. Li, J. Huang and R. B. Kaner, Acc. Chem. Res., 2008, 42, 135 CrossRef PubMed.
- I. Dumitrescu, P. R. Unwin and J. V. Macpherson, Chem. Commun., 2009, 6886 RSC.
- L. Wei and G. Yushin, Nano Energy, 2012, 1, 552 CrossRef CAS.
- H. D. Tran, D. Li and R. B. Kaner, Adv. Mater., 2009, 21, 1487 CrossRef CAS.
- K. S. Park, S. B. Schougaard and J. B. Goodenough, Adv. Mater., 2007, 19, 848 CrossRef CAS.
- C. Su, Y. Ye, L. Xu and C. Zhang, J. Mater. Chem., 2012, 22, 22658 RSC.
- M. Aydın, B. Esat, C. Kılıc, M. E. Kose, A. Ata and F. Yılmaz, Eur. Polym. J., 2011, 47, 2283 CrossRef.
- M. Zhou, J. Qian, X. Ai and H. Yang, Adv. Mater., 2011, 23, 4913 CrossRef CAS PubMed.
- R. D. McCullough and S. P. Williams, J. Am. Chem. Soc., 1993, 115, 11608 CrossRef CAS.
- G. Liu, S. Xun, N. Vukmirovic, X. Song, O.-P. Velasco, H. Zheng, V. S. Battaglia, L. Wang and W. Yang, Adv. Mater., 2011, 23, 4679 CrossRef CAS PubMed.
- C. Y. Wang, A. M. Ballantyne, S. B. Hall, C. O. Too, D. L. Officer and G. G. Wallace, J. Power Sources, 2006, 156, 610 CrossRef CAS.
- Y. Liao, V. Strong, W. Chian, X. Wang, X. G. Li and R. B. Kaner, Macromolecules, 2012, 45, 1570 CrossRef CAS.
- H. R. Ghenaatian, M. F. Mousavi, S. H. Kazemi and M. Shamsipur, Synth. Met., 2009, 159, 1717 CrossRef CAS.
- H. R. Ghenaatian, M. F. Mousavi and M. S. Rahmanifar, Electrochim. Acta, 2012, 78, 212 CrossRef CAS.
- L. J. Bian, F. Luan, S. S. Liu and X. X. Liu, Electrochim. Acta, 2012, 64, 17 CrossRef CAS.
- S. N. Patel, A. E. Javier, K. M. Beers, J. A. Pople, V. Ho, R. A. Segalman and N. P. Balsara, Nano Lett., 2012, 12, 4901 CrossRef CAS PubMed.
- S. N. Patel, A. E. Javier, G. M. Stone, S. A. Mullin and N. P. Balsara, ACS Nano, 2012, 6, 1589 CrossRef CAS PubMed.
- Z. F. Li, H. Zhang, Q. Liu, Y. Liu, L. Stanciu and J. Xie, ACS Appl. Mater. Interfaces, 2014, 6, 5996 CAS.
- C. Ban, Z. Li, Z. Wu, M. J. Kirkham, L. Chen, Y. S. Jung, E. A. Payzant, Y. Yan, M. S. Whittingham and A. C. Dillon, Adv. Energy Mater., 2011, 1, 58 CrossRef CAS.
- J. S. Lee, S. Tai Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34 CrossRef CAS.
- J. F. Mike and J. L. Lutkenhaus, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 468 CrossRef CAS.
- Z. Liu, S. Poyraz, Y. Liu and X. Zhang, Nanoscale, 2012, 4, 106 RSC.
- J. M. Pringle, O. Winther-Jensen, C. Lynam, G. G. Wallace, M. Forsyth and D. R. MacFarlane, Adv. Funct. Mater., 2008, 18, 2031 CrossRef CAS.
- X. Feng, H. Huang, Q. Ye, J. J. Zhu and W. Hou, J. Phys. Chem. C, 2007, 111, 8463 CAS.
- D. Munoz-Rojas, J. Oro-Sole, O. Ayyad and P. J. Gomez-Romero, Mater. Chem., 2011, 21, 2078 RSC.
- Z. Liu, Y. Liu, L. Zhang, S. Poyraz, N. Lu, M. Kim, J. Smith, X. Wang, Y. Yu and X. Zhang, Nanotechnology, 2012, 23, 335603 CrossRef PubMed.
- M. K. Song, S. Park, F. M. Alamgir, J. Cho and M. Liu, Mater. Sci. Eng., R, 2011, 72, 203 CrossRef.
- F. Leroux, G. Goward, W. P. Power and L. F. Nazar, J. Electrochem. Soc., 1997, 144, 3886 CrossRef CAS.
- F. Huguenin, R. M. Torresi and D. A. Buttry, J. Electrochem. Soc., 2002, 149, A546 CrossRef CAS.
- S. L. Chou, X. W. Gao, J. Z. Wang, D. Wexler, Z. X. Wang, L. Q. Chend and H. K. Liu, Dalton Trans., 2011, 40, 12801 RSC.
- S. R. Sivakkumar and D. W. Kim, J. Electrochem. Soc., 2007, 154, A134 CrossRef CAS.
- D. Zhang, Y. Yin, C. Liu and S. Fan, Chem. Commun., 2015, 51, 322 RSC.
- W. Xiao, J. S. Chen, Q. Lu and X. W. Lou, J. Phys. Chem. C, 2010, 114, 12048 CAS.
- L. Yang, S. Wang, J. Mao, J. Deng, Q. Gao, Y. Tang and O. G. Schmidt, Adv. Mater., 2013, 25, 1180 CrossRef CAS PubMed.
- Y. G. Wang, Y. R. Wang, E. J. Hosono, K. X. Wang and H. S. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461 CrossRef CAS PubMed.
- Z. Ding, B. Yao, J. Feng and J. Zhang, J. Mater. Chem. A, 2013, 1, 11200 CAS.
- L. Yue, S. Wang, X. Zhao and L. Zhang, J. Mater. Chem., 2012, 22, 1094 RSC.
- C. Arbizzani, M. Mastragostino and M. Rossi, Electrochem. Commun., 2002, 4, 545 CrossRef CAS.
- H. Wang, Y. Zeng, K. Huang, S. Liu and L. Chen, Electrochim. Acta, 2007, 52, 5102 CrossRef CAS.
- Y. G. Wang, W. Wu, L. Cheng, P. He, C. X. Wang and Y. Y. Xia, Adv. Mater., 2008, 20, 2166 CrossRef CAS.
- L. Mai, X. Xu, C. Han, Y. Luo, L. Xu, Y. A. Wu and Y. Zhao, Nano Lett., 2011, 11, 4992 CrossRef CAS PubMed.
- I. Boyano, M. Bengoechea, I. de Meatza, O. Miguel, I. Canterob, E. Ochoteco, J. Rodriguez, M. L. Cantu and P. G. Romero, J. Power Sources, 2007, 166, 471 CrossRef CAS.
- Z. P. Guo, J. Z. Wang, H. K. Liu and S. X. Dou, J. Power Sources, 2005, 146, 448 CrossRef CAS.
- X. Y. Zhou, J. Tang, J. Yang, Y. L. Zou, S. C. Wang, J. Xie and L. L. Ma, Electrochim. Acta, 2012, 70, 296 CrossRef CAS.
- C. X. Guo, M. Wang, T. Chen, X. W. Lou and C. M. Li, Adv. Energy Mater., 2011, 1, 736 CrossRef CAS.
- Y. Chen, S. Zeng, J. Qian, Y. Wang, Y. Cao, H. Yang and X. Ai, ACS Appl. Mater. Interfaces, 2014, 6, 3508 CAS.
- M. V. Reddy, B. L. W. Wen, K. P. Loh and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 7777 CAS.
- Z. P. Guo, J. Z. Wang, H. K. Liu and S. X. Dou, J. Power Sources, 2005, 146, 448 CrossRef CAS.
- S. Kuwabata, S. Masui and H. Yoneyama, Electrochim. Acta, 1999, 44, 4593 CrossRef CAS.
- J. K. Kim, J. Manuel, M. H. Lee, J. Scheers, D. H. Lim, P. Johansson, J. H. Ahn, A. Matic and P. Jacobsson, J. Mater. Chem., 2012, 22, 15045 RSC.
- Y. H. Wang, H. Liu, D. Zhu, Z. P. Guo, H.-K. Liu and S. X. Dou, Trans. Nonferrous Met. Soc. China, 2011, 21, 1303 CrossRef CAS.
- H. Zhao, A. Yuan, B. Liu, S. Xing, X. Wu and J. Xu, J. Appl. Electrochem., 2012, 42, 139 CrossRef CAS.
- I. Boyano, M. Bengoechea, I. de Meatza, O. Miguel, I. Cantero, E. Ochoteco, J. Rodriguez, M. Lira-Cantu and P. Gomez-Romero, J. Power Sources, 2007, 166, 471 CrossRef CAS.
- O. Y. Posudievsky, O. A. Kozarenko, V. S. Dyadyun, S. W. Jorgensen, J. A. Spearot, V. G. Koshechko and V. D. Pokhodenko, Electrochim. Acta, 2011, 58, 442 CrossRef CAS.
- H. C. Dinh, S. I. Mho and I. H. Yeo, Electroanalysis, 2011, 23, 2079–2086 CrossRef CAS.
- A. H. Gemeay, H. Nishiyama, S. Kuwabata and H. Yoneyama, J. Electrochem. Soc., 1995, 142, 4190 CrossRef CAS.
- S. Kuwabata, S. Masui and H. Yoneyama, Electrochim. Acta, 1999, 44, 4593 CrossRef CAS.
- I. Boyano, M. Bengoeche, I. de Meatza, O. Miguel, I. Cantero, E. Ochoteco, H. Grande, M. L. Cantuc and P. G. Romero, J. Power Sources, 2007, 174, 1206 CrossRef CAS.
- X. Xia, Z. Wang and L. Chen, Electrochem. Commun., 2008, 10, 1442 CrossRef CAS.
- D. Lepage, C. Michot, G. Liang, N. Gauthier and S. B. Schougaard, Angew. Chem., Int. Ed., 2011, 50, 6884 CrossRef CAS PubMed.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed.
- B. Liu, P. Soares, C. Checkles, Y. Zhao and G. Yu, Nano Lett., 2013, 13, 3414 CrossRef CAS PubMed.
- L. J. Fu, H. Liu, C. Li, Y. P. Wu, E. Rahm, R. Holze and H. Q. Wu, Prog. Mater. Sci., 2005, 50, 881 CrossRef CAS.
- X. Ren, C. Shi, P. Zhang, Y. Jiang, J. Liu and Q. Zhang, Mater. Sci. Eng., B, 2012, 177, 929 CrossRef CAS.
- F. Huguenin, E. M. Girotto, R. M. Torresi and D. A. Buttry, J. Electroanal. Chem., 2002, 536, 37 CrossRef CAS.
- F. Huguenin, E. A. Ticianelli and R. M. Torresi, Electrochim. Acta, 2002, 47, 3179 CrossRef CAS.
- M. L. Cantu and P. G. Romero, J. Solid State Chem., 1999, 147, 601 CrossRef.
- M. L. Cantu and P. G. Romero, J. Electrochem. Soc., 1999, 146, 2029 CrossRef.
- M. G. Kanatzidis, C. R. Kannewurf, H. O. Marcy and C. G. Wu, J. Am. Chem. Soc., 1989, 111, 4139 CrossRef CAS.
- K. I. Park, H. M. Song, Y. Kim, S. I. Mho, W. I. Cho and I. H. Yeo, Electrochim. Acta, 2010, 55, 8023 CrossRef CAS.
- H. Ze-qiang, X. Li-zhi, C. Shang, W. Xian-ming, L. Wen-ping and H. Ke-long, Trans. Nonferrous Met. Soc. China, 2010, 20, s262 CrossRef.
- Y. H. Huang, K. S. Park and J. B. Goodenough, J. Electrochem. Soc., 2006, 153, A2282 CrossRef CAS.
- A. V. Murugan, Electrochim. Acta, 2005, 50, 4627 CrossRef CAS.
- Q. Gong, Y. S. He, Y. Yang, X. Z. Liao and Z. F. Ma, J. Solid State Electrochem., 2012, 16, 1383 CrossRef CAS.
- Z. Du, S. Zhang, Y. Liu, J. Zhao, R. Lin and T. Jiang, J. Mater. Chem., 2012, 22, 11636 RSC.
- J. B. Bates, N. J. Dudney, D. C. Lubben, G. R. Gruzalski, B. S. Kwak, X. Yu and R. A. Zuhr, J. Power Sources, 1995, 54, 58 CrossRef CAS.
- N. J. Dudney, J. Electrochem. Soc. Interface, 2008, 3, 44 Search PubMed.
- S. W. Song, H. Choi, H. Y. Park, G. B. Park, K. C. Lee and H. J. Lee, J. Power Sources, 2010, 195, 8275 CrossRef CAS.
- J. Z. Wang, S. L. Chou, J. Chen, S. Y. Chew, G. X. Wang, K. Konstantinov, J. Wu, S. X. Dou and H. K. Liu, Electrochem. Commun., 2008, 10, 1781 CrossRef CAS.
- X. H. Huang, J. P. Tu, X. H. Xia, X. L. Wang and J. Y. Xiang, Electrochem. Commun., 2008, 10, 1288 CrossRef CAS.
- X. H. Huang, J. P. Tu, X. H. Xia, X. L. Wang, J. Y. Xiang and L. Zhang, J. Power Sources, 2010, 195, 1207 CrossRef CAS.
- J. Z. Wang, S. L. Chou, J. Chen, S. Y. Chew, G. X. Wang, K. Konstantinov, J. Wu, S. X. Dou and H. K. Liu, Electrochem. Commun., 2008, 10, 1781 CrossRef CAS.
- G. X. Wang, L. Yang, Y. Chen, J. Z. Wang, S. Bewlay and H. K. Liu, Electrochim. Acta, 2005, 50, 4649 CrossRef CAS.
- Y. H. Huang, K. S. Park and J. B. Goodenough, J. Electrochem. Soc., 2006, 153, A2282 CrossRef CAS.
- X. Y. Zhou, J. J. Tang, J. Yang, Y. L. Zou, S. C. Wang, J. Xie and L. L. Ma, Electrochim. Acta, 2012, 70, 296 CrossRef CAS.
- Y. Yao, N. Liu, M. T. McDowell, M. Pasta and Y. Cui, Energy Environ. Sci., 2012, 5, 7927 CAS.
- Y. Wang, Y. Wang, J. Tang, Y. Xia and G. Zheng, J. Mater. Chem. A, 2014, 2, 20177 CAS.
- D. Kundu, F. Krumeich and R. Nesper, J. Power Sources, 2013, 236, 112 CrossRef CAS.
- L. Shao, J. W. Jeon and J. L. Lutkenhaus, Chem. Mater., 2012, 24, 181 CrossRef CAS.
- W. M. Chen, Y. H. Huang and L. X. Yuan, J. Electroanal. Chem., 2011, 660, 108 CrossRef CAS.
- N. H. Idris, J. Wang, S. Chou, C. Zhong, M. M. Rahman and H. Liu, J. Mater. Res., 2011, 26, 860 CrossRef CAS.
- G. J. Wang, L. C. Yang, Q. T. Qu, B. Wang, Y. P. Wu and R. Holze, J. Solid State Electrochem., 2010, 14, 865 CrossRef CAS.
- W. Tang, L. Liu, Y. Zhu, H. Sun, Y. Wu and K. Zhu, Energy Environ. Sci., 2012, 5, 6909 CAS.
- W. Tang, X. W. Gao, Y. S. Zhu, Y. B. Yue, Y. Shi, Y. P. Wu and K. Zhu, J. Mater. Chem., 2012, 22, 20143 RSC.
- L. L. Liu, X. J. Wang, Y. S. Zhu, C. L. Hu, Y. P. Wu and R. Holze, J. Power Sources, 2013, 224, 290 CrossRef CAS.
- F. Tian, L. Liu, Z. Yang, X. Wang, Q. Chen and X. Wang, Mater. Chem. Phys., 2011, 127, 151 CrossRef CAS.
- A. E. Javier, S. N. Patel, D. T. Hallinan Jr, V. Srinivasan and N. P. Balsara, Angew. Chem., Int. Ed., 2011, 50, 9848 CrossRef CAS PubMed.
- Z. J. Zhang, J. Z. Wang, S. L. Chou, H. K. Liu, K. Ozawa and H. J. Li, Electrochim. Acta, 2013, 108, 820 CrossRef CAS.
- Y. Chen, S. Zeng, J. Qian, Y. Wang, Y. Cao, H. Yang and X. Ai, ACS Appl. Mater. Interfaces, 2014, 6, 3508 CAS.
- F. Han, D. Li, W. C. Li, C. Lei, Q. Sun and A. H. Lu, Adv. Funct. Mater., 2013, 23, 1692 CrossRef CAS.
- L. Cui, J. Shen, F. Cheng, Z. Tao and J. Chen, J. Power Sources, 2011, 196, 2195 CrossRef CAS.
- L. Yuan, J. Wang, S. Y. Chew, J. Chen, Z. P. Guo, L. Zhao, K. Konstantinov and H. K. Liu, J. Power Sources, 2007, 174, 1183 CrossRef CAS.
- S. Y. Chew, Z. P. Guo, J. Z. Wang, J. Chen, P. Munroe, S. H. Ng, L. Zhao and H. K. Liu, Electrochem. Commun., 2007, 9, 941 CrossRef CAS.
- Z. Q. He, L. Z. Xiong, W. P. Liu, X. M. Wu, S. Chen and K. L. Huang, J. Cent. South Univ. Technol., 2008, 15, 214 CrossRef CAS.
- Q. G. Shao, W. M. Chen, Z. H. Wang, L. Qie, L. X. Yuan, W. X. Zhang, X. L. Hu and Y. H. Huang, Electrochem. Commun., 2011, 13, 1431 CrossRef CAS.
- R. Liang, H. Cao, D. Qian, J. Zhang and M. Qu, J. Mater. Chem., 2011, 21, 17654 RSC.
- Y. Zhao, J. Li, N. Wang, C. Wu, G. Dong and L. Guan, J. Phys. Chem. C, 2012, 116, 18612 CAS.
- C. Lai, H. Z. Zhang, G. R. Li and X. P. Gao, J. Power Sources, 2011, 196, 4735 CrossRef CAS.
- C. Lai, G. R. Li, Y. Y. Dou and X. P. Gao, Electrochim. Acta, 2010, 55, 4567 CrossRef CAS.
- F. Zhang, H. Cao, D. Yue, J. Zhang and M. Qu, Inorg. Chem., 2012, 51, 9544 CrossRef CAS PubMed.
- Z. Yin, Y. Ding, Q. Zheng and L. Guan, Electrochem. Commun., 2012, 20, 40 CrossRef CAS.
- R. Vidu and P. Stroeve, Ind. Eng. Chem. Res., 2004, 43, 3314 CrossRef CAS.
- N. H. Idris, J. Wang, S. Chou, C. Zhong, M. M. Rahman and H. Liu, J. Mater. Res., 2011, 26, 860 CrossRef CAS.
- C. J. Cui, G. M. Wu, H. Y. Yang, S. F. She, J. Shen, B. Zhou and Z. H. Zhang, Electrochim. Acta, 2010, 55, 8870 CrossRef CAS.
- H. Wang, K. Huang, Y. Zeng, F. Zhao and L. Chen, Electrochem. Solid-State Lett., 2007, 10, A199 CrossRef CAS.
- A. Fedorkova, A. N. Alejos, P. G. Romero, R. Orinakovac and D. Kaniansky, Electrochim. Acta, 2010, 55, 943 CrossRef CAS.
- Y. Fu and A. Manthiram, Chem. Mater., 2012, 24, 3081 CrossRef CAS.
- J. J. Cai, P. J. Zuo, X. Q. Cheng, Y. H. Xu and G. P. Yin, Electrochem. Commun., 2010, 12, 1572 CrossRef CAS.
- E. A. Ponzio, T. M. Benedetti and R. M. Torresi, Electrochim. Acta, 2007, 52, 4419 CrossRef CAS.
- K. S. Hwang, C. W. Lee, T. H. Yoon and Y. S. Son, J. Power Sources, 1999, 79, 225 CrossRef CAS.
- T. A. Kerr, H. Wu and L. F. Nazar, Chem. Mater., 1996, 8, 2005 CrossRef CAS.
- G. T. Gomez, E. M. T. Rosales and P. G. Romero, Chem. Mater., 2001, 13, 3693 CrossRef.
- X. W. Zhang, C. Wang, A. J. Appleby and F. E. Little, J. Power Sources, 2002, 109, 136 CrossRef CAS.
- G. Liu, S. Xun, N. Vukmirovic, X. Song, P. O. Velasco, H. Zheng, V. S. Battaglia, L. Wang and W. Yang, Adv. Mater., 2011, 23, 4679 CrossRef CAS PubMed.
- S. Xun, X. Song, V. Battaglia and G. Liu, J. Electrochem. Soc., 2013, 160, A849 CrossRef CAS.
- M. Gu, X. C. Xiao, G. Liu, S. Thevuthasan, D. R. Baer, J. G. Zhang, J. Liu, N. D. Browning and C. M. Wang, Sci. Rep., 2014, 4, 3684 Search PubMed.
- M. Wu, X. Xiao, N. Vukmirovic, S. Xun, P. K. Das, X. Song, P. O. Velasco, D. Wang, A. Z. Weber, L. W. Wang, V. S. Battaglia, W. Yang and G. Liu, J. Am. Chem. Soc., 2013, 135, 12048 CrossRef CAS PubMed.
- D. Chao, X. Xia, J. Liu, Z. Fan, C. F. Ng, J. Lin, H. Zhang, Z. X. Shen and H. J. Fan, Adv. Mater., 2014, 26, 5794 CrossRef CAS PubMed.
- S. Wang, L. Hu, Y. Hu and S. Jiao, Mater. Chem. Phys., 2014, 146, 289 CrossRef CAS.
- F. Wu, J. Chen, L. Li, T. Zhao, Z. Liu and R. Chen, ChemSusChem, 2013, 6, 1438 CrossRef CAS PubMed.
- Y. H. Huang and J. B. Goodenough, Chem. Mater., 2008, 20, 7237 CrossRef CAS.
- Z. Chen, J. W. F. To, C. Wang, Z. Lu, N. Liu, A. Chortos, L. Pan, F. Wei, Y. Cui and Z. Bao, Adv. Energy Mater., 2014, 4, 1400207 Search PubMed.
- G. Ma, Z. Wen, J. Jin, Y. Lu, X. Wu, C. Liu and C. Chen, RSC Adv., 2014, 4, 21612 RSC.
- M. Pasta, C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2012, 1149, 3 Search PubMed.
- Z. W. Seh, H. Wang, P. Hsu, Q. Zhang, W. Li, G. Zheng, H. Yao and Y. Cui, Energy Environ. Sci., 2014, 7, 672 CAS.
- H. Chen, W. Dong, J. Ge, C. Wang, X. Wu, W. Lu and L. Chen, Sci. Rep., 2013, 3, 1910 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |