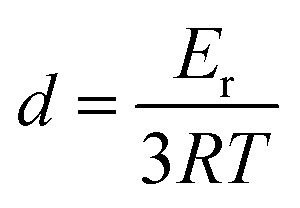DOI:
10.1039/C5PY00172B
(Paper)
Polym. Chem., 2015,
6, 3046-3053
High strain epoxy shape memory polymer
Received
4th February 2015
, Accepted 28th February 2015
First published on 2nd March 2015
Abstract
Epoxy polymers represent a recently emerged class of thermoset shape memory polymers with superior thermo-mechanical endurance and excellent processability. However, the strains at break are typically low for epoxy shape memory polymers. This severely limits their potential applications. In this article, we report a two component epoxy-amine shape memory polymer system with tunable Tg (between 40 °C and 80 °C) and excellent shape memory properties in terms of shape fixity, shape recovery ratios, and cycling stability. Importantly, its values of strain at break above Tg and at the Tg peak reach 111% and 212%, respectively. We anticipate that such a high strain epoxy system will significantly broaden the opportunities for shape memory device applications.
1. Introduction
Shape memory polymers (SMPs), which are capable of memorizing temporary shapes and recovering to their original forms upon external stimulations, have been highly attractive to the scientific community.1–8 The recovery can be triggered by heat,9 light,10–13 electric current,14,15 magnetic fields,16 moisture,17,18 or radiofrequency waves.19 Despite the diversity of triggering stimuli, heat (either directly or indirectly applied) remains the overwhelmingly popular stimulus for actuating SMPs. Thus, the shape memory transition temperature (Ttrans, typically Tg or Tm) is one of the most critical parameters for an SMP.20 Recent advances in the SMP field have led to the emergence of a variety of new and exciting applications, including biomedical devices,21–23 biomimetic reversible adhesives24–26 and programmable optical devices,27,28 to name just a few. Besides the shape memory transition temperature, each application typically demands an SMP with a particular set of properties such as biodegradability, strain at break (εb), or maximum recovery stress (σmax). Amongst all these performance parameters, Ttrans and εb are particularly of general relevance.
Thus far, a wide variety of polymers (both thermoplastic and thermoset) have been found with shape memory properties. Many thermoplastic SMPs possess large εbs,21 but their shape recovery is often less than ideal (with some exceptions29). In contrast, thermoset SMPs typically exhibit excellent shape fixing and recovery behaviors. Thermoset SMPs can be created by either introducing crosslinking in thermoplastic polymers or direct curing of liquid monomers with crosslinkers. The latter has two notable advantages over the former: more flexible structural tuning through comonomer and crosslinker selections, and easier processing into complex devices owing to the liquid nature of the precursors. Because of these, thermoset SMPs obtained from the direct curing of liquid precursors have become the focus of many studies. This type of thermoset SMPs include (meth)acrylate-based systems,30–32 thiol–ene systems33,34 and epoxy-based systems.35–42 For (meth)acrylate-based systems, their εb can be tuned to an extremely high level while still maintaining high shape fixity and shape recovery.31 This type of system, however, often suffers from several processing issues: the volatility of some (meth)acrylate monomers, high curing shrinkage, and oxygen inhibition during curing. Amongst them, high curing shrinkage is particularly cumbersome to deal with if devices with high precision microstructures are to be produced. Thiol–ene systems,33,34 on the other hand, exhibit much lower volume shrinkage and their curing is not inhibited by oxygen. However, the values of εb for thiol–ene systems are not typically reported in the literature.
Epoxy SMPs have gained significant attention lately. Their increasing popularity stems from many attributes including the versatility of curing chemistry, the availability of a wide variety of non-volatile monomers, the low curing shrinkage, and the excellent thermo-mechanical stability of the cured polymers.20,35–41 This has led to epoxy SMPs with highly tunable Tgs.20 Of equal importance is that they can also be easily processed into attractive SMP devices25,43,44 as well as self-healing systems45,46 and triple-shape composites.47,48 Despite the advantages of epoxy SMPs, they typically suffer from low εb values. A protypical example of an epoxy SMP system20 was obtained by curing between an aliphatic diamine and a mixture of an aromatic diepoxide and an aliphatic diepoxide. While maintaining the overall stoichiometry between the amine and epoxy functional groups, a series of SMPs were obtained by changing the ratio between the rigid aromatic diepoxy and the flexible aliphatic diepoxide in the formulations. This allowed adjusting the overall network flexibility, thus arbitrarily tuning the Tg in a wide temperature range between 25 °C and 100 °C. However, the εb values for such an epoxy system were no greater than 30% due to the inherent high crosslinking densities. Although various low strain SMP applications have recently emerged, it is highly desirable to extend epoxy SMP systems to a much higher εb range to fully take advantage of their easy processability for a much wider range of applications. A physical strategy to increase the εb of a particular SMP material as pioneered by Gall's group was to deform it around the onset of the Tg transition, instead of above its Tg.49 This strategy was adopted by Rousseau et al. for an epoxy SMP and its εb was indeed improved from 30% to ca. 60%.35 However, shape fixity ratios were noticeably compromised under such conditions, which may not be acceptable for many applications. Thus, improving εbs for epoxy SMPs without compromising shape fixing performance is highly desirable. One such effort was reported by Leng et al., who claimed a high strain epoxy SMP system.38 Unfortunately, the detailed SMP formulations were not revealed except that it was known to be a three component system with an epoxy base resin, a hardener, and a linear epoxy monomer. Leonardi et al., by contrast, reported the detailed formulation of an epoxy SMP system composed of an aromatic diepoxide, a long-chain monoamine, and an aromatic diamine as the crosslinking agent.39 The cured network comprises of chemical crosslinking and intriguing physical crosslinking owing to the tail-to-tail association of the long alkyl chains. εb as high as 75% was achieved, but only for one single formulation with a Tg around 40 °C. This example nevertheless represents the current state of art high strain epoxy SMP with a precisely known chemical composition. With the practical need for Tg tunable epoxy SMPs with even higher εb, we set to develop epoxy SMPs using the most commonly available commercial epoxy components. This effort is reported hereafter in this paper.
2. Experimental section
2.1. Materials
The epoxy monomer E44 (molecular weight ∼450 g mol−1 and epoxy equivalent weight 210–240 g mol−1) was purchased from China Petrochemical Corporation. The curing agent poly(propylene glycol)bis(2-aminopropyl)ether (Jeffamine D230, abbreviated as D230 hereafter) was purchased from Sigma-Aldrich. All chemicals were used as received.
2.2. Sample preparation
In a typical curing experiment, E44 was weighed into a glass bottle and melted by heating in an oven at 60 °C. Afterwards, weighed Jeffamine D-230 was added into the bottle, which was vigorously shaken by hand for complete mixing. The mixture was poured into an aluminum pan. Curing was conducted thermally at 100 °C for 1 h followed by a postcure at 130 °C for 1 h. Finally, the cured sample was demolded and cut into dog bones with a laser cutter for further testing.
2.3. Thermal analyses
Differential scanning calorimetry (DSC) measurements were conducted using DSC Q200 (TA instruments) at a cooling and heating rate of 10 °C min−1. Dynamic mechanical (DMA) analyses were conducted using DMA Q800 (TA instruments). The conditions for the DMA experiments were: tensile, “multi-frequency, strain” mode at 1 Hz, 0.2% strain, and a heating rate of 3 °C min−1. Samples of 1 mm thickness and 2.5 mm width were used.
2.4. Shape memory properties
For qualitative shape memory demonstrations, samples of rectangular shapes (1 × 5 × 2.5 mm) were used. In a typical experiment, the sample was first immersed in a water bath preset above its Tg for 5 s. It was then taken out of the bath and immediately deformed into a temporary shape. With the deformation force maintained on the sample, it was cooled at room temperature. Subsequent release of the deformation force after cooling completed the shape fixing step. Shape recovery was accomplished by immersing the deformed sample into the same water bath.
Quantitative shape memory performance was evaluated using DMA Q800 in a “controlled force” mode. The heating rate and cooling rate were 10 °C min−1 and 5 °C min−1, respectively. Shape fixity (Rf) and shape recovery (Rr) were calculated using the following equations as defined in ref. 1.
| Rf = εd/εdload and Rr = (εd − εrec)/εd, |
where
εdload and
εrec respectively represent the maximum strain under load and the recovered strain, and
εd is the fixed strain after cooling and load removal.
2.5. Measurements of strains at break (εbs)
ε
b values were measured using a universal material testing machine (Zwick/Roell Z005) equipped with a thermal chamber. The experiments were conducted in a tensile mode at a crosshead speed of 10 mm min−1 and a heating rate of 20 °C min−1. εb was typically recorded as the maximum strain prior to the sample break at its rubbery state. Samples in dog bone shapes with the neck dimensions of 1.2 × 4.5 mm were used. At least five specimens were tested for each sample to obtain statistically meaningful data.
3. Results and discussion
A rule of thumb to increase the εb of an amorphous thermoset SMP is to reduce the crosslinking density whereas Tg is affected by both the crosslinking density and the chain flexibility. Such a design principle has been applied quite successfully to (meth)acrylate SMPs.30–32 Whereas the rule may also be applicable to epoxy SMPs, there appear to be additional factors that should be considered. For instance, we reported a thermoset epoxy SMP system based on the curing between an aromatic diepoxide and an aliphatic diamine. Replacing the aliphatic diamine with a mono-amine (decylamine) while maintaining the overall stoichiometric balance between the amine and the epoxide led to a series of SMPs with tunable Tgs.20 However, the SMPs with more decylamine (thus lower crosslinking densities) in the formulation did not show a noticeable improvement in εb. An intriguing comparison can be made with the high strain example mentioned in the introduction in which the epoxy system was obtained by curing n-dodecylamine, an aromatic diamine crosslinker, and an aromatic diepoxide.39 The formulations in these two examples appear quite similar, yet the outcome in terms of εb is drastically different. This emphasizes the network design in which the former network simply has dangling decyl chains20 whereas the longer dodecyl chains in the latter39 possess the tail-to-tail association that is apparently important. With all these considerations in mind, we herein aimed to simplify the epoxy network design to achieve a tunable crosslinking density without involving the potentially non-beneficial dangling chains. Our network design started with a previously reported base epoxy SMP formulation obtained by curing an aromatic diepoxide (EPON 826 with an epoxy equivalent weight of 180) and an aliphatic diamine (D230).20 This particular SMP has a Tg around 100 °C and εb below 30% owing to its high crosslinking density. Two approaches were combined to reduce the crosslinking density. First, EPON 826 was replaced by another diepoxide (E44) with a much higher epoxy equivalent weight of 225 (Fig. 1). Second, while we still employed the same D230 (also in Fig. 1) as the crosslinker, the ratio between E44 and D230 was shifted away from their stoichiometry with D230 in excess. This led to the network structure in Fig. 1, in which some of the hydrogen atoms on NH groups remain unreacted. This conceptual structure is based on the assumption that primary amine hydrogens are preferentially consumed due to their higher reactivity than secondary amine hydrogens.50–52 Thus, this would statistically leave unreacted hydrogens mostly in the secondary amino form. We further note that actual reaction and gelation are highly complex in crosslinked systems. It is outside the scope of the current study to investigate such aspects and we simply used the somewhat idealized structure in Fig. 1 for qualitative guidance only. In its stoichiometric state, each hydrogen atom on the amine group serves as a crosslinking site. Thus, the excess amines in the formulation left unreacted amine hydrogens that led to the reduction of the network crosslinking density. An additional important consideration here is that, after complete curing with the epoxy groups in the formulation, the remaining unreacted amino hydrogens are not expected to undergo additional reactions under conditions SMPs are typically used. This is necessary for the durability of the SMPs.
 |
| | Fig. 1 Chemical structures of the epoxy precursors and cured networks. | |
Accordingly, four epoxy samples (Epon1 to Epon4 in Table 1) were prepared by varying the ratio between E44 and D230. The crosslinking density for each sample, which can be calculated (eqn (1)) based on its rubbery modulus reported later in the context (Fig. 5), is included in Table 1.
| | 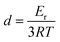 | (1) |
where
d represents the crosslinking density per unit volume (mol cm
−3),
Er is rubbery modulus (MPa),
R and
T are the gas constant and the absolute temperature, respectively.
Table 1 Compositions and shape memory performances of the epoxy samples
| Samples |
E44 (mol) |
D230 (mol) |
d (10−3 g mol−1) |
R
f (%) |
R
r (%) |
|
Epon1
|
1 |
1 |
2.3 |
99.8 |
98.1 |
|
Epon2
|
1.33 |
1 |
3.5 |
99.5 |
97.4 |
|
Epon3
|
1.67 |
1 |
6.1 |
99.4 |
98.2 |
|
Epon4
|
2 |
1 |
6.3 |
99.5 |
99.4 |
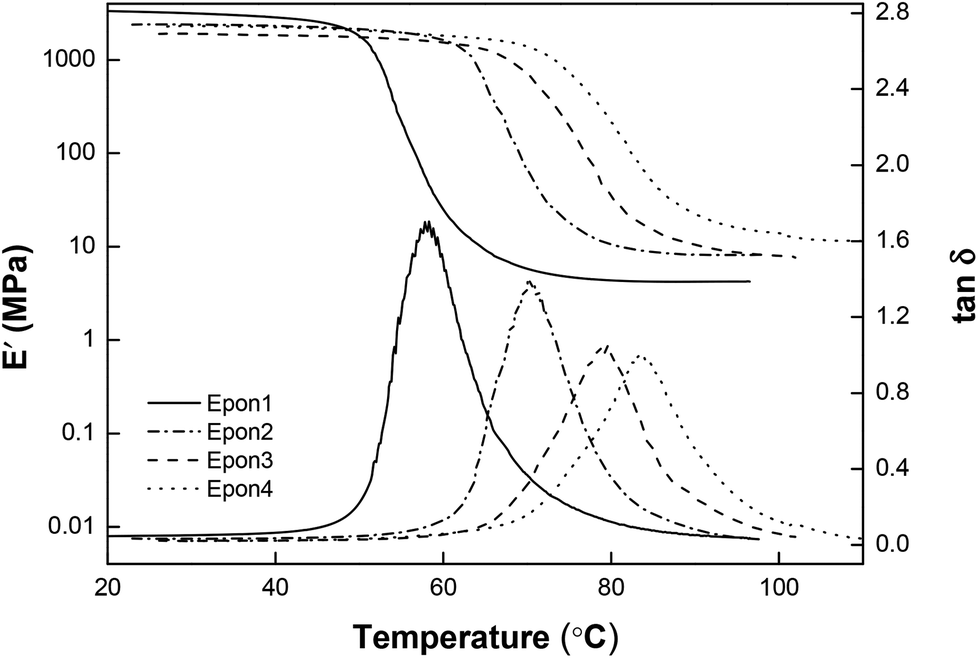
As the ratio between D230 and E44 increases from Epon1 to Epon4, the crosslinking density goes up dramatically. Attempts were also made to prepare a sample with a molarity ratio of 0.98 (between E44 and D230). This sample failed to cure into a solid, implying that Epon1 had already approached the lowest achievable crosslinking density for the current two component epoxy system. The DSC curves of the cured epoxy samples are shown in Fig. 2. All the samples showed readily observable Tg transitions between 40 °C and 80 °C, with increasing Tg from Epon1 to Epon4. DMA curves for these samples (Fig. 3) show the same trend except that the Tg transitions appear to be higher, as judged from their respective temperature corresponding to the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ peaks. A closer examination of the DMA curves in Fig. 3 revealed that all the samples show a relatively constant storage modulus in their glass state. Importantly, they all possess a nice and flat rubbery plateau, a good indication of their crosslinked nature and their potential shape memory functions. The rubbery modulus increases from Epon1 to Epon4, which is consistent with the increasing crosslinking density. The Tg's obtained from DSC and DMA are quantitatively compared in Fig. 4. The Tg and the molar ratio between E44 and D230 show an almost linear relationship at molar ratios below 1.67. Above this ratio, the slope of the Tg increase appears to decrease, indicating that when the crosslink density reaches a specific value, the change in the formulation has less impact on the Tg. Overall, the continuous change in the Tg with the molar ratio suggests that any Tg in these temperature ranges can be obtained.
δ peaks. A closer examination of the DMA curves in Fig. 3 revealed that all the samples show a relatively constant storage modulus in their glass state. Importantly, they all possess a nice and flat rubbery plateau, a good indication of their crosslinked nature and their potential shape memory functions. The rubbery modulus increases from Epon1 to Epon4, which is consistent with the increasing crosslinking density. The Tg's obtained from DSC and DMA are quantitatively compared in Fig. 4. The Tg and the molar ratio between E44 and D230 show an almost linear relationship at molar ratios below 1.67. Above this ratio, the slope of the Tg increase appears to decrease, indicating that when the crosslink density reaches a specific value, the change in the formulation has less impact on the Tg. Overall, the continuous change in the Tg with the molar ratio suggests that any Tg in these temperature ranges can be obtained.
 |
| | Fig. 2 DSC curves of the epoxy samples. | |
 |
| | Fig. 3 DMA curves of the epoxy samples. | |
 |
| | Fig. 4 Glass transition temperatures of the epoxy samples. | |
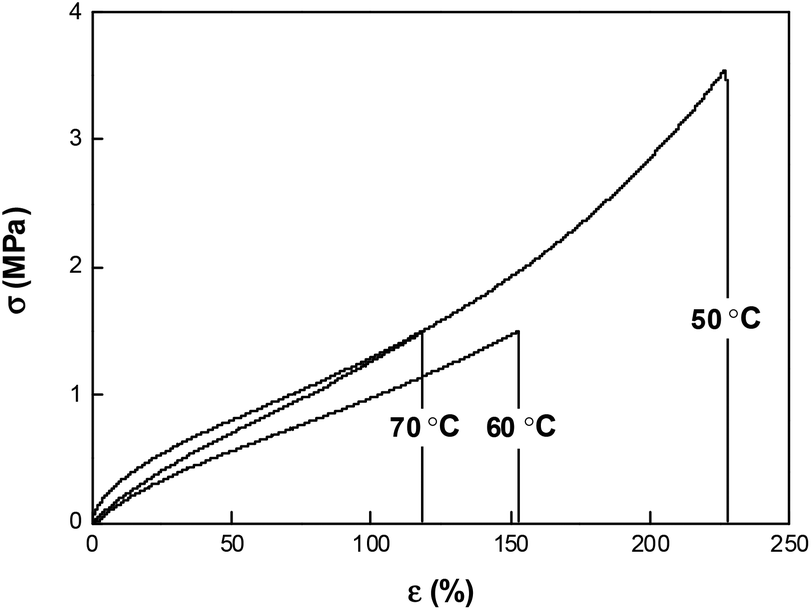
The εb and Er (rubbery modulus) values for the different samples, as measured using a tensile tester, are summarized in Fig. 5. Epon4, with a molar ratio of 2 between E44 and D230, shows a low εb around 40% and a high Er of 14 MPa. This is not surprising due to the high crosslinking nature. Epon3, which has a lower molar ratio of 1.67 between E44 and D230, has similar εb and Er values. This appears to be consistent with the Tg trend shown in Fig. 4, that is, the change in the molar ratio from 2 to 1.67 does not seem to impact much the material properties. Below a molar ratio of 1.67, however, the εb value significantly increases with the decrease in the molar ratio and Er follows an opposite trend. Impressively, Epon1, with the lowest crosslink density and lowest Er of 2.1 MPa, reaches the highest εb value of 111%. Here in Fig. 5, the εb value for each sample was obtained above its Tg, i.e., at a temperature in the rubbery plateau region. It is known in the literature that εb values can be positively impacted by deforming an SMP within the glass transition.36,39 With this in mind, Epon1 was subjected to uniaxial tests at different temperatures around its Tg transition and the obtained stress–strain curves are shown in Fig. 6. As can be seen from this figure, the εb value indeed increased when the test temperature decreased. Impressively, the εb value when tested at 50 °C reached 212% (average over five tests).
 |
| | Fig. 5
ε
b and Er of the epoxy SMPs. | |
 |
| | Fig. 6 Typical uniaxial stress–strain curves at different temperatures for Epon1. | |
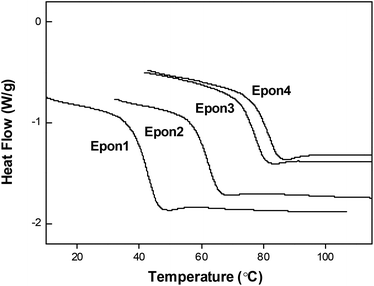
The fact that the epoxy samples all display Tg transitions and rubbery plateaus in their DMA curves indicates that they should possess shape memory behaviors. Qualitatively, a visual demonstration of their shape memory performance is illustrated in Fig. 7. Samples from Epon1 to Epon4 (in rectangular original shapes) were arranged in such an order as in Fig. 7a. They were deformed above their respective Tgs into twisted temporary shapes. Next, immersing in a 60 °C water bath, Epon1 recovered in 5 seconds (Fig. 6c). Epon2 recovered until the temperature of the water bath rose above 70 °C. As the temperature further increased to 80 °C, Epon3 recovered in 5 seconds and Epon4 also recovered at this temperature albeit requiring a longer immersion time of 20 seconds. The shape memory performances of all the samples were further evaluated quantitatively and the results are summarized in Table 1. As can be seen, all the samples show a Rf above 99% and a Rr above 97%. The quantitative shape memory cycle for a representative sample Epon1 (with the largest εb) is plotted in Fig. 8, showing near perfect shape fixing (Rf> 99%) and recovery (Rr> 98%) behaviors, despite its very low crosslinking density.
 |
| | Fig. 7 Visual demonstration of the shape memory performance of all epoxy samples (from Epon1 to Epon4). (a) Original shapes; (b) fixed temporary shapes; (c) T = 60 °C, Epon1 recovered; (d) T = 70 °C, Epon2 recovered; (e) T = 80 °C, recovered shapes for all the samples. | |
 |
| | Fig. 8 Shape memory cycle for Epon1. | |
Whereas Fig. 8 shows the excellent shape fixity and shape recovery, its high strain capability is difficult to demonstrate with a DMA tester due to the upper limit of the sample displacement within the instrument. A εb value, on the other hand, is not necessarily equivalent to a maximum recoverable strain even though these two terms are often used interchangeably in the literature. The differences between the two are: (1) it is difficult to approach the εb in an actual shape memory test without the risk of breaking the sample; (2) shape fixity may not be close to 100% under conditions in which the highest εb value is observed. With these considerations in mind, a separate quantitative shape memory cycle test involving a very high strain was run without using a DMA tester. The experiments are shown in Fig. 9. Epon1 in a dog bone shape was marked by two black lines (distance: 1.5 cm) between the necks. The sample was stretched at 50 °C and the stretching was fixed by cooling. The distance between the two black lines became 4.2 cm, corresponding to a strain of ca. 180%. Importantly, this strain can be fully recovered after reheating. The strain of 180% thus represents the highest recoverable strain achieved for Epon1, even though theoretically it may be possible to obtain values closer to its εb of 212%.
 |
| | Fig. 9 Demonstration of the high strain shape memory performance for Epon1. | |
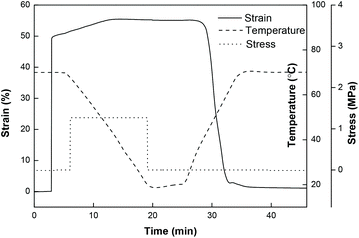
To investigate the cycling stability of the high strain epoxy SMP, Epon1 was subjected to consecutive shape memory cycles with the strain above 70%. As shown in Fig. 10, the cycle to cycle performance appears to be highly consistent and reproducible with only a slight deviation in the maximum strain from 75.8% to 74.8% after seven cycles. This suggests that, despite the very low crosslinking density, the polymer does possess a homogeneous and robust network.
 |
| | Fig. 10 Consecutive shape memory cycles for Epon1. | |
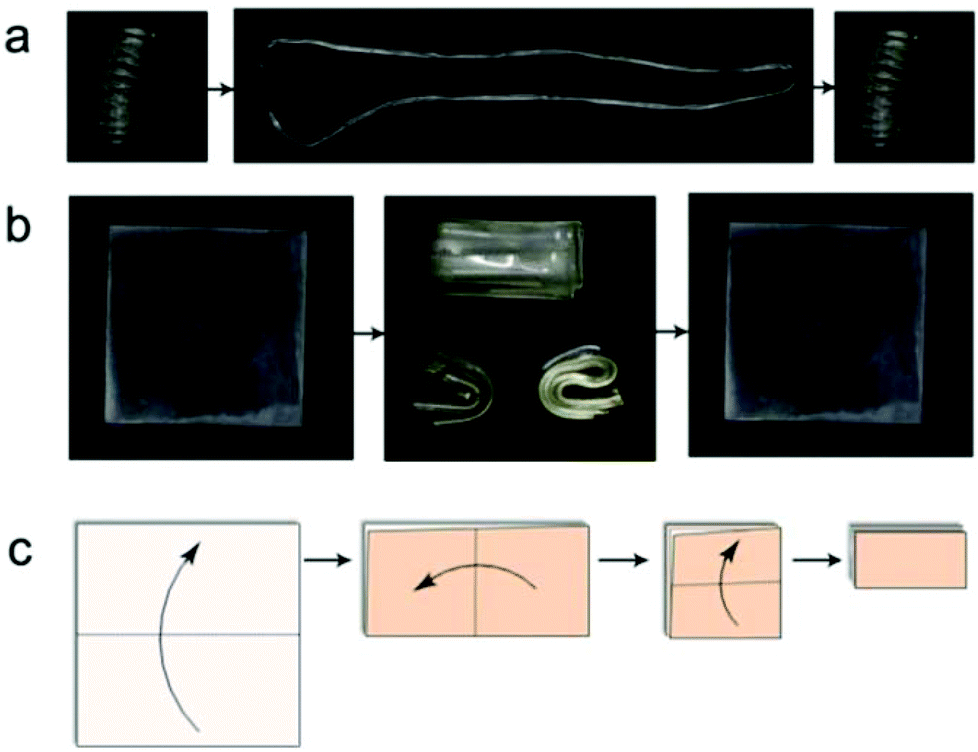
The advantages of the high strain shape memory performance of Epon1 are illustrated in Fig. 11. In one case, a sample was molded into an original tight spiral, which could be straightened into a temporary shape and recovered to the original one (Fig. 11a). Here, the liquid nature of the precursors permitted the use of a liquid molding technique using a simple twisted rubber tube as the mold to make a sophisticated shape (i.e. the spiral). In the second case, a square sample could be tightly folded once in the center line, folded the second time in the perpendicular center line, and folded the third time (Fig. 11b). Fig. 11c shows the folding steps of the square sample. In this latter case, the temporary shapes involved large strains, yet no fracture was observed and the samples could be completely recovered. These experiments and the experiments in Fig. 7 were repeated again 2 months later and no obvious difference in shape memory performance was observed. This confirms the durability of the current SMP system, at least within the timescale of the investigation.
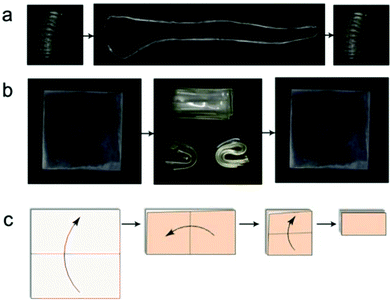 |
| | Fig. 11 Visual demonstration of the high strain capability for Epon1. (a) The shape memory and recovery steps of a tight spiral; (b) a square sample was folded three times and visualized in different directions; (c) the folding steps of the square sample. | |
4. Conclusion
A two component epoxy SMP system was developed via curing between an aromatic diepoxide and an aliphatic diamine. By varying the molar ratio between the two components, the crosslink density and the glass transition temperature (from 40 °C to 80 °C) can be tuned. This led to a high strain epoxy SMP with εb values of 110%. (70 °C, above its Tg) and 212% (50 °C, within its Tg transition). A recoverable strain of 180% can be achieved with this epoxy SMP. In addition to its high strain capability, the SMP also shows excellent shape fixity, shape recovery, and cycling stability. The robustness of the material makes it an ideal candidate for potential SMP device applications.
Acknowledgements
We would like to thank the following programs for their financial support: the National Key Basic Research Program of China (grant no. 2015CB351903), National Natural Science Foundation of China (grant no. 21474084); the Chinese central government's Recruitment Program of Global Experts; and the 985 program for the startup funding.
References
- T. Xie, Recent Advances in Polymer Shape Memory, Polymer, 2011, 52, 4985–5000 CrossRef CAS PubMed.
- J. L. Hu, Y. Zhu, H. H. Huang and J. Lu, Recent Advances in Shape-Memory Polymers: Structure, Mechanism, Functionality, Modeling and Applications, Prog. Polym. Sci., 2012, 37, 1720–1763 CrossRef CAS PubMed.
- P. T. Mather, X. Luo and I. A. Rousseau, Shape memory polymer research, Annu. Rev. Mater. Res., 2009, 39, 445–471 CrossRef CAS.
- M. Behl, M. Y. Razzaq and A. Lendlein, Multifunctional Shape-Memory Polymers, Adv. Mater., 2010, 22, 3388–3410 CrossRef CAS PubMed.
- J. S. Leng, X. Lan, Y. J. Liu and S. Y. Du, Shape-Memory Polymers and Their Composites: Stimulus Methods and Applications, Prog. Mater. Sci., 2011, 56, 1077–1135 CrossRef CAS PubMed.
- L. Sun, W. M. Huang, Z. Ding, Y. Zhao, C. C. Wang, H. Purnawali and C. Tang, Stimulus-Responsive Shape Memory Materials: A Review, Mater. Des., 2012, 33, 577–640 CrossRef CAS PubMed.
- K. M. Lee, T. J. Bunning and T. J. White, Autonomous, Hands-Free Shape Memory in Glassy, Liquid Crystalline Polymer Networks, Adv. Mater., 2012, 24, 2839–2843 CrossRef CAS PubMed.
- Y. Liu, J. K. Boyles, J. Genzer and M. D. Dickey, Self-Folding of Polymer Sheets Using Local Light Absorption, Soft Matter, 2012, 8, 1764–1769 RSC.
- H. Koerner, R. J. Strong, M. L. Smith, D. H. Wang, L. S. Tan, K. M. Lee, T. J. White and R. A. Vaia, Polymer design for high temperature shape memory: Low crosslink density polyimides, Polymer, 2013, 54, 391–402 CrossRef CAS PubMed.
- A. Lendlein, H. Y. Jiang, O. Junger and R. Langer, Light-Induced Shape-Memory Polymers, Nature, 2005, 434, 879–882 CrossRef CAS PubMed.
- X. Z. Zhang, Q. Q. Zhou, H. R. Liu and H. W. Liu, Uv Light Induced Plasticization and Light Activated Shape Memory of Spiropyran Doped Ethylene-Vinyl Acetate Copolymers, Soft Matter, 2014, 10, 3748–3754 RSC.
- H. J. Zhang, H. S. Xia and Y. Zhao, Light-Controlled Complex Deformation and Motion of Shape-Memory Polymers Using a Temperature Gradient, ACS Macro Lett., 2014, 3, 940–943 CrossRef CAS.
- H. J. Zhang, H. S. Xia and Y. Zhao, Optically Triggered and Spatially Controllable Shape-Memory Polymer-Gold Nanoparticle Composite Materials, J. Mater. Chem., 2012, 22, 845–849 RSC.
- J. S. Leng, X. Lan, Y. J. Liu, S. Y. Du, W. M. Huang, N. Liu, S. J. Phee and Q. Yuan, Electrical Conductivity of Thermoresponsive Shape-Memory Polymer with Embedded Micron Sized Ni Powder Chains, Appl. Phys. Lett., 2008, 92 Search PubMed.
- X. F. Luo and P. T. Mather, Conductive Shape Memory Nanocomposites for High Speed Electrical Actuation, Soft Matter, 2010, 6, 2146–2149 RSC.
- R. Mohr, K. Kratz, T. Weigel, M. Lucka-Gabor, M. Moneke and A. Lendlein, Initiation of Shape-Memory Effect by Inductive Heating of Magnetic Nanoparticles in Thermoplastic Polymers, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3540–3545 CrossRef CAS PubMed.
- W. M. Huang, B. Yang, L. An, C. Li and Y. S. Chan, Water-Driven Programmable Polyurethane Shape Memory Polymer: Demonstration and Mechanism, Appl. Phys. Lett., 2005, 86 Search PubMed.
- B. Yang, W. M. Huang, C. Li and L. Li, Effects of Moisture on the Thermomechanical Properties of a Polyurethane Shape Memory Polymer, Polymer, 2006, 47, 1348–1356 CrossRef CAS PubMed.
- Z. He, N. Satarkar, T. Xie, Y.-T. Cheng and J. Z. Hilt, Remote Controlled Multishape Polymer Nanocomposites with Selective Radiofrequency Actuations, Adv. Mater., 2011, 23, 3192–3196 CrossRef CAS PubMed.
- T. Xie and I. A. Rousseau, Facile Tailoring of Thermal Transition Temperatures of Epoxy Shape Memory Polymers, Polymer, 2009, 50, 1852–1856 CrossRef CAS PubMed.
- A. Lendlein and R. Langer, Biodegradable, Elastic Shape-Memory Polymers for Potential Biomedical Applications, Science, 2002, 296, 1673–1676 CrossRef PubMed.
- F. El Feninat, G. Laroche, M. Fiset and D. Mantovani, Shape Memory Materials for Biomedical Applications, Adv. Eng. Mater., 2002, 4, 91–104 CrossRef CAS.
- K. Gall, C. M. Yakacki, Y. P. Liu, R. Shandas, N. Willett and K. S. Anseth, Thermomechanics of the Shape Memory Effect in Polymers for Biomedical Applications, J. Biomed. Mater. Res., 2005, 73A, 339–348 CrossRef CAS PubMed.
- C. M. Chen, C. L. Chiang, C. L. Lai, T. Xie and S. Yang, Buckling-Based Strong Dry Adhesives Via Interlocking, Adv. Funct. Mater., 2013, 23, 3813–3823 CrossRef CAS.
- J. D. Eisenhaure, T. Xie, S. Varghese and S. Kim, Microstructured Shape Memory Polymer Surfaces with Reversible Dry Adhesion, ACS Appl. Mater. Interfaces, 2013, 5, 7714–7717 CAS.
- T. Xie and X. C. Xiao, Self-Peeling Reversible Dry Adhesive System, Chem. Mater., 2008, 20, 2866–2868 CrossRef CAS.
- H. X. Xu, C. J. Yu, S. D. Wang, V. Malyarchuk, T. Xie and J. A. Rogers, Deformable, Programmable, and Shape-Memorizing Micro-Optics, Adv. Funct. Mater., 2013, 23, 3299–3306 CrossRef CAS.
- C. M. Chen, J. C. Reed and S. Yang, Guided Wrinkling in Swollen, Pre-Patterned Photoresist Thin Films with a Crosslinking Gradient, Soft Matter, 2013, 9, 11007–11013 RSC.
- Y. Luo, Y. Guo, X. Gao, B.-G. Li and T. Xie, A General Approach Towards Thermoplastic Multishape-Memory Polymers Via Sequence Structure Design, Adv. Mater., 2013, 25, 743–748 CrossRef CAS PubMed.
- S. Kelch, N. Y. Choi, Z. G. Wang and A. Lendlein, Amorphous, Elastic Ab Copolymer Networks from Acrylates and Poly (L-Lactide)-Ran-Glycolide Dimethacrylates, Adv. Eng. Mater., 2008, 10, 494–502 CrossRef CAS.
- W. Voit, T. Ware, R. R. Dasari, P. Smith, L. Danz, D. Simon, S. Barlow, S. R. Marder and K. Gall, High-Strain Shape-Memory Polymers, Adv. Funct. Mater., 2010, 20, 162–171 CrossRef CAS.
- C. M. Yakacki, R. Shandas, D. Safranski, A. M. Ortega, K. Sassaman and K. Gall, Strong, Tailored, Biocompatible Shape-Memory Polymer Networks, Adv. Funct. Mater., 2008, 18, 2428–2435 CrossRef CAS PubMed.
- D. P. Nair, N. B. Cramer, T. F. Scott, C. N. Bowman and R. Shandas, Photopolymerized Thiol–Ene Systems as Shape Memory Polymers, Polymer, 2010, 51, 4383–4389 CrossRef CAS PubMed.
- A. F. Senyurt, C. E. Hoyle, H. Y. Wei, S. G. Piland and T. E. Gould, Thermal and Mechanical Properties of Cross-Linked Photopolymers Based on Multifunctional Thiol-Urethane Ene Monomers, Macromolecules, 2007, 40, 3174–3182 CrossRef CAS.
- I. A. Rousseau and T. Xie, Shape Memory Epoxy: Composition, Structure, Properties and Shape Memory Performances, J. Mater. Chem., 2010, 20, 3431–3441 RSC.
- D. M. Feldkamp and I. A. Rousseau, Effect of the Deformation Temperature on the Shape-Memory Behavior of Epoxy Networks, Macromol. Mater. Eng., 2010, 295, 726–734 CrossRef CAS.
- X. H. Jing, Y. Y. Liu, Y. X. Liu, Z. G. Liu and H. F. Tan, Toughening-Modified Epoxy-Amine System: Cure Kinetics, Mechanical Behavior, and Shape Memory Performances, J. Appl. Polym. Sci., 2014, 131 Search PubMed.
- J. S. Leng, X. L. Wu and Y. J. Liu, Effect of a Linear Monomer on the Thermomechanical Properties of Epoxy Shape-Memory Polymer, Smart Mater. Struct., 2009, 18(9), 095031 CrossRef.
- A. B. Leonardi, L. A. Fasce, I. A. Zucchi, C. E. Hoppe, E. R. Soule, C. J. Perez and R. J. J. Williams, Shape Memory Epoxies Based on Networks with Chemical and Physical Crosslinks, Eur. Polym. J., 2011, 47, 362–369 CrossRef CAS PubMed.
- Y. Y. Liu, C. M. Han, H. F. Tan and X. W. Du, Thermal, Mechanical and Shape Memory Properties of Shape Memory Epoxy Resin, Mater. Sci. Eng. A, 2010, 527, 2510–2514 CrossRef PubMed.
- R. Biju, C. Gouri and C. P. R. Nair, Shape Memory Polymers Based on Cyanate Ester-Epoxy-Poly (Tetramethyleneoxide) Co-Reacted System, Eur. Polym. J., 2012, 48, 499–511 CrossRef CAS PubMed.
- X. L. Wu, S. F. Kang, X. J. Xu, F. Xiao and X. L. Ge, Effect of the Crosslinking Density and Programming Temperature on the Shape Fixity and Shape Recovery in Epoxy-Anhydride Shape-Memory Polymers, J. Appl. Polym. Sci., 2014, 131 Search PubMed.
- R. M. Wang, X. C. Xiao and T. Xie, Viscoelastic Behavior and Force Nature of Thermo-Reversible Epoxy Dry Adhesives, Macromol. Rapid Commun., 2010, 31, 295–299 CrossRef CAS PubMed.
- J. H. Park and S. C. Jana, Mechanism of Exfoliation of Nanoclay Particles in Epoxy-Clay Nanocomposites, Macromolecules, 2003, 36, 2758–2768 CrossRef CAS.
- X. Luo and P. T. Mather, Shape Memory Assisted Self-Healing Coating, ACS Macro Lett., 2013, 2, 152–156 CrossRef CAS.
- X. Xiao, T. Xie and Y. T. Cheng, Self-healable graphene polymer composites, J. Mater. Chem., 2010, 20, 3508–3514 RSC.
- X. Luo and P. T. Mather, Triple-Shape Polymeric Composites (Tspcs), Adv. Funct. Mater., 2010, 20, 2649–2656 CrossRef CAS.
- T. Xie, X. Xiao and Y. T. Cheng, Achieving triple-shape memory effect in polymer bilayers, Macromol. Rapid Commun., 2009, 30, 1823–1827 CrossRef CAS PubMed.
- C. M. Yakacki, S. Willis, C. Luders and K. Gall, Deformation Limits in Shape-Memory Polymers, Adv. Eng. Mater., 2008, 10, 112–119 CrossRef CAS.
- K. Horie, H. Hiura, M. Sawada, I. Mita and H. Kambe, Calorimetric Investigation of Polymerization Reactions 3 Curing Reaction of Epoxides with Amines, J. Polym. Sci., Part A: Polym. Chem., 1970, 8, 1357–1372 CrossRef CAS.
- D. R. Miller and C. W. Macosko, Substitution Effects in Property Relations for Stepwise Polyfunctional Polymerization, Macromolecules, 1980, 13, 1063–1069 CrossRef CAS.
- L. Matejka, Amine Cured Epoxide Networks: Formation, Structure, and Properties, Macromolecules, 2000, 33, 3611–3619 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.