Site-specific PEGylation of proteins by a Staudinger-phosphite reaction†‡
Remigiusz
Serwa
a,
Paul
Majkut
a,
Benjamin
Horstmann
a,
Jean-Marie
Swiecicki
a,
Michael
Gerrits
b,
Eberhard
Krause
c and
Christian P. R.
Hackenberger
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, 14195, Berlin, Germany. E-mail: hackenbe@chemie.fu-berlin.de; Fax: +0049 30 838 52551; Tel: +0049 30 838 52451
bRiNA GmbH, Takustr. 3, 14195, Berlin, Germany
cLeibniz Institute of Molecular Pharmacology, Robert Rössle Str. 10, 13125, Berlin, Germany
First published on 9th August 2010
Abstract
Current protocols in protein bioengineering allow the site-specific incorporation of chemical reporter moieties. Subsequently, these functional groups can be chemoselectively transformed to decorate proteins with charged and oversized functional units. Based on our recent report on the chemoselective reaction of azides with phosphites, we now apply the Staudinger-phosphite reaction to an efficient and metal-free PEGylation of an azide-containing protein with symmetrical phosphites. Thereby, two types of branched oligoethylene glycol scaffolds are generated, which deliver either a stable or light-cleavable protein-PEG conjugate. Furthermore, we demonstrate that the Staudinger-phosphite reaction is an efficient transformation in both aqueous media as well as in a highly crowded bacterial cell lysate.
Introduction
The past decade brought an unprecedented development in protein engineering raising possibilities for a site-specific functionalization of proteins. The introduction of functional units that alter biophysicochemical properties of proteins may serve either to modulate their biological activities or to allow their imaging.1–7 A variety of protein translation systems has been designed for a site-specific incorporation of unnatural amino acids bearing simple functional moieties.8–11 However, the biochemical incorporation of charged, or oversized modules into polypeptide sequences still represents a major challenge.12–15 An efficient and site-specific incorporation of such functional entities has only been possible by employing proteins that contain unnatural amino acids as chemical reporters, as these moieties can be selectively addressed in a subsequent chemoselective transformation.5,16–21 Amongst chemical reporters, azides have been used most frequently, since these bioorthogonal moieties could easily be introduced into a variety of proteins in vitro and in vivo.8–11,22–24A number of chemoselective reactions of azides has been discovered so far. The copper assisted azide-alkyne cycloaddition reaction,25,26 CuAAC (Scheme 1, path A), was shown to be highly efficient in a variety of in vitro systems,4,5 but it suffers from limited in vivo applicability due to the cytotoxicity of Cu(I) catalysts.27,28 As a metal free alternative to CuAAC, a strain-promoted azide-alkyne cycloaddition was introduced for modifications in vitro and in vivo (Scheme 1, path B), which however introduces a relatively large strained linkage between a polypeptide and a functional module that may influence properties of the modified protein.29–35 The reaction between azides and phosphines, referred to as the Staudinger ligation (Scheme 1, path C), has also been successfully applied for the labeling of biomolecules in vivo,36,37 although here the oxidation of phosphines sometimes limits its utility.16 Given that all of these transformations have their own limitations, a quest for finding new bioorthogonal reaction partners for azides is still of general interest. Recently, we have identified the chemoselective reaction of azides with air-stable phosphites, as an alternative method for a metal-free protein functionalization.38,39
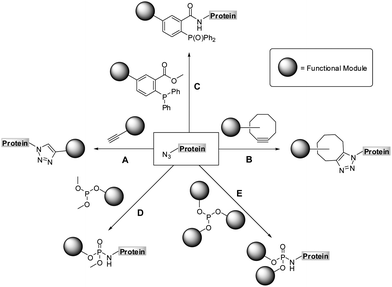 | ||
| Scheme 1 Chemoselective reactions of azides: copper assisted (A) and strain-promoted azide-alkyne cycloaddition (B), Staudinger (C) and Staudinger-phosphite reaction (D and E). | ||
The Staudinger-phosphite reaction delivers phosphoramidate linkages, which mimic natural occurring phosphoric esters and introduce either one (Scheme 1, path D) or two (Scheme 1, path E) functional modules. From the point of view of molecular economy, an application of phosphites bearing a single functional substituent as ligation partners for azides is favourable;38 however, products resulting from reactions between symmetrical phosphites and azides may in some cases profit from the introduction of two functional modules of interest. For instance, symmetrical phosphites bearing polyethylene glycol (PEG) chains would, upon Staudinger reaction, functionalize proteins with stable and light-cleavable branched PEG-scaffolds (Scheme 2). The elements are expected to have important pharmacological implications when presented on therapeutic polypeptides, since it was shown that, in comparison with linear polymers, branched PEG strands possess reduced immunogenicity and decreased ability to accumulate in animal tissues.40–43
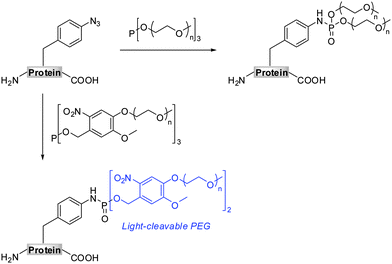
Together with the development of a novel protein PEGylation strategy, we present herein more details on the advances and limitations of the Staudinger-phosphite reaction. In this, we conducted a thorough investigation of the performance of the transformation in aqueous media, taking into account the stability and the reactivity of phosphite modifiers. Additionally, we confirmed that the reaction between azides and phosphites is efficient even in a highly crowded bio-environment, such as an E.coli lysate.
Results and discussion
Modification with small water-soluble PEG-phosphites
As the Staudinger-phosphite reaction has only recently been applied for the modification of polypeptides in aqueous solvents,38,39 only limited information exists about the performance of this transformation in ligation processes.44–46 Therefore, we have decided to investigate issues that may be of key importance for efficient bio-conjugation, which include the stability of the reactants in aqueous media, the reactivity and solubility of different phosphites, the resulting apparent minimal concentration of reagents necessary for a high-yielding modification, and the possibility of conjugation in biological mixtures.We first focused on the modification of a model azido-peptide 1 with water-soluble phosphites in various buffers. During the incubation of peptide 1 with trimethylphosphite (2), a clear trend could be observed: the higher the pH of the buffer was, the more efficient was the formation of phosphoramidate 3, (Table 1, entries 1–3). A similar trend was observed during the conversion of peptide 1 into phosphoramidate 5 in the presence of tris(tetraethylene glycol) phosphite (4), which was prepared according to the procedure adapted from the literature (Table 1, entries 4–7).47
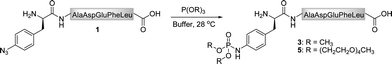
| Entry | Reagent, R = | pH | Product | Yield [%]b |
|---|---|---|---|---|
| a Peptide 1 (0.2 mM) was reacted with phosphites 2 and 4 (20 mM) for 16 h at 28 °C. b Based on the HPLC-UV analysis. c Reagent 4 was added portionwise (4 × 10 mM) during the course of 48 h. | ||||
| 1 | 2, CH3 | 7.4 | 3 | 17 |
| 2 | 2, CH3 | 7.8 | 3 | 28 |
| 3 | 2, CH3 | 8.2 | 3 | 47 |
| 4 | 4, (CH2CH2O)4CH3 | 7.4 | 5 | 31 |
| 5 | 4, (CH2CH2O)4CH3 | 7.8 | 5 | 53 |
| 6 | 4, (CH2CH2O)4CH3 | 8.2 | 5 | 80 |
| 7 | 4, (CH2CH2O)4CH3 | 8.2 | 5 | 98c |
Since phosphites are known to rearrange to phosphonates at low pH in aqueous media,48,49 we proposed that the observed efficiencies of the peptide modification might be associated with the decomposition of phosphites 2 and 4 in the reaction mixtures. In order to evaluate this hypothesis, we monitored the decay of these phosphites in aqueous buffers by 31P-NMR. These measurements confirmed that both compounds disappeared faster at lower pH levels (ESI‡). For instance, after 30 min at 25 °C, 54% of 2 and 17% of 4 decomposed at pH 7.4, but only 21% of 2 and 8% of 4 at pH 8.2, which gave a rational for the conversions presented in Table 1. In addition, tris(tetraethylene glycol) phosphite was found to be far more stable than trimethylphosphite, hence greater conversions of peptide 1 were observed in the presence of this compound. Furthermore, the phosphoramidate formation could be greatly enhanced by dosing the modifier into the reaction mixture (Table 1, entry 7). Finally, the stability studies performed on the Staudinger reaction product 5 indicated that the bis(tetraethylene glycol) phosphoramidate unit was unchanged in Tris buffer (pH 7.4–8.2) after 48 h of incubation at 28 °C.
As phosphite 4 efficiently converted the model peptide into the bis(tetraethylene glycol) phosphoramidate 5 in good yield, we employed this moderate sized PEGylating agent for the conversion of the 18 kDa model protein SecB (6) into the corresponding phosphoramidate 7 (Fig. 1). Protein 6, containing a p-azidophenylalanine at position 156, was prepared using an E. coli derived cell-free translation system depleted from termination factor RF1 and supplemented with enriched fractions of orthogonal amber suppressor tRNA and p-azidophenylalanyl-tRNA synthetase specific for p-azidophenylalanine.8
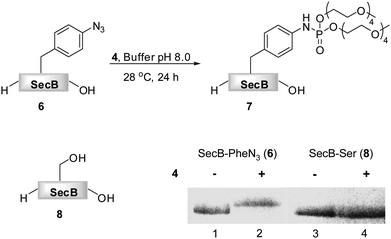 | ||
| Fig. 1 Transformation of azido-protein 6 (5 μM) into 7 with PEG-phosphite 4 (8–25 mM) in phosphate buffer at pH 8.0 for 24 h at 28 °C (Lane 2). 6 (Lane 1) and 8 (Lane 3) in the absence of 4, as well as 8 in the presence of 4 (Lane 4) did not undergo modification as shown by Coomassie stained SDS-PAGE gel. | ||
Phosphite 4 was added portionwise over time to maximize the yield of the transformation. In order to gain further support for the chemoselectivity of the Staudinger-phosphite reaction, a SecB mutant 8, in which the p-azidophenylalanine was replaced by serine, was prepared similarly to 6, using an enzymatically charged Seryl-tRNACUA, and subjected to the analogous reaction conditions. As confirmed by a shift in the protein retention time during SDS-PAGE analysis, the decoration of the azido-protein 6 with tetraethylene glycol strands proceeded quantitatively, whereas no shift was observed upon incubation of protein 8 with phosphite 4 (Fig. 1). Furthermore, MALDI-MS analysis, which was conducted on the protein after isolation from the reaction mixture additionally supported the formation of the ligation product 7 (ESI‡).
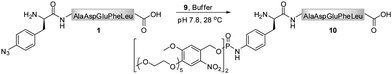
In contrast to the findings in Table 1 for the modification of peptide 1 with phosphites 2 and 4, no pH dependency was observed for the reaction of phosphite 9 to yield a photo-sensitive phosphoramidate 10 (Scheme 3).3931P NMR measurements performed on phosphite 9 revealed that the compound remained stable even after 48 h of incubation in Tris buffer (pH 7.4–8.2) at 28 °C (ESI‡). From a subsequent thermal decomposition study, it was found that compound 9 decayed rapidly only at temperatures greater than 40 °C.
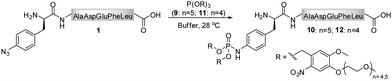
During the search for an optimal reagent for the chemical phosphorylation of proteins,39 we synthesized a homologue 11 of phosphite 9. Although both compounds differed only by three ethylene glycol units, we found that, in contrast to 9, phosphite 11 reacted with peptide 1 to produce phosphoramidate 12 extremely slow (ESI‡).
In addition, we observed that compound 9 (>50 mM) was over 30 times more soluble in water at room temperature than its homologue 11 (1.53 ± 0.03 mM). This finding helps to explain the difference in reactivity of phosphites 9 and 11 towards peptide 1 and is further supported by the observation that addition of DMSO, a solvent in which both 9 and 11 are well soluble, diminished the difference in the conversion of 1. Specifically, it was found that peptide 1 (50 μM) was quantitatively converted with 9 or 11 (15 mM) within 6 h when 50% DMSO was used as a co-solvent.
A chemical functionalization of bio-molecules is frequently performed in the presence of a large excess of modifiers in order to ensure an efficient and fast transformation. Alternatively, as demonstrated for hydrazone and oxime ligations, the reaction rate can be enhanced in the presence of a catalyst, aniline, and nearly stoichiometric quantities of hydrazide and aminoxy modifiers can be used.50,51 We therefore conducted a more detailed investigation on the necessary phosphite/peptide ratio for modifier 9, which has previously been employed in the conversion of peptide 1 into peptide 10 (Scheme 3).39 At millimolar concentrations, it was found that azido-peptide 1 was nearly quantitatively transformed into phosphoramidate 10 in the presence of only 2 equivalents of phosphite 9 within a few hours.52 To estimate the apparent minimal concentration of phosphite 9, at which it efficiently modifies more diluted solutions of azido-polypeptides, peptide 1 (50 μM) was incubated at 28 °C with reagent 9 (300, 150, 75, 38 equivalents) in phosphate buffer for 24 h, and the disappearance of 1 from reaction mixtures was monitored by LC-MS (Table 2). From mixtures containing 9 in concentrations greater or equal to 3.7 mM (75 eq), peptide 1 was depleted within 6 h, whereas a longer incubation time was needed at an initial concentration of 1.9 mM 9 (38 eq). These results imply that stable phosphites are able to modify aryl azide-containing peptides in buffers at relatively low concentrations. Nevertheless, for a rapid bio-conjugation, an increased amount of phosphite, its good solubility and stability may be essential.
Protein modification in bacterial cell lysate
In a recent communication we have presented an efficient transformation of the isolated azido-protein 6 by a Staudinger-phosphite reaction with 9 into phosphoramidate 13.39 We now focused on an analogous transformation in a more crowded environment with a variety of biologically relevant molecules being present (Fig. 2). The importance of such a study was rationalized by the fact that biochemical studies are often executed in whole cell lysates, which are much easier to exploit experimentally in comparison to living cells or animals. In order to visualize the bioorthogonal conversion by the Staudinger-phosphite reaction, we first prepared radioactively labeled protein 6 by cell-free unnatural protein translation in E.coli derived lysates in the presence of 14C-labeled L-leucine. Next to the full length protein (Fig. 2B, Lane 1, top band), which was the major product, the amber terminated protein truncated at position 155 (Fig. 2B, Lane 1, bottom band) was obtained during in vitro translation. After addition of phosphite 9 to the lysate, protein 6, but not the truncated one, underwent an efficient Staudinger-phosphite transformation as verified by a radiogram of SDS-PAGE. The band of the modified protein 13 was shifted upwards compared to 6, whereas no migration of the corresponding p-azidophenylalanine lacking protein occurred (Fig. 2B). This observation confirms that the Staudinger-phosphite reaction can be successfully conducted in whole lysed cell systems.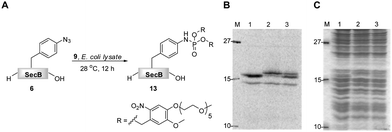 | ||
| Fig. 2 The transformation of 14C-labeled protein 6 (5 μM) into phosphoramidate 13 in the presence of phosphite 9 (5 mM), in E.coli lysate, followed by SDS-PAGE; A. reaction scheme; B. radiogram; C. Coomassie stain; Lane 1. unmodified protein 6; Lane 2. protein 6 incubated with phosphite 9 for 12 h at 28 °C; Lane 3. content of lanes 1 and 2 premixed prior to SDS-PAGE. | ||
Protein PEGylation
Covalent attachment of polyethylene glycol chains to peptides and proteins has gained much attention, especially in the field of medicinal chemistry. This unnatural protein modification was found to reduce immuno- and antigenicity of therapeutic polypeptides, while simultaneously increasing their hydrodynamic size and plasma half life times.41–43 Additionally, PEGylation can be applied to improve the solubility of polypeptides.41,53–55 Early protein PEGylation approaches involved the functionalization of canonical amino acids resulting in the formation of protein mixtures of variable properties.40,42,56,57 The possibility of site-specific incorporation of chemical reporters into polypeptides resulted in the development of a new generation of PEGylating agents that could be applied for targeted ligations.57 These efforts involved alkynyl PEG for copper mediated and strain promoted cycloaddition reactions with azides,30,58,59 as well as phosphine or aminooxyacetyl PEG suitable for Staudinger and oxime ligation, respectively.60–62Encouraged by excellent results for the modification of protein 6 with phosphites 9 and 4, we prepared homologous phosphites 14 and 15 that would deliver longer oligoethylene glycol strands to protein ligation sites. It is important to notice that the PEGylation strategy presented here differs in two aspects from the other previously mentioned chemoselective PEGylation routes.58,60,62 First, the application of symmetrical phosphites (e.g.4, 9, 11, 14, and 15) always results in a functionalization of polypeptides with branched oligoethylene glycol scaffolds. Second, the photo-sensitive phosphites (e.g.9, 11 and 14) allow a removable PEG attachment, since the bis(4-oligo-ethylene glycol-2-nitrobenzyl) phosphoramidates can undergo a light-induced saponification to form non-PEGylated polypeptides.63
Phosphites 14 and 15 were synthesized in good yields in thermally induced reactions between hexamethylphosphorous triamide and the corresponding alcohol precursors (Scheme 4). The precursor for 14 was obtained in a nucleophilic substitution reaction between mPEG750 tosylate and 4-(hydroxymethyl)-2-methoxy-5-nitrophenol, in analogy to the preparation of its homologues utilized for the synthesis of 9 and 11.39 It is important to note that, similarly to 4, phosphite 15 can be easily prepared in one step from inexpensive commercially available reagents.47 Given the fact that mPEG-OH polymers of various molecular weights are readily available, this method could be potentially used for the preparation of other homologues of 15.
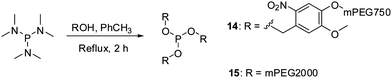 | ||
| Scheme 4 Preparation of reagents 14 and 15 for polypeptide PEGylation. | ||
The reactivities of phosphites 14 and 15 were first assessed in consumption studies with azido-phenylalanine. It was found that the amino acid was depleted from buffered solutions containing an excess of both phosphite reagents after 12 h of incubation at 28 °C (data not shown). In a subsequent experiment 14C-labeled proteins 6 and 8 that were isolated from translation mixtures by their C-terminal His6-Tag, were used. Truncated proteins that lack His6-Tag could not be separated from the full length proteins, due to a strong tendency of SecB to form tetramers. Although, these amber terminated proteins were present in the mixtures, they did not interfere with the chemoselective transformation. The proteins were incubated in phosphate buffer (pH 8.0) in the presence of 14 and 15 and DMSO at 28 °C.64 After 24 h, SecB was separated from modifiers and the reaction progress was analyzed by SDS-PAGE (Fig. 3).
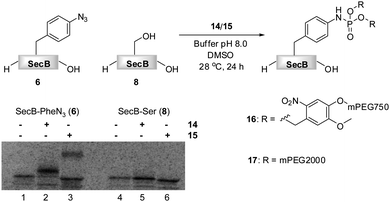 | ||
Fig. 3 PEGylation of 6 (5 μM) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 phosphate buffer pH 8.0/DMSO in the presence of 5 mM 14 (Lane 2) and 3.3 mM 15 (Lane 3).646 (Lane 1) and 8 (Lane 4) in the absence of 14 or 15, and 8 in the presence of 14 or 15 (Lane 4) did not undergo PEGylation as shown on the SDS-PAGE gel. :1 phosphate buffer pH 8.0/DMSO in the presence of 5 mM 14 (Lane 2) and 3.3 mM 15 (Lane 3).646 (Lane 1) and 8 (Lane 4) in the absence of 14 or 15, and 8 in the presence of 14 or 15 (Lane 4) did not undergo PEGylation as shown on the SDS-PAGE gel. | ||
Protein 6 could be efficiently modified with either phosphite 14 or 15 as verified by the SDS-PAGE, in which the bands of the modified proteins 16 and 17 were shifted upwards according to the molecular weight of the modification. A radiation count was employed to estimate 88% conversion of 6 into 16, and 42% conversion into 17. The lower efficiency of the formation of phosphoramidate 17 could be explained on the basis of the partial hydrolytic instability of 15 in aqueous systems, in analogy to its homologue 4. In an additional experiment, 15 was added in portions throughout the incubation period to yield 17 nearly quantitatively (ESI‡). Moreover, as expected, no PEGylation of protein 8 was detected by SDS-PAGE, suggesting the absence of non-specific interactions between PEGylating agents and the protein (Fig. 3).
Finally, we have investigated the efficiency of the light-induced removal of the PEG moiety from protein 16. After the Staudinger-phosphite reaction, SecB was separated from modifier 14, and it was irradiated at 355 nm to yield protein 18.39 The sample was subsequently heated to 40 °C for 20 min in a buffer of an increased acidity (pH 6.8) in order to generate homogeneous p-aminophenylalanine containing protein 19 upon hydrolysis of phosphoramidate 18.65 SDS-PAGE analysis indicated that the photochemical reaction was almost completed after 1 min (Fig. 4). This removal of polymer conjugates could potentially be utilized for a modulation of polypeptide self-assembly. In analogy to the antiimmunogenic function of polyethylene glycol, which is based on the mechanical shielding of proteinous epitops by polymer strains,61 one could envision that by the same principle PEG could shield aggregation prone stretches and thus attenuate or even inhibit the self-assembly of polypeptides. Currently, we are exploring the use of reagent 14 for a temporal control of the aggregation process in a peptide model system.
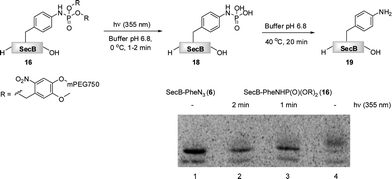 | ||
| Fig. 4 The light induced saponification of PEGylated protein 16 in phosphate buffer pH 6.8 at 355 nm. The radiogram of the SDS-PAGE depicts azido-protein 6 (Lane 1), 16 irradiated for 2 min (Lane 2), 16 irradiated for 1 min (Lane 3), non-irradiated 16. | ||
Conclusions
We have demonstrated that phosphites are useful reagents for aqueous Staudinger-based transformations, in particular for the attachment of PEG-chains to proteins. However, certain structural criteria need to be met in order for them to become efficient modifiers. We have shown that tris(oligoethylene glycol) and the corresponding 2-nitrobenzyl substituted phosphites are relatively stable in aqueous media at physiological pH and thus are able to efficiently modify azides when added portionwise to the reaction medium. In addition to the sufficient stability a high solubility of the phosphites has to be ensured to guarantee efficient ligations in biologically relevant media. The decoration of phosphites with chemically inert solubilizing groups (i.e. oligoethylene glycol) has been shown to greatly enhance the hydrophilicity of these compounds. A previously reported reagent for chemical phosphorylation seems to be a good example of a phosphite possessing such optimized properties. As demonstrated in the present report, this reagent can also be used to efficiently modify azido-polypeptides in a complex E.coli lysate. Additionally, in a series of experiments involving side by side incubations of a model azido-protein and its mutant (PheN3 → Ser) in the presence of symmetrical phosphites, further evidence for the chemoselectivity of the Staudinger-phosphite transformation was presented. Finally, we have demonstrated for the first time the decoration of a protein with branched oligoethylene glycol scaffolds as a result of Staudinger-phosphite reaction, and thus expanded the scope of this modification methodology.In summary, the Staudinger-phosphite reaction appears to be a powerful metal-free method for bio-conjugations. Current efforts in our laboratory are devoted to the preparation of other symmetrical and unsymmetrical phosphites that will be used for further functionalization of azido-proteins.
Experimental
Materials and methods
Peptide 1 was prepared by an automated Fmoc-SPPS utilizing standard coupling (HBTU/HOBT/DIPEA) and deprotection (25% piperidine) conditions. Sec B proteins (6 and 8) were prepared via cell-free protein expression using the EasyXpress Protein Synthesis Kit and purified utilizing Ni-NTA magnetic beads (both Qiagen).39 Radioactively labeled protein was obtained by addition of 14C-Leu (final specific activity 4 dpm/pmol Leu) to the in vitro translation reaction. All buffers were treated with Chelex 100 (Sigma) before use. SDS-PAGE gels (15% and 17%) were run at 180 V until 10 kDa protein marker was eluted out. SDS-PAGE gels containing radioactive proteins were exposed to Storage Phosphor Screens (Molecular Dynamics), and the read out was accomplished with a Typhoon 8600 imager (Molecular Dynamics). Visualization and estimation of the protein conversion was accomplished with Image Quant 5.1 software. Photocleavage of PEG chains from proteins and peptides was performed by irradiation with a Laser at 355 nm (Spectron Laser Systems, settings: 10 Hz, 10 ns and 50 mJ/pulse). LC-UV-MS was performed on an Agilent 6210 TOF LC/MS system, Agilent Technologies (Santa Clara, CA). Spray voltage was set to 4 kV, and drying gas flow rate was set to 25 psi. Separation of samples was performed on a single Luna 5μ C18(2) 100 A column (5 μm, 4.6 × 150 mm) at a flow rate of 0.6 mL min−1, utilizing gradients of MeCN in H2O contained 1% AcOH.Modification of peptide 1 with phosphites in buffers
To a buffered solution (1.0 mL) of azido-peptide 1 (200 μM) was added a phosphite (20 μmol; 2.5 mg of 2; 13.8 mg of 4) and the mixture was stirred for 16 h at 28 °C. In a separate experiment, phosphite 4 was added in 4 portions (each: 10 μmol, 6.9 mg) at t = 0, 16, 24, 40 h. Following the incubation, reaction mixtures were immediately analyzed (injection volume: 50 μL) by RP-HPLC connected in line to a UV detector (for quantitative analysis) and an ESI-TOF detector (for qualitative analysis). Identities of products were supported by HRMS data, 3: m/z = 864.3573 [M + H]+, calcd.: 864.3593; 5: 1216.5690 [M + H]+, calcd.: 1216.5636, whereas the conversion of substrate 1 into products 3 and 5 was calculated based on UV240nm absorbance values measured for the corresponding peaks.Incubation of proteins 6 and 8 in the presence of phosphite 4
To a suspension of protein 6 (20 μM) or 8 (20 μM) in phosphate buffer pH 8.0 (20μL) at 28 °C was added a solution of 4 in the same buffer (50 mM) over time (4 μL/2 h, 5×), and the sample was shaken (2000 RPM) for an additional 14 h at 28 °C. Following the incubation, the mixture was diluted with water (60 μL) and 4 × sample loading buffer (35 μL), and it was incubated at 35 °C (10 min) prior to loading it (40 μL) on a 17% SDS-PAGE gel. Upon electrophoresis, the gel was stained with Coomassie Brilliant Blue and photographed.Modification of protein 6 with phosphite 9 in E.coli lysate
To E.coli lysate (25 μL) containing 14C-labeled protein 6 (5 μM) was added a solution of phosphite 9 in 1 × PBS (15 μL, 20 mM), and the sample was incubated for 12 h at 28 °C. Following the incubation, aliquots of the mixture (20 μL) were diluted with water (100 μL) and precipitated with ice cold acetone (300 μL) for 15 min. Precipitates were collected via centrifugation (12000 RPM, 10 min) followed by decantation; then they were dried with SpeedVac (40 °C, 10 min). 1 × Sample loading buffer (20 μL) was added to the precipitates, and proteins were denatured at 90 °C (5 min) prior to loading them on a 17% SDS-PAGE gel. Upon electrophoresis, the gel was dried and exposed to a Storage Phosphor Screen for filmless autoradiography for 72 h.Incubation of proteins 6 and 8 in the presence of phosphites 14 and 15
To a suspension of 14C-labeled protein 6 (5 μM) or 8 (5 μM) in phosphate buffer pH 8.0 (10 μL) was added a solution of 14 in DMSO (10 μL of 10 mM) or phosphite 15 (0.39 mg, 65 nmol) and DMSO (10 μL), and the sample was shaken (2000 RPM) for 24 h at 28 °C. Following the transformation, the samples were incubated for 15 min in a mixture of water (50 μL) and acetone (150 μL) at 0 °C. The protein was isolated by centrifugation, it was taken up in 1× sample loading buffer (50 μL), and it was incubated at 35 °C (10 min) prior to loading it (25 μL) on a 15% SDS-PAGE gel. Upon electrophoresis, the gel was dried and exposed to a Storage Phosphor Screen for filmless autoradiography for 72 h.The light induced saponification of protein 16
Following the acetone precipitation, the protein was taken up in phosphate buffer pH 6.8 (40 μL) containing SDS (1%) and β-mercaptoethanol (1%). The sample was divided into two equal parts, which were irradiated with a laser at 355 nm for 1 or 2 min at 0 °C. Glycerol (2 μL) was added to the samples, and they were incubated for 20 min at 40 °C (in order to convert relatively labile phosphoramidate 18 into a stable product 19) prior to loading them on a 15% SDS-PAGE gel. Upon electrophoresis, the gel was dried and exposed to a Storage Phosphor Screen for filmless autoradiography for 72 h.Acknowledgements
The authors acknowledge financial support from the German Science Foundation (DFG, Emmy-Noether program, HA 4468/2-1), the SFB 765, the Max-Buchner Foundation and the Fonds der chemischen Industrie (FCI). We also thank Iris Claußnitzer, Henrike Roterberg, Heike Stephanowitz, Malgorzata Broncel, Cathleen Schlesener and Dr Andreas Schäfer for their help.Notes and references
- B. G. Davis, Pure Appl. Chem., 2009, 81, 285–298 CrossRef CAS.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS.
- N. Jessani and B. F. Cravatt, Curr. Opin. Chem. Biol., 2004, 8, 54–59 CrossRef CAS.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- P. F. van Swieten, M. A. Leeuwenburgh, B. M. Kessler and H. S. Overkleeft, Org. Biomol. Chem., 2005, 3, 20–27 RSC.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS.
- K. L. Kiick and D. A. Tirrell, Tetrahedron, 2000, 56, 9487–9493 CrossRef CAS.
- J. Xie and P. G. Schultz, Nat. Rev. Mol. Cell Biol., 2006, 7, 775–782 CrossRef CAS.
- T. Arslan, S. V. Mamaev, N. V. Mamaeva and S. M. Hecht, J. Am. Chem. Soc., 1997, 119, 10877–10887 CrossRef CAS.
- A. Nguyen, D. M. Rothman, J. Stehn, B. Imperiali and M. B. Yaffe, Nat. Biotechnol., 2004, 22, 993–1000 CrossRef CAS.
- G. F. Short III, A. L. Laikhter, M. Lodder, Y. Shayo, T. Arslan and S. M. Hecht, Biochemistry, 2000, 39, 8768–8781 CrossRef.
- D. M. Rothman, E. J. Petersson, M. E. Vázquez, G. S. Brandt, D. A. Dougherty and B. Imperiali, J. Am. Chem. Soc., 2005, 127, 846–847 CrossRef CAS.
- N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644–648 CrossRef CAS.
- T. K. Tiefenbrunn and P. E. Dawson, Biopolymers, 2010, 94, 95–106 CrossRef CAS.
- A. J. Link, M. L. Mock and D. A. Tirrell, Curr. Opin. Biotechnol., 2003, 14, 603–609 CrossRef CAS.
- M. B. Francis, in Chemical Biology, ed. S. L. Schreiber, T. M. Kapoor, G. Wess, Wiley-VCH Verlag Gmbh & Co. KGaA, Weinheim, Germany, 2007, vol. 2, pp. 593–634 Search PubMed.
- M. D. Gieselman, Y. Zhu, H. Zhou, D. Galonic and W. A. van der Donk, ChemBioChem, 2002, 3, 709–716 CrossRef CAS.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS.
- I. C. Tanrikulu, E. Schmitt, Y. Mechulam, W. A. Goddard III and D. A. Tirrell, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15285–15290 CrossRef CAS.
- A. J. Link, M. K. S. Vink and D. A. Tirrell, J. Am. Chem. Soc., 2004, 126, 10598–10602 CrossRef CAS.
- N. Budisa, Angew. Chem., Int. Ed., 2004, 43, 6426–6463 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS . For seminal contributions, see: R. Huisgen, G. Szeimies and L. Moebius, Chem. Ber., 1967, 100, 2494–2507 Search PubMed.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS.
- T. Ramenda, T. Kniess, R. Bergmann, J. Steinbach and F. Wuest, Chem. Commun., 2009, 7521–7523 RSC.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS.
- M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC.
- X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS.
- J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Berozzi, J. Am. Chem. Soc., 2008, 130, 11486–11493 CrossRef CAS.
- E. Kaya, K. Gutsmiedl, M. Vrabel, M. Müller, P. Thumbs and T. Carell, ChemBioChem, 2009, 10, 2858–2861 CrossRef CAS.
- D. P. Nguyen, H. Lusic, H. Neumann, P. B. Kapadnis, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2009, 131, 8720–8721 CrossRef CAS.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS.
- J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873–877 CrossRef CAS.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141 CrossRef CAS.
- V. Boehrsch, R. Serwa, P. Majkut, E. Krause and C. P. R. Hackenberger, Chem. Commun., 2010, 46, 3176–3178 RSC.
- R. Serwa, I. Wilkening, G. Del Signore, M. Mühlberg, I. Claußnitzer, C. Weise, M. Gerrits and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2009, 48, 8234–8239 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS.
- F. M. Veronese and A. Mero, BioDrugs, 2008, 22, 315–329 Search PubMed.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS.
- P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261–1277 CrossRef CAS.
- R. L. Letsinger and G. A. Heavner, Tetrahedron Lett., 1975, 11, 147–150 CrossRef.
- J. Xue, J. Wu and Z. Guo, Org. Lett., 2004, 6, 1365–1368 CrossRef CAS.
- A. Zidani, R. Carrie and M. Vaultier, Bull. Soc. Chim. France, 1992, 129, 71–75 CAS.
- K. Skopek and J. A. Gladysz, J. Organomet. Chem., 2008, 693, 857–866 CrossRef CAS.
- A. E. Arbuzov and M. G. Imaev, Dokl. Akad. Nauk SSSR, 1957, 112, 856–859 CAS.
- S. K. McIntyre and T. M. Alam, Magn. Reson. Chem., 2007, 45, 1022–1026 CrossRef CAS.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS.
- A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS.
- Peptide 1 (2 mM) was transformed (>98%) into phosphoramidate 10 in the presence of phosphite 9 (4 mM) in 1M Tris buffer pH 8.2 at 28 °C within 4 h (LC-MS). After 6–8 h, peptide 1 could not be detected anymore by LC-MS.
- A. Basu, K. Yang, M. Wang, S. Liu, R. Chintala, T. Palm, H. Zhao, P. Peng, D. Wu, Z. Zhang, J. Hua, M.-C. Hsieh, J. Zhou, G. Petti, X. Li, A. Janjua, M. Mendez, J. Liu, C. Longley, Z. Zhang, M. Mehlig, V. Borowski, M. Viswanathan and D. Filpula, Bioconjugate Chem., 2006, 17, 618–630 CrossRef CAS.
- L.-C. Chang, H.-F. Lee, M.-J. Chung and V. C. Yang, Bioconjugate Chem., 2005, 16, 147–155 CrossRef CAS.
- G. Pasut and G. F. Veronese, Progr. Polym. Sci., 2007, 32, 933–961 CrossRef CAS.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 RSC.
- L. A. Canalle, D. W. P. M. Löwik and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 329–353 RSC.
- A. Deiters, T. A. Cropp, D. Summerer, M. Mukherji and P. G. Schultz, Bioorg. Med. Chem. Lett., 2004, 14, 5743–5745 CrossRef CAS.
- B. Peschke, M. Zundel, S. Bak, T. R. Clausen, N. Blume, A. Pedersen, F. Zaragoza and K. Madsen, Bioorg. Med. Chem., 2007, 15, 4382–4395 CrossRef CAS.
- C. S. Cazalis, C. A. Haller, L. Sease-Cargo and E. L. Chaikof, Bioconjugate Chem., 2004, 15, 1005–1009 CrossRef CAS.
- L. P. Miranda, H. Shao, J. Williams, S.-Y. Chen, T. Kong, R. Garcia, Y. Chinn, N. Fraud, B. O'Dwyer, J. Ye, J. Wilken, D. E. Low, E. N. Cagle, M. Carnevali, A. Lee, D. Song, A. Kung, J. A. Bradburne, X. Paliard and G. G. Kochendoerfer, J. Am. Chem. Soc., 2007, 129, 13153–13159 CrossRef CAS.
- R. A. Scheck and M. B. Francis, ACS Chem. Biol., 2007, 2, 247–251 CrossRef CAS.
- In a previous study (ref. 39) it was reported that peptide 10 remained stable for 72 h in buffers (pH 7.4–8.2) in the absence of light. However, 10 could be readily saponified upon irradiation at 355 nm. Similarly, protein 12 was also efficiently saponified upon irradiation.
- The reaction of protein 6 with 14 and 15 in plain buffer led to very low levels of PEGylation . An addition of GdnCl or DMSO significantly enhanced the formation of PEGylated protein 17 (ESI‡). Since 6 has a tendency to aggregate and precipitate from the solution upon storage, it is reasonable to expect that high molecular weight phosphites 14 and 15 may have a limited access to the modification site. Addition of denaturing agents, such as GdnCl or DMSO, was most likely responsible for protein solubilization, thus promoting the Staudinger-phosphite transformation. Furthermore, we have observed both 14 and 15 being able to efficiently modify water soluble peptides in buffers in the absence of denaturing agents or co-solvents.
- Under these conditions an undecapeptide containing the free phosphoramidate moiety (generated upon light induced saponification) was hydrolyzed completely to the corresponding aniline. HRMS was used to monitor the conversion and to confirm the structures of the saponified phosphoramidate (m/z = 1327.5517 [M + H]+, calcd.: 1327.5567) and the hydrolyzed aniline peptide (m/z = 1247.5940 [M + H]+, calcd.: 1247.5903). For the sequence of the undecapeptide see: R. A. Serwa, J.-M. Swiecicki, D. Homann and C. P. R. Hackenberger, J. Pept. Sci., 2010 DOI:10.1002/psc.1236.
Footnotes |
| † This work is dedicated to Prof. Rolf Huisgen on the occasion of his 90th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthetic protocols and additional analytical data. See DOI: 10.1039/c0sc00324g |
| This journal is © The Royal Society of Chemistry 2010 |