Polymers of intrinsic microporosity (PIMs): robust, solution-processable, organic nanoporous materials
Peter M.
Budd
,
Bader S.
Ghanem
,
Saad
Makhseed
,
Neil B.
McKeown
*,
Kadhum J.
Msayib
and
Carin E.
Tattershall
Department of Chemistry, University of Manchester, Manchester, UK M13 9PL. E-mail: neil.mckeown@man.ac.uk; Fax: 0161 275 4598; Tel: 0161 275 4710
First published on 5th December 2003
Abstract
Microporous materials can be derived directly from soluble polymers whose randomly contorted shapes prevent an efficient packing of the macromolecules in the solid state.
Nanoporous materials (i.e. microporous, pore size < 2 nm, and/or mesoporous, pore size 2–50 nm) are of great technological importance for adsorption, separation and heterogeneous catalysis due to their large and accessible surface areas (typically 300–1500 m2 g−1).1 The two main classes of microporous materials widely used in industry are the zeolites (aluminosilicates) and activated carbons. The former are crystalline with well-defined surface composition whereas the latter are ill-defined both in structure and surface chemistry.2 At present, there is a vibrant international effort to produce organic–inorganic hybrid materials that mimic the structure of zeolites.3,4 As an alternative, we have designed organic materials that possess a similar amorphous nanoporous structure to activated carbons but which offer high surface areas which are chemically well-defined. Using this concept, microporous networks composed of planar functional units such as phthalocyanine,5 porphyrin6 and hexaazatrinaphthylene7 have been prepared. These microporous network polymers possess open structures due to a rigid spirocyclic molecular scaffold preventing aggregation of the rigid, planar components. Here we demonstrate that a network of covalent bonds is not a requirement for microporous organic materials. A family of non-network polymers is described that form microporous solids simply because their highly rigid and contorted molecular stuctures cannot fill space efficiently. The profound significance of these polymers of intrinsic microporosity (PIMs) is that, unlike conventional microporous materials, they are soluble and can be processed readily using solvent-based techniques.
The molecular scaffold used for the preparation of the polymer microporous networks is derived from the inexpensive bis-catechol 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spirobisindane (Scheme 1: A1) and is assembled using dibenzodioxane formation. Network polymers occur due to the number of spirocyclic linkages attached to each planar functional unit being greater than two. However, the same efficient dibenzodioxane-forming reaction (i.e. aromatic nucleophilic substitution) between the aromatic tetrol monomers A1–A3 with the appropriate fluorine-containing compounds B1–B3 gave soluble PIMs 1–6 (Scheme 1 and Table 1). With the exception of the readily prepared 1,2,4,5-tetrahydroxybenzene (A3),8 the monomers are all commercially available. The efficiency of formation of the dibenzodioxane linking group was assessed by model reactions between catechol and monomers B1–B3. In each case, the expected low molar mass products were prepared rapidly and in high yield. This confirmed that the fluorinated aromatics B1–B3 act as efficient di-reactive monomers in dibenzodioxane-based step-growth polymerisations. With the exception of PIM 6, which is soluble only in acidic solvents (e.g. TFA), the polymers are freely soluble in polar aprotic solvents (e.g. THF, DMAc) and this allows their average molecular mass to be estimated by gel permeation chromatography (GPC) relative to polystyrene standards (Table 1). The experimental conditions required to optimise the average molecular mass were determined for PIM 1, whereas the molecular masses reported for the other polymers are not optimised values. PIMs 1–5 are purified by repeated precipitation from THF solution into methanol and when collected by filtration give fluorescent yellow (PIMs 1 and 4) or white (PIMs 2, 3, 5 and 6) free-flowing powders. Elemental analysis and spectroscopic analysis of the polymers are consistent with their proposed structures. Thermal analysis of each of the polymers shows no glass transition or melting point below the temperature of thermal decomposition, which is above 370 °C for PIMs 1–3 and above 450 °C for PIMs 4–6. The enhanced thermal stability of the latter polymers is due to their lack of aliphatic ring components.
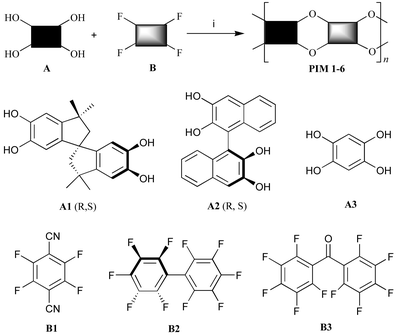 | ||
| Scheme 1 The preparations of PIMs 1–6. Reagents and conditions: (i) K2CO3, DMF, 60–120 °C. | ||
Surface area determination using nitrogen adsorption (multipoint BET calculation) demonstrates that PIMs 1–6 are microporous with high surface areas (Table 1). The nitrogen adsorption/desorption isotherm for PIM 1 is shown in Fig. 1. Micropore analysis (Horvath–Kawazoe method)9 indicates a significant proportion of micropores with dimensions in the range 0.4–0.8 nm. There is also evidence of some mesoporosity. The marked hysteresis at low pressures may be attributed to pore network effects (for example, mesopores accessible only through micropores). The total pore volume, estimated from the amount adsorbed at p/po = 0.98, is 0.78 cm3 g−1.
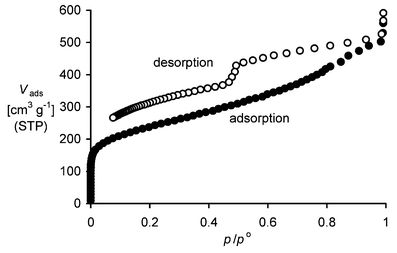 | ||
| Fig. 1 The nitrogen adsorption/desorption isotherm for a powder sample of PIM 1 (i.e. the volume of nitrogen adsorbed, Vads, versus relative pressure, p/po). BET analysis gave a surface area of 850 m2 g−1. | ||
The microporosity of the PIMs arises from their high rigidity combined with a randomly contorted shape (Fig. 2), which prevents an efficient packing of the macromolecules in the solid state. Monomers A1, A2 and B2 provide a ‘site of contortion’ due either to a spiro-centre (A1) or a covalent bond about which there is restricted rotation (A2 and B2). However, monomers A3, B1 and B3 do not possess a site of contortion and, therefore, the two polymers derived from the reaction of A3 and B1 or A3 and B3 are likely to be linear (dibenzodioxane is planar)10 and thus able to pack efficiently. Indeed, these polymers proved to be highly insoluble, non-porous materials (BET surface area < 20 m2 g−1).8
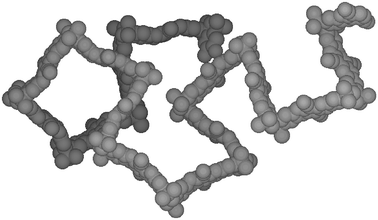 | ||
| Fig. 2 Space-filling molecular model of a fragment of PIM 1 showing its rigid, randomly contorted structure. | ||
The microporosity of PIMs 1–6 is termed intrinsic as it arises solely from their molecular structures and is not dependent on the thermal or processing history of the material. For example, free-standing cast films of PIM 1 also demonstrate high surface area (> 600 m2 g−1). In addition, samples of powdered material heated to below their decomposition temperatures (300 °C for 24 hours) or left for prolonged periods of time under ambient conditions (> one year) display a similar surface area to a freshly precipitated sample. The combination of physical stability and processability offered by PIMs make them particularly attractive as materials for the fabrication of separation membranes.
Generally, polymers exist as non-porous solids because their molecular flexibility ensures space-efficient packing. Although there are many ways to induce porosity within polymers by processing or preparation within a colloidal system (e.g. high internal phase emulsions), most polymers cannot be considered as materials of intrinsic microporosity. However, all non-crystalline (i.e. glassy) polymers contain some void space; usually termed free volume (Vf). This is typically less than 5% of the total volume but for some glassy polymers, specifically those with a rigid molecular structure, it is possible to ‘freeze-in’ additional free volume (up to 20%) by rapid cooling of the molten state below the glass transition temperature. Alternatively, additional free volume can be induced by the rapid removal of a solvent from a swollen glassy polymer. Such high free volume polymers (e.g. polyimides, polyphenyleneoxides, polysulfones) are used in industrial membranes due to the voids assisting the transport of gas or liquid across the material. These materials differ from conventional microporous materials in that the voids are not interconnected and thus the accessible surface area, as measured by gas adsorption, is low.
However in addition to the materials presented here, there is a family of substituted polyacetylenes containing bulky substituents, best represented by poly(1-trimethylsilyl-1-propyne) (PTMSP), that has been classified as microporous – due to exceptionally high gas permeabilities, which can be 2–3 orders of magnitude higher than those displayed by typical high free volume polymers. The large amount of free volume found in freshly prepared PTMSP (∼30%) is interconnected, thus allowing the rapid diffusion of gas. Masuda first described PTMSP in 198311 and since that time there have been approx. 150 papers and 300 patents relating to this superpermeable polymer.12 In particular, the ability of PTMSP to separate organic compounds from permanent gases or water has caused particular interest. However, the technological potential of PTMSP is severely limited due to its rapid loss of microporosity and lack of stability towards heat/oxygen/radiation/UV light or combinations of these factors.13† A direct comparison of the microporosity of our PIMs with that of PTMSP is difficult, as a detailed nitrogen adsorption isotherm of PTMSP has never been published, however its BET surface area has been quoted as 550 m2 g−1 in reviews.12 Therefore, it seems likely that some of the PIMs offer greater microporosity than PTMSP in addition to far greater thermal and chemical stability.14 Studies to assess the potential of PIMs for adsorption and separation processes are in progress.
We acknowledge funding from EPSRC for this research.
Notes and references
- Handbook of Porous Solids, ed. F. Schüth, K. Sing and J. Weitkamp, Wiley-VCH, Berlin, 2002 Search PubMed.
- T. J. Barton, L. M. Bull, W. G. Klemperer, D. A. Loy, B. McEnaney, M. Misono, P. A. Monson, G. Pez, G. W. Scherer, J. C. Vartuli and O. M. Yaghi, Chem. Mater., 1999, 11, 2633 CrossRef CAS.
- S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keefe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef.
- N. B. McKeown, S. Makhseed and P. M. Budd, Chem. Commun., 2002, 2780 RSC.
- N. B. McKeown, S. Hanif, K. Msayib, C. E. Tattershall and P. M. Budd, Chem. Commun., 2002, 2782 RSC.
- P. M. Budd, B. Ghanem, K. Msayib, N. B. McKeown and C. E. Tattershall, J. Mater. Chem., 2003, 13, 2727 RSC.
- R. Wolf and C. S. Marvel, J. Polym. Sci., Polym. Chem. Ed., 1969, 7, 2481 CrossRef CAS.
- G. Horvath and K. Kawazoe, J. Chem. Eng. Jpn., 1983, 16, 470 Search PubMed.
- A. W. Cordes and C. K. Fair, Acta Crystallogr., Sect. B, 1974, 30, 1621 CrossRef.
- T. Masuda, E. Isobe, T. Higashimura and K. Takada, J. Am. Chem. Soc., 1983, 7473 CrossRef CAS.
- K. Nagai, T. Masuda, T. Nakagawa, B. D. Freeman and I. Pinnau, Prog. Polym. Sci., 2001, 26, 721 CrossRef CAS.
- R. E. Kesting and A. K. Fritzsche, Polymeric Gas Separation Membranes, Wiley-Interscience, New York, 1993 Search PubMed.
- UK Patent Appl. 0317557.7, filed 16/07/2003.
Footnote |
| † The questions as to whether PTMSP should be classified as a ‘PIM’ can only be answered when the cause of its rapid loss of microporosity is established – if it is due to physical relaxation rather than degradation then it should instead be referred to as a high free volume polymer. |
| This journal is © The Royal Society of Chemistry 2004 |