First linear alignment of five C–Se⋯O⋯Se–C atoms in anthraquinone and 9-(methoxy)anthracene bearing phenylselanyl groups at 1,8-positions†
Warô
Nakanishi
*,
Satoko
Hayashi
and
Norio
Itoh
Department of Material Science and Chemistry, Faculty of Systems Engineering, Wakayama University, 930 Sakaedani, Wakayama, 640-8510, Japan. E-mail: nakanisi@sys.wakayama-u.ac.jp; Fax: +81 73 457 8253; Tel: +81 73 457 8252
First published on 25th November 2002
Abstract
Five Ci–Se⋯O⋯Se–Ci atoms in anthraquinone and 9-(methoxy)anthracene bearing phenylselanyl groups at 1,8-positions align linearly, the origin of which is shown to be a non-bonded 5c–6e interaction of the five atoms.
Non-bonded interactions1 are of great interest. This is especially so if they lead to linear bonds longer than the three center–four electron bond (3c–4e) through direct orbital overlaps containing group 16 elements.2 Naphthalene 1,8-positions supply a good system to study such non-bonded interactions, containing 2c–4e, 3c–4e and 4c–6e interactions.2–5 However, the nature of the 5c–6e interaction is as yet not well understood. Anthracene 1,8,9-positions also serve as a good system for such interactions.6
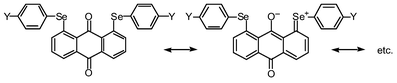
Five C–Se⋯O⋯Se–C atoms are shown to align linearly for 1,8-bis(phenylselanyl)anthraquinone (1)† and 9-(methoxy)-1,8-bis(phenylselanyl)anthracene (2).†The linear alignment can be analyzed by the 5c–6e model. The structure of 1,8-bis(phenylselanyl)anthracene (3)†is also investigated for convenience of comparison, in which five C–Se⋯H⋯Se–C atoms are not aligned linearly.
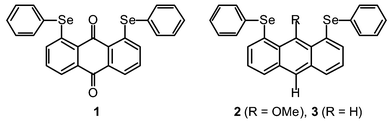
Figs. 1–3 ‡ show the structures of 1–3, respectively.7 Conformations around the two Se atoms are both type B8 (1 (BB)). Consequently, the five C–Se⋯O⋯Se–C atoms align linearly with Se(1)–O(1)–Se(2) 153°. The slightly bent alignment is a reflection of the differences in the r(C,O) and r(C,Se) values. The structure of 1 is close to C2 symmetry. The two phenyl planes in 1 are perpendicular to the anthraquinone plane. Conformations around the Se atoms of 2 are also both type B8 (2 (BB)). The five atoms in 2 also align linearly with Se(1)–O(1)–Se(2) 148°. Indeed, the structure of 2 is very similar to that of 1, but is closer to Cs symmetry. However, the structure of 3 is very different from those of 1 and 2. The conformations around the Se atoms are both type A8 (3 (AA)). Two phenyl groups are located at the same sides of the plane (3 (AA-cis)). It is well demonstrated that the double type A structure in 3 changes dramatically to the double type B in 1 and 2 with each O atom at the 9-position.
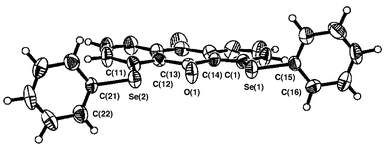 | ||
| Fig. 1 Structure of 1. Selected bond lengths (Å), angles (°) and torsion angles (°): Se(1)–C(1), 1.922(7), Se(1)–C(15) 1.924(6), Se(2)–C(11) 1.917(6), Se(2)–C(21) 1.927(6), C(13)–O(1) 1.225(7), Se(1)–O(1) 2.688(4), Se(2)–O(1) 2.673(4); C(1)–Se(1)–C(15) 98.5(3), C(11)–Se(2)–C(21) 100.2(3), Se(1)–O(1)–Se(2) 152.5(2); C(14)–C(1)–Se(1)–C(15) −172.8(6), C(12)–C(11)–Se(2)–C(21) −171.3(5), C(1)–Se(1)–C(15)–C(16) 90.5(6), C(11)–Se(2)–C(21)–C(22) 103.5(6). | ||
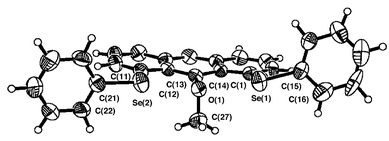 | ||
| Fig. 2 Structure of 2. Selected bond lengths (Å), angles (°) and torsion angles (°): Se(1)–C(1) 1.930(4), Se(1)–C(15) 1.921(5), Se(2)–C(11) 1.932(5), Se(2)–C(21) 1.938(5), C(13)–O 1.387(5), C(27)–O 1.436(6), Se(1)–O 2.731(3), Se(2)–O 2.744(3); C(1)–Se(1)–C(15) 99.2(2), C(11)–Se(2)–C(21) 99.9(2), C(13)–O–C(27) 111.7(4), Se(1)–O–Se(2) 147.9(1); C(14)–C(1)–Se(1)–C(15) –163.1(4), C(12)–C(11)–Se(2)–C(21) 175.8(4), C(1)–Se(1)–C(15)–C(16) –104.2(5), C(11)–Se(2)–C(21)–C(22) 88.3(5), C(14)–C(13)–O–C(27) 89.1(5). | ||
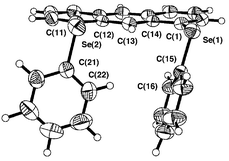 | ||
| Fig. 3 Structure of 3. Selected bond lengths (Å), angles (°) and torsion angles (°): Se(1)–C(1) 1.923(3), Se(1)–C(15) 1.923(3), Se(2)–C(11) 1.943(3), Se(2)–C(21) 1.931(3); C(1)–Se(1)–C(15) 100.3(1), C(11)–Se(2)–C(21) 98.3(1); C(14)–C(1)–Se(1)–C(15) 72.6(3), C(12)–C(11)–Se(2)–C(21) –103.0(3), C(1)–Se(1)–C(15)–C(16) 6.7(3), C(11)–Se(2)–C(21)–C(22) 85.8(3). | ||
The p-type lone pair orbitals of the O atoms (npx(O)) in 1 and 2 extend toward the Se atoms (the axes in 1 (and 2) are defined in Fig. 4). Observed non-bonded Se⋯O distances of 2.67–2.74 Å are about 0.7 Å shorter than the sum of their van der Waals radii (3.40 Å).9 Therefore, npx(O) directly overlaps with the σ*(Se–C) orbitals at both sides of npx(O). If the two non-bonded O⋯Se–C 3c–4e interactions are connected effectively through the central npx(O), the non-bonded σ*(C–Se)⋯npx(O)⋯σ*(Se–C) 5c–6e arrangement is formed. The 5c–6e interaction must play an important role in the double type B structures of 1 and 2.
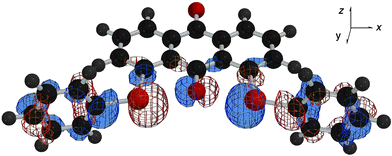 | ||
Fig. 4 HOMO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −2 drawn on the optimized structure of 1 (BB). −2 drawn on the optimized structure of 1 (BB). | ||
Quantum chemical calculations are performed on 1–3, together with 1-(phenylselanyl)anthraquinone (4) and 9-(methoxy)-1-(phenylselanyl)anthracene (5), at the DFT (B3LYP) level.10 Conformers, AA, AB and BB, are optimized to be stable for 1 and 2, which correspond to 3c–6e, 4c–6e and 5c–6e, respectively. The results are collected in Table 1.11 Energies evaluated for each process from 1 (AA) to 1 (BB) (32–29 kJ mol−1) are very close to that in 4.12 The average value from 2 (AA) to 2 (BB) (18 kJ mol−1) is close to that in 5.12These results demonstrate that the non-bonded σ*(C–Se)⋯npx(O)⋯σ*(Se–C) 5c–6e interaction is effectively present in 1 and 2.13 The two non-bonded 3c–4e interactions are well connected through the central npx(O) orbital. Some MOs in 1 and 2 extend over the five C–Se⋯O⋯Se–C atoms as shown in Fig. 4, exemplified by HOMO−2 in 1 (BB),14 which supports the above discussion.
It is worthwhile to comment on the through π-bond interactions between npz(Se) and npz(O) via the π-framework of anthracene in 1. Since the carbonyl group acts as a good electron acceptor, electron densities at O and Se atoms in 1 become larger and smaller, relative to those without such interactions, respectively. This will create advantageous conditions for the non-bonded 5c–6e interaction, since the character of CT is of the type σ*(C–Se)←npx(O)→σ*(Se–C). The rigid structure in 1, except for the rotation around the Se–C bonds, must be advantageous for the π-conjugation. Almost equal stabilization energies calculated in each process from 1 (AA) to 1 (BB) must arise from the rigid structure. Lack of the effective π-conjugation between npz(Se) and np(O) through the π-framework in 2 must be responsible for the smaller stabilization energies evaluated for the corresponding processes relative to those in 1. The additional flexibility around C–O bonds in 2 would also play an important role in its characteristic energy profile.
This work was supported by a Grant-in-Aid for Scientific Research on Priority Areas (A) (Nos. 11120232, 11166246, and 12042259) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, by a Grant-in-Aid for Encouragement of Young scientists (No. 13740354) from Japan Society for Promotion of Science, and by the Hayashi Memorial Foundation for Female Natural Scientists.
Notes and references
- ed. S. Scheiner, Molecular Interactions. From van der Waals to Strongly Bound Complexes, Wiley, New York, 1997 Search PubMed; K. D. Asmus, Acc. Chem. Res., 1979, 12, 436–442 Search PubMed; W. K. Musker, Acc. Chem. Res., 1980, 13, 200–206 CrossRef CAS.
- W. Nakanishi, S. Hayashi and S. Toyota, J. Org. Chem., 1998, 63, 8790–8800 CrossRef CAS; W. Nakanishi, S. Hayashi and T. Uehara, J. Phys. Chem. A, 1999, 103, 9906–9912 CrossRef CAS; S. Hayashi and W. Nakanishi, J. Org. Chem., 1999, 64, 6688–6696 CrossRef CAS; W. Nakanishi, S. Hayashi, A. Sakaue, G. Ono and Y. Kawada, J. Am. Chem. Soc., 1998, 120, 3635–3646 CrossRef CAS; W. Nakanishi and S. Hayashi, J. Org. Chem., 2002, 67, 38–48 CrossRef CAS; W. Nakanishi, S. Hayashi and A. Arai, Chem. Commun., 2002, 2416–2417 RSC.
- R. S. Glass, S. W. Andruski, J. L. Broeker, H. Firouzabadi, L. K. Steffen and G. S. Wilson, J. Am. Chem. Soc., 1989, 111, 4036–4045 CrossRef CAS; R. S. Glass, L. Adamowicz and J. L. Broeker, J. Am. Chem. Soc., 1991, 113, 1065–1072 CrossRef CAS.
- F. B. Mallory, C. W. Mallory and M.-C. Fedarko, J. Am. Chem. Soc., 1974, 96, 3536–3542 CrossRef CAS; F. B. Mallory, C. W. Mallory, K. B. Butler, M. B. Lewis, A. Q. Xia, E. D. Luzic, L. E. Fredenburgh, M. M. Ramanjulu, Q. N. Van, M. M. Francl, D. A. Freed, C. C. Wray, C. Hann, M. Nerz-Stormer, P. J. Carroll and L. E. Chirlian, J. Am. Chem. Soc., 2000, 122, 4108–4116 CrossRef CAS.
- H. Fujihara, H. Ishitani, Y. Takaguchi and N. Furukawa, Chem. Lett., 1995, 571–572 CAS; H. Fujihara, M. Yabe, J.-J. Chiu and N. Furukawa, Tetrahedron Lett., 1991, 32, 4345–4348 CrossRef CAS; H. Taka, A. Matsumoto, T. Shimizu and N. Kamigata, Heteroat. Chem., 2001, 12, 227–237 CrossRef CAS.
- For example, Akiba et al. reported the 1,8-dimethoxy-9-dimethoxymethylanthracene monocation as a model of the transition state of the SN2 reaction: K-y. Akiba, M. Yamashita, Y. Yamamoto and S. Nagase, J. Am. Chem. Soc., 1999, 121, 10644–10645 Search PubMed ; see also: M. Yamashita, Y. Yamamoto, K-y. Akiba and S. Nagase, Angew. Chem., Int. Ed., 2002, 39, 4055–4058 CrossRef CAS.
- The structures of p,p′-dichloro derivatives of 1 and 3 are substantially the same as those of 1 and 3, respectively.
- Structures of the naphthalene system, 8-G-1-(ArSe)C10H6, are well classified using type A, type B and type C, where the Se–CAr bond is placed almost perpendicular to the naphthyl plane in type A, the bond is located on the plane in type B and type C is intermediate between type A and type B. The notation is applied to the structures of 1–3. See ref. 2 and W. Nakanishi and S. Hayashi, Eur. J. Org. Chem., 2001, 20, 3933–3943 Search PubMed.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, New York, 3rd edn., 1960, ch. 7 Search PubMed.
- Gaussian 98, Revision A.9 is employed for the calculations (J. A. Pople et al., Gaussian, Inc., Pittsburgh PA, 1998).11 The 6-311+G(d) basis sets are employed for Se and O atoms and the 6-31G(d) basis sets for C and H atoms..
- Details are shown in the ESI†.
- 4 (B) and 5 (B) are more stable than 4 (A) and 5 (A) by 30.5 and 14.7 kJ mol−1, respectively..
- Model calculations are also performed on model a (Ha′Hb′BSe⋯(H2C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) )O⋯ASeHaHb) and model b (Ha′Hb′BSe⋯(H2)O⋯ASeHaHb) with the B3LYP/6-311++G(3df,2pd) method. The non-bonded Se⋯O distances in the models are fixed at 2.658 Å, observed in the
phenyl p,p′-dichloro derivative of 1. No noticeable saturation is predicted in the energy lowering effect in each conformational change from type A to type B in the models. The results also support the 5c–6e nature of the σ*(H–Se)⋯npx(O)⋯σ*(Se–H) interaction in the models..
)O⋯ASeHaHb) and model b (Ha′Hb′BSe⋯(H2)O⋯ASeHaHb) with the B3LYP/6-311++G(3df,2pd) method. The non-bonded Se⋯O distances in the models are fixed at 2.658 Å, observed in the
phenyl p,p′-dichloro derivative of 1. No noticeable saturation is predicted in the energy lowering effect in each conformational change from type A to type B in the models. The results also support the 5c–6e nature of the σ*(H–Se)⋯npx(O)⋯σ*(Se–H) interaction in the models.. - The MacSpartan Plus program Ver. 1.0 is used (H. J. Hehre, Wavefunction Inc., Irvine, CA 92612, USA).
Footnotes |
| † Electronic supplementary information (ESI) available: experimental procedures, analytical and spectroscopic data for 1–3. See http://www.rsc.org/suppdata/cc/b2/b209261a/ |
‡
Crystal data: for 1: monoclinic, space group P21/n (#14), a = 7.412(2), b = 16.002(2), c = 17.682(2) Å, β = 93.58(2)°, V = 2092.9(7) Å3, Z = 4. For 2: triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2), a = 10.930(3), b = 13.673(4), c = 8.052(2) Å, α = 96.64(2), β = 102.20(3), γ = 106.48(2)°, V = 1107.9(6) Å3, Z = 2. For 3: triclinic, space group P (#2), a = 10.598(3), b = 11.575(3), c = 9.947(3) Å, α = 113.59(2), β = 95.42(2), γ = 109.39(2)°, V = 1017.6(6) Å3, Z = 2. Rigaku AFC5R four-circle diffractometer, Mo-Kα radiation (λ = 0.71069 Å). The structure analyses are based on 2756 observed reflections with I > 1.50σ(I) for 1, on 2795 for 2 and on 3395 for 3 and 271 variable parameters for 1, 271 for 2 and 253 for 3. The structures of 1–3 were solved by heavy-atom Patterson methods (PATTY) and expanded using Fourier techniques (DIRDIF94) and refined by full-matrix least squares on |F|2. R = 0.046, Rw = 0.029, GOF = 1.51 for 1, R = 0.040, Rw = 0.030, GOF = 1.59 for 2 and R = 0.034, Rw = 0.026, GOF = 2.46 for 3. CCDC reference numbers 175767 (1), 175768 (2) and 175769 (3). See http://www.rsc.org/suppdata/cc/b2/b209261a/ for crystallographic data in CIF or other electronic format. (#2), a = 10.930(3), b = 13.673(4), c = 8.052(2) Å, α = 96.64(2), β = 102.20(3), γ = 106.48(2)°, V = 1107.9(6) Å3, Z = 2. For 3: triclinic, space group P (#2), a = 10.598(3), b = 11.575(3), c = 9.947(3) Å, α = 113.59(2), β = 95.42(2), γ = 109.39(2)°, V = 1017.6(6) Å3, Z = 2. Rigaku AFC5R four-circle diffractometer, Mo-Kα radiation (λ = 0.71069 Å). The structure analyses are based on 2756 observed reflections with I > 1.50σ(I) for 1, on 2795 for 2 and on 3395 for 3 and 271 variable parameters for 1, 271 for 2 and 253 for 3. The structures of 1–3 were solved by heavy-atom Patterson methods (PATTY) and expanded using Fourier techniques (DIRDIF94) and refined by full-matrix least squares on |F|2. R = 0.046, Rw = 0.029, GOF = 1.51 for 1, R = 0.040, Rw = 0.030, GOF = 1.59 for 2 and R = 0.034, Rw = 0.026, GOF = 2.46 for 3. CCDC reference numbers 175767 (1), 175768 (2) and 175769 (3). See http://www.rsc.org/suppdata/cc/b2/b209261a/ for crystallographic data in CIF or other electronic format. |
| This journal is © The Royal Society of Chemistry 2003 |