
How is CO2 hydrogenated to ethanol on metal–organic framework HKUST-1? Microscopic insights from density-functional theory calculations†
Bikun
Zhang
 and
Jianwen
Jiang
*
and
Jianwen
Jiang
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, 117576, Singapore. E-mail: chejj@nus.edu.sg
First published on 31st December 2024
Abstract
Thermocatalytic hydrogenation of CO2 to multi-carbon chemicals (C2+) has received considerable interest to reduce CO2 footprint and mitigate global warming. Comprising Cu paddle-wheel clusters, a metal–organic framework (MOF) namely HKUST-1 has been experimentally reported as a promising catalyst for CO2 hydrogenation to ethanol under ambient conditions with the assistance of non-thermal plasma (NTP). Yet, there lacks microscopic understanding of the active center, reaction pathway and product selectivity. In this study, we conduct density-functional theory calculations to quantitatively and explicitly elucidate the fundamental mechanism involved. NTP is revealed to be responsible for H2 dissociation, while the defective HKUST-1 with exposed Cu atoms accounts for highly selective CO2 hydrogenation to ethanol via facile *CHOH–CO coupling, with *CHOH adsorbed on the Cu atoms and CO from the gas phase. The strong binding between the carbonyl C atoms in C2 intermediates and Cu atoms, and the high stability of *CH3CHOH intermediate, contribute to the higher selectivity of ethanol over acetaldehyde and ethylene, respectively. From bottom-up, this computational study provides deep microscopic insights into the catalytic mechanism of CO2 hydrogenation to C2 products on HKUST-1, and it will facilitate the design of new MOFs for efficient CO2 conversion and other important chemical transformations.
1. Introduction
Mitigation of CO2 emissions has become a gigantic challenge in the 21st century due to the increasing combustion of fossil fuels driven by modern industry and transportation.1–3 Direct CO2 hydrogenation to high value-added products is regarded as an efficient solution, as it reduces the carbon footprint and meanwhile produces renewable fuels and chemicals.4–7 Compared with mono-carbon (C1) chemicals such as carbon monoxide, methane and methanol, multi-carbon (C2+) chemicals possess a higher economic value and energy density.8–10 Specifically, higher alcohols (i.e., alcohols containing two or more carbon atoms) have a greater variety of applications as liquid fuels, fuel additives and reaction intermediates to synthesize commodity and specialty products in the chemical, pharmaceutical and energy sectors.8,11–13 Among higher alcohols, ethanol, as an important solvent, an industrial building block and a promising renewable fuel, has attracted great interest.Direct production of ethanol from CO2 and H2 is challenging, as CO2 is chemically inert with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonding energy of 806 kJ mol−1 (ref. 14) and it is also thermodynamically very stable with formation enthalpy of −394 kJ mol−1.15 In addition, CO2 hydrogenation requires the availability of H atoms from H2 dissociation.9 Another obstacle for C2+ production from CO2 is that it involves uncontrollable C–C bond formation with a high energy barrier. Furthermore, many bifurcation routes are involved, which are much more complicated than C1 production. As a consequence, CO2 hydrogenation to ethanol is mostly conducted at a high temperature and always ends up with a low selectivity.16–18
O bonding energy of 806 kJ mol−1 (ref. 14) and it is also thermodynamically very stable with formation enthalpy of −394 kJ mol−1.15 In addition, CO2 hydrogenation requires the availability of H atoms from H2 dissociation.9 Another obstacle for C2+ production from CO2 is that it involves uncontrollable C–C bond formation with a high energy barrier. Furthermore, many bifurcation routes are involved, which are much more complicated than C1 production. As a consequence, CO2 hydrogenation to ethanol is mostly conducted at a high temperature and always ends up with a low selectivity.16–18
Experimentally, non-thermal plasma (NTP) is an effective tool in thermocatalysis, as it facilitates activating source gases and overcoming both kinetic and thermodynamic limitations of chemical reactions to produce desired products.19,20 Especially, NTP has been utilized for CO2 conversion due to the intrinsic obstacles mentioned above. With a mean electron energy between 1 and 10 eV,21,22 the high-energy electrons generated by NTP can excite CO2 and H2 molecules to activate or even break their chemical bonds, because only 5.5 eV is required to break a CO bond22 and the H–H bond energy is even lower than the CO bond energy.23 Therefore, NTP contributes to both CO2 activation and H2 dissociation, thus promoting thermocatalytic CO2 hydrogenation.
Metal–organic frameworks (MOFs), assembled from metal clusters and bridging organic linkers, have served as efficient catalysts for many reactions including CO2 hydrogenation due to their porous structures, large surface areas and tunable compositions and functionalities.21,24–32 However, most of the current studies for CO2 hydrogenation using MOFs have been focused on C1 products, and successful CO2 hydrogenation to ethanol requires complicated structural modification to construct active sites.30–32 Recently, Zou et al. reported successful NTP-assisted ethanol production by direct CO2 hydrogenation with high selectivity on a Cu-MOF under ambient conditions.21 This Cu-MOF was synthesized by partially reducing HKUST-1 (Cu3(BTC)2, BTC = 1,3,5-benzene tricarboxylate) with CuII paddle-wheel clusters to generate CuI atoms. However, the mechanism behind improvement of CO2 conversion, C–C coupling and origin of high ethanol selectivity is still unclear. Since Cu paddle-wheel clusters are very common in MOFs and can be conveniently modified for property manipulation,33–36 deep understanding of their catalytic performance is of great significance for the broader application of MOFs on CO2 hydrogenation and other important reactions.
In this study, we conduct in-depth theoretical investigation into the fundamental mechanism of CO2 hydrogenation to ethanol on HKUST-1 using density functional theory (DFT) calculations. Following the computational methods briefly described below, we first present microscopic insight into the improvement of CO2 conversion by untreated HKUST-1 with CO as the main product; then different intermediates from CO along C1 pathways are examined; next, possible C–C coupling pathways are explored on the defective HKUST-1; finally, the origin of the higher selectivity of ethanol over other C2 products (e.g., acetaldehyde and ethylene) is discussed. The findings from this work provide quantitative and explicit elucidation on the important mechanism that was elusive in the experiment, thus being conducive to the development of new MOFs for efficient CO2 conversion.
2. Computational methods
Experimentally, divalent Cu atoms in HKUST-1 were partially reduced to monovalent by thermal treatment at 200 °C in the presence of methanol,21 which is a typical defect engineering approach to remove organic linkers in MOFs.36,37 This thermal treatment could remove one BTC linker and the exposed Cu atom might coordinate with a H atom or methoxy group due to the existence of methanol. On this basis, we built four clusters to mimic the plausible catalysts, as shown in Fig. 1, including the original paddle-wheel (o_PW), defective paddle-wheel (d_PW), hydrogen-coordinated paddle-wheel (H_PW) and methoxy-coordinated paddle-wheel (CH3O_PW). The natural population analysis (NPA) charges of two Cu atoms are approximately identical: 0.924/0.925 on o_PW, and a decrease to 0.858/0.878 on d_PW upon removing one BDC linker. After being coordinated with one H atom, both Cu atoms possess less NPA charge (0.788/0.800). On CH3O_PW, one Cu atom is oxidized by methoxy group and its NPA charge increases to 0.935, while the other one is almost unchanged in NPA charge (0.837). Therefore, all three modified clusters (d_PW, H_PW and CH3O_PW) could exist after thermal treatment21 and are thus considered here. To saturate the cleaved bonds, CN groups were added and fixed on all the clusters during DFT calculations.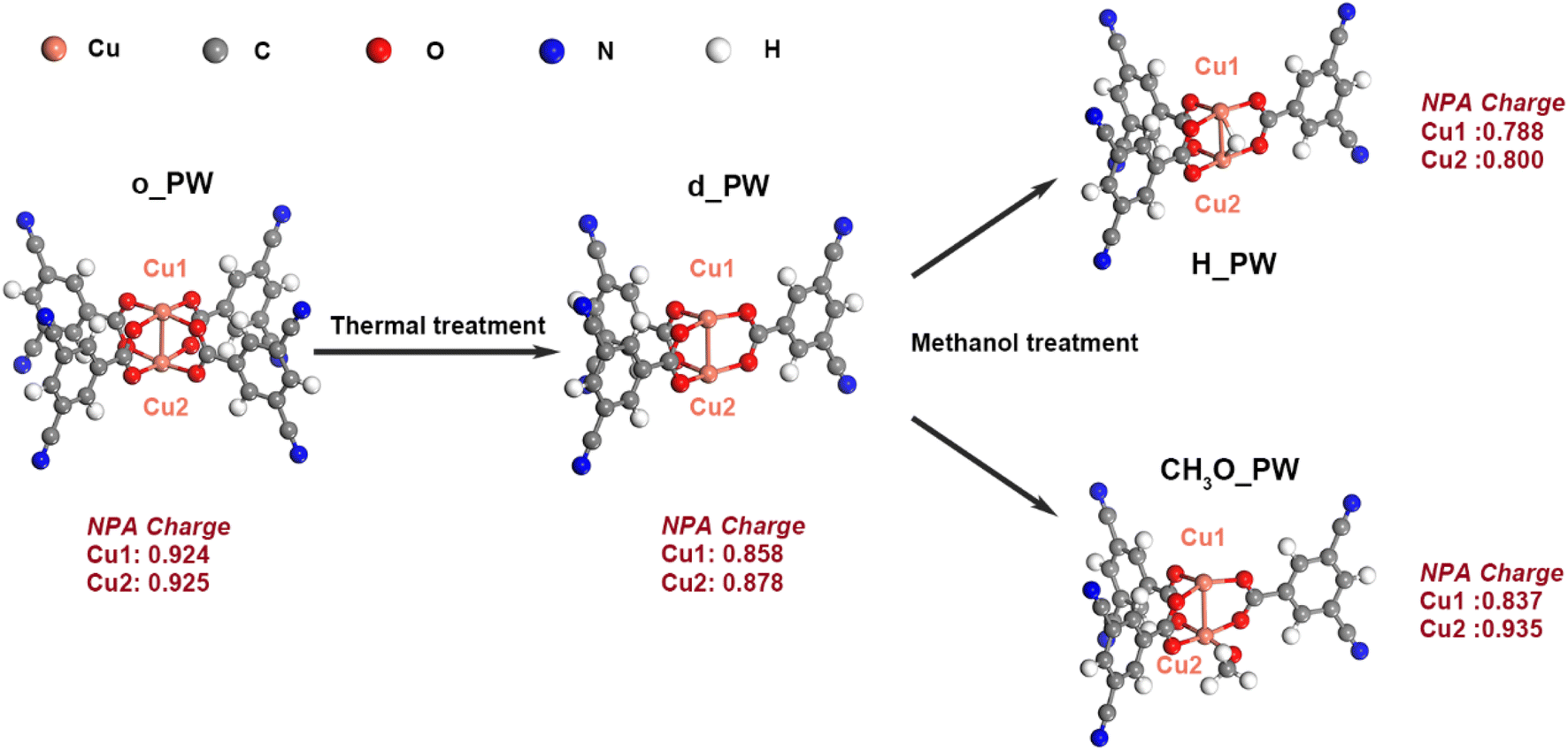 | ||
| Fig. 1 Original, defective, H-coordinated and methoxy-coordinated Cu paddle-wheel clusters (o_PW, d_PW, H_PW and OH_PW), where the NPA charges of Cu atoms are indicated. | ||
DFT calculations were performed using Gaussian 16.38,39 All the clusters were optimized at the B3LYP hybrid functional in conjunction with Grimme's D3BJ dispersion correction.40 The main group atoms including C, N, O and H were described by using the def2-SVP basis set41 and the Stuttgart-Dresden (SDD) pseudopotential was used for Cu. Vibrational frequencies were calculated at the same level of theory as optimizations, in order to provide thermal corrections and verify the nature of stationary points (no imaginary frequency for an optimized intermediate and only one imaginary frequency for a transition state). To verify a transition state connecting directly to reactant and product, intrinsic reaction coordinate (IRC) calculations were conducted. The electronic energies and NPA charges were estimated through natural bond orbital (NBO) calculations using the M06L hybrid functional.42 The Gibbs energies (G) were estimated from
| G = Eele + Gcor | (1) |
3. Results and discussion
3.1. Improvement of CO2 conversion
In experiments, CO2 hydrogenation was feasible with the assistance of NTP in the absence of any catalyst and CO was found to be the main product with selectivity of 77.6%.21 After the untreated original HKUST-1 (i.e., o_PW cluster) was placed in an NTP discharge zone, CO2 conversion improved greatly from 11.9% to 32.3%, indicating a great contribution of o_PW to the improvement of CO2 conversion. Consequently, we consider the possibility of o_PW to facilitate H2 dissociation or CO2 activation. As evidenced by the positive adsorption Gibbs energy ΔGads in Fig. S1,† CO2 cannot adsorb stably on the Cu atom of o_PW. This agrees well with the low CO2 adsorption capacity experimentally reported on HKUST-1.43,44 In addition, CO2 activation is hindered on o_PW, indicated by the trivial NPA charge of CO2 (0.053) on o_PW. For H2 adsorption, ΔGads is even more positive, and furthermore the negligible NPA charge of H2 (0.050) indicates no activation of the H–H bond. Apart from this, due to the lack of adjacent exposed Cu atom, H2 dissociation is not feasible. On the other hand, direct CO2 hydrogenation by H2 to generate formic acid (HCOOH) on o_PW would overcome a high Gibbs energy barrier (ΔGb) of 76.80 kcal mol−1 (Fig. S2†), almost the same as 78.28 kcal mol−1 in the gas phase (Fig. 2a and S3†). Therefore, the experimentally observed improvement of CO2 conversion by o_PW is not accomplished through promoting H2 dissociation or CO2 activation. Given that H2 can be readily dissociated into H atoms by NTP-generated high energy electrons, the initial CO2 hydrogenation is significantly accelerated with ΔGb of only 23.68 kcal mol−1 for CO2 + H → HCOO without a catalyst, as shown in Fig. 2a, which is much lower than that for direct CO2 hydrogenation by H2 gas. This hydrogenation is further boosted on o_PW with a much lower ΔGb of only 8.36 kcal mol−1. Therefore, although o_PW cannot promote direct CO2 hydrogenation by H2, it significantly decreases the activation barrier of CO2 + H → HCOO, leading to the great improvement of CO2 conversion as observed in experiments.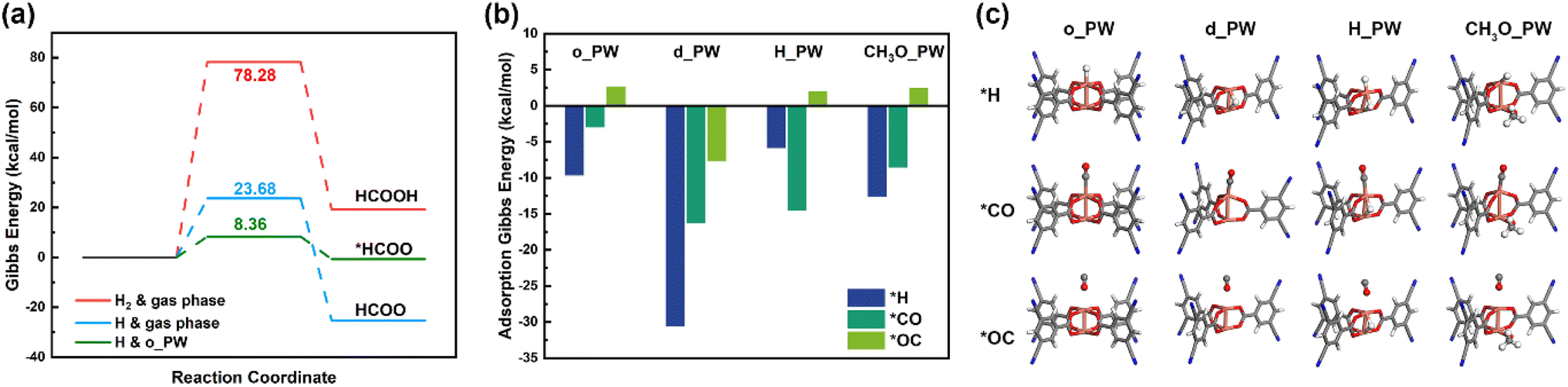 | ||
| Fig. 2 (a) Gibbs energy profile of initial CO2 hydrogenation under different conditions (H2 & gas phase: direct hydrogenation by H2 in the gas phase; H & gas phase: hydrogenation by atomic H in the gas phase; H & o_PW: hydrogenation by atomic H on the original HKUST-1); (b) adsorption Gibbs energies and (c) structures of *H, *CO and *OC on the four paddle-wheel clusters. | ||
3.2. C1 pathways from CO
The presence of original HKUST-1 greatly improved CO2 conversion; however, CO was the main product like the situation in non-catalytic reaction and the production of ethanol was negligible.21 To elucidate the mechanism, we examine different C1 intermediates (i.e., C1 pathways) from CO on the four clusters. As illustrated in Fig. 2b and c, the adsorption sites for CO and H on o_PW are the same, and H adsorption is stronger than CO adsorption with a negative ΔGads of −9.66 kcal mol−1. In addition, *CO hydrogenation to either *CHO or *COH is not favorable because *CO strongly repels H atom from the gas phase and no rational kinetic pathway exists (Fig. S4†). Therefore, CO hydrogenation on o_PW is difficult to proceed. Moreover, we also consider CO dimerization on o_PW. Although there are two open Cu atoms on o_PW, their open directions are opposite, making C–C coupling via the Langmuir–Hinshelwood mechanism45 impossible. Therefore, the Eley–Rideal mechanism46 is the only possibility for C–C coupling on o_PW, where only one CO molecule is adsorbed on the Cu atom and then coupled with another CO molecule from the gas phase. However, we find that the adsorbed *CO intermediate strongly repels the gaseous CO molecule, indicating that CO dimerization cannot be realized on o_PW. Therefore, CO adsorption, hydrogenation and dimerization are all impeded on o_PW, and the untreated original HKUST-1 cannot provide a catalytic effect on CO conversion. Consequently, CO is the main product on o_PW and its further reaction is very unfavorable.Subsequently, we turn our attention to d_PW, H_PW and CH3O_PW clusters. First, H and CO adsorption is examined. From Fig. 2c, it is seen that H adsorption on d_PW is extremely favorable, with ΔGads of −30.55 kcal mol−1, much lower than CO adsorption with both *CO (Cu–C interaction, −16.27 kcal mol−1) and *OC (Cu–O interaction, −7.65 kcal mol−1) configurations. After H adsorption on d_PW, the cluster is identical to H_PW, on which the additional H adsorption is less favorable than CO adsorption with *CO configuration (ΔGads = −14.54 kcal mol−1), while formation of the *OC configuration is endothermic. As a result, the exposed Cu atoms can bind with CO molecules, avoiding blockage by additional H atoms. Since d_PW is extremely easy to hydrogenate to H_PW in an H-rich environment, we take H_PW as the hydrogenated d_PW and thereafter combine the study of d_PW and H_PW. On CH3O_PW, H adsorption is also stronger than CO adsorption with *CO configuration, but the difference is relatively smaller, resulting in competition of adsorption site. Since the unoccupied Cu atom is highly exposed, the co-adsorption of H and CO is plausible. After H adsorption on CH3O_PW, ΔGads for additional CO adsorption on the Cu atom is −12.78 kcal mol−1. However, the Cu–C distance is 2.54 Å (Fig. S5†), much longer than that in the *CO + *H co-adsorption structure on d_PW (2.05 Å), indicating that the existence of methoxy group reduces CO attraction to the Cu atom.
Next, we investigate CO hydrogenation on d_PW, where the isomers of several intermediates are considered from both thermodynamic and kinetic perspectives. As shown in Fig. 3, starting from *H + *CO, a surmountable ΔGb of 22.87 kcal mol−1 needs to be overcome for *H–*CO coupling to *CHO_1, where the C atom is coordinated to one Cu atom and the O atom faces outward. Since the C atom is closer to *H, formation of *COH is apparently more difficult than *CHO_1 from *H + *CO and thus excluded. We further examine the possibility of *CO hydrogenation by atomic H via the Eley–Rideal mechanism. However, this is endothermic with a Gibbs reaction energy (ΔGr) of 18.33 kcal mol−1, and *COH + *H is much less stable than *CHO + *H by 36.67 kcal mol−1, as shown in Fig. S6.† Therefore, only *CHO is considered in the following discussion. After the formation of *CHO_1, the unoccupied Cu atom is not well coordinated and primed to adsorb another H atom, which is spontaneous with ΔGads of −19.47 kcal mol−1. However, the adjacent *H and *CHO_1 strongly repel each other and thus cannot couple to form *CH2O. Instead, *CHO_1 is hydrogenated to *CHOH_1 via the Eley–Rideal mechanism with ΔGb of 13.91 kcal mol−1. On the other hand, there exists a more stable isomer of *CHO, where the H atom faces outward, denoted as *CHO_2. The transformation from *CHO_1 to *CHO_2 requires a 180° rotation with a negligible energy barrier of only 3.14 kcal mol−1. The further H adsorption is also spontaneous (−17.59 kcal mol−1) and the subsequent *H–*CHO_2 coupling via the Langmuir–Hinshelwood mechanism only needs to overcome a very low ΔGb of 4.12 kcal mol−1 to generate *CHOH_2, which is thermodynamically more stable than *CHOH_1. Therefore, we identify *CHO_2 and *CHOH_2 as dominant configurations for *CHO and *CHOH, respectively. Theoretically, *CHOH_2 can be hydrogenated to *CH2OH via the Eley–Rideal mechanism, which is the key intermediate for methanol production. However, the C atom in *CHOH_2 repels the H atom and no rational kinetic pathway is found to form either *CH2OH or *CH by further dehydration. Therefore, further hydrogenation of *CHOH_2 is suppressed on d_PW, which accounts for the experimental observation that both CH4 and CH3OH as C1 products exhibited extremely low selectivity.21Fig. 3 summarizes the evolution of Gibbs energy and intermediate structures along C1 pathways on d_PW, and the detailed ΔGr and ΔGb of the elementary steps are listed in Table 1.
 | ||
| Fig. 3 Evolution of Gibbs energy and intermediate structures along C1 pathways on d_PW. | ||
| *H + *CO → *CHO_1 | *CHO_1 + H(g) → *CHOH_1 | *CHO_1 → *CHO_2 | *CHO_2 + H(g) → *CHOH_2 | |
|---|---|---|---|---|
| ΔGr (kcal mol−1) | 1.12 | −6.11 | −0.57 | −11.86 |
| ΔGb (kcal mol−1) | 22.87 | 13.91 | 3.14 | 4.12 |
We also investigate the C1 pathway on CH3O_PW by exploring *CHO formation from *H + *CO co-adsorption structure. However, *CHO is found to be unstable, as the H atom tends to be attracted by the methoxy group rather than *CO. As a result, the methoxy group couples with *H to form CH3OH, and in the end *CHO + CH3O_PW evolves into CH3OH + CO + d_PW, as illustrated in Fig. S7.† This is because CO adsorption on the Cu atom is oxidation via electron transfer from CO to Cu. Compared with CO in the gas phase, the NPA charge of the C atom increases by 0.136 and 0.082 in the *H + *CO co-adsorption structure on d_PW and CH3O_PW, respectively, which results in stronger CO adsorption on d_PW than CH3O_PW, as discussed above. For the C atom, *CO hydrogenation to *CHO is a reduction process and its higher oxidation state leads to stronger attraction to *H. Apparently, CH3O_PW is unstable in CO hydrogenation because the methoxy group tends to detach from the Cu atom and finally both Cu atoms are exposed, just as d_PW. Therefore, after the analysis of all four clusters, we conclude that only the defective Cu-paddle wheel (d_PW) is stable and serves as an active catalyst for C–C coupling, as further considered below.
3.3. C–C coupling pathways
In their experimental study, Zou et al. assumed that C–C coupling might occur between adsorbed CH2O and CO,21 where the two intermediates were bound to Cu atoms via Cu–O interaction parallelly along with *OCH2–*OC coupling, as illustrated in Fig. 4a; however, there was no solid proof and further validation. Therefore, we evaluate the possibility of this coupling pathway first. The geometry of CH2O is trigonally planar, where the C atom is sp2 hybridized and less favorable to be attracted to the Cu atom than the carboxyl O atom. As shown in Fig. S8a,† CH2O binds to Cu (i.e., *OCH2) via Cu–O interaction rather than Cu–C interaction, which would cause a problem. That is, the Cu–C bond needs to be broken to realize conversion from *CHO to *OCH2, which undergoes a drastic structural change with a huge energy obstacle for HCHO formation. The difficult formation of *OCH2 also accounts for why no formaldehyde (HCHO) was found experimentally.21 On the other hand, Cu–O interaction is much weaker than Cu–C interaction as illustrated in Fig. 2b. Therefore, CO is less likely to bind with Cu than *OC to enable *OCH2–*OC coupling. To validate our analysis, we investigated *OCH2 + *OC co-adsorption structure and found that the CO molecule cannot adsorb on d_PW after optimization; instead, it stays in the gas phase and is located far away from *OCH2 with C–C distance over 5.53 Å (Fig. 4c). This result is quite different from the assumption by Zou et al.21 On this basis, we infer that *OCH2–*OC coupling is not likely to occur and thus exclude it. | ||
| Fig. 4 Schematics of (a) *OCH2–*OC and (b) *CO–*CO coupling pathways; co-adsorption structures of (c) *OCH2 + *OC and (d) *CO + *CO on d_PW. | ||
As one of the most common C–C coupling pathways on Cu-based catalysts, CO dimerization is then investigated. On d_PW, it is expected that two CO molecules co-adsorb on the two exposed Cu atoms separately and stand side by side with an appropriate C–C distance for CO dimerization, as shown in Fig. 4b. However, the C–C distance in the optimized co-adsorption structure is 6.13 Å (Fig. 4d), which is too long for CO dimerization, and the adsorption of the second CO molecule is relatively weak with ΔGads of only −2.00 kcal mol−1. On the other hand, as the adsorption of H atom is inevitable with highly negative adsorption energy (Fig. 2b), we further examine CO dimerization on d_PW with the existence of *H. As shown in Fig. S8b,† after geometry optimization, two *CO stay in an ‘end to end’ position and even farther apart. Consequently, *CO–*CO coupling is not practically feasible on d_PW and also excluded.
As discussed in Section 3.2, *CHO and *CHOH intermediates can be generated easily on d_PW, therefore, *CHO–*CO and *CHOH–*CO coupling might be possible pathways. Since there are two exposed Cu atoms on d_PW, we first consider the Langmuir–Hinshelwood mechanism, where one Cu atom is bound with *CHO or *CHOH and the other Cu atom with *CO before coupling. However, the situation for *CHO + *CO or *CHOH + *CO co-adsorption is similar to that for *CO + *CO, where *CHO or *CHOH and *CO are bound to different Cu atoms and too far apart with C–C distance over 5 Å, as shown in Fig. 5a and 6a. Therefore, C–C coupling via the Langmuir–Hinshelwood mechanism is unlikely to proceed on d_PW. Next, we consider the Eley–Rideal mechanism, where *CHO or *CHOH intermediate is bound to one Cu atom while a CO molecule approaches them from the gas phase. As shown in Fig. 5e, *CHO–CO coupling is slightly endothermic under ambient conditions with ΔGb of 18.17 kcal mol−1. Fig. 5b–d show the structures of the initial state, transition state and final state along the *CHO–CO coupling pathway, respectively. It can be seen that during the coupling, the Cu atom attracts the CO molecule to form a new Cu–C bond, while the Cu–C bond between Cu and *CHO breaks, followed by *CHO moving towards *CO to form a C–C bond. However, compared with *CHO–CO coupling, the exothermic *CHO hydrogenation (*CHO + *H → *CHOH) is more favorable with a lower ΔGb of 13.91 kcal mol−1 (Table 1). Therefore, *CHO–CO coupling is suppressed by hydrogenation.
 | ||
| Fig. 5 Structures of (a) *CHO + *CO co-adsorption, and (b) initial state, (c) transition state and (d) final state along the *CHO–CO coupling pathway. (e) Total energy profile along the *CHO–CO coupling pathway. | ||
 | ||
| Fig. 6 Structures of (a) *CHOH + *CO co-adsorption, and (b) initial state, (c) transition state and (d) final state along the *CHOH–CO coupling pathway. (e) Total energy profile along the *CHOH–CO coupling pathway. | ||
From the discussion in Section 3.2, *CHOH hydrogenation is unfavorable due to the repelling of atomic H in the gas phase, which is conducive to *CHOH–CO coupling. Similar to *CHO–CO coupling, *CHOH–CO coupling leads to the formation of a new Cu–C bond between Cu and CO, while the Cu–C bond between Cu and *CHOH breaks (Fig. 6b–d). As shown in Fig. 6e, *CHOH–CO coupling is exothermic with ΔGb of 11.18 kcal mol−1, which is lower than that in *CHO–CO coupling. Therefore, *CHOH–CO coupling is more favorable than *CHO–CO coupling both thermodynamically and kinetically, and thus we consider *CHOH–CO coupling as the dominant C–C coupling mechanism. *CHOHCO forms as a result of *CHOH–CO coupling. Interestingly, *CHOCO can be easily hydrogenated to *CHOHCO, where *H is formed spontaneously upon H adsorption on the exposed Cu atom, with ΔGb of only 7.19 kcal mol−1. Therefore, both *CHO–CO and *CHOH–CO coupling pathways lead to the formation of *CHOHCO.
3.4. Selectivity of C2 products
Theoretically, hydrogenation along different pathways may occur after the formation of *CHOHCO, ending up with different C2 products including ethanol (CH3CH2OH). Especially, acetaldehyde (CH3CHO) and ethylene (CH2CH2) are two main byproducts in ethanol production from CO/CO2 hydrogenation.47–50 However, only CH3CH2OH was detected in the experiment by Zou et al.21 Therefore, it is instructive to investigate why the byproducts were suppressed. First, we calculated the desorption Gibbs energies (ΔGdes) and the numbers of electrons (|Ne|) transferred between adsorbates and d_PW for CH3CHO, CH3CH2OH and CH2CH2. As shown in Fig. 7a, all three C2 products are weakly adsorbed on d_PW, with ΔGdes lower than 15.59 kcal mol−1 and the electron transfer is negligible with |Ne| smaller than 0.13, indicating that they are easy to desorb from d_PW. Therefore, the suppression of CH3CHO and CH2CH2 occurs before the formation of corresponding intermediates. There are two possible pathways for CH3CHO formation: *CH2CHO + H → *CH3CHO and *CH3CO + H → *CH3CHO. We find *CH3CO is much more stable than *CH2CHO by 24.56 kcal mol−1, indicating the higher possibility of the latter pathway. However, *CH3CO hydrogenation tends to form intermediate *CH3COH, which only needs to overcome a low ΔGb of 11.30 kcal mol−1 (see Fig. 7b), while the carbonyl C atom repels H atom (i.e., the carbonyl C atom is difficult to be hydrogenated). Such repulsion was also observed when we performed transition state calculations from *CHOHCO to *CHOHCHO, *CH2OHCO to *CH2OHCHO, and *CH2CO to *CH2CHO. Accordingly, we conclude that the carbonyl C atom is stably bound to the Cu atom and more difficult to hydrogenate than the carbonyl O atom, resulting in the suppression of CH3CHO formation. For competition between CH3CH2OH and CH2CH2, the selectivity is determined by *CH2CH2OH, which is the precursor of both CH3CH2OH and CH2CH2; while its isomer *CH3CHOH cannot convert to CH2CH2. Thermodynamically, *CH2CH2OH is less stable than *CH3CHOH by 18.85 kcal mol−1, and the conversion from *CH3CHOH to *CH2CH2OH needs to overcome a high ΔGb of 36.18 kcal mol−1 (see Fig. 7c). Therefore, the stabilization of *CH3CHOH over *CH2CH2OH results in a higher selectivity of CH3CH2OH than CH2CH2. | ||
| Fig. 7 (a) Desorption Gibbs energies (ΔGdes) and the numbers of electrons transferred (|Ne|) between adsorbates (CH3CHO, CH3CH2OH and CH2CH2) and d_PW; (b) Gibbs energy profile of *CH2CHO, and of the initial state (IS), transition state (TS) and final state (FS) in *CH3CO + H → *CH3COH; (c) Gibbs energy profile of the IS, TS and FS in *CH3CHOH → *CH2CH2OH. | ||
3.5. Discussion
In experiments, CH4 and CH3OH could be produced from CO2 hydrogenation with the assistance of NTP.21 Thus, we also consider the possible coupling between CO and *CH2/*CH3/*CH2OH on d_PW. The ΔGb values in *CH2–CO, *CH3–CO and *CH2OH–CO coupling are 14.77, 15.66 and 15.76 kcal mol−1, respectively (Fig. S9†), comparable to that in *CHOH–CO coupling. These reactions are in the premise that CH2, CH3 and CH2OH groups can be formed in the gas phase; however, their formation is suppressed on d_PW, as discussed in Section 3.2, and they need being adsorbed on d_PW in order to couple with CO. These processes involve much more uncertainty than the facile adsorption of CO/H and the formation of *CHOH on d_PW. In addition, as the exposed Cu atoms on d_PW are largely occupied by *CO and *H with highly negative ΔGads, the adsorption of CH2/CH3/CH2OH groups is suppressed. Therefore, we infer that *CHOH–CO is the dominant C–C coupling on d_PW.In this work, we provide a guideline and fundamental understanding in CO2 hydrogenation to ethanol on Cu paddle-wheel clusters, with the primary focus on the C–C coupling mechanism. However, the practical experimental conditions are complicated and the structural complexity of HKUST-1 is difficult to be fully captured by small clusters. Large clusters incorporating structural complexity are highly desired to gain better understanding of reaction mechanisms. During reaction, *CHOHCO hydrogenation to ethanol, especially the removal of OH group, is possible and further investigation is needed. Nevertheless, such investigation involves over 20 different intermediates and requires considering a quite large number of elementary steps, with prohibitively high computational cost. In this context, experimental characterization is instrumental to identify key C2 intermediates, effectively narrow down calculation tasks and greatly facilitate catalyst design.
It is interesting to note that while HKUST-1 is a commonly examined MOF, a portion of MOFs containing Cu paddle-wheel (Cu2(COO)4) clusters are experimentally available. These Cu-MOFs are expected to have comparable catalytic activity to HKUST-1 for CO2 hydrogenation. In addition, there exist Cu-MOFs with different configurations surrounding Cu2(COO)4 clusters. For example, two pyridine groups in Cu-HMOF51 are bound to a Cu2(COO)4 cluster, and three neighboring Cu2(COO)4 clusters in Cu2PDC2-MOF are connected by one 3,5-pyridyl group.52 The chemical coordination environment of Cu2(COO)4 clusters is crucial to the formation, adsorption and stabilization of intermediates and transition states, thus these Cu-MOFs might exhibit unique catalytic activity towards CO2 hydrogenation. In the literature, certain other Cu-MOFs are also catalytically active for CO2 hydrogenation. Bimetallic CuI2 centres of Zr12-bpdc-Cu MOF30 were found to catalyse direct C–C coupling between CHO and methanol. Furthermore, inspired by the successful CO2 hydrogenation to ethanol in CuII(HxPO4)y@Ru-UiO MOF,31 where the valence state of Cu was modulated by Ru, introducing a different metal to Cu-MOFs37 may serve as a new strategy for further development of MOFs for CO2 hydrogenation.
4. Conclusions
Through DFT calculations, we have unraveled the microscopic mechanism for ethanol production by CO2 hydrogenation on HKUST-1. H2 dissociation is accomplished with the assistance of NTP, which generates high-energy electrons to break H–H bond. Although untreated HKUST-1 cannot catalyze H2 dissociation, it greatly facilitates CO2 hydrogenation to *HCOO with the existence of NTP-induced H atoms. With exposed Cu atoms, defective HKUST-1 is catalytically active for CO hydrogenation and C–C coupling. *CHOH–CO coupling via the Eley–Rideal mechanism is identified as the dominant C–C coupling pathway, leading to ethanol production on the defective HKUST-1. The production of acetaldehyde and ethylene is suppressed due to difficult hydrogenation of carbonyl C atoms in C2 intermediates and the higher stability of *CH3CHOH over *CH2CH2OH, respectively. Through comprehensive theoretical investigation, the fundamental mechanism of CO2 hydrogenation to ethanol on HKUST-1 is well elucidated. We hope the bottom-up insights would be useful for the design of new MOF-based catalysts for CO2 conversion into multi-carbon chemicals.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the A*STAR LCER-FI project (LCERFI01-0033 U2102d2006), the Ministry of Education of Singapore and the National University of Singapore (C-261-000-207-532/C-261-000-777-532 and R-279-000-574-114) for financial support.References
- S. Saeidi, S. Najari, V. Hessel, K. Wilson, F. J. Keil, P. Concepción, S. L. Suib and A. E. Rodrigues, Prog. Energy Combust. Sci., 2021, 85, 100905 CrossRef.
- S. Shafiee and E. Topal, Energy Policy, 2009, 37, 181–189 CrossRef.
- V. G. Dovì, F. Friedler, D. Huisingh and J. J. Klemeš, J. Clean. Prod., 2009, 17, 889–895 CrossRef.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed.
- S. Saeidi, S. Najari, F. Fazlollahi, M. K. Nikoo, F. Sefidkon, J. J. Klemeš and L. L. Baxter, Renew. Sustain. Energy Rev., 2017, 80, 1292–1311 CrossRef CAS.
- X. Xiaoding and J. A. Moulijn, Energy Fuels, 1996, 10, 305–325 CrossRef CAS.
- B. B. Asare Bediako, Q. Qian and B. Han, Acc. Chem. Res., 2021, 54, 2467–2476 CrossRef CAS PubMed.
- Y. Sheng, M. V. Polynski, M. K. Eswaran, B. Zhang, A. M. H. Lim, L. Zhang, J. Jiang, W. Liu and S. M. Kozlov, Appl. Catal., B, 2024, 343, 123550 CrossRef CAS.
- B. Zhang and J. Jiang, Energy Environ. Mater., 2024, 7, e12738 CrossRef CAS.
- R. G. Herman, Catal. Today, 2000, 55, 233–245 CrossRef CAS.
- H. T. Luk, C. Mondelli, D. C. Ferré, J. A. Stewart and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- V. R. Surisetty, A. K. Dalai and J. Kozinski, Appl. Catal., A, 2011, 404, 1–11 CAS.
- S. Zhang, Z. Xia, Y. Zou, F. Cao, Y. Liu, Y. Ma and Y. Qu, J. Am. Chem. Soc., 2019, 141, 11353–11357 CrossRef CAS PubMed.
- P. J. Linstrom and E. W. G. Mallard, NIST Chemistry WebBook, NIST Standard Reference Database, National Institute of Standards and Technology, Gaithersburg MD, 20899, 2024 Search PubMed.
- S. Zhang, X. Liu, Z. Shao, H. Wang and Y. Sun, J. Catal., 2020, 382, 86–96 CrossRef CAS.
- S. S. Ali, S. S. Ali and N. Tabassum, J. Environ. Chem. Eng., 2022, 10, 106962 CrossRef CAS.
- L. Wang, L. Wang, J. Zhang, X. Liu, H. Wang, W. Zhang, Q. Yang, J. Ma, X. Dong, S. J. Yoo, J.-G. Kim, X. Meng and F.-S. Xiao, Angew. Chem., Int. Ed., 2018, 57, 6104–6108 CrossRef CAS PubMed.
- E. C. Neyts, K. Ostrikov, M. K. Sunkara and A. Bogaerts, Chem. Rev., 2015, 115, 13408–13446 CrossRef CAS PubMed.
- A. Bogaerts, X. Tu, J. C. Whitehead, G. Centi, L. Lefferts, O. Guaitella, F. Azzolina-Jury, H.-H. Kim, A. B. Murphy, W. F. Schneider, T. Nozaki, J. C. Hicks, A. Rousseau, F. Thevenet, A. Khacef and M. Carreon, J. Phys. D: Appl. Phys., 2020, 53, 443001 CrossRef CAS.
- N. Zou, J. Chen, T. Qiu and Y. Zheng, J. Mater. Chem. A, 2023, 11, 10766–10775 RSC.
- B. Ashford and X. Tu, Curr. Opin. Green Sustainable Chem., 2017, 3, 45–49 CrossRef.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC press, 2004 Search PubMed.
- S.-L. Hou, J. Dong, X.-Y. Zhao, X.-S. Li, F.-Y. Ren, J. Zhao and B. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202305213 CrossRef CAS PubMed.
- S. Shao, C. Cui, Z. Tang and G. Li, Nano Res., 2022, 15, 10110–10133 CrossRef CAS.
- R. Krishnan, K. Yang, K. Hashem and J. Jiang, Mol. Catal., 2022, 527, 112407 CrossRef CAS.
- K. Yang and J. Jiang, J. Mater. Chem. A, 2020, 8, 22802–22815 RSC.
- B. Rungtaweevoranit, J. Baek, J. R. Araujo, B. S. Archanjo, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2016, 16, 7645–7649 CrossRef CAS PubMed.
- Z. Li, T. M. Rayder, L. Luo, J. A. Byers and C.-K. Tsung, J. Am. Chem. Soc., 2018, 140, 8082–8085 CrossRef CAS PubMed.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS.
- L. Zeng, Z. Wang, Y. Wang, J. Wang, Y. Guo, H. Hu, X. He, C. Wang and W. Lin, J. Am. Chem. Soc., 2020, 142, 75–79 CrossRef CAS PubMed.
- S.-C. Qi, Z.-H. Yang, R.-R. Zhu, X.-J. Lu, D.-M. Xue, X.-Q. Liu and L.-B. Sun, J. Mater. Chem. A, 2021, 9, 24510–24516 RSC.
- S. Bureekaew, S. Amirjalayer and R. Schmid, J. Mater. Chem., 2012, 22, 10249–10254 RSC.
- V. Paredes-García, R. C. Santana, R. Madrid, A. Vega, E. Spodine and D. Venegas-Yazigi, Inorg. Chem., 2013, 52, 8369–8377 CrossRef PubMed.
- S. J. Jenniefer and P. T. Muthiah, Chem. Cent. J., 2013, 7, 35 CrossRef PubMed.
- K. Hashem, R. Krishnan, K. Yang, B. A. Anjali, Y. Zhang and J. Jiang, Phys. Chem. Chem. Phys., 2024, 26, 7109–7123 RSC.
- I. Agirrezabal-Telleria, I. Luz, M. A. Ortuño, M. Oregui-Bengoechea, I. Gandarias, N. López, M. A. Lail and M. Soukri, Nat. Commun., 2019, 10, 2076 CrossRef PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView 6.0, Semichem Inc., Shawnee Mission KS, 2016 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03. Wallingford, CT, 2016 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- M. Mohamedali, A. Henni and H. Ibrahim, Microporous Mesoporous Mater., 2019, 284, 98–110 CrossRef CAS.
- C. Zhu, Z. Zhang, B. Wang, Y. Chen, H. Wang, X. Chen, H. Zhang, N. Sun, W. Wei and Y. Sun, Microporous Mesoporous Mater., 2016, 226, 476–481 CrossRef CAS.
- L. R. L. Ting, O. Piqué, S. Y. Lim, M. Tanhaei, F. Calle-Vallejo and B. S. Yeo, ACS Catal., 2020, 10, 4059–4069 CrossRef CAS.
- Y. Wang, B. Li, B. Xue, N. Libretto, Z. Xie, H. Shen, C. Wang, D. Raciti, N. Marinkovic, H. Zong, W. Xie, Z. Li, G. Zhou, J. Vitek, J. G. Chen, J. Miller, G. Wang and C. Wang, Sci. Adv., 2023, 9, eade3557 CrossRef CAS PubMed.
- Y. Wang, H. Luo, D. Liang and X. Bao, J. Catal., 2000, 196, 46–55 CrossRef CAS.
- C. Jia, J. Gao, Y. Dai, J. Zhang and Y. Yang, J. Energy Chem., 2016, 25, 1027–1037 CrossRef.
- W.-F. Xiong, D.-H. Si, H.-F. Li, X. Song, T. Wang, Y.-B. Huang, T.-F. Liu, T. Zhang and R. Cao, J. Am. Chem. Soc., 2024, 146, 289–297 CrossRef CAS PubMed.
- Y. Ouyang, L. Shi, X. Bai, C. Ling, Q. Li and J. Wang, ACS Catal., 2023, 13, 15448–15456 CrossRef CAS.
- Z.-X. Xu, X. Wu, J. Liu, Y. Kang and J. Zhang, CrystEngComm, 2015, 17, 6107–6109 RSC.
- T. Furuta, I. Kanoya, H. Sakai and M. Hosoe, Polymers, 2011, 3, 2133–2141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08052a |
| This journal is © The Royal Society of Chemistry 2025 |