
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lipoic acid/ethyl lipoate as cleavable comonomers for synthesis of degradable polymer networks†
Frances
Dawson
,
Gavin
Irvine
 and
Maciej
Kopeć
*
and
Maciej
Kopeć
*
Department of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: mk2297@bath.ac.uk
First published on 8th May 2025
Abstract
α-Lipoic acid, and its derived ester ethyl lipoate, can copolymerise with n-butyl acrylate to install labile disulfide bonds within the polymer backbone. Covalently crosslinked gel networks containing these comonomers were synthesised by conventional radical polymerisation (FRP) and reversible addition fragmentation chain transfer (RAFT) polymerisation. Gels synthesised by both methods and using both comonomers could be degraded by thiol-disulfide exchange or heating in DMF to form soluble polymer fragments. The critical comonomer loading for degradation was lower for RAFT-synthesised gels due to their more homogenous network structure. As these fragments were thiol functional, they could be oxidised in air with a base catalyst to reform a solid network. However, the presence of the carboxylic acid and the relatively low dispersity of the fragments act to prevent regelation. Therefore, only the gels containing the minimum amount of ethyl lipoate synthesised by RAFT could successfully regel as these fragments had no acid functionality and the highest dispersity value. Furthermore, cleavage of the trithiocarbonate end-groups aids reformation due to the presence of additional thiols. We suggest that uniform comonomer incorporation leading to lower dispersity of the degraded fragments can be detrimental for the efficient reformation of the degraded network. However, the large amounts of the lipoate comonomer allow the dynamic exchange properties of the disulfide bonds within the polymer backbone, in the presence of DBU catalyst, to impart the networks with self-healing ability with no external pressure or heat.
Introduction
Crosslinked polymer networks are inherently difficult to recycle but are widely used in a variety of applications due to their superior mechanical properties and thermal stability.1–4 Efforts to improve the sustainability of polymer materials has increased research into modifying these materials to be more readily degraded and/or recycled. One strategy that has been explored for both crosslinked and non-crosslinked materials is the incorporation of chemically labile bonds into the polymer chains, which can be cleaved by some external stimulus, breaking the polymer down into smaller fragments.5–8 A theoretical and experimental study from Johnson et al.,8,9 as well as our recent work,10–12 have shown that networks with cleavable units within the polymer strands (i.e., the backbone) are more readily degradable than when the cleavable units are within the crosslinks. Not only are the presence of these labile bonds beneficial for recycling processes, but they also could be useful in biomedical applications such as drug delivery13 or for self-healing materials.14–16An effective method to install labile bonds into the polymer backbone is by radical ring-opening (co)polymerisation (rROP) of vinyl monomers with cyclic comonomers6,17,18 such as cyclic ketene acetals,19,20 allyl sulfide lactones21 and thionolactones such as dibenzo[c,e]-oxepane-5-thione (DOT).9,22–25 The ring opening of the DOT comonomer implements a labile thioester bond which can be cleaved by aminolysis, and the copolymerisation is compatible with both conventional free radical polymerisation (FRP) and reversible deactivation radical polymerisation techniques (RDRP). We have previously shown that DOT can be copolymerised with n-butyl acrylate and a crosslinker using both FRP11 and reversible addition fragmentation chain transfer polymerisation (RAFT)10 to synthesise degradable crosslinked networks. Upon treatment with isopropylamine, macroscopic degradation (i.e., full solubilisation) of the gel occurred at a critical DOT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :crosslinker loading of 4:1 for FRP-made networks, which decreased to 1.5:1 for RAFT-made networks, highlighting the more homogeneous structure of networks synthesised using RDRP.
:crosslinker loading of 4:1 for FRP-made networks, which decreased to 1.5:1 for RAFT-made networks, highlighting the more homogeneous structure of networks synthesised using RDRP.
Although effective at affording degradability to both linear and crosslinked polymers at low comonomer loadings, one disadvantage of DOT, as well as other comonomers classes, is the multi-stage synthesis required to produce the comonomer, sparking interest in more sustainable alternatives.23,26 The dietary supplement α-lipoic acid (LA) has been considered as attractive alternative due to its commercial availability, low cost and biocompatibility. LA can undergo rROP through the scission of the disulfide bond present within the 1,2-dithiolane motif, and the carboxylic acid group can act as a functionalisation site allowing for a variety of modifications.27,28 The polymerisation of LA can be triggered by heat14,29–31 or UV light,15,32,33 either directly or through an external source of radicals.27,28,34,35 This reaction will produce polymers containing dynamic disulfide bonds, which can be cleaved by reduction,35 thiol-disulfide exchange10,36 or directly depolymerised.37
The polymerisation of LA has previously been used to synthesise polymer networks. For example, Sieredzinska et al. grafted LA to pendant amines on a polydimethylsiloxane (PDMS), which was then cured with UV to achieve crosslinking through the photopolymerisation of the 1,2-dithiolane group without a photoinitator present.33,37 Pendant 1,2-dithiolane groups have also been utilised in the formation of dynamic hydrogels from ABA triblock copolymers. With the addition of a dithiol, the pendant 1,2-dithiolane and thiols can reversibly react to form dynamic crosslinks, allowing the gel to be self -healable and shear thinning.38 Choi et al. used PDMS-LA macromonomers to synthesise soft bottle-brush elastomers through photopolymerisation, with these networks having a full poly(disulfide) backbone.15 This allowed for efficient self-healing under UV light and degradation of the network through heating at 180 °C, as well as via base or reducing agent. This highlights the low ceiling temperature of LA based polymers, also reported by Tsarevsky and Nicolay for the esterified form of lipoic acid, ethyl lipoate (ELp).27 Similarly, Zheng et al. also synthesised bottlebrush elastomers using a LA-PDMS macromonomer, with these networks synthesised through thermal self-polymerisation of the macromonomer.30 In a study from Alraddadi et al., crosslinked films were created using a difunctional lipoic acid-derived monomer and a tri-thiol crosslinker, which could react by base-mediated thiol-disulfide exchange.39 Both photo and thermal polymerisation of LA groups have been utilised in resins suitable for 3D printing.15,31,37,40 By either using a functionalised LA monomer,16,31 crosslinker40 or both,37 disulfide bonds were inserted into these resins, which could demonstrate reprocessability, closed loop recycling and self-healing.
When LA or its derivatives copolymerise with vinyl monomers, the labile disulfide bond that imparts degradability to the polymer is only formed when two LA units polymerise sequentially, forming a dyad (Scheme 1). Tang and Tsarevsky reported the copolymerisation of ELp with ethyl acrylate, with the presence of disulfide bonds confirmed by the reduction in the molecular weight of the polymer under reductive conditions.41 They continued to show the homopolymerisation of a vinyl functional LA derivative to form degradable branched polymers. In their study of ELp and LA-based pressure sensitive adhesives, Albanese et al. reported that in the copolymerisation of ELp with n-BA, dyad formation can be promoted by low temperature and high [M] reaction conditions due to the reversible propagation the ELp monomer.28 This work showed both LA and ELp could polymerise with n-BA using conventional FRP with a thermal radical initiator. After light crosslinking, these high molecular weight copolymers produced comparable pressure sensitive to acrylic acid/butyl acrylate-based analogues. These materials were degradable under reducing conditions and the fragments were successfully recycled using oxidation with I2/pyridine. Albanese et al. also showed the compatibility of LA copolymerisation with acrylate and acrylamide monomers using RAFT polymerisation to synthesise degradable, linear polymers.35 They found that no copolymerisation occurred with methacrylate, a result that Endo et al. also reported in their early work on the polymerisation of lipoamide with vinyl monomers.42 Interestingly, Endo et al. reported that lipoamide did not homopolymerise with AIBN, whereas Raeisi and Tsarevsky27 showed this is possible for ELp, which indicates that the functionalisation of LA derivatives would impact their polymerisation kinetics.
 | ||
| Scheme 1 Copolymerisation of lipoic acid with n-butyl acrylate by radical ring-opening polymerisation. Two sequential additions of lipoic acid to the polymer forms a disulfide bond within the chain. | ||
In this work, the LA and the esterified form ELp are used as cleavable comonomers with n-butyl acrylate to form covalently crosslinked gel networks with 0–30 mol% LA or ELp (Scheme 2). Degradation of these gels by thiol-disulfide exchange and heating in DMF is investigated to find the critical comonomer loadings for macroscopic degradation to soluble fragments. Both FRP and RAFT polymerisation were implemented to illustrate how network topology would affect the degradation of these materials. The self-healing and recycling ability is also explored.
 | ||
| Scheme 2 (A) Synthesis of PBA networks containing LA or ELp comonomer synthesised by either RAFT polymerisation (DDMAT present) or FRP (no DDMAT). (B) Degradation of synthesised gel networks by thiol-disulfide exchange using 2,2′-(ethylenedioxy)diethanethiol (EDDET) with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) catalyst. | ||
Results and discussion
Kinetics of lipoic acid/ethyl lipoate RAFT copolymerisation with n-butyl acrylate
The copolymerisation of n-butyl acrylate and lipoic acid has been shown to be compatible with both conventional FRP and RAFT polymerisation, using a trithiocarbonate RAFT agent 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT).43 Lipoic acid (LA) was functionalised with ethanol by Steglich esterification to form the comonomer ethyl lipoate (ELp, Fig. S1 and S2†). Before synthesising crosslinked networks containing these two comonomers, the RAFT polymerisation kinetics for comparable non-crosslinked reactions were studied. In this case a targeted degree of polymerisation of 150 was chosen with [BA]:[LA or ELp]:[AIBN]:[CTA] of 120:30:0.4:1, and anisole as a solvent (1:1 v/v with BA). BA and comonomer conversions were monitored by 1H NMR (Fig. S3, S4, S5A, B and S6A, B†). The reaction kinetics of BA with both comonomers were very similar, with the comonomer being incorporated only slightly faster into the polymer than the BA, suggesting that the comonomer, and thus the disulfide bonds, are incorporated in a uniform distribution throughout the polymer chains. This directly contrasts the copolymerisation of DOT and BA, where the DOT was incorporated significantly faster into the polymer than BA, leading to a gradient microstructure of the polymer chains.10,11,23,25,44 The differing distribution of cleavable sites within the polymers will affect the structure of the products of degradation, which was modelled in recent work from Lundberg et al.45
Both the BA-LA and BA-ELp copolymerisation reached ∼80% conversion in 120 minutes, with the comonomer functionality having little effect on the reaction kinetics. Samples were also analysed by GPC (Fig. S7, S8 and Tables S1, S2†), and the measured Mn values were close to the calculated theoretical values from the conversion measurements, although the dispersity values were slightly elevated, with both systems approaching Đ = 1.40 at high monomer conversions (Fig. S5 and S6C, D†). This is most likely due to increased termination from the slightly higher [AIBN] than used in other literature studies, which was required to produce fully developed gel networks under these conditions (vide infra). The PBA-LA system showed better correlation to theoretical Mn values than the PBA-ELp system; however, this could be due to how closely the polystyrene calibration correlates to the copolymer.
Thermomechanical properties of PBA-LA and PBA-ELp gels
Gel networks were synthesised by both FRP and RAFT with varying amounts of LA or ELp between 0–30 mol%, with anisole as a solvent. When synthesis of crosslinked networks with relatively low radical initiator amount ([BA]:[AIBN] = 100:0.1) was attempted by RAFT polymerisation, the gels produced were very soft and not suited for mechanical analysis due to the slow reaction rate. Therefore, the amount of initiator was elevated ([BA]:[AIBN] = 100:0.4) to make defined gels on a practical timescale, which led to a slight loss of control at high monomer conversions in reactions without crosslinkers (Fig. S5 and S6C, D†).
When designing a degradable polymer network, the aim is to install degradable units into the network without compromising its thermomechanical properties. In our previous work on PBA-DOT gels, the small amount of additive had no effect on the ESRs, gel fractions or storage modulus of the gels.10,11 In contrast, significantly more LA or ELp would be required to impart similar degradability as two units of comonomer are required to form a single disulfide linkage. Therefore, these physical properties may be more affected by the comonomer addition.
The equilibrium swelling ratio (ESR) and gel fraction % were measured in THF for the PBA-LA (Table 1 and Fig. S9†) and PBA-ELp gels (Table 2 and Fig. S10†). When synthesised using FRP, the ESR does not significantly change with addition of either comonomer, and the gel fraction remains at >90%. For the RAFT synthesised gels, the addition of both comonomers reduces the gel fraction and increases the ESR. The ESR increases from 8.7 ± 0.7 to 13.5 ± 0.3 for 30 mol% LA sample and to 13.0 ± 0.2 for ELp. It is commonly observed that gels synthesised by RAFT polymerisation display higher ESR values and lower gel fraction percentages compared to gels synthesised by FRP.10,36 This is due to the very different mechanism of network formation, with RAFT polymerisation producing homogenous networks containing primary chains with lower molecular weights.4,46,47 Indeed, increasing the crosslinker loading slightly (2 mol% to 3.3 mol% relative to total monomer amount), did improve the gel fractions of RAFT-made networks (Table 1, entries 13–15, and Fig. S11†).
| Entry | Sample | Crosslinker loadinga (mol%) | ESRb | Gel fractionc (%) | Macroscopic degradationd | Mass losse (%) |
|---|---|---|---|---|---|---|
| a Relative to total monomer amount (molBA + molLA). b Determined gravimetrically by swelling in excess THF, average over 3 replicates. c Determined gravimetrically upon washing in THF, average over 3 replicates % GF = mgel/(mgel + msol) × 100%. d Macroscopic degradation determined visually when no solids remain. Degradation by treatment with EDDET and DBU in THF. e Measured gravimetrically by washing and drying remaining solids after degradation. When full macroscopic degradation was observed, the mass loss was assumed to be 100%. f 1 sample out of 3 replicates macroscopically degraded. | ||||||
| 1 | PBA-0% LA-RAFT | 2 | 8.7 ± 0.7 | 88 ± 0.6 | No | 0.45 ± 0.7 |
| 2 | PBA-10% LA-RAFT | 2 | 9.9 ± 0.3 | 86 ± 0.3 | No | 16.9 ± 0.6 |
| 3 | PBA-15% LA-RAFT | 2 | 10.6 ± 0.2 | 83 ± 1.3 | No | 74.9 ± 1.1 |
| 4 | PBA-20% LA-RAFT | 2 | 11.1 ± 0.1 | 82 ± 0.5 | Yes | 100 |
| 5 | PBA-25% LA-RAFT | 2 | 11.9 ± 0.1 | 80 ± 1.0 | Yes | 100 |
| 6 | PBA-30% LA-RAFT | 2 | 13.5 ± 0.3 | 73 ± 1.3 | Yes | 100 |
| 7 | PBA-0% LA-FRP | 2 | 6.4 ± 0.2 | 93 ± 2.8 | No | −1.32 ± 0.9 |
| 8 | PBA-10% LA-FRP | 2 | 7.0 ± 0.74 | 98 ± 0.9 | No | 21.7 ± 11.3 |
| 9 | PBA-15% LA-FRP | 2 | 6.7 ± 0.3 | 94 ± 2.9 | No | 26.6 ± 0.6 |
| 10 | PBA-20% LA-FRP | 2 | 6.8 ± 0.1 | 95 ± 0.2 | No | 68.9 ± 8.5 |
| 11 | PBA-25% LA-FRP | 2 | 7.5 ± 0.2 | 92 ± 0.7 | Yes | 100 |
| 12 | PBA-30% LA-FRP | 2 | 7.4 ± 0.9 | 90 ± 1.6 | Yes | 100 |
| 13 | PBA-10% LA-RAFT | 3.3 | 6.4 ± 0.03 | 93 ± 1.5 | No | 10.7 ± 0.5 |
| 14 | PBA-20% LA-RAFT | 3.3 | 7.0 ± 0.1 | 82 ± 0.4 | Nof | 88.3 ± 8.4 |
| 15 | PBA-30% LA-RAFT | 3.3 | 8.2 ± 0.07 | 86 ± 0.5 | Yes | 100 |
| Entry | Sample | Crosslinker loadinga (mol%) | ESRb | Gel fractionc (%) | Macroscopic degradationd | Mass losse (%) |
|---|---|---|---|---|---|---|
| a Relative to total monomer amount (molBA+molLA). b determined gravimetrically by swelling in excess THF, average over 3 replicates. c Determined gravimetrically upon washing in THF, average over 3 replicates % GF = mgel/(mgel + msol) × 100%. d Macroscopic degradation determined visually when no solids remain. Degradation by treatment with EDDET and DBU in THF. e Measured gravimetrically by washing and drying remaining solids after degradation. When full macroscopic degradation was observed, the mass loss was assumed to be 100%. | ||||||
| 1 | PBA-0% ELp-RAFT | 2 | 8.7 ± 0.7 | 88 ± 0.6 | No | 0.45 ± 0.7 |
| 2 | PBA-10% ELp-RAFT | 2 | 11.9 ± 0.4 | 79 ± 4.1 | No | 69 ± 7.3 |
| 3 | PBA-15% ELp-RAFT | 2 | 14.2 ± 0.6 | 66 ± 1.5 | Yes | 100 |
| 4 | PBA-20% ELp-RAFT | 2 | 12.3 ± 1.23 | 74 ± 2.6 | Yes | 100 |
| 5 | PBA-25% ELp-RAFT | 2 | 13.0 ± 0.2 | 73 ± 0.5 | Yes | 100 |
| 6 | PBA-0% ELp-FRP | 2 | 6.4 ± 0.2 | 93 ± 2.8 | No | −1.32 ± 0.9 |
| 7 | PBA-10% ELp-FRP | 2 | 6.1 ± 0.2 | 99 ± 0.5 | No | 19.1 ± 0.78 |
| 8 | PBA-15% ELp-FRP | 2 | 7.3 ± 0.7 | 93 ± 2.8 | No | 62.1 ± 3.2 |
| 9 | PBA-20% ELp-FRP | 2 | 9.5 ± 0.1 | 89 ± 0.8 | Yes | 100 |
| 10 | PBA-25% ELp-FRP | 2 | 7.4 ± 0.1 | 92 ± 0.4 | Yes | 100 |
As-synthesised gel samples were analysed by oscillatory rheology (Fig. 1 and Fig. S12, S13†) and showed the apparent difference between gels synthesised by RAFT and FRP. As commonly observed, RAFT-synthesised gels have lower and more frequency-dependent storage moduli compared to FRP-synthesised gels.36,48 Although all the phase angles recorded are <45° indicating the gels behave more elastically than viscously overall, the RAFT-synthesised gels consistently show greater phase angle values than the FRP synthesised gels, indicating that there is a larger contribution from viscous flow for these samples (Fig. S12†). The polymer chains for the RAFT-made gels are much shorter than for the FRP gels, and likely below the critical molecular weight of entanglement. This leads to the lower G′ value as the contribution from entanglements to the overall modulus in negligible.49 The frequency dependence displayed is likely due to gradual stress relaxation of the high concentration of dangling ends and higher sol fraction in these gels compared to those made by FRP.50 Additionally, FRP networks contain spatial inhomogeneities in the form of densely crosslinked microclusters, which can further influence the network's mechanical properties.12,51–53 For FRP-synthesised gels with no comonomer added, the storage modulus (G′) value is 7.3 kPa at 1 rad s−1 which rises to 26.2 kPa and 12.2 kPa with 25 mol% LA and ELp loadings respectively. Previous reports on materials based on homopolymerised LA show the presence of hydrogen bonding between the pendant carboxylic acid groups, which act as non-covalent crosslinks between chains.14,54 The large increase in G′ value for the PBA-LA gels could be due to these additional physical crosslinks contributing to the elastic response of the material, and the smaller increase in G′ for PBA-ELp gels could be from less significant polar interaction from the disulfides and ester groups. For the RAFT-synthesised gels, the effect of the comonomer on G′ is much less apparent, especially for the PBA-ELp gels, despite the ESRs increasing with comonomer addition. The contribution to the storage modulus from the additional comonomer may be competing with the effect of the increased sol fraction for gels with high comonomer loading. When a higher crosslinker amount was used for PBA-LA-RAFT gels, the value of the storage modulus increased as expected. The frequency dependence of G′ also seemed lessened, possibly due to the higher gel fraction of these gels. This is also apparent in the much lower phase angle for these gels, due to less contribution of flow from sol and/or network strands (Fig. S13†).
 | ||
| Fig. 1 Oscillatory rheology frequency sweep of (A and B) PBA-LA and (C and D) PBA-ELp gels synthesised by FRP (A and C) and RAFT (B and D). Closed symbols represent storage modulus (G′) open symbols represent loss modulus (G′′). | ||
The thermal properties of the PBA networks are also affected by the addition of LA/Elp comonomer. Through thermogravimetric analysis (TGA) the thermal degradation profiles were measured for PBA networks containing no comonomer, 25% LA and 25% ELp synthesised by both RAFT and FRP (Fig. S14 and Table S3†). Addition of either comonomer reduced the degradation onset and the peak rate of degradation temperatures of the networks, due to the presence of weaker disulfide and thioether bonds within the polymer backbone. This reduction of thermal stability is consistent with other reports of disulfide-containing polymer materials.55,56 Through differential scanning calorimetry (DSC) the glass transition temperatures (Tg) of these networks were also measured (Fig. S15 and Table S4†). Compared to an unmodified PBA network (Tg = −40 °C for PBA-0%LA-RAFT), addition of 25 mol% ELp comonomer slightly reduced the measured Tg of the material (Tg = −47 °C) and LA addition significantly elevated the Tg (Tg = −23 °C). This increase is expected due to the presence of hydrogen bonding between lipoic acid groups reducing mobility of polymer chains leading to an increase in Tg.57 Addition of both comonomers affect the thermal stability of the networks, and LA addition significantly impacts the Tg. In contrast, we have previously shown that using DOT comonomer does not affect thermal properties,11 so it is important that these effects are noted when using comonomers in certain applications.
Degradation of lipoic acid/ethyl lipoate containing gels
Dried network samples were degraded using 2,2′-(ethylenedioxy)diethanethiol (EDDET, 3 equiv. to comonomer) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 1 equiv. to comonomer) in THF at room temperature (Scheme 2B and Tables 1, 2). Both FRP and RAFT gels successfully degraded into soluble fragments at some LA or ELp loading, indicating a significant number of disulfide bonds incorporated into the polymer structure. We have previously shown that for FRP-made PBA gels, placing disulfide bonds within the crosslinks did not produce fully degradable materials.10,12 Here the disulfide bonds are found within the polymer backbone, which confirms that the position of the labile bonds is important when considering degradable networks.8 Generally, the critical comonomer loading required for full degradation was 5 mol% (vs. monomer) lower for ELp compared to LA, and it was also 5 mol% lower for RAFT gels compared to FRP. It is consistent that RAFT networks require fewer cleavable bonds present to fully degrade, due to their more homogenous network structure. The gel sample that fully degraded with the lowest comonomer loading was the 15 mol% ELp gel synthesised by RAFT polymerisation. This is still significantly higher than the critical DOT loading we have previously reported10 for a comparable gel (3 mol%) which is expected due to the reversibility of the LA/ELp propagation and the necessity to form dyads to produce disulfide linkages. When a higher crosslink density is used (i.e., 3.3 mol% vs. monomer instead of 2 mol%), it is expected that more comonomer would be required to afford full degradability to the network.8 Whereas the PBA-20%LA-RAFT sample did macroscopically degrade at the lower crosslinker loading (Table 1, entry 4), when the crosslinker content was elevated, the sample with corresponding LA loading did not (Table 1, entry 14). However, the measured mass loss was very high (88.3 ± 8.4%) and 1 of the 3 triplicate measurements did macroscopically degrade to soluble fragments, suggesting that the critical lipoic acid loading for full degradation is only slightly higher than 20 mol%. Indeed, a PBA-30% LA-RAFT sample did macroscopically degrade (Table 1, entry 15). This indicates that well developed networks with higher gel fractions can be synthesised by RAFT polymerisation and still degrade with comparable comonomer loadings.The fragments of the fully degraded PBA-LA and PBA-ELp gels were analysed by 1H NMR (Fig. S16†) and GPC (Fig. 2). The absence of a peak at 3.5 ppm, corresponding to a proton within the dithiolane ring of the monomer, suggests that depolymerisation back to LA or ELp comonomer is not occurring. GPC analysis was only possible for gels synthesised by RAFT polymerisation as when FRP was used the fragments were too large to be filtered for analysis. The GPC traces were broad and multimodal, indicating significant branching. The molecular weights decrease with increasing comonomer loading, which is expected for gels with higher numbers of labile bonds within the backbone. The dispersity of these fragments also decreases with increasing comonomer loading and seems to approach Đ = 2, reflecting the uniform incorporation of both comonomers seen in kinetics experiments. Indeed, the degradation of polymer networks with cleavable comonomers was modelled by Johnson et al. who found that for the ideally random copolymerisation of a cleavable comonomer into a crosslinked network the dispersity of the degraded fragments were ∼2.8 Compared to PBA-DOT gels synthesised by RAFT polymerisation,10 the degraded fragments for the PBA-LA and PBA-ELp gels were also less multimodal and their measured dispersities were significantly lower. Whereas the DOT-BA copolymerisation formed a distinct gradient structure, the degraded fragment analysis corroborates the linear polymerisation kinetics study to suggest that the LA/ELp dyads are distributed more evenly throughout the network.
 | ||
| Fig. 2 GPC traces of the degraded gel fragments for (A) PBA-LA gels and (B) PBA-ELp gels synthesised by RAFT polymerisation. | ||
Interestingly, a few recent studies reported efficient degradation/depolymerisation of polydisulfides in common solvents. Machado et al. synthesised resins with a full polydisulfide backbone form lipoic-acid-derived monomers/crosslinker, which could be thermally depolymerised by refluxing the resin in DMF for 2 hours.37 Similarly, Okayama et al. reported the effective degradation of a copolymer of ELp and BA, containing only 5 mol% ELp, by heating in a polar solvent like DMF in air.58 Therefore, we tested the degradation of the PBA-LA and PBA-ELp networks in DMF, heating the samples at 100 °C with the vial open to air (Tables S5 and S6†). While this method of degradation was effective for LA-containing gels, it was less successful for the ELp-containing gels. This is likely due to better compatibility of PBA-LA networks with DMF due to LA being more polar. For PBA-LA gels, the comonomer loading required for full degradation was reduced compared to when EDDET/DBU was used for degradation (10% compared to 20% for RAFT-made gels and 20% compared to 25% for FRP-made gels). This corroborates the previous suggestion that thioether C–S bonds could be being cleaved in addition to the disulfides.58 The degraded fragments were analysed by 1H NMR, and no monomer peaks were present indicating no depolymerisation was occurring (Fig. S17†). Since depolymerisation of LA occurs by backbiting of terminal thiyl/thiolate to a disulfide, this is not likely to be occurring due to the low comonomer loading and the low probability of forming a dyad at the end of a chain. This indicates that for both degradation methods the breakdown of the network is due to cleavage of disulfides to thiols, and so reoxidation of these groups could lead to reformation of networks, rather than explicit repolymerisation of cyclic monomer as seen in closed-loop recycling of poly(LA)-based materials.31,37,59
Regelation
We have previously shown that thiol-functional degraded gel fragments can be oxidised in air in the presence of base to reform disulfide linkages and reform a solid network.10 The polymer fragments formed upon degradation of these PBA-LA and PBA-ELp networks also are thiol functional, and Albanese et al. have also shown the repolymerisation of linear PBA-ELp using thiol oxidation.28 Therefore, reformation of these networks after degradation should be possible. After degradation with a small amount of EDDET (1.5 equiv. to comonomer) and DBU (1 equiv. to comonomer) in THF, AcOH (1.5 equiv.) was added to quench the thiol groups on the fragments. The fragments were precipitated in methanol/water 90:10 and then transferred to a small vial. In order to facilitate disulfide formation, additional pyridine (5 equiv. to comonomer) and DMF were added to the degraded fragments before heating in air, as polar aprotic solvent and base are known to promote thiol oxidation in air.60 However, since heating in DMF did induce degradation at 100 °C, we first tested whether heating at 35 °C for the regelation process would act to degrade the network rather than reform it. A representative selection of networks were heated in DMF at 35 °C, and none of them macroscopically degraded with minimal mass losses, except for the PBA-25%ELp-RAFT which showed ca. 15% mass loss (Table S7†). Even though DMF did not degrade our gels at ambient temperatures, a control series of regelation experiments were also conducted in anisole. Degradation-regelation procedure was attempted for all samples listed in Tables 1 and 2 which macroscopically degraded. The samples were left for 1 week to (re)form the maximum number of disulfides.
In our previous study on PBA-DOT networks, the regelation of gels with 3–5 mol% DOT synthesised by RAFT was relatively easy, whereas gels made by FRP the regelation could not occur. A similar result was observed here, where neither FRP-made gel could successfully form a solid network. Surprisingly however, reformation of the PBA-LA and PBA-ELp gels synthesised by RAFT polymerisation also was difficult to accomplish. All attempts led to an increase in fragment viscosity to varying extents, however the only successful sample to fully regel to a solid network was the PBA-15%ELp-RAFT gel (Fig. 3 and Fig. S18†). Likely, there are two reasons for this behaviour. First, as the oxidation of thiols is base-mediated, reaction with the acid groups in the LA-containing gels may be competing with the thiol oxidation reaction, making the PBA-ELp gels more suitable for regelation. Furthermore, the structure of the fragments is clearly affecting the ease of relation. Notably, the PBA-15% ELp fragments had a significantly higher dispersity than any other sample (Đ = 16.59, Fig. 5B), which indicates that fragment dispersity is key to reforming networks. The other fragments have much lower dispersity values, which were also lower than any of the PBA-DOT gel fragments which successfully regelled in our previous study.10 These results suggest that the regelation success depends on the presence of acid groups (chemical contribution), and the dispersity of the fragments (structural contribution). Interestingly, this would indicate that uniform distribution of the cleavable groups in the backbone, which is typically a sought-after feature that improves degradation by producing low dispersity fragments, might be detrimental if the network is designed to also reform after deconstruction.
 | ||
| Fig. 3 Degradation and regelation of PBA-15% ELp RAFT gel. Images of sample with DMF used as regelation solvent. | ||
While this effect may at first seem counterintuitive, it can be rationalised by referring to the Flory-Stockmayer theory, where larger dispersity of crosslinking polymer chains results in lower number of crosslinks needed to reach the gel point.48,61 On the other hand, low dispersity enhances degradation, whether through cleavable comonomers/strands8 or crosslinks,62 and therefore the opposite relationship should be expected when reforming the degraded network.
Nevertheless, regelation of the PBA-15%ELp-RAFT sample with both solvents led to a well-defined material that could be analysed by swelling and oscillatory rheology (Fig. 4). For the DMF regelation, there was only a very slight increase in ESR value from 14 to 16 before and after the degradation/regelation process suggesting small reduction in network connectivity. This was confirmed by oscillatory rheology which showed a slight decrease in G′ value compared to the original sample, indicating that the disulfide reformation, while still high, was not 100% complete. For the anisole regelation, the increase in ESR and decrease in G′ compared to the original gel was much more significant, suggesting that the network was not reformed to the same extent as when DMF was used. This shows that although DMF at high temperatures acts to degrade the network, at lower temperatures the polarity of the solvent aids reformation of the disulfide bonds.
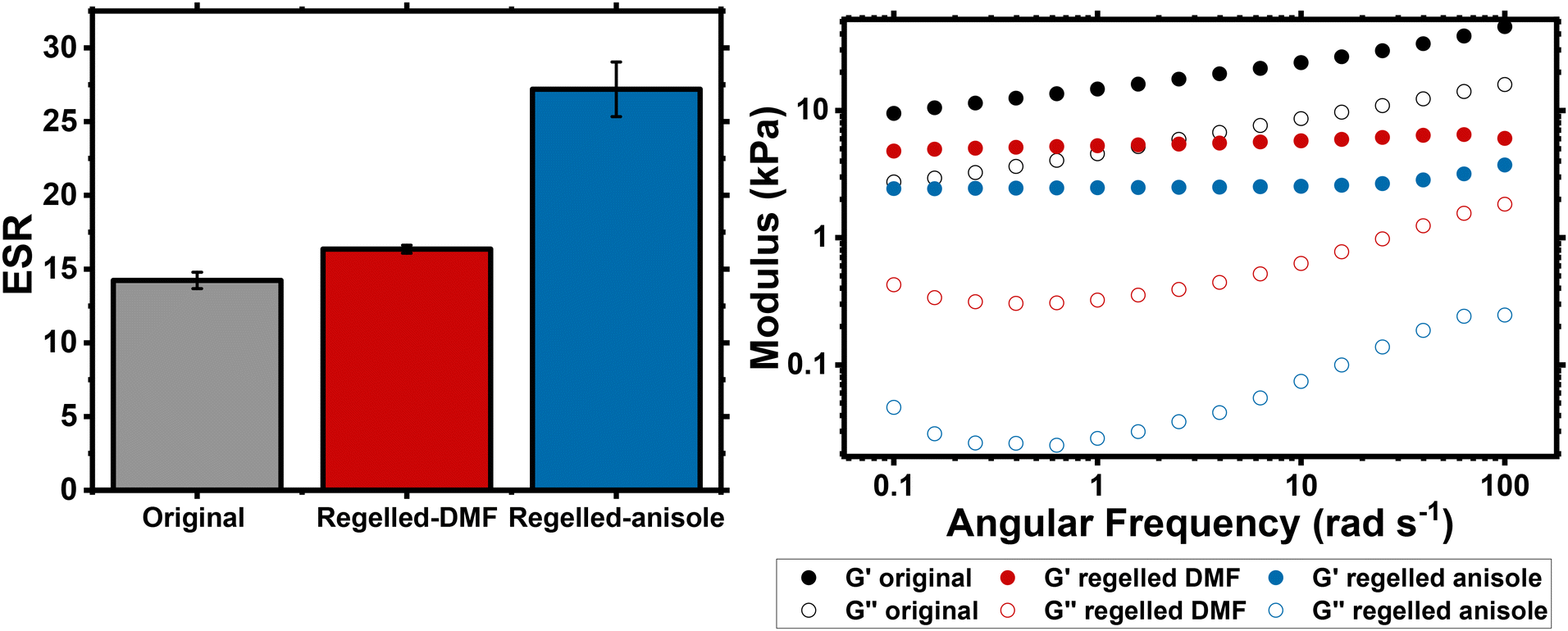 | ||
| Fig. 4 (left) Equilibrium swelling ratio and (right) oscillatory rheology frequency sweep of PBA-15%ELp gels before and after degradation/regelation process using DMF or anisole as the regelation solvent. For rheological measurements, gels were tested as synthesised. | ||
The reformed material also was significantly less yellow than the original material. Methods of removing RAFT end groups from polymers have been extensively studied to allow for further polymer processing, and has been shown to remove the polymer's bright colour.63–65 The dithiol EDDET combined with DBU would produce thiolate ions, which could act as a nucleophile to cleave the trithiocarbonate end group. Cleavage of the RAFT end group by nucleophiles produces thiols, the reaction of which would assist the reformation of the network, and result in an altered network topology. This would also explain the loss of frequency dependence in the storage modulus and the reduction of loss modulus in the oscillatory rheology data, as the reaction of these end-group thiols would lower the number of dangling chains. The reformed network was also degraded again by treatment with EDDET/DBU showing full solubilisation (i.e. 100% mass loss), indicating that disulfide formation is the mechanism for regelation.
Self-healing
The presence of dynamic disulfide bonds in a polymer network is often utilised for some form of self-healing behaviour.14–16,54,66 Usually this is in response to heat, light or other stimulus but can also be spontaneous if the concentration of disulfide is high enough. When the network is cut, the disulfide bonds can rearrange across the fracture. Choi et al. produced crosslinked 3D-printed objects from the photopolymerisation of a PEG-LA macromonomer to form super-soft bottlebrush networks.16 The dynamic exchange of the network was tested by crushing the objects and reprocessing under pressure with heat or light. It was also shown that cut portions of the material could be reattached upon the addition of DBU, acting as a strong base to facilitated disulfide rearrangement. A similar approach was very recently utilised by Han et al. for healing of 3D-printed resins containing disulfide bonds within the network's backbone and crosslinks.40 This method was used to test the PBA-LA and PBA-ELp gels, to see if a similar healing behaviour could be observed in materials with significantly fewer disulfide linkages.Gels with 25 mol% comonomer loading were tested, with the PBA-25%ELp-RAFT gel healing enough to hold the disc together, even with light amounts of pulling apart, after 24 hours which improved after 5 days (Fig. 5). Once again, the ELp gels were more successful than the LA gels due to the competing reaction of the DBU with the carboxylic acid groups. The RAFT-made gels were also more successful as they were softer and, as the previous rheology data shows (Fig. 1 and S12†), have more viscous flow behaviour due to shorter chains and higher sol fraction. Since the gel pieces are placed next to each other with no external pressure, this flow allows for greater contact across the fracture allowing for disulfide exchange to occur between chains within the two pieces. If the comonomer loading was increased, this healing behaviour possibly would become more effective, however at the expense of influencing other properties of the gel.
 | ||
| Fig. 5 Healing of cut PBA-25% ELp-RAFT gel using DBU catalyst. | ||
Conclusions
We have shown that α-lipoic acid and its ester ethyl lipoate can be used as a comonomer to impart macroscopic degradability to poly(n-butyl acrylate) crosslinked networks, at loadings as low as 15 mol% (vs. monomer). Degradation can be induced by treatment with dithiol or DMF at 100 °C, and degradation conditions should be chosen to suit the polarity of the polymer for most efficient degradation. Disulfide bonds are evenly distributed throughout the chains where two comonomer units copolymerise sequentially to form dyads which can be cleaved by thiol-disulfide exchange. Whereas networks synthesised by RAFT have been shown before to readily degrade using both cleavable comonomers and cleavable crosslinkers, gels synthesised by FRP are not as easily degraded. Here, LA and ELp comonomers successfully produced degradable PBA networks synthesised by FRP, requiring slightly higher loadings of comonomer than their corresponding RAFT-synthesised networks. It is possible to reform networks after degradation, however due to competing reaction with acid groups and low fragment dispersity this is difficult. Reformation was only possible when the networks are made using RAFT polymerisation, possibly due better network homogeneity and/or the presence of additional thiol groups formed by cleavage of the trithiocarbonate end group. Low dispersity of the polymer chains has been shown to make degradation more efficient, however here we show that this can negatively impact network reformation. These relationships could be utilised to control the degradation/regelation properties of a material, modifying the dispersity to make a material moderately degradable but easily reprocessable. The dynamic disulfide bonds located in the backbones also allow these networks to heal in the presence of DBU catalyst, at comonomer loadings of only 25 mol%. Lipoic acid comonomers could be utilised in making degradable and healable crosslinked materials for use in medical or cosmetic products due to its non-toxicity and biocompatibility.Data availability
Raw data associated with this manuscript will be made freely available through the University of Bath Research Data Archive at https://researchdata.bath.ac.uk/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC New Investigator Award Grant No. EP/W034778/1. Analytical equipment was provided by the Chemical Characterisation Facility at the University of Bath. The authors thank Dr Martin Levere for help with the GPC measurements.References
- E. Morici and N. T. Dintcheva, Polymers, 2022, 14, 4153 CrossRef CAS PubMed.
- W. Post, A. Susa, R. Blaauw, K. Molenveld and R. J. I. Knoop, Polym. Rev., 2020, 60, 359–388 CrossRef CAS.
- Y. Gu, J. Zhao and J. A. Johnson, Trends Chem., 2019, 1, 318–334 CrossRef CAS.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 5022–5049 CrossRef CAS PubMed.
- C. Lefay and Y. Guillaneuf, Prog. Polym. Sci., 2023, 147, 101764 CrossRef CAS.
- S. Ma and D. C. Webster, Prog. Polym. Sci., 2018, 76, 65–110 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Nature, 2020, 583, 542–547 CrossRef CAS PubMed.
- G. R. Kiel, D. J. Lundberg, E. Prince, K. E. L. Husted, A. M. Johnson, V. Lensch, S. Li, P. Shieh and J. A. Johnson, J. Am. Chem. Soc., 2022, 144, 12979–12988 CrossRef CAS PubMed.
- F. Dawson, T. Kazmi, P. J. Roth and M. Kopeć, Polym. Chem., 2023, 14, 5166–5177 RSC.
- H. Elliss, F. Dawson, Q. un-Nisa, N. M. Bingham, P. J. Roth and M. Kopeć, Macromolecules, 2022, 55, 6695–6702 CrossRef CAS.
- G. Irvine, F. Dawson, A. George and M. Kopeć, Eur. Polym. J., 2024, 213, 113089 CrossRef CAS.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS PubMed.
- Q. Zhang, C.-Y. Shi, D.-H. Qu, Y.-T. Long, B. L. Feringa and H. Tian, Sci. Adv., 2018, 4, eaat8192 CrossRef CAS PubMed.
- C. Choi, J. L. Self, Y. Okayama, A. E. Levi, M. Gerst, J. C. Speros, C. J. Hawker, J. Read de Alaniz and C. M. Bates, J. Am. Chem. Soc., 2021, 143, 9866–9871 CrossRef CAS PubMed.
- C. Choi, Y. Okayama, P. T. Morris, L. L. Robinson, M. Gerst, J. C. Speros, C. J. Hawker, J. Read de Alaniz and C. M. Bates, Adv. Funct. Mater., 2022, 32, 2200883 CrossRef CAS.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- T. Pesenti and J. Nicolas, ACS Macro Lett., 2020, 9, 1812–1835 CrossRef CAS PubMed.
- S. Agarwal, Polym. Chem., 2010, 1, 953–964 RSC.
- A. W. Jackson, S. Reddy Mothe, L. R. Chennamaneni, A. van Herk and P. Thoniyot, Materials, 2020, 13, 2325 CrossRef CAS PubMed.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805–9812 CrossRef CAS PubMed.
- N. Gil, C. Thomas, R. Mhanna, J. Mauriello, R. Maury, B. Leuschel, J.-P. Malval, J.-L. Clément, D. Gigmes, C. Lefay, O. Soppera and Y. Guillaneuf, Angew. Chem., Int. Ed., 2022, 61, e202117700 CrossRef CAS PubMed.
- N. M. Bingham and P. J. Roth, Chem. Commun., 2018, 55, 55–58 RSC.
- R. A. Smith, G. Fu, O. McAteer, M. Xu and W. R. Gutekunst, J. Am. Chem. Soc., 2019, 141, 1446–1451 CrossRef CAS PubMed.
- Q. Un Nisa, W. Theobald, K. S. Hepburn, I. Riddlestone, N. M. Bingham, M. Kopeć and P. J. Roth, Macromolecules, 2022, 55, 7392–7400 CrossRef CAS.
- E. A. Prebihalo, A. M. Luke, Y. Reddi, C. J. LaSalle, V. M. Shah, C. J. Cramer and T. M. Reineke, Chem. Sci., 2023, 14, 5689–5698 RSC.
- M. Raeisi and N. V. Tsarevsky, J. Polym. Sci., 2021, 59, 675–684 CrossRef CAS.
- K. R. Albanese, Y. Okayama, P. T. Morris, M. Gerst, R. Gupta, J. C. Speros, C. J. Hawker, C. Choi, J. R. de Alaniz and C. M. Bates, ACS Macro Lett., 2023, 787–793 CrossRef CAS PubMed.
- R. C. Thomas and L. J. Reed, J. Am. Chem. Soc., 1956, 78, 6148–6149 CrossRef CAS.
- S. Zheng, H. Xue, J. Yao, Y. Chen, M. A. Brook, M. E. Noman and Z. Cao, ACS Appl. Mater. Interfaces, 2023, 15, 41043–41054 CrossRef CAS PubMed.
- G. Zhu, N. von Coelln, Y. Hou, C. Vazquez-Martel, C. A. Spiegel, P. Tegeder and E. Blasco, Adv. Mater., 2024, 36, 2401561 CrossRef CAS PubMed.
- G. M. Scheutz, J. L. Rowell, S. T. Ellison, J. B. Garrison, T. E. Angelini and B. S. Sumerlin, Macromolecules, 2020, 53, 4038–4046 CrossRef CAS.
- B. Sieredzinska, Q. Zhang, K. J. van den Berg, J. Flapper and B. L. Feringa, Chem. Commun., 2021, 57, 9838–9841 RSC.
- K. R. Albanese, J. Read de Alaniz, C. J. Hawker and C. M. Bates, Polymer, 2024, 304, 127167 CrossRef CAS.
- K. R. Albanese, P. T. Morris, J. Read de Alaniz, C. M. Bates and C. J. Hawker, J. Am. Chem. Soc., 2023, 145, 22728–22734 CrossRef CAS PubMed.
- J. Cuthbert, S. V. Wanasinghe, K. Matyjaszewski and D. Konkolewicz, Macromolecules, 2021, 54, 8331–8340 CrossRef CAS.
- T. O. Machado, C. J. Stubbs, V. Chiaradia, M. A. Alraddadi, A. Brandolese, J. C. Worch and A. P. Dove, Nature, 2024, 1–6 Search PubMed.
- G. A. Barcan, X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2015, 137, 5650–5653 CrossRef CAS PubMed.
- M. A. Alraddadi, V. Chiaradia, C. J. Stubbs, J. C. Worch and A. P. Dove, Polym. Chem., 2021, 12, 5796–5802 RSC.
- S. Han, V. A. Bobrin, M. Michelas, C. J. Hawker and C. Boyer, ACS Macro Lett., 2024, 1495–1502 CrossRef CAS PubMed.
- H. Tang and N. V. Tsarevsky, Polym. Chem., 2015, 6, 6936–6945 RSC.
- T. Suzuki, Y. Nambu and T. Endo, Macromolecules, 1990, 23, 1579–1582 CrossRef CAS.
- K. R. Albanese, P. T. Morris, J. Read de Alaniz, C. M. Bates and C. J. Hawker, J. Am. Chem. Soc., 2023, 145, 22728–22734 CrossRef CAS PubMed.
- N. M. Bingham and P. J. Roth, Chem. Commun., 2019, 55, 55–58 RSC.
- D. J. Lundberg, K. Ko, L. J. Kilgallon and J. A. Johnson, ACS Macro Lett., 2024, 521–527 CrossRef CAS PubMed.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- A. Bagheri, C. M. Fellows and C. Boyer, Adv. Sci., 2021, 8, 2003701 CrossRef CAS PubMed.
- F. Dawson, H. Jafari, V. Rimkevicius and M. Kopeć, Macromolecules, 2023, 56, 2009–2016 CrossRef CAS PubMed.
- G. Irvine, K. Myronidis, F. Pinto and M. Kopeć, Angew. Chem., Int. Ed., 2025, e202421970 CAS.
- M. Rubinstein and R. Colby, Polymer Physics, Oxfor University Press, 2003 Search PubMed.
- S. Seiffert, Polym. Chem., 2017, 8, 4472–4487 RSC.
- F. Di Lorenzo, J. Hellwig, R. von Klitzing and S. Seiffert, ACS Macro Lett., 2015, 4, 698–703 CrossRef CAS PubMed.
- S. Seiffert, Prog. Polym. Sci., 2017, 66, 1–21 CrossRef CAS.
- J. Zhu, S. Zhao, J. Luo, W. Niu, J. T. Damron, Z. Zhang, M. A. Rahman, M. A. Arnould, T. Saito, R. Advincula, A. P. Sokolov, B. G. Sumpter and P.-F. Cao, CCS Chem., 2022, 5, 1841–1853 CrossRef.
- J. Wang, P. Sun, Z. Zheng, F. Wang and X. Wang, Polym. Degrad. Stab., 2012, 97, 2294–2300 CrossRef CAS.
- L. Zhou, M. Chen and X. Zhao, Polymer, 2017, 120, 1–8 CrossRef CAS.
- S. A. Jenekhe and M. F. Roberts, Macromolecules, 1993, 26, 4981–4983 CrossRef CAS.
- Y. Okayama, P. Morris, K. Albanese, S. Olsen, A. Mori, J. R. de Alaniz, C. M. Bates and C. J. Hawker, J. Polym. Sci., 2025, 63, 1345–1351 CrossRef CAS.
- Q. Zhang, Y. Deng, C.-Y. Shi, B. L. Feringa, H. Tian and D.-H. Qu, Matter, 2021, 4, 1352–1364 CrossRef CAS.
- J. L. G. Ruano, A. Parra and J. Alemán, Green Chem., 2008, 10, 706–711 RSC.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, 1953 Search PubMed.
- T. Shimizu, R. Whitfield, G. R. Jones, I. O. Raji, D. Konkolewicz, N. P. Truong and A. Anastasaki, Chem. Sci., 2023, 14, 13419–13428 RSC.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- A. B. Lowe, in Thiol-X Chemistries in Polymer and Materials Science, ed. A. Lowe and C. Bowman, The Royal Society of Chemistry, 2013, pp. 28–58 Search PubMed.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- C.-Y. Shi, Q. Zhang, B.-S. Wang, M. Chen and D.-H. Qu, ACS Appl. Mater. Interfaces, 2021, 13, 44860–44867 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00379b |
| This journal is © The Royal Society of Chemistry 2025 |