
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDistorted 2H/1T MoS2 nanostructures with improved field emission and sodium-ion battery performance†
Ujjwala
Chothe
*a,
Priyanka
Sumbe
b,
Mahendra A.
More
 b,
Murthy
Chavali
a,
Milind V.
Kulkarni
c and
Bharat B.
Kale
*ac
b,
Murthy
Chavali
a,
Milind V.
Kulkarni
c and
Bharat B.
Kale
*ac
aDr. Vishwanath Karad MIT World Peace University (MIT-WPU), Kothrud, Pune 411038, Maharashtra, India. E-mail: k.ujjwala@yahoo.com; bbkale1@gmail.com
bCentre for Advanced Studies in Materials Science and Condensed Matter Physics, Department of Physics, Savitribai Phule Pune University, Pune 411007, Maharashtra, India
cCentre for Materials for Electronics Technology (C-MET), Ministry of Electronics and Information Technology (MeitY), Panchavati, Off Pashan Road, Pune, 411008, Maharashtra, India
First published on 16th April 2025
Abstract
The present investigation demonstrates a facile and scalable synthesis strategy for the preparation of 2H and 1T phases of MoS2 using a solid-state method. Our findings reveal that MoS2 exhibits a distorted octahedral coordination structure, formed owing to the thiourea concentration. The structural transitions from the semiconducting 2H phase to the metallic 1T phase have been confirmed by XRD and Raman spectroscopy. The resulting MoS2 (1T) can provide more active sites by forming an ultrathin monolayer with a thickness of a few nanometers, enhancing the electrical conductivity. Field electron emission (FEE) behavior was investigated at a base pressure of 1 × 10−8 mbar. The utmost emission current density i.e. 820 μA cm−2 was observed for the 1T MoS2 emitter at an applied field of 4.56 V μm−1, in comparison to 2H MoS2, which showed 542 μA cm−2 at 4.85 V μm−1. Considering the good electrical conductivity and layered ultrathin sheet structure, 1T MoS2 has been explored as an anode for SIBs. Interestingly, it delivered a reversible capacity of 870 mA h g−1 at 100 mA g−1, and good cycling performance at 200 mA g−1 with 81% retention after 300 cycles. By integrating MoS2 electrodes with glyme electrolytes, a synergistic effect is achieved, leading to stable electrochemical performance of SIBs. The existence of an active 1T-MoS2 basal plane compared to 2H-MoS2 is responsible for the noticeable field electron emission and SIB application. It is noteworthy that MoS2 without any modifications (such as doping, carbon integration or composite formation) showed excellent performance in both applications. This paper underscores MoS2's versatility and promising avenues for revolutionizing electronic devices and energy storage technologies.
1. Introduction
Layered transition metal dichalcogenides (TMDs) have emerged as key materials in the advancement of novel nanomaterials for a wide range of technological applications.1–3 Among these, molybdenum disulfide (MoS2) dichalcogenide, with its weak van der Waals forces, has garnered significant attention due to its unique properties, including the layered structure, low cost, mechanical strength, and chemical stability.4–7 More significantly, the 1T phase of molybdenum disulfide represents a distinct structural configuration that has brought significant attention in the field of materials science and nanotechnology. Unlike the more common 2H phase, the 1T phase exhibits unique electronic and mechanical properties, making it particularly attractive for various applications.5,8–10 The 1T MoS2 phase adopts a distorted octahedral coordination geometry, resulting in a metallic behavior with enhanced conductivity (∼105 times that of the 2H phase) compared to the semiconducting nature of the 2H phase.11,12 This metallic character arises from the breaking of the inversion symmetry and the absence of a bandgap, offering opportunities for applications in electronic devices. Moreover it is a promising candidate for energy conversion and storage applications. Researchers have employed various synthesis techniques, including chemical vapour deposition (CVD), hydrothermal methods, and solvothermal approaches, exfoliation strategies to produce the 1T-MoS2 phase with controlled morphology, crystallinity, and surface properties.9,13–17In the field emission domain, MoS2 demonstrates remarkable electron-emission properties, making it suitable for use in field emission displays, electron sources for microscopy, and other vacuum electronic devices.6,18–20 Its high aspect ratio, tunable electronic properties, and robustness under high electric fields make MoS2 a promising candidate for enhancing the efficiency and performance of field emission-based devices.21 Researchers have explored different synthesis methods to tailor the nanostructures of 2H/1T-MoS2, enhancing its field emission properties such as emission current density, turn-on field, and emission stability of 1T-MoS2 emitters.6,19,22
Furthermore, MoS2 has emerged as a promising electrode material for SIBs. As the demand for energy storage solutions continues to rise globally, efforts toward ecofriendly and economical battery technologies have been in progress.23–27 MoS2 offers several advantages as an electrode material for SIBs, including its high theoretical capacity, abundant availability, and ability to accommodate Na ions during charge–discharge cycles.28–31 Additionally, the layered structure of MoS2 facilitates the diffusion of Na ions, thereby enhancing the battery's rate capability and cycling stability.4 The 1T phase of MoS2, with its distinct Mo–S atom coordination and superior electronic conductivity compared to the 2H phase, has garnered significant research interest in SIB electrodes.14 Experimental work has shown promising results, indicating that 1T-MoS2 exhibits high capacity, good cycling stability, and improved rate performance compared to other materials.24,32,33 Efforts have been made to boost the performance of MoS2, which include nanostructuring, surface modification, doping, and hybridization with other materials to improve ion diffusion kinetics, enhance electronic conductivity, and mitigate volume changes during cycling.32,34–37 Moreover, combining MoS2 with glyme-based (ether-based) electrolytes has synergistic effects, further improving battery performance.38 Ether-based electrolytes have garnered significant attention from SIBs due to their superior stability against reduction. This stability results in a higher initial coulombic efficiency and the formation of a thinner solid-electrolyte interphase (SEI) on the anode compared to traditional ester-based electrolytes.39 Glyme-based electrolytes offer several advantages, including high ionic conductivity, good compatibility with sodium metal anodes, and improved safety compared to conventional carbonate electrolytes.
Overall, MoS2 dichalcogenide exhibits tremendous potential, and this paper offers an in-depth exploration of its applications in field emission and SIB technologies. This study examines the impact of 2H and 1T phases of MoS2 on field emission performance. The structural and electronic differences between these phases significantly influence emission characteristics. Moreover, this paper provides a synergistic effect achieved by integrating MoS2 as anode electrodes with glyme-based electrolytes in SIBs. It examines the electrochemical behavior of MoS2, focusing on key parameters such as capacity, cycling stability, and rate capability. Moreover, the mechanisms governing the sodium ion insertion/extraction processes within MoS2 electrodes in glyme electrolytes are elucidated to provide insights into the enhanced battery performance. This straightforward and scalable approach is promising for addressing current challenges, advancing SIB technologies, and enhancing field emission applications with improved performance characteristics.
2. Experimental section
2.1 Synthesis of MoS2
In a typical synthesis, molybdenum sulfide (MoS2) was prepared using the solid-state method. Analytical grade ammonium heptamolybdate ((NH4)6Mo7O24·4H2O) and thiourea were combined, and the ratios of Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S were 1:4 and 1:8. The precursors were thoroughly ground with a mortar and pestle, and the mixture was heated at 270 °C in an air atmosphere for 12 hours. Once cooled, the resulting powder was ground again with a mortar and pestle. This powder was then mixed with distilled water and stirred for 3–4 hours. Following this, the mixture was filtered and dried in an oven at 100 °C. The dried powder was further calcined at 400 °C under an argon atmosphere for 3 hours. The molybdenum sulfide samples, synthesized with thiourea contents of 1:4 and 1:8, were designated as MS-H and MS-T, respectively.
:S were 1:4 and 1:8. The precursors were thoroughly ground with a mortar and pestle, and the mixture was heated at 270 °C in an air atmosphere for 12 hours. Once cooled, the resulting powder was ground again with a mortar and pestle. This powder was then mixed with distilled water and stirred for 3–4 hours. Following this, the mixture was filtered and dried in an oven at 100 °C. The dried powder was further calcined at 400 °C under an argon atmosphere for 3 hours. The molybdenum sulfide samples, synthesized with thiourea contents of 1:4 and 1:8, were designated as MS-H and MS-T, respectively.
2.2 Materials characterization
Phase identification and crystal structure analysis were performed using X-ray diffraction (XRD) on a Bruker-D8 Advance diffractometer equipped with Cu-Kα radiation, spanning a 2θ range of 5–80°. The surface morphology and microstructural features were analyzed using a Field Emission Scanning Electron Microscope (FESEM; CLARA, TESCAN, Czech Republic) operating at 5 kV. Transmission electron microscopy (TEM) studies, including high-resolution TEM (HRTEM), selected area electron diffraction (SAED), energy-dispersive X-ray spectroscopy (EDAX), and electron mapping, were carried out on a JEOL JEM 2200FS electron microscope operating at 200 kV. Micro Raman scattering at room temperature was measured using an HR 800 Raman spectrometer.2.3 Field electron emission measurements
The field electron emission experiments, dealing with current density versus electric field (J–E) and emission current stability over time (I–t) characteristics, were conducted in a planar diode configuration inside a fully metallic chamber evacuated at ∼1 × 10−8 mbar. The parallel diode FEE setup comprises an emitter held in close proximity to an anode (a semitransparent ITO glass coated with a cathodoluminescent phosphor, diameter ∼5 cm) The emitter was prepared by depositing a small amount of as-synthesized powder onto a piece of electroconductive carbon tape (dimensions ∼0.5 cm × 0.3 cm), which was subsequently placed on a copper stub. Field electron emission current (I) versus applied voltage (V) measurements were carried out using a Keithley 6514 electrometer and a Spellman high-voltage DC power supply (0–40 kV). Before recording the I–V characteristics, the cathode was conditioned at ∼500 V relative to the anode for 30 minutes to remove surface asperities and/or contaminants via in situ ion bombardment. Current stability measurements were performed using a digital multimeter integrated into a data acquisition system (sampling interval of 10 seconds). To ensure reproducibility and reliability, the FEE measurements were performed on at least two samples prepared under identical experimental conditions, characterized by similar morphologies.2.4 Electrode fabrication and electrochemical measurement
Electrochemical measurements for Na-ion batteries were conducted using 2032-type coin cells. These cells were assembled inside an argon-filled glove box (MTI, USA) to prevent contamination. Metallic sodium foil was used as the counter electrode, while quartz filter paper served as the separator. The electrolyte comprised 1 M NaPF6 dissolved in diglyme. The MoS2 electrodes were fabricated by blending the active material (80 wt%), conductive carbon (10 wt%), and polyvinylidene fluoride (PVDF, 10 wt%) in N-methyl-2-pyrrolidone (NMP) to form a uniform slurry. This mixture was then coated onto aluminum foil and dried in a vacuum oven at 120 °C for 12 hours. After drying, the coated aluminum foil was punched into 16 mm diameter disks to serve as the working electrodes, with an active material mass loading of 2–3 mg. Galvanostatic charge–discharge cycling was evaluated at room temperature using an MTI battery analyzer (vs. Na/Na+). Cyclic voltammetry (CV) of the half-cells was performed using an Autolab potentiostat/galvanostat (Metrohm Autolab) over a voltage range of 0.01–3 V. Electrochemical impedance spectroscopy (EIS) was carried out with a 5 mV amplitude across a frequency range from 0.1 Hz to 1 MHz.3. Results and discussion
The XRD patterns (Fig. 1a) are examined to identify the crystal structure and phase purity of the as-synthesized MoS2 (MS-H and MS-T). For 2H-MoS2, characteristic diffraction peaks appear at specific angles corresponding to the hexagonal crystal structure. The main peaks typically occur at 2θ of around 13.1°, 33.02°, 39.40°, 58.9°, and 69.2°, corresponding to the (002), (101), (103), (110), and (006) planes of MoS2 (a = b = 0.316 nm, c = 1.230 nm, JCPDS 37-1492), respectively. The (002) peak observed for 2H MoS2 corresponds to the periodic arrangement along the c-axis of the MoS2 structure, reflecting the ordered stacking of its 2D layers.9,40 The (002) peak of MS-H is shifted to lower angles, indicating an expansion of the lattice along the c-axis. Interestingly, the (002) peak disappears with increasing the thiourea concentration during the synthesis of the MS-T structure. This indicates that the few-layered 2H MoS2 structure was effectively exfoliated into predominantly single-layered MoS2 sheets. Furthermore, the broadening of the diffraction peaks suggests that the synthesized MoS2 exhibits poor crystallinity.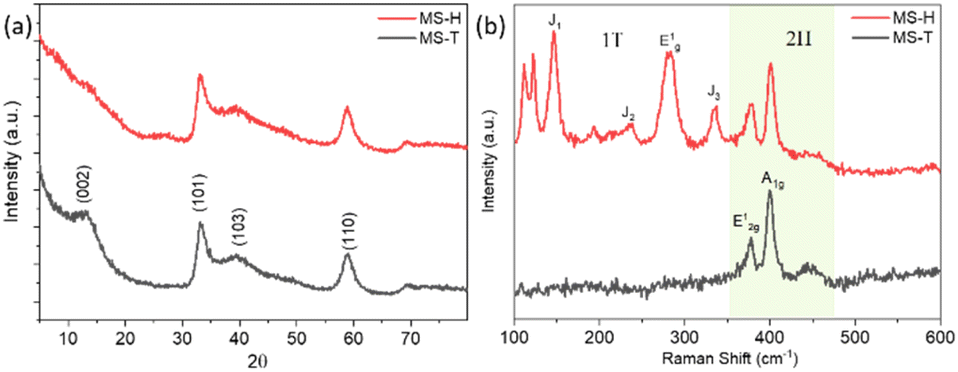 | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra of 2H MoS2 (MS-H) and 1T MoS2 (MS-T). | ||
Raman spectroscopy was used to characterize the 2H (hexagonal) and 1T (trigonal) phases of molybdenum disulfide (MoS2) by distinguishing their different vibrational modes. Raman spectra of the samples MS-H and MS-T are shown in Fig. 1b. The Raman spectrum of MS-H typically exhibits several characteristic peaks at around 376, 399, and 447 cm−1 that are indicative of its structural properties. The most prominent peaks, observed at 376 and 399 cm−1, correspond to the E12g and A1g vibration modes.41,42 The E12g peak is associated with the in-plane vibration mode of Mo and S atoms in the 2H layer, while the A1g peak involves the molecular vibration mode of S atoms along the c-axis, exhibiting symmetry outside the layer.43,44 In the Raman spectrum of MS-T, there are noticeable differences observed compared to the MS-H phase. In the case of the MS-T sample, new bands appeared in the as-prepared MoS2 phase, which clearly shows the formation of the 1T MoS2 phase. Additionally, other peaks associated with out-of-plane vibrations also emerge, with their frequencies due to the specific structure and strain present in the 1T phase.
MS-T has six significant peaks located at 112, 123, 147, 194, 238, 283, 334, 378, and 402 cm−1, respectively. The strong peaks at 147, 238, 283 and 334 cm−1 could be assigned to the stretching vibration of the Mo–Mo bonds and the phonon modes of 1T-MoS2, which confirm the presence of 1T-MoS2.5,7,45 Meanwhile, the E12g and A1g modes show a slight frequency shift due to the lattice structure's distortion, which is generally observed close to the frequency of 2H-MoS2. The peaks at 378, and 402 cm−1 are attributed to the typical E12g and A1g modes of MS-H, indicating the co-existence of 2H-MoS2. These differences observed in Raman analysis underscore the distinct structural characteristics of 1T-MoS2 compared to its 2H counterpart.
Fig. 2 presents the Field Emission Scanning Electron Microscopy (FESEM) analysis, which was used to examine the morphology and particle size of the synthesized samples. Fig. 2a and b display FESEM images of the MS-H sample, revealing irregularly shaped (elongated and nano/microspheres) particles in the range of 0.1 to 1 μm. In contrast, the MS-T sample, synthesized with increased thiourea concentration, exhibits interconnected MoS2 sheets, as shown in Fig. 2c and d.
 | ||
| Fig. 2 FESEM images of MoS2 samples: (a and b) MS-H and (c and d) MS-T. | ||
A detailed microstructural analysis of the samples was carried out using high-resolution transmission electron microscopy (HRTEM). Fig. 3 and 4 show representative HRTEM images of the MS-H and MS-T samples, respectively. As observed in the FESEM images, Fig. 3a and b illustrate the formation of MoS2 microspheres with an approximate size of ∼0.5 μm, while Fig. 4a and b, reveal that MoS2 is composed of ultrathin sheets with a few nanometers in thickness. Consistent with the findings from XRD analysis, a higher thiourea concentration facilitates the exfoliation of MoS2, leading to the development of thin, layered structures. The high-resolution TEM images (Fig. 3b and 4b) clearly show well-defined lattice fringes for the MS-H and MS-T samples. These fringes correspond to a d-spacing of 0.71 nm and 0.78 nm, associated with the (002) lattice plane of layered MoS2 (JCPDS: 37-1492). The observed d-spacing is greater than 0.61 nm, aligned with the XRD results, which show a peak shift towards a lower angle. The increased thiourea concentration induces a transformation of MoS2 microspheres from multilayered structures to monolayers. Furthermore, the continuous MoS2 nanosheets are fragmented into smaller pieces, as indicated by the arrows in the images. This exfoliation process enhances the exposure of abundant active sites, which could be useful for certain applications. The insets of Fig. 3b and 4b display the selected area electron diffraction (SAED) patterns, confirming the semicrystalline nature of MoS2. Fig. 3c–e and 4c–e show elemental mapping images, demonstrating the uniform molybdenum (Mo) and sulfur (S) distribution within the observed structures.
 | ||
| Fig. 3 (a) TEM image of the MS-H sample, (b) HR-TEM image of lattice fringe spacing and the SAED pattern (inset in (b)), and (c–e) STEM elemental mapping of the MoS2 sample: (c) greyscale image, (d) Mo-Kα, and (e) S-Kα of MS-T. | ||
 | ||
| Fig. 4 (a) TEM image of the MS-T sample, (b) HR-TEM image of lattice fringe spacing and the SAED pattern (inset in (b)), and (c–e) STEM elemental mapping of the MoS2 sample: (c) greyscale image, (d) Mo-Kα, and (e) S-Kα of MS-T. | ||
4 Formation mechanism
Fig. 5 illustrates a schematic representation of the preparation process for MoS2, highlighting the synthesis of the 2H and 1T phases by varying the concentration of thiourea. The synthesis procedure involved thoroughly mixing ammonium molybdate with thiourea in different ratios (1:4 and 1:8) and subjecting the mixture to a two-step calcination process. During the initial calcination at 270 °C, diammonium molybdate dissociated, releasing NH3 and water vapor and forming a molybdenum radical as an intermediate. Concurrently, thiourea decomposes to produce sulfur species (H2S), which serve as sulfurizing agents, and release ammonia. H2S further decomposed to yield sulfur, which was adsorbed onto the surface of molybdenum radicals. The sulfur species (H2S and elemental sulfur) react with molybdenum radicals and form MoS2.| (NH4)6Mo7O24 + nCH4N2S → 7MoS2 + nNH3 + nH2O + nCO2 + nH2S | (1) |
 | ||
| Fig. 5 Schematic illustration of the formation of 2H and 1T MoS2. | ||
The product was cooled after 12 hours, ground using a mortar and pestle, and then dispersed in deionized water. Stirring for 6 hours helped remove impurities and byproducts. X-ray diffraction (XRD) analysis of the initial material indicated its amorphous nature. To improve crystallinity, the washed and dried powder underwent a second calcination step at 400 °C, leading to the formation of 2H phase MoS2 microspheres. When the concentration of thiourea was increased, the synthesis dynamics shifted. Higher thiourea concentration is likely to generate more H2S and ammonia during decomposition. These gases diffused into the amorphous MoS2 layers, preventing nanosheet agglomeration and facilitating the exfoliation of MoS2 into the 1T phase. The sulfur-rich environment and thermal energy create lattice strain and induce sulfur vacancies. Additionally, the excess sulfur incorporation between the layers destabilizes the 2H phase. Electron redistribution occurs as the Mo atoms gain electrons, weakening the trigonal prismatic coordination in the 2H phase. This leads to the formation of the 1T phase with exfoliated nanosheets along with a cracked monolayer. The lower thiourea concentration favored the formation of 2H phase microspheres, while higher thiourea concentration promoted the formation of 1T phase exfoliated nanosheets with abundant active sites. This might be beneficial for various applications.
Thiourea is widely used as a sulfur source in MoS2 synthesis due to its controlled sulfur release, dual role as a sulfur donor and reducing agent, and ability to influence morphology. It decomposes at moderate temperatures, releasing H2S, thereby enabling uniform sulfidation of molybdenum precursors. Thiourea promotes phase purity, preventing unwanted impurities and facilitating the synthesis of 2H or 1T-MoS2. Its cost-effectiveness, water solubility, and safer handling compared to H2S gas make it an ideal choice for MoS2 synthesis.
Here, a solid state synthesis method was utilized, which offers high purity, scalability, and precise control over stoichiometry, making it ideal for producing stable and well-crystallized materials. Additionally, it avoids the need for solvents, reducing contamination and making it an environmentally friendly and cost-effective method.
5. Field emission study
Fig. 6a depicts the J–E characteristics of the pristine MS-H and MS-T emitters. The data reveal that the current density initially increases gradually, followed by a rapid rise, displaying an exponential behavior over the entire range of the applied electric field, consistent with the Fowler–Nordheim (F–N) theory.46 Herein, values of the turn-on field and threshold field are defined at emission current densities of ∼1 and ∼10 μA cm−2, respectively. From the observed J–E curves, the turn-on field values are estimated to be ∼1.84 and 2.07 V μm−1 and threshold field values are ∼2.61 and 2.98 V μm−1, for the MS-T and MS-H emitters, respectively. Furthermore, the MS-T emitter delivered a maximum current density of ∼830 μA cm−2 at a relatively lower applied field of 4.37 V μm−1, in contrast to ∼551 μA cm−2 at 5.15 V μm−1 from the MS-H emitter. These values indicate that the MS-T nanosheet emitters show superior FEE behavior compared to the MS-H emitter. The turn-on field of MS emitters could not be further lowered, which may be attributed to the noticeable screening effect due to many closely packed emission sites.47 | ||
| Fig. 6 (a) Plot of field emission current density as a function of the applied electric field for the as-synthesized MS-H and MS-T emitters. (b) Corresponding Fowler–Nordheim (F–N) plots. (c and d) Long-term emission current stability plots of MS-H (c) and MS-T (d). The insets show typical field emission images recorded during the measurements. | ||
In this study, the applied electric field (E) is defined as E = V/d, where V is the applied voltage and d is the distance between the anode and cathode, approximately 1.5 mm. The emission current density (J) is calculated using J = I/A, where I represents the emission current and A is the total area of the emitter. To analyze the J–E characteristics, a graph of ln(J/E2) versus (1/E), known as a Fowler–Nordheim (F–N) plot, was drawn. The corresponding F–N plots, as displayed in Fig. 6b, are characterized by non-linear behaviour, which may be attributed to the semiconducting nature of the emitters. The traditional F–N theory predicts the ‘linear’ behaviour of the F–N plot. There are various parameters responsible for the observed non-linear behaviour, and accordingly, modifications to the original F–N theory (for semiconducting emitters) have been discussed in the literature.48,49 The modified F–N equation is as follows,
| J = aφ−1E2β2 exp{−bφ3/2|βE} | (2) |
 | (3) |
From an application point of view, it is expected that the emitter should have excellent emission stability. Fig. 6c and d show the emission current stability of the emitters tested at a pre-set value of ∼10 μA over 3-hour duration. Both the emitters demonstrate fairly good stability at 10 μA, with slight current fluctuations with few excursions. A closer examination reveals that these excursions are accompanied by “spikes.” The primary causes of these spike-like fluctuations are various atomic scale processes such as adsorption, migration, and/or desorption of the residual gaseous species, as well as in situ ion bombardment. During adsorption/desorption events, slight variations in the local work function of emission site(s) occur due to the electropositive/electronegative nature of the adsorbed atoms/molecules. Moreover, the in situ ion bombardment due to the presence of a high electrostatic field may result in mechanical damage, creating and destroying emission sites on the emitter surface. These processes, in turn, lead to fluctuations and/or excursions in the emission current. The insets of Fig. 6c and d show typical field emission images recorded during the stability measurements, showcasing several emission sites (bright spots), which are indicative of the presence of nanometric sharp protrusions on the emitter surface.
The benefits of 1T MoS2 include its superior electrical conductivity compared to its 2H counterpart, making it highly advantageous for field emission applications. The work function of the 1T phase is generally lower than that of the 2H phase. A lower work function means that less energy is required to remove an electron from the material, making the 1T phase more efficient for electron emission. The metallic nature of 1T MoS2 facilitates efficient electron transport, which enhances the overall emission current. Additionally, the increased density of states near the Fermi level in 1T MoS2 contributes to improved field emission performance. The presence of abundant edge sites and defects in 1T MoS2 further enhances its field emission characteristics by providing numerous active sites for electron emission. To highlight the technical significance of the observed FEE characteristics, FEE characteristics of similar 2D layered emitters are compared and shown in Table S1.† According to these reports, the observed FEE performance (turn-on and maximum current density) is comparable to other 2D layered emitters, such as WS2, VS2, ReS2, and MoS2.6,51–56
6. Electrochemical evaluation
The electrochemical performance of MS-H and MS-T electrodes was evaluated using a 2032 half-cell configuration with sodium metal as a counter/reference electrode. Fig. 7a demonstrates the rate performance of the as-synthesized MoS2 (MS-H and MS-T). The MS-T cell delivered reversible capacities of 870, 506, 484, 381 and 312 mA h g−1 at 100, 200, 400, 800, and 1600 mA g−1, respectively, which are significantly higher than those of the MS-H cell, namely, 730, 363, 296, 111 and 68 mA h g−1, respectively. The optimized MS-T underwent a transformation from the original micron-sized spheres to ultra-fine nanosheets during synthesis. This structural modification enhances the utilization of electrode materials, which significantly shortens the ion transport path and hence exhibits better performance. The MS-H cell demonstrated initial discharge and charge capacities of 1202 mA h g−1 and 730 mA h g−1, respectively, with an initial coulombic efficiency (CE) of 60%. Subsequently, MS-T cells exhibited discharge and charge capacities of 1180 mA h g−1 and 870 mA h g−1, respectively, with an improved CE of 73%. The lower coulombic efficiency (CE) observed during the initial cycles is attributed to the poor reversibility of the transition from Mo and Na2S to MoS2. This is primarily because of the initial process of solid-electrolyte interface (SEI) formation.57 The MoS2 electrodes are used without doping, carbon integration, or composite formation. It takes a long time to stabilize the SEI layer, hence it showed a gradual capacity decline during the initial 8 to 10 cycles. Furthermore, the capacity stabilized and remained consistent for the subsequent cycles at a higher rate. Also, the CE remained higher than 98%, indicating a good electrochemical reversibility of the electrode. | ||
| Fig. 7 Electrochemical properties: (a) the rate performance of all samples at different current densities, (b) cycling stability, (c) the discharge–charge profiles of MS-T at different cycles, (d) cyclic voltammetry curves of MS-T at a scan rate of 0.1 mV s−1, (e) Nyquist plots of MS-H and MS-T, and (f) the relationship between Z′ and ω−1/2 in the low-frequency region. | ||
To confirm the reversibility, we disassembled the cells and characterized them using XRD and Raman analysis. Fig. S1(a) and (b)† present the XRD patterns of the MS-H and MS-T phases, respectively. Fig. S1(a)† shows higher-intensity peaks due to the presence of Al foil. In contrast, Fig. S1(b)† provides a zoomed-in view of the XRD graph, demonstrating that after rate capability cycles, MoS2 recovers its original phase. The MS-H phase exhibits the (002) peak at 2θ around 12.09°, which shifts to a lower angle due to an expansion in d-spacing, attributed to Na intercalation. This expansion facilitates the smooth insertion and extraction of Na+ ions during cycling. Additionally, XRD analysis was conducted on electrodes using a slow scan, revealing that the MS-T phase retains the (002) peak at 2θ around 9.07°. This clearly indicates the exfoliation of the MS-T phase, as discussed earlier. Fig. S1(c)† presents the Raman analysis of the disassembled electrodes, revealing the characteristic peaks of the MS-H and MS-T phases.
Therefore, the XRD and Raman analyses confirm the reversibility of the MoS2 phases after rate capability cycling, demonstrating the recovery of the original structure. The observed peak shifts indicate successful Na intercalation and exfoliation, ensuring smooth ion transport during cycling.
The MS-T sample, exhibiting better performance, was further evaluated for cycling stability, as shown in Fig. 7b. The long-term stability of the electrochemical performance of MS-T was assessed by applying a current rate of 200 mA g−1. Initially, the MS-T electrode was discharged at 100 mA g−1 for the first cycles, and then at 200 mAg−1 for the following 300 cycles. Fig. 7b shows that when discharged at 200 mA g−1, the MS-T electrode has a reversible capacity of approximately 673 mA h g−1 during the first cycle and retains a capacity of 615 mA h g−1 after 10 cycles, with 91% retention. Afterwards, the cell exhibits a capacity of around 501 mA h g−1 after the 300th cycle with 81% retention. The coulombic efficiency consistently remains at ∼98% throughout the cycles. The slightly higher rate capability and capacity of MS-T nanosheets are attributed to the 1T phase of MoS2. The synthesized MoS2 was explored without any modifications such as doping, carbon integration or composite formation. However, the capacity obtained is higher than that of the majority of the reported MoS2, as shown in Table S2.†5,35,38,57,58
The typical galvanostatic charge–discharge (GCD) profile of the MS-T electrode, including the initial discharge and the 1st, 2nd, 10th, and 50th cycles, is shown in Fig. 7c. As noted in the rate capability discussion, the coulombic efficiency (CE) for the initial discharge and charge is 73%, attributed to the formation of a solid-electrolyte interface (SEI) film. The corresponding GCD curve also evolves from a sloped shape in the 5th cycle to a step-like profile with distinct voltage plateaus after several cycles, further indicating the phase transformation of the electrode throughout the charge–discharge process. By the 50th cycle, certain notches in the profile suggest the intercalation of Na ions. The inset of Fig. 7c displays the GCD profiles for the 100th, 150th, 200th, 250th, and 300th cycles, further confirming the intercalation of Na ions within the MS-T structure. Following the 10th cycle, the CE increased and remained above 98%, indicating excellent electrochemical reversibility of the electrode. The superior performance of ether-based electrolytes, particularly because of diglyme as a solvent for electrolytes, is observed. As is known, diglyme has a unique ability to intercalate solvated molecules, i.e. coordination complexes with Na+ ions, into layered electrode structures. This intercalation significantly lowers the energy barrier for ion migration and enhances electron transport within the electrode. Consequently, these properties improve reversible capacity and cycling stability compared to traditional ester-based electrolytes.39
Cyclic voltammetry was performed to precisely examine the redox behaviour. Fig. 7d shows the CV curves of MS-T electrodes, recorded at a scan rate of 0.1 mV s−1 over a potential window of 0.01–3 V. In the cathodic sweep, MoS2 shows two reduction peaks at around 1.71 and 0.28 V owing to the insertion of Na+ into the MoS2 interlayer to form NaxMoS2, and the further transformation of NaxMoS2 into Mo and Na2S, respectively.59 However in the anodic sweep, corresponding oxidation peaks appeared at around 1.90, 0.94 and 0.45 V. These peaks correspond to the removal of Na+ and the formation of NaxMoS2 from Mo and Na2S, and the further desodiation of NaxMoS2 into MoS2, respectively.60 As observed in the discharge and charge plateaus, the stability of CV peaks in both cycles demonstrates a highly reversible and stable sodiation and desodiation process. Consequently, according to the reports, the overall reaction can be described as follows:38,61
| MoS2 + xNa+ + xe− = NaxMoS2 (x < 2) | (4) |
| NaxMoS2 + (4 − x)Na+ + (4 − x)e− = 2Na2S + Mo | (5) |
The kinetics of the MS-H and MS-T samples were thoroughly investigated using electrochemical impedance spectroscopy (EIS). As depicted in Fig. 7e, the Nyquist plots feature a semicircle at high to medium frequencies, followed by a sloping line at low frequencies. The charge transfer resistance (Rct) values were determined to be 757 Ω for MS-H and 381 Ω for MS-T. The lower Rct of 1T-MoS2 compared to 2H-MoS2 is due to its metallic nature, which provides higher electrical conductivity and faster charge transfer at the electrode–electrolyte interface. Fig. 7f illustrates the relationship between Z′ and ω−1/2 in the low-frequency region, which is used to determine the Na-ion diffusion kinetics based on the equations provided below:61,62
 | (6) |
 | (7) |
Overall, the excellent electrochemical performance of the MS-T sample is attributed to the distinct advantages of the 1T phase over the 2H phase. The 1T phase features more active sites and expanded interlayer spacing, enabling more efficient sodium ion intercalation and deintercalation. This contributes to a superior sodium ion storage capacity, making the 1T phase particularly effective for SIBs. Additionally, the flexible structure of the 1T phase is sustainable for the accommodation of ions and the volume changes that occur during the insertion and extraction of sodium ions. This flexibility helps to retain the structural integrity of the electrode material throughout multiple charge–discharge cycles, thereby enhancing the rate capability and lifespan. Furthermore, the increased chemical reactivity and larger surface area of the 1T phase improve its interaction with the electrolyte and sodium ions, resulting in enhanced electrochemical performance. Overall, the MS-T phase of MoS2 offers several benefits that make it a more favorable option for sodium-ion batteries compared to the 2H phase. Our investigation reveals that 1T MoS2 has great potential as an anode material in SIBs.
7. Conclusions
In a nutshell, we have demonstrated the synthesis of 2H and 1T MoS2 using a scalable solid-state method, influenced by thiourea concentration. The study also demonstrates the impact of 2H and 1T phases of MoS2 on field emission and SIB performance. The field emission study shows that turn-on fields for the MS-T and MS-H emitters are approximately 1.84 and 2.07 V μm−1, while the threshold fields are around 2.61 and 2.98 V μm−1, respectively, based on emission current densities of ∼1 and ∼10 μA cm−2. In SIBs, 2H and 1T MoS2 electrodes deliver capacities of 363 and 506 mA h g−1 at a current rate of 200 mA g−1, with good cycling performance and capacity retention. The 1T phase of MoS2, with its lower work function and metallic nature, enhances electron transport and field emission performance. Its increased density of states, abundant edge sites, and defects further improve electron emission. Additionally, the expanded interlayer spacing and more active sites enable efficient sodium-ion intercalation, making the 1T phase highly effective for sodium-ion batteries. Overall, the research highlights the potential of 1T MoS2 in field emission and SIBs, offering a cost-effective and eco-friendly alternative with abundant resources.Data availability
The data supporting the findings of this study are included in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author would like to thank the Department of Chemistry, Dr Vishwanath Karad MIT World Peace University (MIT-WPU) for providing research facilities. UC would like to acknowledge the financial support received as D.S. Kothari Post-Doctoral Fellowship (DSKPDF), University Grant Commission (UGC), New Delhi. The field emission work was carried out as a part of the DSKPDF research project. Mahendra More would like to acknowledge the financial support from CEPIFRA, DST, Govt. of India, (Project 62T8-1). The authors would like to thank the Centre for Materials for Electronics Technology (C-MET) for providing battery research facilities.References
- S. R. Kadam, D. J. Late, R. P. Panmand, M. V. Kulkarni, L. K. Nikam, S. W. Gosavi, C. J. Park and B. B. Kale, J. Mater. Chem. A, 2015, 3, 21233–21243 Search PubMed.
- L. Yin, Z. Zhang, Z. Li, F. Hao, Q. Li, C. Wang, R. Fan and Y. Qi, Adv. Funct. Mater., 2014, 24, 4176–4185 Search PubMed.
- X. Wang, Q. Weng, Y. Yang, Y. Bando and D. Golberg, Chem. Soc. Rev., 2016, 45, 4042–4073 RSC.
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 12794–12798 CrossRef CAS PubMed.
- J. Wu, J. Liu, J. Cui, S. Yao, M. Ihsan-Ul-Haq, N. Mubarak, E. Quattrocchi, F. Ciucci and J.-K. Kim, J. Mater. Chem. A, 2020, 8, 2114–2122 RSC.
- D. J. Late, P. A. Shaikh, R. Khare, R. V. Kashid, M. Chaudhary, M. A. More and S. B. Ogale, ACS Appl. Mater. Interfaces, 2014, 6, 15881–15888 Search PubMed.
- Y. Jiao, A. Mukhopadhyay, Y. Ma, L. Yang, A. M. Hafez and H. Zhu, Adv. Energy Mater., 2018, 8, 1702779 Search PubMed.
- X. Geng, Y. Zhang, Y. Han, J. Li, L. Yang, M. Benamara, L. Chen and H. Zhu, Nano Lett., 2017, 17, 1825–1832 CrossRef CAS PubMed.
- Y. Li, L. Wang, S. Zhang, X. Dong, Y. Song, T. Cai and Y. Liu, Catal. Sci. Technol., 2017, 7, 718–724 RSC.
- P. Kiran, K. Kumar, N. Pandit, S. Indupuri, R. Kumar, V. Wagh, A. Islam and A. Keshri, Adv. Funct. Mater., 2024, 34, 2316266 CrossRef CAS.
- G. Wen, S. Zhong, X. Zhang, Z. Shi, B. Wang, Y. Sui, J. Zeng, Z. Zhang and L. Wu, Ceram. Int., 2022, 48, 9781–9787 CrossRef CAS.
- S. Shi, Z. Sun and Y. H. Hu, J. Mater. Chem. A, 2018, 6, 23932–23977 RSC.
- S. Duraisamy, A. Ganguly, P. K. Sharma, J. Benson, J. Davis and P. Papakonstantinou, ACS Appl. Nano Mater., 2021, 4, 2642–2656 Search PubMed.
- D. Sun, D. Huang, H. Wang, G.-L. Xu, X. Zhang, R. Zhang, Y. Tang, D. Abd Ei-Hady, W. Alshitari, A. Saad Al-Bogami, K. Amine and M. Shao, Nano Energy, 2019, 61, 361–369 Search PubMed.
- R. Somphonsane, T. Chiawchan, W. Bootsa-ard and H. Ramamoorthy, Materials, 2023, 16, 4817 CrossRef CAS PubMed.
- R. I. Romanov, I. V. Zabrosaev, M. G. Kozodaev, D. I. Yakubovsky, M. K. Tatmyshevskiy, A. A. Timofeev, N. V. Doroshina, S. M. Novikov, V. S. Volkov and A. M. Markeev, ACS Omega, 2023, 8, 16579–16586 CrossRef CAS PubMed.
- X. Guo, E. Song, W. Zhao, S. Xu, W. Zhao, Y. Lei, Y. Fang, J. Liu and F. Huang, Nat. Commun., 2022, 13, 5954 CrossRef PubMed.
- F. Giubileo, A. Grillo, M. Passacantando, F. Urban, L. Iemmo, G. Luongo, A. Pelella, M. Loveridge, L. Lozzi and A. Di Bartolomeo, Nanomaterials, 2019, 9, 717 CrossRef CAS PubMed.
- A. Pelella, A. Grillo, F. Urban, F. Giubileo, M. Passacantando, E. Pollmann, S. Sleziona, M. Schleberger and A. Di Bartolomeo, Adv. Electron. Mater., 2021, 7, 2000838 CrossRef CAS.
- P. K. Bankar, M. A. More and D. J. Late, in Science and Technology of 2D MoS2, ed. D. J. Late and C. S. Rout, Springer Nature Singapore, Singapore, 2024, pp. 179–204, DOI:10.1007/978-981-97-7367-1_11.
- P. K. Bankar, B. R. Thombare, D. S. Gavhane, S. Kulkarni, G. S. Lole, K. D. Daware, M. M. More, S. I. Patil and P. R. Dusane, J. Mater. Sci.: Mater. Electron., 2024, 35, 1214 CrossRef CAS.
- R. S. Devan, V. P. Thakare, V. V. Antad, P. R. Chikate, R. T. Khare, M. A. More, R. S. Dhayal, S. I. Patil, Y.-R. Ma and L. Schmidt-Mende, ACS Omega, 2017, 2, 2925–2934 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- T. S. Sahu and S. Mitra, Sci. Rep., 2015, 5, 12571 CrossRef CAS PubMed.
- J. Cui, S. Yao and J.-K. Kim, Energy Storage Mater., 2017, 7, 64–114 Search PubMed.
- M. Liu, Y. Liu, Y. Yan, F. Wang, J. Liu and T. Liu, Chem. Commun., 2017, 53, 9097–9100 RSC.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- Q. Yun, Q. Lu, X. Zhang, C. Tan and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 626–646 CrossRef CAS PubMed.
- Q. Yun, L. Li, Z. Hu, Q. Lu, B. Chen and H. Zhang, Adv. Mater., 2020, 32, 1903826 CrossRef CAS PubMed.
- M. Minakshi, D. R. G. Mitchell, A. R. Munnangi, A. J. Barlow and M. Fichtner, Nanoscale, 2018, 10, 13277–13288 Search PubMed.
- M. Minakshi, D. R. G. Mitchell, C. Baur, J. Chable, A. J. Barlow, M. Fichtner, A. Banerjee, S. Chakraborty and R. Ahuja, Nanoscale Adv., 2019, 1, 565–580 RSC.
- X. Yue, J. Wang, Z. Xie, A. M. Patil, T. Yu, X. Du, Z. Wang, X. Hao, A. Abudula and G. Guan, J. Mater. Sci., 2020, 55, 14389–14400 CrossRef CAS.
- W. Tang, X. Wang, D. Xie, X.-h. Xia, C. Gu and J. Tu, J. Mater. Chem. A, 2018, 6, 18318–18324 Search PubMed.
- P. Li, Y. Yang, S. Gong, F. Lv, W. Wang, Y. Li, M. Luo, Y. Xing, Q. Wang and S. Guo, Nano Res., 2019, 12, 2218–2223 CrossRef CAS.
- R. Wang, S. Gao, K. Wang, M. Zhou, S. Cheng and K. Jiang, Sci. Rep., 2017, 7, 7963 CrossRef PubMed.
- G. S. Bang, K. W. Nam, J. Y. Kim, J. Shin, J. W. Choi and S.-Y. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 7084–7089 CrossRef CAS PubMed.
- H. Zhang, S. Zhang, B. Guo, L. J. Yu, L. Ma, B. Hou, H. Liu, S. Zhang, J. Wang, J. Song, Y. Tang and X. Zhao, Angew Chem. Int. Ed. Engl., 2024, 63, e202400285 CrossRef CAS PubMed.
- Y. Zhu, H. Li, Y. Wu, L. Yang, Y. Sun, G. Chen, Y. Liu, Z. Wu, C. Zhang and X. Guo, RSC Adv., 2021, 11, 28488–28495 RSC.
- C. Zheng, Y. Chen, X. Xu, Q. Lin, H. Wang, Q. Xue, B. Jian, Z. Guo and W. Lv, J. Power Sources, 2022, 526, 231098 CrossRef CAS.
- J. Zheng, H. Zhang, S. Dong, Y. Liu, C. Tai Nai, H. Suk Shin, H. Young Jeong, B. Liu and K. Ping Loh, Nat. Commun., 2014, 5, 2995 CrossRef PubMed.
- J. Wu, Z. Lu, K. Li, J. Cui, S. Yao, M. Ihsan-ul Haq, B. Li, Q.-H. Yang, F. Kang, F. Ciucci and J.-K. Kim, J. Mater. Chem. A, 2018, 6, 5668–5677 Search PubMed.
- X. Xie, T. Makaryan, M. Zhao, K. L. Van Aken, Y. Gogotsi and G. Wang, Adv. Energy Mater., 2016, 6, 1502161 CrossRef.
- H. Wei, A. Tan, W. Liu, J. Piao, K. Wan, Z. Liang, Z. Xiang and Z. Fu, Catalysts, 2022, 12, 947 Search PubMed.
- Y. Shi, Y. Zhou, D.-R. Yang, W.-X. Xu, C. Wang, F.-B. Wang, J.-J. Xu, X.-H. Xia and H.-Y. Chen, J. Am. Chem. Soc., 2017, 139, 15479–15485 Search PubMed.
- B. Yu, Y. Chen, Z. Wang, D. Chen, X. Wang, W. Zhang, J. He and W. He, J. Power Sources, 2020, 447, 227364 CrossRef CAS.
- R. H. Fowler and L. Nordheim, Proc. R. soc. Lond. Ser. A-Contain. Pap. Math. Phys. Character, 1928, 119, 173–181 CAS.
- I. Chakraborty and P. Ayyub, Nanotechnology, 2012, 23, 015704 Search PubMed.
- R. G. Forbes, Nanotechnology, 2012, 23, 095706 CrossRef PubMed.
- R. G. Forbes, Appl. Phys. Lett., 2016, 110, 133109 Search PubMed.
- J. Joe, C. Bae, E. Kim, T. A. Ho, H. Yang, J. H. Park and H. Shin, Catalysts, 2018, 8, 580 CrossRef.
- K. Lobo, P. R. Sumbe, M. A. More, D. J. Late and H. S. S. Ramakrishna Matte, Dalton Trans., 2024, 53, 3465–3469 RSC.
- C. S. Rout, P. D. Joshi, R. V. Kashid, D. S. Joag, M. A. More, A. J. Simbeck, M. Washington, S. K. Nayak and D. J. Late, Sci. Rep., 2013, 3, 3282 CrossRef PubMed.
- C. Rout, R. Khare, R. Kashid, D. Joag, M. More, N. Lanzillo, M. Washington, S. Nayak and D. Late, Eur. J. Inorg. Chem., 2014, 2014 Search PubMed.
- C. D. Mistari and M. A. More, Nano Express, 2021, 2, 020018 CrossRef CAS.
- Q. Zhang, K. Yu, B. Zhao, Y. Wang, C. Song, S. Li, H. Yin, Z. Zhang and Z. Zhu, RSC Adv., 2013, 3, 10994–11000 RSC.
- R. V. Kashid, D. J. Late, S. S. Chou, Y.-K. Huang, M. De, D. S. Joag, M. A. More and V. P. Dravid, Small, 2013, 9, 2730–2734 Search PubMed.
- X. Xu, Y. Qiu, Z. Len, Z. Chen, W. Zhu, W. Zhao, Y. Dai, L. Cao and H. Geng, J. Colloid Interface Sci., 2024, 656, 252–261 CrossRef CAS PubMed.
- J. Han, W. Xu, Z. Liu, Z. Gao, S. Tao, H. Min, H. Yang and J. Wang, J. Alloys Compd., 2023, 957, 170282 Search PubMed.
- W. Ren, H. Zhang, C. Guan and C. Cheng, Adv. Funct. Mater., 2017, 27, 1702116 CrossRef.
- G. Dong, H. Yu, L. Li, R. Zhang, X. Yang, K. Zhu, G. Wang and D. Cao, J. Colloid Interface Sci., 2023, 647, 395–405 CrossRef CAS PubMed.
- U. Chothe, C. Ugale, M. Kulkarni and B. Kale, Crystals, 2021, 11, 660 CrossRef CAS.
- K. K. Halankar, B. P. Mandal and A. K. Tyagi, Carbon Trends, 2021, 5, 100103 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5na00004a |
| This journal is © The Royal Society of Chemistry 2025 |