
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Xenon adsorption-induced flexibility of the zeolitic imidazolate framework ZIF-4 observed by in situ 129Xe NMR spectroscopy
Tobias Bodea,
Wen-Long Xue b,
Yutong Wangc,
Karen M. Garcia Alvarezc,
Silvia Paascha,
Andreas Schneemannc,
Sebastian Henkeb and
Eike Brunner*a
b,
Yutong Wangc,
Karen M. Garcia Alvarezc,
Silvia Paascha,
Andreas Schneemannc,
Sebastian Henkeb and
Eike Brunner*a
aChair of Bioanalytical Chemistry, Faculty of Chemistry and Food Chemistry, TU Dresden, 01062 Dresden, Germany. E-mail: eike.brunner@tu-dresden.de
bInorganic Chemistry, Department of Chemistry & Chemical Biology, TU Dortmund, 44227 Dortmund, Germany
cChair of Inorganic Chemistry I, Faculty of Chemistry and Food Chemistry, TU Dresden, 01062 Dresden, Germany
First published on 24th September 2025
Abstract
In situ 129Xe NMR spectroscopy reveals an unexpected, xenon adsorption-induced flexibility of ZIF-4. Beyond a certain threshold pressure, xenon induces a structural transition at low temperature. This behaviour is in contrast to CO2, where the material remains rigid, as shown by in situ 13C NMR spectroscopy. Adsorbed gas species are significantly confined in the narrow and anisotropic pore system with restricted atomic/molecular reorientation.
Flexibility of some metal–organic frameworks (MOFs) is an important property with strong impact on applications such as gas adsorption and separation.1–4 Flexible MOFs undergo adaptive structural transitions when triggered by external stimuli like electromagnetic radiation,5 mechanical pressure,6,7 temperature,8 electric fields,9 and adsorption of guest molecules.2,10,11 Gas-responsive flexibility is often characterized by the switching between distinct states, including closed or narrow pore states, intermediate states, and open pore states. ZIF-8 (ZIF: zeolitic imidazolate framework) opens the pore apertures, thus enabling gas molecules to enter the pores at a certain pressure depending on the kinetic diameter of the gas.12 Both, ZIF-8 and ZIF-4, consist of divalent metal ions interconnected by imidazolate derivatives.13 During the transition, ZIF-8 maintains its space group because only the 2-methylimidazolate linkers rotate to open the pore apertures. The structural flexibility of ZIF-4 was studied previously.6,8,14 It undergoes pore closure from an open to a closed phase under mechanical pressure at 280 bar as well as at low temperatures at 140 K.6,8 Here, we report a xenon adsorption-induced structural transition from a low-pressure (lp) phase to a high-pressure (hp) phase at ca. 8 bar and 240 K. In contrast to the closed pore phase formed under hydrostatic pressure or at cryogenic temperatures (<140 K), the material remains xenon-accessible in both phases but the adsorption capacity is significantly lower in the lp phase. The structural transitions are monitored by in situ 129Xe NMR spectroscopy of adsorbed xenon, a favourable probe for surface NMR spectroscopy.15–20 In situ 13C NMR spectroscopy of 13C-enriched CO2 exploits the residual chemical shift anisotropy (CSA), which depends on the framework structure.2,21–24
In situ 129Xe NMR spectra of ZIF-4 (Fig. 1) are measured at 240 K as a function of pressure and plotted at variable relative xenon pressure p/p0 (boiling pressure: p0 = 19 bar). The weak signal below 25 ppm represents the free xenon gas inside the sample cell which is not adsorbed in the MOF particles (Fig. 1, top). Its chemical shift increases with the gas density due to increasing Xe–Xe interactions.19 At the lowest applied relative pressure of p/p0 = 0.05, the slightly asymmetric signal of adsorbed xenon occurs at 93 ppm. Its chemical shift as well as the xenon uptake increase slightly with raising pressure up to p/p0 = 0.4 following a linear trend (Fig. 1, bottom). This suggests that the increase is mainly due to the continuously increasing xenon uptake leading to stronger Xe–Xe interactions.19 The simultaneous decrease of the total linewidth and disappearance of the asymmetry of the signal (Fig. 1, top, signal highlighted in light blue) suggest that the mobility increases with the pressure. The pore system of ZIF-4 consists of cages with 4.7 Å diameter connected by windows of 2.4 Å diameter, much less than the kinetic diameter of xenon (4.4 Å).26 Nonetheless, the pores are expected to be xenon accessible because DMF with a kinetic diameter of 4.9 Å27 is able to pass the pore apertures during solvent removal after synthesis.8 Consequently, xenon can only be adsorbed in the cages of the low-pressure (lp) phase for p/p0 < 0.4. This is confirmed by the xenon uptake which corresponds to approximately one xenon per cavity for the lp phase (Fig. 1, bottom). It appears that the lp phase corresponds to the crystal structure of the open pore phase at zero loading.26 Unfortunately, in situ PXRD is not applicable to the xenon-loaded sample because xenon heavily absorbs X-rays. However, the above-described observations strongly suggest that the pores are accessible in the lp phase. The high xenon uptake strongly contradicts the alternative explanation that xenon is adsorbed in defect pores at low pressures.
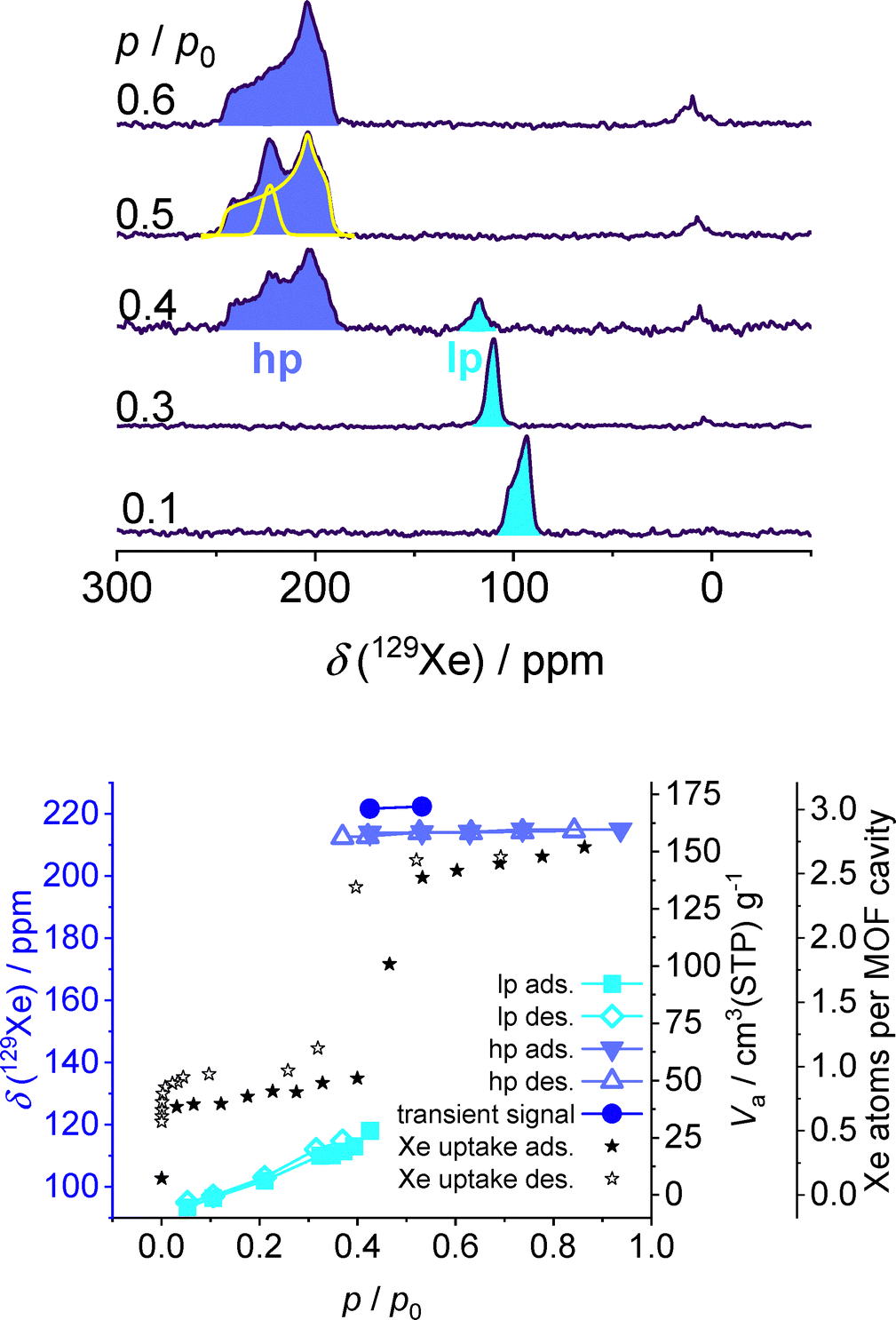 | ||
| Fig. 1 Top: In situ 129Xe NMR spectra of ZIF-4 at 240 K. Line shape decomposition (yellow lines) was carried out using the dmfit software.25 Bottom: chemical shift isotherm of adsorbed xenon (left vertical axis for blue/cyan symbols) and volumetric sorption isotherm (right vertical axis for black stars). | ||
At p/p0 = 0.43, the signal of adsorbed xenon occurs at 118 ppm with a significantly reduced intensity compared to lower pressures. Additionally, a new signal appears at δ > 180 ppm. The sudden increase of chemical shift is caused by the collective structural transition schematically shown in SI, Fig. S7. This is accompanied by a stronger confinement and higher adsorbed amount leading to stronger Xe-wall and Xe–Xe interactions. The signal at high chemical shift contains two contributions (Fig. 1, top): a Gaussian line at 221 ppm isotropic chemical shift and a signal with the shape resembling a chemical shift anisotropy (CSA) tensor, i.e., the orientational dependence of the chemical shift with respect to the magnetic field. The isotropic chemical shift δiso = 214 ppm of the latter is obtained from the principal values of the slightly non-axially symmetric CSA tensor δ⊥1 = 193 ppm, δ⊥2 = 205 ppm and δ‖ = 246 ppm. The residual CSA amounts to 47 ppm. The presence of a CSA seems counterintuitive because xenon is a single atom with a spherically symmetric electron distribution. However, xenon atoms in pore networks with narrow channels are known to exhibit CSAs because their confinement leads to an ellipsoidal electron cloud deformation.28–30 Therefore, xenon atoms in channels aligned perpendicular to the external magnetic field B0 axis have another chemical shift δ⊥ than xenon in channels parallel to B0 (δ‖).31 Moreover, the presence of three different principal values yields an asymmetry parameter of η = 0.35. That means, the pore network is also anisotropic in the plane perpendicular to B0 implying an ellipticity of channel cross-sections.31,32 We thus conclude that xenon contributing to the CSA-broadened signal at high isotropic chemical shift is strongly confined in narrow pores. While the electron cloud of xenon adsorbed in the lp phase was spherically symmetric, it must be deformed leading to the assumption that the material adopts a new phase with smaller average cage diameter. Since this transition is accompanied by an increase of the adsorbed amount (Fig. 1, bottom), we hypothesize the formation of the high-pressure (hp) phase with increasing window size and decreasing cage diameter. This may involve rotation of the imidazolate linkers thus enabling xenon atoms to better surpass the cage-connecting windows, whereas the cage-diameter reduces. Therefore, xenon can also occupy the space in the cage-connecting windows leading to approximately 2.5 xenon atoms per cavity in the hp phase (see schematic in SI, Fig. S6). A similar behaviour has been reported for xenon in ZIF-4 at room temperature but with less significant increase of the adsorbed amount.26 It was attributed to the formation of energetically favourable Xe–π interactions. Due to the high degree of confinement suggested by the CSA tensor, a simple pore opening as in the case of ZIF-8 can be excluded. The Gaussian line at 221 ppm is tentatively assigned to a transiently present pore type disappearing at increasing pressure.
To characterize exchange processes, 2D EXSY (exchange spectroscopy) spectra (Fig. 2) were recorded at different mixing times. For the lp phase (Fig. 2, left), cross-peaks appear at mixing times one order of magnitude lower than in the hp phase, indicating faster chemical exchange (Fig. 2, right). Note that the observed time regime of tens or even hundreds of milliseconds is characteristic of inter-particle exchange for both phases, lp and hp.33,34 The very slow chemical exchange in the hp phase, however, reflects the strong immobilization of xenon, the strong confinement in the hp phase, and the interpretation of the line shape of the signal by a CSA tensor. The lp phase signal could be explained by a narrower CSA tensor. It can, however, not fully be excluded that superimposed lines cause the asymmetric line shape in the lp phase. Taken together, the framework undergoes a structural transition from an lp phase to an hp phase during adsorption at p/p0 > 0.4 and 240 K. Note that our observations cannot simply be explained by increasing xenon–xenon interactions during adsorption. If this would be the case, a continuous increase of chemical shift and xenon uptake should be observed for increasing p/p0 and the line would still resemble a CSA tensor in the low-pressure range but with opposite sign.20 Moreover, the two signals at 118 and 214 ppm would not co-exist in the spectrum. At a relative pressure of p/p0 = 0.63, the transformation seems to be completed since the adsorbed amount, the shift δiso of 214 ppm, and the principal values of the CSA tensor remain constant during further pressure increase. During desorption, the properties of the CSA tensor as well as the signal intensity remain almost constant down to p/p0 = 0.32. In contrast to the adsorption branch, the phase transition is shifted to lower relative pressures as reflected by the hysteresis in the chemical shift adsorption/desorption isotherm as well as in the volumetric isotherm, a behaviour which is frequently observed in flexible porous materials. Moreover, the additional signal at 221 ppm is not observable during desorption.
 | ||
| Fig. 2 In situ 129Xe EXSY (exchange spectroscopy) NMR spectra measured at 240 K in the lp phase at p/p0 = 0.11 (left) and in the hp phase at p/p0 = 0.68 (right) at different mixing times as displayed in the figure. Centre glitches (artifacts) are marked with an asterisk. | ||
We also recorded time-dependent in situ 129Xe NMR spectra during desorption at p/p0 = 0.32 over 300 min with 5 min acquisition time for one spectrum to investigate the coexistence of both pore types during the phase transition in more detail (Fig. 3).
 | ||
| Fig. 3 In situ 129Xe NMR spectra during desorption measured at 240 K and p/p0 = 0.32 over 300 min with 5 min acquisition time per spectrum (left). Integrated intensities of the signals as a function of the total amount adsorbed at the beginning of measurement at p/p0 = 0.32, i.e., t0 = 0. | ||
Immediately after pressure adjustment to p/p0 = 0.32, only the signal of the hp phase is detected, whereas the signal of the lp phase at 115 ppm starts to appear after 15 min. The hp phase signal continuously decreases over time. After 300 min, the signal of the hp phase is almost undetectable, i.e., the transition is almost complete. During this period, the total adsorbed amount reduces significantly and the final lp signal intensity represents only about 1/3 of the initial hp phase signal intensity (Fig. 3, right). This observation suggests that the pore windows narrow during the 300 minutes experiment time. Xenon is thus released from the pore system and remains only adsorbed in the cage centres (see above).
To evaluate the dependence of this special behaviour of ZIF-4 on the temperature, we also carried out in situ 129Xe NMR spectroscopy at 167 K (see SI, Fig. S5). An analogous behaviour is observed, but the lp-hp transition is significantly shifted to lower relative pressure (p/p0 = 0.08 compared to p/p0 = 0.43 at 240 K). During the transition, two signals are again present, namely the lp signal at 90 ppm and the hp signal at 200 ppm.
In order to demonstrate the dependence of the structural transition on the gas type, we carried out comparative in situ 13C NMR studies with 13C-enriched CO2 at 240 K (Fig. 4, left).
 | ||
| Fig. 4 In situ 13C NMR spectra of ZIF-4 of 13CO2 measured during adsorption at 240 K and different relative pressures (left) and volumetric adsorption isotherms of CO2 under identical conditions (right). | ||
The spectrum of adsorbed 13CO2 at p/p0 = 0.08 consists of a line with characteristic shape of an axially symmetric CSA tensor, i.e., only two principal components are distinguishable. However, the residual CSA of only 9 ppm is rather low compared to fully immobilized solid CO2 with 335 ppm CSA. This implies a high degree of mobility of CO2 in the pores.35,36 CO2 as a linear molecule is presumably able to easily surpass the pore apertures and – similar to xenon – is mainly adsorbed in the cage centres at low pressure providing sufficient space for reorientation. At higher pressures, the residual anisotropy increases (>20 ppm) and the CSA tensor becomes asymmetric with an asymmetry parameter of η = 0.75. The isotropic chemical shift of the signals is constant for all pressures under consideration in agreement with the fact that the δiso of adsorbed CO2 is rather insensitive.35 The increasing CSA at higher loading is explained by an increasing fraction of CO2 molecules adsorbed in the cage-connecting narrow windows. The molecular reorientation is then restrained giving rise to larger CSA (see schematic in SI, Fig. S7). Fast exchange of CO2 molecules between the cage centres and the narrow windows gives rise to only one signal with averaged CSA as observed. However, the structural transition induced by xenon (see above) does not occur as can be seen from the volumetric adsorption/desorption isotherm (Fig. 4, right) showing the characteristic shape of non-flexible microporous materials.21
In summary, we discovered a xenon-induced structural transition of ZIF-4 by in situ NMR methods. ZIF-4 undergoes an lp-hp phase transition during xenon adsorption accompanied by a steeply increasing adsorption capacity. This transition was studied at different temperatures and variable gas pressure. Most prominently observed at 240 K, xenon is strongly confined in the pores thus exhibiting a pronounced chemical shift anisotropy. During desorption, the transition is reversed but a hysteresis of the transition pressure occurs. By comparison with in situ 13C NMR spectroscopy of adsorbed 13CO2, we demonstrated that this structural transition strongly depends on the gas type. CO2 continuously enters the pore system over the full pressure range. Hence, the flexibility of ZIF-4 strongly depends on properties like the kinetic diameter of the adsorbed gas. The reported unexpected behaviour may be important for applications such as gas separation.
T. B. – investigation, formal analysis, visualization, writing; W.-L. X. – investigation, formal analysis; Y. W. – investigation; K. M. G. A. – investigation; S. P. – supervision; A. S. – supervision; S. H. – conceptualization, supervision, funding acquisition, E. B. – conceptualization, supervision, funding acquisition, writing.
Financial support from the Deutsche Forschungsgemeinschaft (DFG), grant number BR 1278/36-1 is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc04620c.References
- C. Bachetzky, J. L. Fiorio, M. Maliuta, T. Bode, J.-O. Joswig, I. Senkovska, T. Heine, S. Kaskel and E. Brunner, J. Phys. Chem. C, 2024, 128, 6997–7006 CrossRef CAS.
- K. Roztocki, M. Rauche, V. Bon, S. Kaskel, E. Brunner and D. Matoga, ACS Appl. Mater. Interfaces, 2021, 13, 28503–28513 CrossRef CAS.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS.
- C. Gu, N. Hosono, J. J. Zheng, Y. Sato, S. Kusaka, S. Sakaki and S. Kitagawa, Science, 2019, 363(6425), 387–391 CrossRef CAS.
- Y.-X. Shi, W.-H. Zhang, B. F. Abrahams, P. Braunstein and J.-P. Lang, Angew. Chem., Int. Ed., 2019, 58, 9453–9458 CrossRef CAS.
- S. Henke, M. T. Wharmby, G. Kieslich, I. Hante, A. Schneemann, Y. Wu, D. Daisenberger and A. K. Cheetham, Chem. Sci., 2018, 9, 1654–1660 RSC.
- P. G. Yot, Q. Ma, J. Haines, Q. Yang, A. Ghoufi, T. Devic, C. Serre, V. Dmitriev, G. Férey, C. Zhong and G. Maurin, Chem. Sci., 2012, 3, 1100 RSC.
- M. T. Wharmby, S. Henke, T. D. Bennett, S. R. Bajpe, I. Schwedler, S. P. Thompson, F. Gozzo, P. Simoncic, C. Mellot-Draznieks, H. Tao, Y. Yue and A. K. Cheetham, Angew. Chem., Int. Ed., 2015, 54, 6447–6451 CrossRef CAS.
- A. Ghoufi, K. Benhamed, L. Boukli-Hacene and G. Maurin, ACS Cent. Sci., 2017, 3, 394–398 CrossRef CAS PubMed.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357–361 CrossRef CAS PubMed.
- S. Krause, J. D. Evans, V. Bon, I. Senkovska, F.-X. Coudert, G. Maurin, E. Brunner, P. L. Llewellyn and S. Kaskel, Chem. Soc. Rev., 2025, 54, 1251–1267 RSC.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS.
- P. Vervoorts, C. L. Hobday, M. G. Ehrenreich, D. Daisenberger and G. Kieslich, Z. Anorg. Allg. Chem., 2019, 645, 970–974 CrossRef CAS.
- T. Ito and J. Fraissard, J. Chem. Phys., 1982, 76, 5225–5229 CrossRef CAS.
- T. Ito, L. C. de Menorval, E. Guerrier and J. P. Fraissard, Chem. Phys. Lett., 1984, 111, 271–274 CrossRef CAS.
- J. Demarquay and J. Fraissard, Chem. Phys. Lett., 1987, 136, 314–318 CrossRef CAS.
- A. K. Jameson, C. J. Jameson and H. S. Gutowsky, J. Chem. Phys., 1970, 53, 2310–2321 CrossRef CAS.
- C. J. Jameson, A. K. Jameson and S. M. Cohen, J. Chem. Phys., 1973, 59, 4540–4546 CrossRef CAS.
- C. J. Jameson and A. C. de Dios, J. Chem. Phys., 2002, 116, 3805–3821 CrossRef CAS.
- M. Sin, N. Kavoosi, M. Rauche, J. Pallmann, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Langmuir, 2019, 35, 3162–3170 CrossRef CAS PubMed.
- V. J. Witherspoon, J. Xu and J. A. Reimer, Chem. Rev., 2018, 118, 10033–10048 CrossRef CAS PubMed.
- S. Chen, B. E. Lucier, P. D. Boyle and Y. Huang, Chem. Mater., 2016, 28, 5829–5846 CrossRef CAS.
- A. C. Forse, M. I. Gonzalez, R. L. Siegelman, V. J. Witherspoon, S. Jawahery, R. Mercado, P. J. Milner, J. D. Martell, B. Smit, B. Blümich, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2018, 140, 1663–1673 CrossRef CAS PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- B. Liu, X. Wu, Q. Liu, Y. Gong, S. Qi, S. Dong, Z. Mao, S. Xiong and S. Hu, Sep. Purif. Technol., 2025, 354, 128868 CrossRef CAS.
- A. U. Rehman, D. Arepalli, S. F. Alam, M.-Z. Kim, J. Choi and C. H. Cho, Nanomaterials, 2021, 11, 2327 CrossRef CAS PubMed.
- C. D. Keenan, M. M. Herling, R. Siegel, N. Petzold, C. R. Bowers, E. A. Rössler, J. Breu and J. Senker, Langmuir, 2013, 29, 643–652 CrossRef CAS.
- M. A. Springuel-Huet and J. Fraissard, Chem. Phys. Lett., 1989, 154, 299–302 CrossRef CAS.
- M.-A. Springuel-Huet, A. Nossov, Z. Adem, F. Guenneau, C. Volkringer, T. Loiseau, G. Férey and A. Gédéon, J. Am. Chem. Soc., 2010, 132, 11599–11607 CrossRef CAS PubMed.
- T. Meersmann and E. Brunner, New Developments in NMR: Hyperpolarized Xenon-129 Magnetic Resonance, Concepts, Production, Techniques and Applications, The Royal Society of Chemistry, Cambridge, 2015 Search PubMed.
- V. V. Terskikh, I. L. Moudrakovski, H. Du, C. I. Ratcliffe and J. A. Ripmeester, J. Am. Chem. Soc., 2001, 123, 10399–10400 CrossRef CAS.
- I. L. Moudrakovski, C. I. Ratcliffe and J. A. Ripmeester, Appl. Magn. Reson., 1995, 8, 385–399 CrossRef CAS.
- R. Giovine, C. Volkringer, M.-A. Springuel-Huet, A. Nossov, F. Blanc, J. Trébosc, T. Loiseau, J.-P. Amoureux, O. Lafon and F. Pourpoint, J. Phys. Chem. C, 2017, 121, 19262–19268 CrossRef CAS.
- H. Omi, T. Ueda, K. Miyakubo and T. Eguchi, Appl. Surf. Sci., 2005, 252, 660–667 CrossRef CAS.
- V. Bon, J. Pallmann, E. Eisbein, H. C. Hoffmann, I. Senkovska, I. Schwedler, A. Schneemann, S. Henke, D. Wallacher, R. A. Fischer, G. Seifert, E. Brunner and S. Kaskel, Microporous Mesoporous Mater., 2015, 216, 64–74 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |