
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Group 13 Lewis acid-mediated formation of 5-oxazolone derivatives from tert-butyl isocyanoacetate†
Zhou
He‡
ab,
Shaoying
Ju‡
 b,
Ting
Chen
b,
Xinghua
Zhang
*ab,
Douglas W.
Stephan
*c and
Yile
Wu
*bd
b,
Ting
Chen
b,
Xinghua
Zhang
*ab,
Douglas W.
Stephan
*c and
Yile
Wu
*bd
aSchool of Chemical and Environmental Engineering, Shanghai Institute of Technology, Shanghai 201418, China. E-mail: xhzhang@sit.edu.cn
bInstitute of Drug Discovery Technology, Ningbo University, Ningbo 315211, Zhejiang, China. E-mail: wuyile@nbu.edu.cn
cDepartment of Chemistry, University of Toronto, Toronto, 80 St. George Street, Ontario M5S 3H6, Canada. E-mail: douglas.stephan@utoronto.ca
dQian Xuesen Collaborative Research Center of Astrochemistry and Space Life Sciences, Ningbo University, Ningbo 315211, Zhejiang, China
First published on 12th June 2025
Abstract
tert-Butyl isocyanoacetate 1 reacted with B(C6F5)3 to give a Lewis acid–base adduct 2. GaCl3 and GaI3 promoted cyclization affording the N-bound Lewis acid adducts of the cyclized product 5-oxazolone derivatives 3 and 4, resulting from isocyano insertion into the ester C–O bond, with the loss of isobutylene. In contrast, the reactions with InBr3 and InI3 afforded the analogous adducts of 5(4H)-oxazolone derivatives 5 and 6, without the loss of the tert-butyl group. A proposed reaction mechanism is provided for these reactions of 1.
Isocyanides and their derivatives are valuable synthetic intermediates and are widely used in pharmaceuticals,1 materials science,2,3 and organic synthesis.4–7 They are frequently used in multicomponent reactions (e.g., Passerini, Ugi, Barton–Zard and Van Leusen reactions),8–10 which exploit the acidity of the α-C–H bonds to enable efficient cyclization or coupling reactions.11 Additionally, isocyanides can insert into C–X (X = halogen, C, O, S and H)12–14 or metal–carbon bonds12,15,16, mediated or catalyzed by transition metal complexes (e.g. Pd and base metals).
Group 13 Lewis acids have been widely exploited as reagents or catalysts in organic synthesis,17–22 as Lewis acid activation can induce the transformation of small molecules and functional groups.23–28 For example, the Lewis acids AlCl3 and GaCl3 are known to mediate the Friedel–Crafts reactions29,30 as well as cyclocondensations.31–33 In recent years, highly active group 13 Lewis acids such as B(C6F5)3 and Al(C6F5)3 have attracted considerable attention.27,28,34,35 Perhaps most notably, such Lewis acids generate “encounter complexes” with bulky Lewis bases prompting frustrated Lewis pair (FLP) chemistry of a variety of small molecules and affording new reaction types.36–41 While these group 13 Lewis acids have been used to effect cyclization reactions of alkynes,42–48 the corresponding reactions of isocyanides have not been reported. However, reactions of a phosphorus/boron FLP with isocyanides have been reported to generate a heterocyclic zwitterionic product of 1,1-addition of the FLP to the terminal carbon atom (Scheme 1).49
 | ||
| Scheme 1 Group 13 Lewis acid-mediated isocyanide activation. | ||
Despite the presence of a triple bond in both alkynes and isocyanides, the latter are strong sigma donors due to the lone pair of electrons on the terminal carbon atom. As a consequence, these species form stable adducts with Lewis acids, although group 13 Lewis acids have been used to mediate the insertion of isocyanide groups into heterocycles containing C–O bonds, effecting ring expansion (Scheme 1).50–52 In the present study, we probe the reactivity of tert-butyl isocyanoacetate with various group 13 Lewis acids. The findings demonstrate that, depending on the Lewis acid used, the reaction pathway can provide Lewis acid–base adducts, effect intramolecular cyclization and in some cases also induce degradation of the tert-butyl group and transfer of the hydrogen atom to the isocyanide carbon atom, thus providing facile and unique access to 5-oxazolone derivatives53 (Scheme 1).
Initially, density functional theory (DFT) calculations for the molecular orbitals of tert-butyl isocyanoacetate 1 were performed at the B3LYP-D3/def2-TZVP level of theory. The lone pairs of electrons at the carbonyl oxygen atom contribute to the highest occupied molecular orbital (HOMO) of 1, while the HOMO−1 of 1 is mainly associated with the lone pair at the terminal carbon atom of the isocyanide group (Fig. 1a). Interestingly, computations for the corresponding interactions of B(C6F5)3 with 1 showed that isocyanide-binding to B(C6F5)3 is energetically favoured (−91.3 kJ mol−1) over interaction with the carbonyl group. This may be in part attributed to the steric hindrance between B(C6F5)3 and the tert-butyl group, but the focused directionality of the lone pair at the terminal carbon atom of the isocyanide group may also be important here.
 | ||
| Fig. 1 (a) HOMO and HOMO−1 of 1 and (b) different coordination modes of 1 with B(C6F5)3, each accompanied by its gas-state single-point energy. | ||
To probe this experimentally, a mixture of 1 and B(C6F5)3 in toluene solution was stirred at ambient temperature for 1 hour (Scheme 2). This afforded a new set of 19F NMR resonances at −132.9, −154.9 and −162.7 ppm, as well as a new singlet at −21.2 ppm in the 11B NMR spectrum. These data are suggestive of a tetracoordinate boron center.54 Colourless crystals of 2 suitable for X-ray diffraction (XRD) analysis were obtained from toluene solution at −30 °C. The structural solution affirmed the tetracoordinated nature of the boron center and the formulation of 2 as (tBuO2CCH2NC)B(C6F5)3 (Fig. 2). The binding of the isocyanide to boron gave a B–Cisocyano bond length of 1.613(5) Å. This value is very close to the B–Cisocyano bond length found for the (Ph3PNNC)B(C6F5)3 adduct,55 while the N–C bond length in 2 (1.133(4) Å) of the isocyano group is consistent with a triple bond.
 | ||
| Scheme 2 Synthesis of 2–6. | ||
 | ||
| Fig. 2 POV-ray image of 2. Solvents are omitted for clarity. H: white, C: black, F: hot pink, N: blue, O: red, and B: yellow-green. | ||
In contrast, treating 1 with one equivalent of BCl3 or BF3(OEt2) gave complex and inseparable mixtures of products. On the other hand, a reaction with GaCl3 in a toluene solution (Scheme 2) led to a new singlet in the 1H NMR spectrum at 6.36 ppm. The singlet at 164.80 ppm in the 13C NMR spectrum was assigned to the carbonyl group, while no methyl resonances were observed in the 1H or 13C NMR spectra. Colourless crystals of product 3 were isolated from a toluene solution after standing at −30 °C overnight. The molecular formulation of 3 (Fig. 3a) was crystallographically determined to be (CH2CO2CHN)GaCI3 featuring a five-membered 5(4H)-oxazolone ring coordinated to a GaCl3 moiety via the N atom. This was consistent with the degradation of the t-butyl group. The Ga–N bond (1.978(6) Å) and three Ga–Cl bonds (2.1349(19), 2.1562(18) and 2.158(2) Å) complete the pseudo-tetrahedral coordination spheres of the Ga atom. The C3NO ring is nearly planar with a C–N–C angle of 107.0(6)°. The two N–C bonds with lengths of 1.260(9) and 1.465(9) Å can be attributed to a N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and a N–C single bond, respectively. The IR spectrum of 3 features a strong resonance around 1733 cm−1, which was attributed to the carbonyl moiety.
C double bond and a N–C single bond, respectively. The IR spectrum of 3 features a strong resonance around 1733 cm−1, which was attributed to the carbonyl moiety.
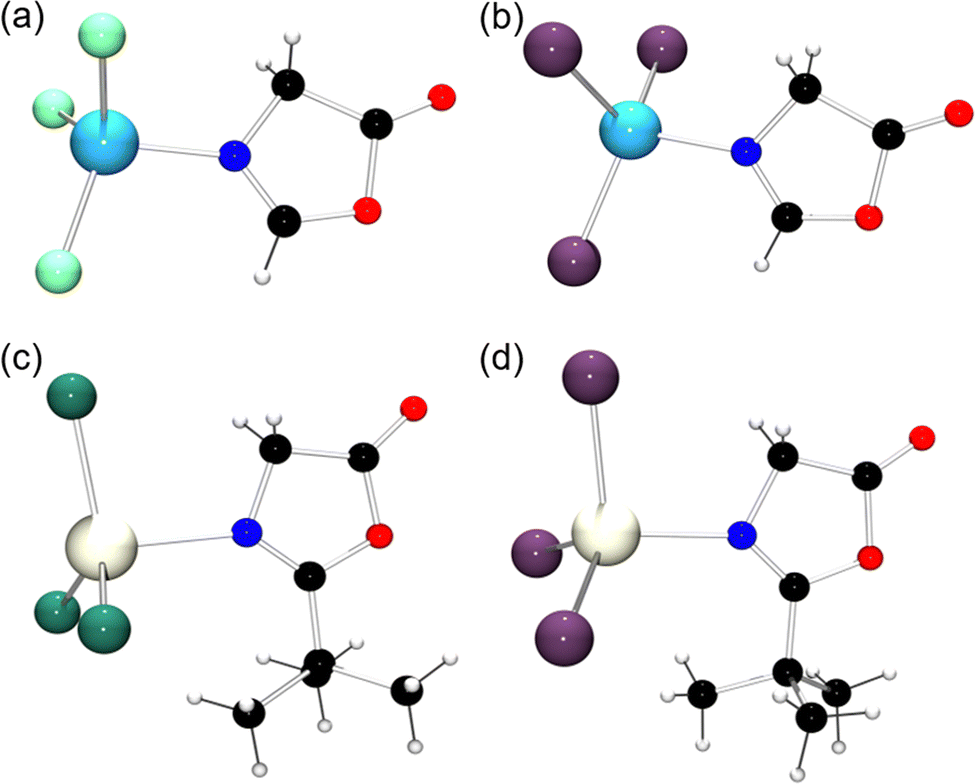 | ||
| Fig. 3 POV-ray images of (a) 3, (b) 4, (c) 5 and (d) 6. Solvents and disorders are omitted for clarity. H: white, C: black, N: blue, O: red, Ga: sky blue, In: wheat, and I: violet. | ||
The analogous reaction of 1 with GaI3 in a toluene solution results in the formation of compound 4 (Scheme 2). The singlet 1H and 13C NMR spectra were similar to those observed for 3 and compound 4 also showed a strong CO resonance peak at 1731 cm−1. Colourless crystals of 4 suitable for single-crystal XRD analysis were obtained after storing the toluene solution at −30 °C overnight. The molecular structure of (CH2CO2CHN)GaI34 (Fig. 3b) is similar to that of 3, with a Ga–N bond length of 2.023(11) Å and three Ga–I bond lengths of 2.5321(18), 2.504(2) and 2.4978(19) Å.
The corresponding reaction of 1 with InBr3 (Scheme 2) proceeded in CH2Cl2 solution at room temperature. In contrast to the Ga products 3 and 4, a new singlet was observed at 1.57 ppm in the 1H NMR spectrum and a corresponding singlet at 26.31 ppm in the 13C NMR spectrum. These were attributed to a tert-butyl group in the product. The IR spectrum of product 5 showed a strong absorption peak around 1904 cm−1, again consistent with the presence of a carbonyl fragment. Single-crystal X-ray crystallographic analysis of 5 (Fig. 3c) confirmed the formulation ((tBu)COC(O)CH2N)InBr3. In this case, analogous cyclization and binding of the Lewis acid to the oxazolone ring is observed; however, the C-tBu moiety is retained. The In–N bond length was determined to be 2.315(4) Å.
Using InI3, the related adduct ((tBu)COC(O)CH2N)InI36 showed similar spectral data and its formulation was confirmed by crystallographic data (Fig. 3d). The planar five-membered 5(4H)-oxazolone ring was bound to InI3 with an In–N bond length of 2.280(6) Å, while the carbonyl fragment exhibited an IR absorption peak around 1901 cm−1.
The formation of these oxazolone products 2–6 is thought to be initiated via initial coordination of 1 to the group 13 Lewis acids (Scheme 3). The steric demands of B(C6F5)3 result in a preference for interaction with the HOMO−1 of 1 located on the isocyanide fragment, affording the isolation of adduct 2. In contrast, binding of gallium and indium Lewis acids to the HOMO on oxygen induces cyclization, with concurrent migration of the tert-butyl group to the isocyanide carbon as well as migration of the Lewis acid to the more basic imine nitrogen atom (Scheme 2) as in the indium derivatives 5 and 6. However, in the Ga reactions, the resulting steric congestion between the GaX3 and tert-butyl fragments induces degradation of the tert-butyl group, resulting in the loss of isobutylene leaving a C–H bond on the heterocycle providing gallium derivatives 3 and 4. Similar degradation of tert-butyl ester fragments has been reported for the reactions of di-tert-butyl diazodicarboxylate with B(C6F5)356 or BF3.57
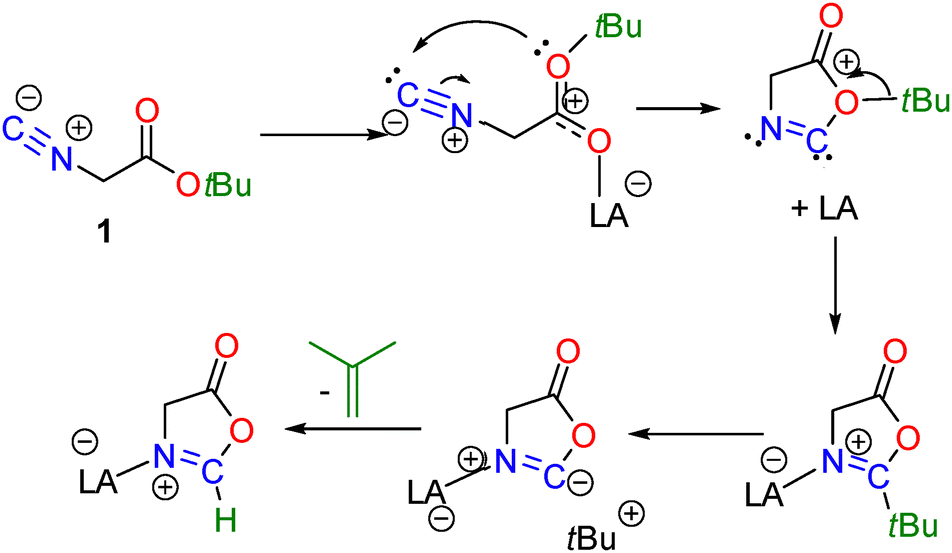 | ||
| Scheme 3 Proposed reaction mechanism for the reactions of 1 with group 13 Lewis acids. | ||
In conclusion, we have demonstrated the varying reactivities of tert-butyl isocyanoacetate 1 with a series of group 13 Lewis acids. The reaction of 1 with B(C6F5)3 gave the classic Lewis acid–isocyanate adduct 2, while reactions employing gallium and indium Lewis acids induced cyclizations, yielding 5(4H)-oxazolone derivatives 3–6. It is further proposed that the steric congestion of adjacent tert-butyl and GaX3 fragments results in the elimination of isobutylene, affording the C–H fragments in 3 and 4. It is interesting to note that the present study illustrates that variations of the Lewis acidity and steric properties of group 13 Lewis acid centers can be exploited in the synthesis of 5(4H)-oxazolone derivatives. Moreover, the variation of the Lewis acids affords products that model the various stages of the reaction pathway. We are continuing to explore the utility of group 13 Lewis acid mediated transformation in our effort to uncover novel strategies to heterocyclic species.
Z. H. performed the experiments. S. J. carried out the DFT calculations. T. C. assisted Z. H. with data collection. X. Z., D. W. S. and Y. W. prepared the manuscript and managed the project.
The National Natural Science Foundation of China (No. 22401163, 42388101 and 92256203) and Ningbo Natural Science Foundation (No. 2022J108) are acknowledged for financial support. Y. W. thank the Technology and Engineering Center for Space Utilization, the Chinese Academy of Sciences (No. KJZ-YY-NSM0406) and the Ningbo Top Talent Project (215-432094250) for financial support. We also acknowledge the Analysis Center of Institute of Drug Discovery Technology for collecting spectral data.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- A. Massarotti, F. Brunelli, S. Aprile, M. Giustiniano and G. C. Tron, Chem. Rev., 2021, 121, 10742–10788 CrossRef CAS PubMed.
- G. D. Sutton, M. E. Olumba, Y. H. Nguyen and T. S. Teets, Dalton Trans., 2021, 50, 17851–17863 RSC.
- Z. Cai, Y. Ren, X. Li, J. Shi, B. Tong and Y. Dong, Acc. Chem. Res., 2020, 53, 2879–2891 CrossRef CAS PubMed.
- S. Lang, Chem. Soc. Rev., 2013, 42, 4867–4880 RSC.
- T. Vlaar, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Angew. Chem., Int. Ed., 2013, 52, 7084–7097 CrossRef CAS PubMed.
- M. Giustiniano, A. Basso, V. Mercalli, A. Massarotti, E. Novellino, G. C. Tron and J. Zhu, Chem. Soc. Rev., 2017, 46, 1295–1357 RSC.
- L. Reguera and D. G. Rivera, Chem. Rev., 2019, 119, 9836–9860 CrossRef CAS PubMed.
- J. C. Flores-Reyes, A. Islas-Jácome and E. González-Zamora, Org. Chem. Front., 2021, 8, 5460–5515 RSC.
- A. V. Gulevich, A. G. Zhdanko, R. V. A. Orru and V. G. Nenajdenko, Chem. Rev., 2010, 110, 5235–5331 CrossRef CAS PubMed.
- T. Kaur, P. Wadhwa, S. Bagchi and A. Sharma, Chem. Commun., 2016, 52, 6958–6976 RSC.
- M. Gao, S. Lu and B. Xu, Chem. Soc. Rev., 2024, 53, 10147–10170 RSC.
- V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698–2779 CrossRef CAS PubMed.
- G. Qiu, Q. Ding and J. Wu, Chem. Soc. Rev., 2013, 42, 5257–5269 RSC.
- J. W. Collet, T. R. Roose, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Angew. Chem., Int. Ed., 2020, 59, 540–558 CrossRef CAS PubMed.
- S. J. Tereniak and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 14533–14541 CrossRef CAS PubMed.
- Y.-X. Xue, Y.-Y. Zhu, L.-M. Gao, X.-Y. He, N. Liu, W.-Y. Zhang, J. Yin, Y. Ding, H. Zhou and Z.-Q. Wu, J. Am. Chem. Soc., 2014, 136, 4706–4713 CrossRef CAS PubMed.
- M. Shibasaki, K.-I. Yamada and N. Yoshikawa, Lewis Acids in Organic Synthesis, ed. H. Yamamoto, Wiley-VCH, Weinheim, 2000 Search PubMed.
- A. Corma and H. García, Chem. Rev., 2003, 103, 4307–4366 CrossRef CAS PubMed.
- Y. Ma, S. Zhang, C.-R. Chang, Z.-Q. Huang, J. C. Ho and Y. Qu, Chem. Soc. Rev., 2018, 47, 5541–5553 RSC.
- P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, Chem. Soc. Rev., 2015, 44, 7025–7043 RSC.
- L. L. Liu and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3454–3463 RSC.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC.
- H. Yamamoto and K. Ishihara, Acid catalysis in modern organic synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- M. M. Alharbi, Y. van Ingen, A. Roldan, T. Kaehler and R. L. Melen, Dalton Trans., 2023, 52, 1820–1825 RSC.
- J. F. Kögel, A. Y. Timoshkin, A. Schröder, E. Lork and J. Beckmann, Chem. Sci., 2018, 9, 8178–8183 RSC.
- Y. Ma, S.-J. Lou and Z. Hou, Chem. Soc. Rev., 2021, 50, 1945–1967 RSC.
- S. Basak, L. Winfrey, B. A. Kustiana, R. L. Melen, L. C. Morrill and A. P. Pulis, Chem. Soc. Rev., 2021, 50, 3720–3737 RSC.
- J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC.
- R. Sunke, S. B. Nallapati, J. S. Kumar, K. Shiva Kumar and M. Pal, Org. Biomol. Chem., 2017, 15, 4042–4057 RSC.
- M. K. Gupta and T. P. O'Sullivan, RSC Adv., 2013, 3, 25498–25522 RSC.
- V. Vinayagam, S. K. Karre, S. R. Kasu, R. Srinath, H. S. Naveen Babu Bathula and S. K. Sadhukhan, Org. Lett., 2022, 24, 6142–6147 CrossRef CAS PubMed.
- Y. Miyahara and Y. N. Ito, J. Org. Chem., 2014, 79, 6801–6807 CrossRef CAS PubMed.
- W.-C. Gao, Y.-F. Cheng, H.-H. Chang, X. Li, W.-L. Wei and P. Yang, J. Org. Chem., 2019, 84, 4312–4317 CrossRef CAS PubMed.
- M. Hong, J. Chen and E. Y. X. Chen, Chem. Rev., 2018, 118, 10551–10616 CrossRef CAS PubMed.
- M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. W. Stephan, Chem, 2020, 6, 1520–1526 CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- L. C. Wilkins, J. R. Lawson, P. Wieneke, F. Rominger, A. S. K. Hashmi, M. M. Hansmann and R. L. Melen, Chem. Eur. J., 2016, 22, 14618–14624 CrossRef CAS PubMed.
- J. Zhang and Z. Xie, Chem. Sci., 2021, 12, 1745–1749 RSC.
- A. Ullah and G.-Q. Chen, Org. Chem. Front., 2022, 9, 4421–4425 RSC.
- J. Cui and T. Wang, Chem. Commun., 2023, 59, 10279–10282 RSC.
- C. Chen, M. Harhausen, R. Liedtke, K. Bussmann, A. Fukazawa, S. Yamaguchi, J. L. Petersen, C. G. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 5992–5996 CrossRef CAS PubMed.
- J. Möbus, G. Kehr, C. G. Daniliuc, C. Mück-Lichtenfeld and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 12366–12369 CrossRef PubMed.
- J. Li, P. Wu, W. Jiang, B. Li, B. Wang, H. Zhu and H. W. Roesky, Angew. Chem., Int. Ed., 2020, 59, 10027–10031 CrossRef CAS PubMed.
- O. Ekkert, G. G. Miera, T. Wiegand, H. Eckert, B. Schirmer, J. L. Petersen, C. G. Daniliuc, R. Fröhlich, S. Grimme, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 2657–2664 RSC.
- T. Saegusa, N. Taka-ishi, M. Takami and Y. Ito, Synth. Commun., 1971, 1, 99–102 CrossRef CAS.
- G. Bez and C.-G. Zhao, Org. Lett., 2003, 5, 4991–4993 CrossRef CAS PubMed.
- S. Yoshioka, M. Oshita, M. Tobisu and N. Chatani, Org. Lett., 2005, 7, 3697–3699 CrossRef CAS PubMed.
- D. S. A. Haneen, W. S. I. Abou-Elmagd and A. S. A. Youssef, Synth. Commun., 2021, 51, 215–233 CrossRef CAS.
- D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed.
- B. Tang, S. Ju, W. Yan, T. Chen, D. W. Stephan and Y. Wu, Chem. Commun., 2024, 60, 12932–12935 RSC.
- Z. Hussain, Y.-a Luo, Y. Wu, Z.-w Qu, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2023, 145, 7101–7106 CrossRef CAS PubMed.
- V. Bedi, D. Mandal, Z. Hussain, S.-M. Chen, Y. Wu, Z.-W. Qu, S. Grimme and D. W. Stephan, Dalton Trans., 2024, 53, 439–443 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and calculation details, spectral data, X-ray data, and CIF files. CCDC 2448214–2448218. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02623g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |