
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Five key concepts linking vacancies, structure, and oxygen evolution reaction activity in cobalt-based electrocatalysts
Kenneth
Crossley
 a,
Thomas J.
Schmidt
ab and
Emiliana
Fabbri
*a
a,
Thomas J.
Schmidt
ab and
Emiliana
Fabbri
*a
aPSI Center for Energy and Environmental Science, 5232 Villigen PSI, Switzerland. E-mail: emiliana.fabbri@psi.ch
bInstitute for Molecular Physical Science, ETH Zürich, CH-8093 Zürich, Switzerland
First published on 16th July 2025
Abstract
Focusing on five key concepts, we review the roles of cation and oxygen vacancies in determining the surface reconstruction pathway, reaction mechanism, and ultimate activity of cobalt-based oxygen evolution reaction (OER) electrocatalysts. Cation and oxygen vacancies can initiate reactant adsorption, facilitating active surface reconstruction, and can switch the dominant mechanism from the adsorbate evolution mechanism (AEM) to the lattice oxygen evolution mechanism (LOEM). However, these effects are facet-dependent. Rigorous oxygen vacancy quantification promises to identify the OER mechanism steering thresholds and unlock the full potential of vacancy engineering. Finally, oxygen vacancy quantification strategies are critically examined to facilitate this goal.
 Kenneth Crossley | Kenneth Crossley previously studied physics (BA, Colorado College) and materials science/chemistry through the SERP+ Erasmus Mundus program (MS, University of Genoa/Université Paris-Saclay). He is currently a doctoral candidate at ETH Zürich focused on next generation oxygen evolution reaction electrocatalysts in the Electrocatalysis and Interfaces group at the Paul Scherrer Institute (PSI). Outside the lab he enjoys fly fishing, rock climbing, drawing, skiing, and cycling. |
 Thomas J. Schmidt | Prof. Thomas J. Schmidt is a Professor and Chair for Electrochemistry, ETH Zürich and Head of the PSI Center for Energy & Environmental Research at PSI. He is also the Director of the Swiss Center of Excellence on Netzero Emissions. He graduated with a PhD in chemistry at the University of Ulm, Germany and then continued his career as a postdoc and a scientist at Lawrence Berkeley National Laboratory and at PSI. He spent eight years in industrial fuel cell research (BASF Fuel Cell) as Director of R&D. Between 2014 and 2021, he also acted a Director of the Swiss Competence Center for Energy Research (SCCER) Heat & Electricity Storage. His work is focused on all aspects of electrochemical energy conversion and storage. |
 Emiliana Fabbri | Dr Emiliana Fabbri received her PhD in Materials Science from the University of Rome Tor Vergata. In 2009 she was appointed tenured scientist at the National Institute for Materials Science (NIMS), Japan. Since January 2012, she has been working at PSI in Switzerland on materials for electrochemical energy storage and conversion. To gain a fundamental understanding of electrochemical reaction mechanisms and catalytic activity descriptors, she is particularly interested in the surface chemistry and electronic structure of catalysts studied by operando X-ray synchrotron techniques. Emiliana Fabbri was awarded an SNSF PRIMA grant in 2020 and since 2024 she is co-leader of the Electrocatalysis and Interfaces group of the Electrochemistry Laboratory of PSI. |
Green hydrogen, produced via water electrolysis, is a promising next-generation energy vector. The oxygen evolution reaction (OER) is the rate limiting half-reaction for water electrolysis. OER electrocatalyst cost and performance are important factors impeding global water electrolysis scale up. In an acidic environment, typical industrial OER electrocatalysts are precious metal-based materials (IrOx, RuOx). In an alkaline environment, less expensive and more abundant transition metal (hydr)oxides based on Fe, Co, and Ni are becoming competitive. Herein we focus on Co-based electrocatalysts, but the conclusions should generally be applicable to other Fe- and Ni-based materials.
The prevailing aspiration to increase the activity of Co-based (hydr)oxides is to selectively switch the OER mechanism. The prototypical adsorbate evolution mechanism (AEM)—exhibiting concerted proton/electron transfer—has a fundamental activity limit imposed by universal adsorption energy scaling relationships.1 Conversely, the lattice oxygen evolution reaction (LOER) breaks these activity limitations via non-concerted proton/electron transfer and involves an O–O coupling step between the surface lattice oxygen and adsorbed oxygen species.2,3 It is suggested that LOER activation requires a covalent M–O electronic structure with O 2p character near the Fermi level.4 The LOER cannot be activated without accompanying dissolution and surface reconstruction under applied potential,5,6 but a dynamic equilibrium can be established to balance activity and durability.7–9
Multiple LOER mechanisms (LOEMs) have been proposed—including intramolecular nucleophilic attack,10 Mars van Krevelen,11 and lattice oxygen mechanism (LOM)4,12,13—all of which dynamically create/fill oxygen vacancies (VO in the Kröger–Vink notation). The oxide path mechanism (OPM) has also been proposed to occur via O–O radical coupling between adjacent metal adsorbate sites when the metal–metal distance is short enough, although it is more common in acidic conditions.14 The AEM, LOEM, and OPM are depicted in Scheme 1. Presently, it remains difficult to experimentally differentiate between, and selectively activate, these non-AEM mechanisms (see Concept 3).
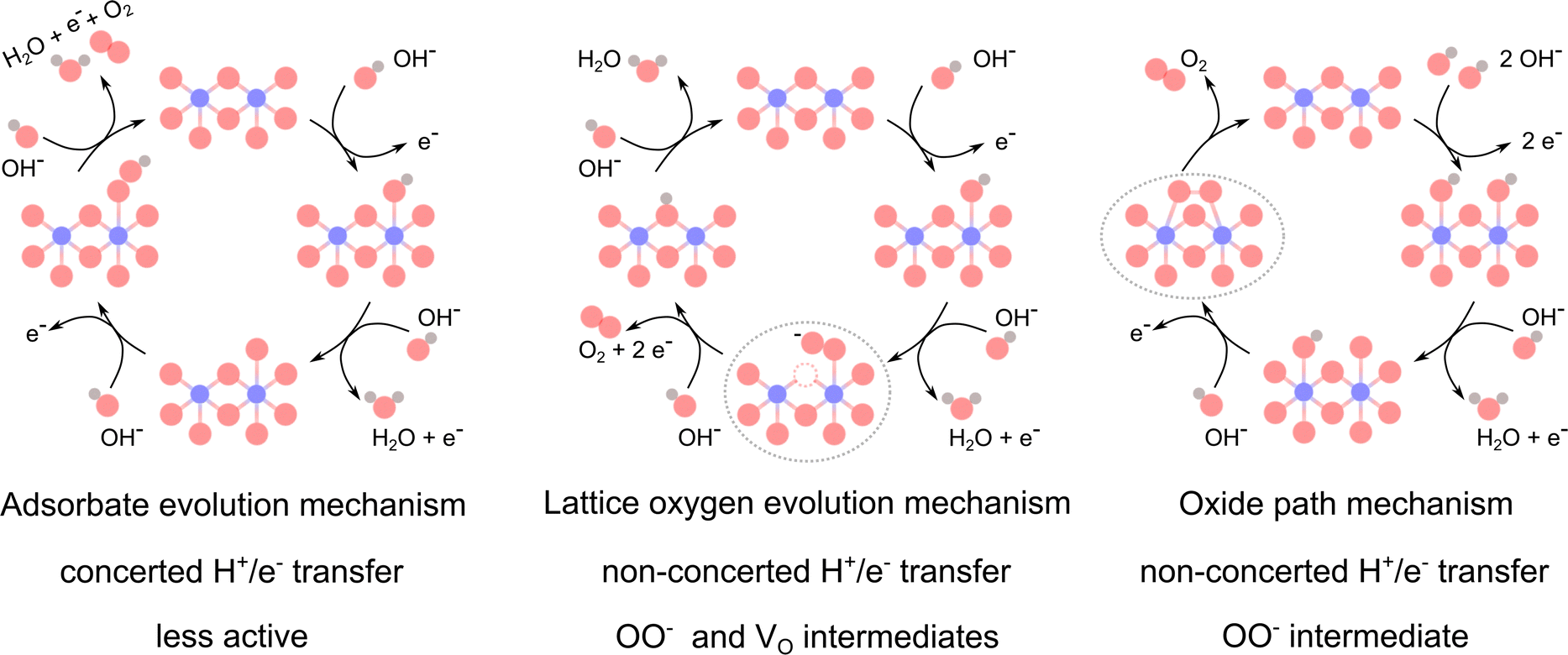 | ||
| Scheme 1 Proposed mechanisms for the alkaline OER. Co atoms are shown in blue, O in red, H in grey, and oxygen vacancies (VO) in dashed red circles. The characteristic steps with superoxide OO− intermediates are indicated with grey dashed ellipses. Scheme inspired by Rong et al.14 | ||
In this review, we highlight recent works which indicate key variables to control surface reconstruction pathways, active site geometry, and selective mechanism activation. We structure these works around five key concepts with a view towards fundamental/mechanistic OER understanding.
1 Vacancies do more than dangle bonds
Recent works on amorphous OER catalysts/amorphous surface layers often attribute increased activity to flexible structure, increased surface area, or dangling bonds.15–17 While these geometric factors certainly contribute, the effects of oxygen vacancies are far more nuanced, can be disentangled far more rigorously, and do not universally improve catalytic activity.The primary effects of oxygen vacancies are modulating the adsorption site density and free energies of adsorption. As clearly investigated and summarized by Tao et al., introducing surface oxygen vacancies to p-type semiconductors shifts the hybridized O 2p–M 3d antibonding states closer to the Fermi level and increases the metal site adsorption energy of O intermediates on pristine precatalysts.18 Note that density functional theory (DFT) calculations of adsorption energies on the actual reconstructed active surfaces remain elusive. Moreover, high bulk oxygen vacancy concentrations can down-shift the O 2p band and reduce the catalytic activity.19 Particularly in double perovskites such as PrBaCo2O6−δ, high oxygen deficiency (δ ≈ 0.5) can lead to bulk ordering of oxygen vacancies, decreasing conductivity, and a detrimental Co3+ high-to-low spin state transition.20 Cheng et al. examined a La1−xSrxCoO3−δ series, a LaMO3−δ series (M = Cr, Mn, Fe, Co, Ni), Ba0.5Sr0.5Co0.8Fe0.2O3−δ and PrBaCo2O6−δ perovskites and found δ > 0.2 to be desirable for OER applications.21
Intuitively, the OH− and H2O species can also adsorb to oxygen vacancies. Oxygen vacancies and OH− adsorption have been linked to initiating surface reconstruction in many Co-based electrocatalysts including CoSn(OH)6,22 CoMoO4/CoWO4/Co2VO4,23 Co(OH)2,24 and Co3O4.25 The roles of these adsorption sites and species in surface reconstruction are examined in more depth in Concept 4.
Cation vacancies can also positively affect the OER activity. Chen et al. used Ar plasma treatment on Co0.9Fe0.1Sn(OH)6−δ to selectively form Sn and O vacancies.26 The cation vacancy selectivity was achieved by a relative difference in M–OH bond strengths between the cations. Their defective material showed increased hydrophilicity, a Co/Fe-rich amorphous surface layer, and enhanced electrochemical activity compared to the untreated parent material. Their DFT results suggest that the selective Sn–O vacancies decreased the coordination number and free energy of O adsorption (ΔG0(O*) = 3.69 vs. 2.49 eV) at the Co sites, leading to a lower Tafel slope (77 vs. 42 mV dec−1) and a shift in the rate determining step compared to the pristine material. Zhang et al. have also demonstrated beneficial Co vacancies in Co3−xO4 derived from glycerolatocobalt(II) pyrolysis.27,28 The Co vacancies were confirmed combining X-ray diffraction (XRD), transmission electron microscopy (TEM) energy dispersive spectroscopy, and positron annihilation lifetime spectroscopy measurements. Their DTF results suggest that the Co vacancies introduce a high density of unoccupied states above the Fermi level and increase electron delocalization. These combined effects yielded a turnover frequency an order of magnitude higher than that of pristine Co3O4.
The extent of dissolution-derived Co vacancies during alkaline OER depends on the precatalyst structure, the presence of other metal ions, the pH, and the applied potential. Moysiadou and Hu used operando electrochemical quartz crystal microbalance and inductively coupled plasma optical emission spectroscopy (ICP-OES) to determine the dissolution rates of amorphous electrodeposited CoOx, CoFeOx, and CoFeNiOx in 1 M KOH at 1.58 V vs. RHE (reversible hydrogen electrode).29 The CoFeNiOx mass remained constant, but CoFeOx and CoOx showed 20–30% mass loss in the first 6 h before equilibrating with trace Fe adsorption from the electrolyte. In contrast, crystalline Co3O4 and CoFe2O4 (111) epitaxial thin films remain stable except for sub-nanometer dissolution and reconstruction at the surface.30,31 Lopez et al. coupled rotating disk electrode and inductively coupled plasma mass spectrometry (ICP-MS) characterizations to evaluate operando dissolution in La1−xSrxCoO3 nanoparticles.32 La1−xSrxCoO3 showed increasing Co dissolution with increasing Sr content, but negligible Co dissolution above 1.5 V vs. RHE in pristine 0.1 M KOH and at all tested potentials when 1 ppm Fe was introduced into the electrolyte. Conversely, CoOOH on Pt showed an increasing rate of Co dissolution in the Fe contaminated electrolyte above 1.4 V vs. RHE and a 3× lower stability factor. Mn incorporation has also been shown to help stabilize Co dissolution.33,34 Overall, it is becoming clear that a dynamic equilibrium must be achieved between the catalyst bulk, reconstructed surface, and transition metal ions in the double layer (see Concept 4).7,8
Vacancies created by introducing sacrificial cations in the structure can also selectively influence the active site population, stability, and geometry. Menezes et al. demonstrated that selective Zn etching from ZnCo2O4 preferentially exposes octahedral Co sites at the reconstructed interface.35 Ca- and Fe-dominated dissolution in brownmillerite-type Ca2FeCoO5 drives the transformation to an amorphous CoOOH structure which is stable for at least 4 weeks under OER conditions.36 Wei et al. investigated a La0.3Sr0.7Co1−xAlxO3−δ material series and concluded that Al3+ dissolution initiated surface reconstruction via oxygen vacancy formation, but that the equilibrium state down-shifted the O 2p band and prevented continued bulk reconstruction.37 Liu et al. found that this dynamic equilibrium of Al3+ dissolution/Al(OH)n− adsorption was responsible for improved activity, stability, and Cl− repulsion in CoFeAl layered double hydroxides (LDHs) exhibiting high sea water electrolysis performance.9
2 The effect of surface oxygen vacancies is facet-dependent
Using NaBH4 reduction of Co3O4 nanoparticles, Chen et al. introduced surface oxygen vacancies to cubic ((001) facets) and truncated octahedron ((111) majority, (001) minority facets) geometries.38 Both types of defective particles outperformed their pristine parent materials, yet, the defective cubic material showed an order of magnitude higher activity and 2–4× greater pH dependence than the pristine cubic and defective octahedral particles. Their DFT results indicate that the upshift of the O 2p band center for the (001) facet reduced the Co 3d and O 2p interband center gap and increased the overlapping O and Co density of unoccupied states just above the Fermi level. There was little change for the (111) facet. Along with operando Raman spectroscopy and tetramethylammonium (TMA) oxygen radical quenching measurements, these findings point to the selective activation of the LOEM on oxygen deficient Co3O4 (001). This AEM to LOEM shift and the relation to surface reconstruction is further discussed in Concepts 3 and 4. Davis et al.39 and Wei et al.40 have shown that the extent of Co3O4 surface reconstruction is also facet-dependent.Considering other Co3O4 facets, Shojaee et al.'s DFT calculations reveal that it is easier to form oxygen vacancies on the Co3O4 (100) facet than the (110) facet.41 Similarly, the Co3O4 (220) facet has been calculated to have an even lower oxygen vacancy formation energy,42 which could be advantageous to exploit for OER.
This facet dependence is not limited to Co3O4. Co(OH)2 derived β-CoOOH hexagonal nanosheets have been shown to preferentially drive OER on the lateral facets, with the larger area (0001) basal planes being mainly inactive.43,44 Similar lateral facet-dominant activity and facet-dependent reconstruction has also been confirmed for NiOOH nanosheet OER catalysts.45 Introducing oxygen vacancies to the lateral (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) facets slightly reduces the rate determining potential barrier (−0.05 V), whereas a reduction of −0.4 V is seen for the (0001) and (100) facets.44 This large barrier reduction indicates that a combined doping/dissolution plus oxygen vacancies engineering strategy may be sufficient to activate the CoOOH/NiOOH basal (0001) plane for OER.
0) facets slightly reduces the rate determining potential barrier (−0.05 V), whereas a reduction of −0.4 V is seen for the (0001) and (100) facets.44 This large barrier reduction indicates that a combined doping/dissolution plus oxygen vacancies engineering strategy may be sufficient to activate the CoOOH/NiOOH basal (0001) plane for OER.
3 The OER mechanism can be controlled via oxygen vacancy concentrations
Differentiating between the multiple OER mechanisms remains a challenge. The LOEM and OPM show pH-dependent activity, the order of which should be determined with a fixed overpotential to avoid mixed kinetic–thermodynamic reaction order changes. The intermediate O2− radical is present in both the LOEM and OPM, which can be detected via TMA quenching,12,38 the 18O/16O kinetic isotope effect,46 or Fourier transform infrared spectroscopy.38,47,48 Labeling catalysts with 18O isotopes along with online mass spectrometry10,19,49 or operando Raman spectroscopy50 can help differentiate between the AEM, LOEM, and OPM. Operando application of one or more of these techniques aids in the mechanistic interpretation of pH-dependent activity.To decouple the multiple effects of introducing oxygen vacancies by aliovalent cation doping, Lu et al. employed ball milling for incremented times to increase the oxygen vacancy concentrations in a series of LaxSr1−xCoO3−δ (LSCO-δ) materials with fixed cation stoichiometries.19 The activity change of this doping + ball milled oxygen vacancies series is reproduced in Fig. 1. The authors clearly demonstrate that increasing the oxygen deficiency can either deactivate the LOEM (high Sr content, Fig. 1(A)) or activate the LOEM/LOM (high La content, Fig. 1(B) and (C)) during CV cycling depending on the Co 3d and O 2p band alignments. In the case of deactivation, the activity-limiting “lockup effect” reflects decreasing participation of the possible OER active sites as the material's cation reducibility limit is approached with increasing oxygen deficiency.6,19 Beyond pH-dependent activity and DFT calculations, their Raman spectroscopy results also demonstrate switchable surface reconstruction pathways associated with CoOOH formation.
 | ||
| Fig. 1 Patterns in OER mechanism shifts for LaxSr1−xCoO3−δ (LSCO-δ). (A) Increasing oxygen deficiency in the high Sr regime deactivates the LOEM. (B) Increasing oxygen deficiency in the mid Sr/La regime activates and then deactivates the LOEM. (C) Increasing oxygen deficiency in the La majority regime activates the LOEM, but the high La range always follows the AEM. Δj is the normalized current density difference from the initial to the maximum activity cycle. For each material, moving left to right increments the ball milling time (0, 2, 4, 6 h). Reproduced from Lu et al. (CC-BY-NC 4.0).19 | ||
LOER activation does not appear to be dependent on the method of oxygen vacancy introduction. When Chen et al. introduced surface oxygen vacancies to their cubic Co3O4 nanoparticles via NaBH4 reduction, they detected increased OER activity and pH dependence compared to the pristine material which followed the AEM.38 Their operando Raman measurements showed peaks at 1095 and 1125 cm−1, which correspond to Co–O–O–Co vibrational modes and indicate an O2− intermediate formed by a LOEM or OPM. In addition, the activity decreased with TMA–O2− quenching. These results indicate the selective activation of an O2− radical-producing mechanism on Co3O4 (001) facets. Similarly, Zhou et al. created surface oxygen vacancies in Co3O4via chemical reduction with increasing concentrations of NaBH4.10 Their electrochemical, kinetic isotope effect, operando Raman spectroscopy, and online mass spectrometry characterizations indicate that the oxygen vacancies activated the LOER and shifted the mechanism to intramolecular nucleophilic attack (metal-adsorbed O attacking adjacent lattice O). However, as discussed in Concept 4, high oxygen vacancy densities can also introduce quenching mechanisms that limit the OER activity.
4 Vacancies can steer the surface reconstruction pathway and ultimate catalyst activity
Universally, metal (hydr)oxide catalysts have thermodynamically unstable interfaces under OER conditions and will self-reconstruct with varying degrees of dissolution.5 Fabbri et al. first applied operando X-ray absorption spectroscopy (XAS) to observe the surface reconstruction of oxygen deficient Ba0.5Sr0.5Co0.8Fe0.2O3−δ to form a (CoFe)Ox(OH)y layer.7 Using operando and ex situ freeze-quenched XAS, Bergmann et al. demonstrated that CoO (in rock salt and wurtzite structure), Co3O4, and CoOOH all reconstruct to a principally octahedrally-coordinated 3D cross-linked CoOx(OH)y surface layer under neutral and alkaline OER conditions.51 They identified reducible di-μ-oxo-bridged Co3+ ions as the reconstructed active site common to all these materials. Despite a common meta-stable active layer, a wide range of activities are observed for Co-based OER electrocatalysts. The reconstruction pathway is key to determining the final activity.There is mounting evidence that Co (hydr)oxide surfaces with oxygen vacancies take different reconstruction pathways compared with their pristine counterparts. Using operando XAS and X-ray photoelectron spectroscopy (XPS), Xiao et al. demonstrated that Ar plasma-derived oxygen vacancies increase the adsorption of OH groups at lower potentials compared to defect-free Co3O4.25 Their defective Co3O4 displayed a lower charge transfer resistance above 1.15 V vs. RHE and a faster rate of oxidation/deprotonation before 1.45 V vs. RHE. Moreover, Alex et al. have shown that crystalline Co3O4 with oxygen vacancies can have higher reconstructed intrinsic activity and outperform nanocrystalline/amorphous Co3O4.52 Despite minimal long range order and 4.7× more surface area, the nanocrystalline catalyst was reported to have fewer surface oxygen vacancies (based on the troublesome O 1s XPS adsorbed OH peak, see Concept 5) and a lagging Tafel slope of 153 mV dec−1. Conversely, Liu et al. reported that amorphous Co(OH)2 nanocages with abundant oxygen vacancies (detected by electron paramagnetic resonance) outperform crystalline Co3O4 and demonstrate faster oxidation/reduction at lower potentials.24 Overall, it is becoming clear that (oxy)hydroxide species adsorbed on or near oxygen vacancies can reconstruct more easily than on fully ordered facets.
Xiao et al. insightfully utilized operando Raman spectroscopy to observe the surface reconstruction of pristine and oxygen deficient CoMoO4, CoWO4, and Co2VO4.23 They showed that oxygen vacancies accelerate Mo/W/V dissolution, thus exposing more oxygen vacancies and Co sites. Oxygen vacancy-adsorbed OH and H2O then formed hydrated amorphous Co(OH)2 within 5 min at 1.15 V vs. RHE. Within 5 min at 1.2 V vs. RHE, the intercalated amorphous Co(OH)2 converted to CoOOH. Intriguingly, when the defective samples were soaked at open circuit potential in 1 M KOH for 60 min, the oxygen vacancies were filled, and CoOx and crystalline Co(OH)2 formed. Neither the pristine materials nor the 60 min soaked samples displayed the Raman shifts associated with either water adsorption or CoOOH formation at 1.2 V vs. RHE. Both had lower OER activities than the fresh defective sample. Such a defect adsorption/H2O intercalation reconstruction mechanism at least partially explains why CoOOH reconstructed from boride/phosphide/sulfide precatalysts often outperforms directly synthesized CoOOH and Co3O4.53–58 Taken together, these results suggest that H2O adsorption and intercalation into the reconstructed Co(OH)2/CoOOH with an applied potential are necessary for high activity.
Akin to the α-/β-Ni(OH)2 system, Leng et al.59 and Sanchis-Gual et al.60 examined β-Co(OH)2, and anion/H2O intercalated α-Co(OH)2 as OER precatalysts. They found that α-Co(OH)2 exhibited the most reconstruction. Leng et al.59 and Dionigi et al.61 further confirmed that β-/α-Co(OH)2 selectively reconstruct to β-/γ-CoOOH, with γ-CoOOH exhibiting the higher OER activity. Recently, Wang et al. stabilized γ-CoOOH in an alkaline electrolyzer and demonstrated 1 at 1.78 V.62 These results further confirm that the reconstructed CoOOH layers OER activity depends on the reconstruction pathway.
K. Fan et al. have proposed different surface reconstruction pathways for CoOOH-like surfaces.63 As shown in Scheme 2, after oxidation of Co2+ species to β-CoOOH species, a bifurcation occurs. A slow deprotonation and water intercalation step leads to γ-CoOOHx, which can then be further oxidized to the OER active site or quenched by dense oxygen vacancy concentrations. A faster deprotonation pathway to β-CoO2 produces sites with lower intrinsic activity.63 Based on the works previously discussed in this section, we propose an additional pathway mediated by oxygen vacancies and intercalated electrolyte species which proceeds directly to γ-CoOOHx or a Co LDH structure.
 | ||
| Scheme 2 The surface reconstruction pathways followed by Co-based OER catalysts. Oxygen is shown in red, hydrogen in cream, and cobalt in blue. α-Co(OH)2 is intercalated by water and anions (i.e. CO32− with C shown in brown), whereas γ-CoOOHx is intercalated by cations (i.e. K+ shown in purple).61 Co3O4,64 β-Co(OH)2,65 β-CoOOHx66 data accessed via the Crystallography Open Database. Note that the more active intermediate form of γ-CoOOHxvs. a Co layered double hydroxide (LDH) structure is not yet fully clear and may be material specific. | ||
In regions with high oxygen vacancy density, there is evidence suggesting an additional reconstruction pathway which quenches the oxidized Co active sites. For example, Zhou et al.'s highest oxygen vacancy concentration sample (1 M NaBH4 treatment) displayed two distinct electronic environments and a lower turnover frequency than the intermediate vacancy concentration sample.10 Fan et al. proposed the following quenching reaction for high oxygen vacancy density environments which increases the number of reconstructed Co2+ spectator sites.63
| Co4+ + Co–VO + H2O → Co2+ + Co–O + 2H+ | (1) |
We have included the Co4+ species by convention, but the quenched species could also be a Co3+ coordinated with a oxygen ligand electron hole in covalent systems. The authors proposed that amorphous materials are more prone to this quenching pathway, which may contribute to an increasing degree of crystallinity in amorphous catalysts during/post OER. However, such a chemical reaction does not explain the third anodic peak (A2 in Scheme 2) observed between the typical Co2+/3+ (A1) and Co3+/4+ (A3) peaks in some amorphous or high oxygen vacancy materials.10,24 An additional electrochemical reaction which could account for this activity quenching in high oxygen vacancy density regions below the Co3+/4+ oxidation potential follows.
 | (2) |
Regardless of the exact species/mechanism(s), note that such quenching reactions are dynamic competing processes during surface reconstruction, whereas the lockup effect originates from a fundamental charge conservation limit.
5 Oxygen vacancy quantification is complex and demands user knowledge
To determine exact oxygen vacancy thresholds to steer the reaction mechanism and surface reconstruction pathways, we need robust quantification strategies to be widely implemented. However, many of these techniques are misused or misunderstood in the interdisciplinary electrocatalysis literature. The advantages and disadvantages of various oxygen vacancy quantification techniques are summarized in Table 1.| Technique | Accuracy | Advantages | Disadvantages |
|---|---|---|---|
| XPS O 1s 531–532 eV | Low | None | Measures hydroxyls, not VO |
| XPS cation oxidation + normalized lattice O 1s + binding energy | Medium | Quantitative treatment possible | Material specific choices/knowledge requirements |
| H2O2/probe redox + colorimetric detection | Medium | Probes surface VO in a liquid environment | Requires standards, unproven hydroxide catalyst applicability |
| EPR spectroscopy | Medium | Directly detects single electrons trapped at VO | Full quantification requires valence state/magnetic characterization |
| X-ray diffraction | Medium | Low sample masses possible | Less sensitive to light elements |
| Neutron diffraction | High | High sensitivity to light elements | Large masses, specialized facilities |
| X-ray absorption spectroscopy | Medium high | Sensitive to bulk VO | Less sensitive to surface VO |
| Thermogravimetry | Medium high | Directly measures mass loss for thermal/gaseous VO creation | Gas species detection (ICP-MS/OES) required for definitive attribution |
| Iodometric titration | Very high | Standard O quantification | Difficult back calculation for multiple, multi-valent transition metals |
5.1 Near surface techniques
The recent trend of using the fitting area ratio of XPS O 1s peaks at approximately 530 eV (lattice O) and 531–532 eV (often misattributed to oxygen vacancies) to quantify oxygen vacancy densities is fundamentally incorrect.67,68 In the case of the O 1s region, XPS measures electrons ejected from core atomic orbitals; thus, there cannot be a signal from a chemical species defined by a missing O atom. Using in situ XPS, Yamamoto et al. have clearly demonstrated that the 531–532 eV features are characteristic of OH groups formed by the dissociation of water on metal or metal oxide surfaces.69 This process will spontaneously fill surface oxygen vacancies upon contact with ambient water vapor or electrolyte.2,39,67 Although there is a correlation with this surface hydroxyl spectral feature and surface reconstruction/subsequent OER activity,10,25 using the hydroxyl XPS O 1s peak method is not a robust measure of oxygen vacancy concentrations because there are multiple surface hydroxyl adsorption sites70 and spectral overlaps with contaminants (i.e. Na/K/Ca/Mg chlorides {Auger}, (bi)carbonates).70,71Despite this recent trend, it is often possible to quantify surface oxygen vacancies in a more precise manner using XPS. Wang et al. advocate a three-fold quantification strategy using cation valence state peak ratios, lattice O 1s peak ratios, and binding energy shifts.68 For the cation valence state peak ratio approach, one must account for/exclude cation protonation and anion redox. When using the lattice O 1s peak approach, one must first normalize the spectra to the baseline or a redox-inactive cation peak intensity. Then the normalized lattice O peak (A(≈530 eV)) fitting area can be compared to a fully oxidized surface using the following equation.68
 | (3) |
If sample charging, band bending, and space-charge layer effects are minimal, the binding energy shift can be a third strategy to quantify oxygen vacancy concentrations. One must understand the sample to select the appropriate method with XPS.
Alternatively, Li et al. have developed a rapid, inexpensive, and quantitative colorimetric method to detect surface oxygen vacancies in oxide catalysts.72,73 As depicted in Fig. 2, this method uses the surface oxygen vacancy sites of the material to catalyze the decomposition of H2O2 in a pH 4 buffer. The liberated hydroxides subsequently deprotonate the amine groups in the 3,3′,5,5′-tetramethylbenzidine probe. This reaction changes the color from clear to turquoise and is quantified with an optical absorbance measurement. Although this method gives an indirect quantification of surface oxygen vacancies, it has the advantages of background subtraction and dynamically probing the vacancy sites accessible for OER in a liquid environment.
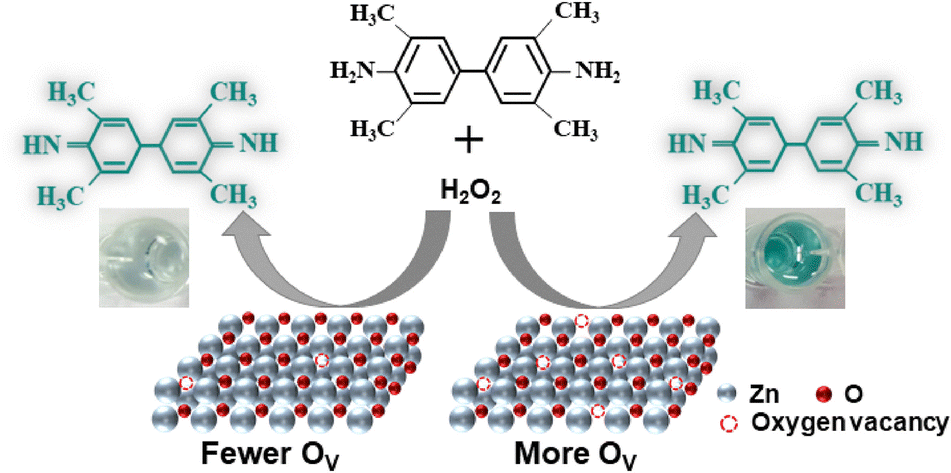 | ||
| Fig. 2 A schematic view of the colorimetric surface oxygen vacancy quantification method demonstrated by Li et al.72 Reprinted with permission from Elsevier, copyright 2025. | ||
5.2 Bulk techniques
Electron paramagnetic resonance (EPR) is also frequently used for semi-quantitative oxygen vacancy characterization. Yet, only single unpaired electrons trapped at oxygen vacancies are directly detected at the common g ≈ 2.002 signal, which represents only a subset of the oxygen vacancy population.74 A rigorous quantification requires analysis of the catalysts valence state (cation reduction) and magnetic structure.74–77Given accurate cation ratio constraints, hard XAS allows bulk oxygen vacancies to be calculated. First, the cation K-shell absorption edge energy is detected. Then, standards of known oxidation state are used to linearly correlate edge energies with the oxidation state. Finally, the oxygen content is determined by charge neutrality of the cation oxidation state. Fitting neutron diffraction data can give more precise information on oxygen vacancies; however, large amounts (≈4 g) and highly crystalline samples are usually required.78 XRD using a synchrotron source can be used for smaller masses, yet the technique is inherently less sensitive than neutron diffraction to light elements and their disorder.
Thermogravimetric analysis (TGA) can be used to directly measure the mass loss associated with the formation of oxygen vacancies; however, one should know the exact off-gassing species (H2O, CO2, O2) to make an accurate back calculation of the O content and origin. TGA coupled with in-line ICP-MS or ICP-OES can simultaneously detect the exhaust gas composition. Our experience suggests that a high purity sample is required to avoid ambiguous signals from incorporated/adsorbed solvents and trace byproducts.
Iodometric titration can give precise oxygen stoichiometry results, but the sample should be a single phase and ideally contain only one metal ion with changing oxidation state for accurate back calculation.78,79 A thorough understanding of the valence states of multimetallic oxides, especially Co and Fe, should be obtained with a complementary technique to avoid ambiguous titration results. Selective complexation can help isolate different ions in some cases.80
6 Summary and future perspectives
In Concept 1, we clarify that the effects of oxygen vacancies include shifting the electronic band alignments, adsorbing reaction intermediates, and initiating surface reconstruction. Cation vacancies can also affect the adsorption energies and surface reconstruction pathway via relative dissolution rates. A dynamic equilibrium between catalyst bulk, surface, and double layer is required for durability. In Concept 2, we establish that the effects of oxygen vacancies on a material's electronic properties and surface reconstruction are facet dependent. Combining doping and selective vacancy strategies is promising for activating CoOOH/NiOOH OER active surfaces. In Concept 3, we show that the precise introduction of oxygen vacancies can selectively change the dominant reaction coordinate of the OER between the AEM and the more active LOM. In Concept 4, we summarize and examine recent operando works on possible surface reconstruction pathways in Co-based OER catalysts. Oxygen vacancy-mediated adsorption of OH− and H2O at low applied potentials is key for a highly active reconstructed CoOOH surface. However, high oxygen vacancy densities can initiate chemical or electrochemical active site quenching. In Concept 5, we highlight a remarkably common oxygen vacancy quantification XPS error (531 eV O 1s) and critically examine more accurate quantification techniques.There is mounting evidence that oxygen vacancies, formed both via precatalyst modification methods and in situ metal dissolution, can determine the dominant surface reconstruction pathway and ultimate electrocatalyst activity. Fully understanding and influencing the surface reconstruction pathway of OER catalysts promises to increase the reconstructed active site density and activity. By continuing to combine operando surface characterization with rigorous oxygen vacancy quantification techniques, we expect that the vacancy thresholds for selectively steering the reaction mechanism and reconstruction pathway will be understood in the near future. Future work should focus on systematically introducing—and thoroughly quantifying— selected oxygen vacancy densities in a range of materials to observe trends in the mechanism steering thresholds. Selectively activating the LOER will enable escape from the universal scaling relationships limiting the activity of catalysts following the AEM.
Author contributions
KC: conceptualization, investigation, visualization, writing – original draft; TJS: supervision, writing – review & editing; EF: conceptualization, funding acquisition, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Acknowledgements
KC and EF gratefully acknowledge the Swiss National Science Foundation through its PRIMA grant (grant no. PR00P2_193111). KC thanks Mikhail Agrachev, Albert Schuler, and Ekaterina Pomjakushina for clarifying discussions about EPR, TGA-ICP-OES, and iodometric titration respectively.References
- I. C. Man, H. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS
.
- J. O. Bockris and T. Otagawa, J. Phys. Chem., 1983, 87, 2960–2971 CrossRef CAS
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628–4636 CrossRef CAS
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed
- T. Binninger, R. Mohamed, K. Waltar, E. Fabbri, P. Levecque, R. Koetz and T. J. Schmidt, Sci. Rep., 2015, 5, 12167 CrossRef CAS PubMed
- X. Rong, J. Parolin and A. M. Kolpak, ACS Catal., 2016, 6, 1153–1158 CrossRef CAS
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schaublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925 CrossRef CAS PubMed
- A. E. Thorarinsdottir, S. S. Veroneau and D. G. Nocera, Nat. Commun., 2022, 13, 1243 CrossRef CAS PubMed
- W. Liu, J. Yu, T. Li, S. Li, B. Ding, X. Guo, A. Cao, Q. Sha, D. Zhou, Y. Kuang and X. Sun, Nat. Commun., 2024, 15, 4712 CrossRef CAS PubMed
- D. Zhou, F. Li, Y. Zhao, L. Wang, H. Zou, Y. Shan, J. Fu, Y. Ding, L. Duan, M. Liu, L. Sun and K. Fan, ACS Catal., 2023, 13, 4398–4408 CrossRef CAS
- J. Ferreira de Araujo, F. Dionigi, T. Merzdorf, H. S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14981–14988 CrossRef CAS PubMed
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS
- A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed
- C. Rong, X. Huang, H. Arandiyan, Z. Shao, Y. Wang and Y. Chen, Adv. Mater., 2025, 37, 2416362 CrossRef CAS PubMed
- L. G. Li, Q. Shao and X. Q. Huang, Chem. – Eur. J., 2020, 26, 3943–3960 CrossRef CAS PubMed
- S. Anantharaj and S. Noda, Small, 2020, 16, 24 CrossRef PubMed
- T. M. Pham, M. Plevova, S. Bartling, N. Rockstroh, A. Springer, A. Slabon, J. Hnat, A.-E. Surkus and R. Francke, J. Catal., 2024, 438, 115675 CrossRef CAS
- H. B. Tao, L. Fang, J. Chen, H. B. Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed
- M. Lu, Y. Zheng, Y. Hu, B. Huang, D. Ji, M. Sun, J. Li, Y. Peng, R. Si, P. Xi and C.-H. Yan, Sci. Adv., 2022, 8, eabq3563 CrossRef CAS PubMed
- X. Miao, L. Wu, Y. Lin, X. Yuan, J. Zhao, W. Yan, S. Zhou and L. Shi, Chem. Commun., 2019, 55, 1442–1445 RSC
- X. Cheng, E. Fabbri, Y. Yamashita, I. E. Castelli, B. Kim, M. Uchida, R. Haumont, I. Puente-Orench and T. J. Schmidt, ACS Catal., 2018, 8, 9567–9578 CrossRef CAS
- F. Song, K. Schenk and X. Hu, Energy Environ. Sci., 2016, 9, 473–477 RSC
- H. Xiao, H. Yin, X. Li, X. Zhou, W. Zhang, F. Pan, J. Zhao, J. Guo, S. Li and L. Qian, Sustainable Mater. Technol., 2024, 40, e00955 CrossRef CAS
- J. Liu, J. Nai, T. You, P. An, J. Zhang, G. Ma, X. Niu, C. Liang, S. Yang and L. Guo, Small, 2018, 14, 1703514 CrossRef PubMed
- Z. Xiao, Y.-C. Huang, C.-L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS PubMed
- D. Chen, M. Qiao, Y.-R. Lu, L. Hao, D. Liu, C.-L. Dong, Y. Li and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 8691–8696 CrossRef CAS PubMed
- R. Zhang, Y.-C. Zhang, L. Pan, G.-Q. Shen, N. Mahmood, Y.-H. Ma, Y. Shi, W. Jia, L. Wang, X. Zhang, W. Xu and J.-J. Zou, ACS Catal., 2018, 8, 3803–3811 CrossRef CAS
- R. Zhang, L. Pan, B. Guo, Z.-F. Huang, Z. Chen, L. Wang, X. Zhang, Z. Guo, W. Xu, K. P. Loh and J.-J. Zou, J. Am. Chem. Soc., 2023, 2271–2281 CrossRef CAS PubMed
- A. Moysiadou and X. Hu, J. Mater. Chem. A, 2019, 7, 25865–25877 RSC
- C. Qiu, F. Maroun, M. Bouvier, I. Pacheco, P. Allongue, T. Wiegmann, C. Hendric Scharf, V. de Manuel-Gonzalez, F. Reikowski, J. Stettner and O. M. Magnussen, ChemCatChem, 2024, 16, e202400988 CrossRef CAS
- E. M. Davis, A. Bergmann, C. Zhan, H. Kuhlenbeck and B. R. Cuenya, Nat. Commun., 2023, 14, 4791 CrossRef CAS PubMed
- P. P. Lopes, D. Y. Chung, X. Rui, H. Zheng, H. He, P. Farinazzo Bergamo Dias Martins, D. Strmcnik, V. R. Stamenkovic, P. Zapol, J. F. Mitchell, R. F. Klie and N. M. Markovic, J. Am. Chem. Soc., 2021, 143, 2741–2750 CrossRef CAS PubMed
- H. He, T. Matsuda, A. Miura, M. Nagao, J. K. Padarti, T. Ohno and S. Hirai, Sustainable Energy Fuels, 2024, 8, 789–796 RSC
- B. He, P. Hosseini, D. Escalera-López, J. Schulwitz, O. Rüdiger, U. Hagemann, M. Heidelmann, S. DeBeer, M. Muhler, S. Cherevko, K. Tschulik and T. Li, Adv. Energy Mater., 2025, 15, 2403096 CrossRef CAS
- P. W. Menezes, A. Indra, A. Bergmann, P. Chernev, C. Walter, H. Dau, P. Strasser and M. Driess, J. Mater. Chem. A, 2016, 4, 10014–10022 RSC
- Y. Sato, Y. Aoki, K. Takase, H. Kiuchi, D. Kowalski and H. Habazaki, ACS Appl. Energy Mater., 2020, 3, 5269–5276 CrossRef CAS
- Y. Wei, Y. Hu, P. Da, Z. Weng, P. Xi and C.-H. Yan, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2312224120 CrossRef CAS PubMed
- X. Chen, X. Xu, C. Shao, Z. Ke, Y. Cheng, H. Jin, Y. Da, D. Liu and W. Chen, ACS Energy Lett., 2024, 9, 2182–2192 CrossRef CAS
- E. M. Davis, A. Bergmann, H. Kuhlenbeck and B. Roldan Cuenya, J. Am. Chem. Soc., 2024, 146, 13770–13782 CrossRef CAS PubMed
- Y. Wei, Z. Zhang, C. Mei, J. Tan, Z. Wang, J. Li and L. Gan, Chem. Mater., 2023, 35, 4461–4470 CrossRef CAS
- K. Shojaee, A. Montoya and B. S. Haynes, Comput. Mater. Sci., 2013, 72, 15–25 CrossRef CAS
- H. Zhu, X. Song, X. Han, X. Zhang, J. Bao, N. Zhang and G. He, Environ. Sci. Technol., 2020, 54, 8601–8611 CrossRef CAS PubMed
- J. T. Mefford, A. R. Akbashev, M. Kang, C. L. Bentley, W. E. Gent, H. D. Deng, D. H. Alsem, Y.-S. Yu, N. J. Salmon, D. A. Shapiro, P. R. Unwin and W. C. Chueh, Nature, 2021, 593, 67–73 CrossRef CAS PubMed
- S. Wang, Q. Jiang, S. Ju, C.-S. Hsu, H. M. Chen, D. Zhang and F. Song, Nat. Commun., 2022, 13, 6650 CrossRef CAS PubMed
- Y. Yao, G. Zhao, X. Guo, P. Xiong, Z. Xu, L. Zhang, C. Chen, C. Xu, T.-S. Wu, Y.-L. Soo, Z. Cui, M. M.-J. Li and Y. Zhu, J. Am. Chem. Soc., 2024, 146, 15219–15229 CrossRef CAS PubMed
- S. Haschke, M. Mader, S. Schlicht, A. M. Roberts, A. M. Angeles-Boza, J. A. C. Barth and J. Bachmann, Nat. Commun., 2018, 9, 4565 CrossRef PubMed
- H. Han and H. Frei, J. Phys. Chem. C, 2008, 112, 16156–16159 CrossRef CAS
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed
- J. F. de Araffljo, F. Dionigi, T. Merzdorf, H.-S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14981–14988 CrossRef PubMed
- S. Lee, K. Banjac, M. Lingenfelder and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 10295–10299 CrossRef CAS PubMed
- A. Bergmann, T. E. Jones, E. Martinez Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Nat. Catal., 2018, 1, 711–719 CrossRef CAS
- C. Alex, S. C. Sarma, S. C. Peter and N. S. John, ACS Appl. Energy Mater., 2020, 3, 5439–5447 CrossRef CAS
- J. N. Hausmann, S. Mebs, H. Dau, M. Driess and P. W. Menezes, Adv. Mater., 2022, 34, 2207494 CrossRef CAS PubMed
- C. L. Farrow, D. K. Bediako, Y. Surendranath, D. G. Nocera and S. J. L. Billinge, J. Am. Chem. Soc., 2013, 135, 6403–6406 CrossRef CAS PubMed
- G. Kwon, H. Jang, J.-S. Lee, A. Mane, D. J. Mandia, S. R. Soltau, L. M. Utschig, A. B. F. Martinson, D. M. Tiede, H. Kim and J. Kim, J. Am. Chem. Soc., 2018, 140, 10710–10720 CrossRef PubMed
- G. Chen, Z. W. Hu, Y. P. Zhu, B. B. Gu, Y. J. Zhong, H. J. Lin, C. T. Chen, W. Zhou and Z. P. Shao, Adv. Mater., 2018, 30, 8 Search PubMed
- X. Ma, W. Zhang, Y. Deng, C. Zhong, W. Hu and X. Han, Nanoscale, 2018, 10, 4816–4824 RSC
- Y. Pan, H. Ren, H. Du, F. Cao, Y. Jiang, H. Du and D. Chu, J. Mater. Chem. A, 2018, 6, 22497–22502 RSC
- X. Leng, K.-H. Wu, Q. Zeng, I. R. Gentle and D.-W. Wang, Asia-Pac. J. Chem. Eng., 2016, 11, 415–423 CrossRef CAS
- R. Sanchis-Gual, D. Hunt, C. Jaramillo-Hernández, A. Seijas-Da Silva, M. Mizrahi, C. Marini, V. Oestreicher and G. Abellán, ACS Catal., 2023, 13, 10351–10363 CrossRef CAS PubMed
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. Araujo, M. Gliech, D. Teschner, J. Zhu, W. X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed
- M. Wang, Z. Wang, Y. Zhang, Y. Shi, T.-S. Chan, S.-C. Haw, J. Wang, H. Wang, S. Wang, H. Fei, R. Liu, T. Liu, C.-F. Yan and J. Wang, ACS Energy Lett., 2024, 9, 5502–5508 CrossRef CAS
- K. Fan, D. Zhou, H. Yang, L. Wang, Y. Shan, M. Wan, A. Zheng and L. Sun, ACS Catal., 2025, 3256–3266 CrossRef CAS
- X. Liu and C. T. Prewitt, Phys. Chem. Miner., 1990, 17, 168–172 CrossRef CAS
- Q. Zhao and H. J. Kulik, J. Chem. Theory Comput., 2018, 14, 670–683 CrossRef CAS PubMed
- M. Deliens and H. Goethals, Mineral. Mag., 1973, 39, 152–157 CrossRef CAS
- H. Idriss, Surf. Sci., 2021, 712, 121894 CrossRef CAS
- J. Wang, D. N. Mueller and E. J. Crumlin, J. Eur. Ceram. Soc., 2024, 44, 116709 CrossRef CAS
- S. Yamamoto, H. Bluhm, K. Andersson, G. Ketteler, H. Ogasawara, M. Salmeron and A. Nilsson, J. Phys.: Condens. Matter, 2008, 20, 184025 CrossRef
- A. Boucly, L. Artiglia, E. Fabbri, D. Palagin, D. Aegerter, D. Pergolesi, Z. Novotny, N. Comini, J. T. Diulus, T. Huthwelker, M. Ammann and T. J. Schmidt, J. Mater. Chem. A, 2022, 10, 2434–2444 RSC
- B. V. Crist, The International XPS Database: Oxygen (O), 2025, https://xpsdatabase.net/oxygen-o-z8/.
- M. Li, L. Zhang, W. Liu, Y. Jin and B. Li, Talanta, 2025, 282, 126969 CrossRef CAS PubMed
- M. Li, L. Zhang, W. Liu, Y. Jin and B. Li, Anal. Chem., 2024, 96, 8999–9006 CrossRef CAS PubMed
-
E. Giamello, M. Chiesa and M. C. Paganini, in Point Defects in Electron Paramagnetic Resonance, ed. J. Jupille and G. Thornton, Springer International Publishing, Cham, 2015, pp. 303–326 Search PubMed
- K. Dyrek and M. Che, Chem. Rev., 1997, 97, 305–332 CrossRef CAS PubMed
- R.-A. Eichel, Phys. Chem. Chem. Phys., 2011, 13, 368–384 RSC
- T. Pinheiro Araújo, C. Mondelli, M. Agrachev, T. Zou, P. O. Willi, K. M. Engel, R. N. Grass, W. J. Stark, O. V. Safonova, G. Jeschke, S. Mitchell and J. Pérez-Ramírez, Nat. Commun., 2022, 13, 5610 CrossRef PubMed
- E. Marelli, J. Gazquez, E. Poghosyan, E. Mueller, D. J. Gawryluk, E. Pomjakushina, D. Sheptyakov, C. Piamonteze, D. Aegerter, T. J. Schmidt, M. Medarde and E. Fabbri, Angew. Chem., Int. Ed., 2021, 60, 14609–14619 CrossRef CAS PubMed
- K. Conder, E. Pomjakushina, A. Soldatov and E. Mitberg, Mater. Res. Bull., 2005, 40, 257–263 CrossRef CAS
- W. M. Chen, W. Hong, J. F. Geng, X. S. Wu, W. Ji, L. Y. Li, L. Qui and X. Jin, Phys. C, 1996, 270, 349–353 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |