
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of ultra-small octahedral PtNiCo skeletons with high activity for the oxygen reduction reaction†
Minseok Ko‡
ad,
Mrinal Kanti Kabiraz‡ b,
Yuseong Heoc,
Daehee Yangc,
Yeongseop Leed,
Sang Hoon Jood,
Young-Min Kimc,
Sang-Il Choi*b and
Won Seok Seo*a
b,
Yuseong Heoc,
Daehee Yangc,
Yeongseop Leed,
Sang Hoon Jood,
Young-Min Kimc,
Sang-Il Choi*b and
Won Seok Seo*a
aDepartment of Chemistry and Institute of Biological Interfaces, Sogang University, Seoul 04107, Korea. E-mail: wsseo@sogang.ac.kr
bDepartment of Chemistry and Green-Nano Materials Research Centre, Kyungpook National University, Daegu 41566, Korea. E-mail: sichoi@knu.ac.kr
cDepartment of Energy Science, Sungkyunkwan University, Suwon 16419, Korea
dDepartment of Chemistry, Seoul National University, Seoul 08826, Korea
First published on 8th July 2025
Abstract
Here, we report the synthesis of a ternary PtNiCo octahedral skeletal catalyst for the oxygen reduction reaction (ORR). A mixed flow of H2 and CO facilitated complete reduction, promoted Pt diffusion, and enabled the formation of octahedral PtNiCo nanoparticles (NPs). The resulting ternary skeletal catalyst, obtained after acid treatment, exhibited a mass activity (MA) of 1.64 A mgPt−1 and a specific activity (SA) of 3.73 mA cm−2, which are 7.5 and 14.3 times higher, respectively, than those of commercial Pt/C.
The development of clean energy technologies is imperative due to concerns over depletion of fossil fuels and the worsening environmental pollution. Among various proposed alternatives, polymer electrolyte membrane fuel cells (PEMFCs) have emerged as particularly promising candidates.1–4 Fuel cells, which generate electricity without emitting greenhouse gases, have the theoretical potential to replace fossil fuels. However, to realize their practical application, it is essential to facilitate fast and efficient electrochemical reactions at both the anode and cathode. In particular, the ORR that occurs at the cathode of PEMFCs suffers from sluggish reaction kinetics, necessitating the use of catalysts.5,6 Platinum (Pt) is widely recognized as the most efficient ORR catalyst. Nevertheless, the high cost and scarcity of Pt limit the economic feasibility of commercial PEMFC systems.7–9 Furthermore, Pt-based catalysts are prone to agglomeration during long-term operation, resulting in a gradual loss of catalytic activity.10–12 To overcome these challenges, extensive efforts have been made to enhance the catalytic performance and durability of Pt-based catalysts. Various strategies have been adopted, including the design of hollow, core–shell, and alloy nanostructures, as well as morphology control to Pt-based NPs.13–16 Among them, bimetallic PtNi NPs with highly exposed (111) facets have demonstrated excellent ORR selectivity.17–21 For example, Zhang et al. introduced a CO atmosphere during the liquid-phase synthesis of PtNi NPs, which suppressed the growth of the (111) planes and enabled the formation of octahedral NPs.22 This shape control was attributed to the interaction between CO and Ni, which retarded the growth of specific crystal facets.23 These octahedral NPs exhibited substantially higher ORR activity compared to their cubic counterparts. Similarly, Oh et al. synthesized PtNi NPs using a liquid-phase method under a CO atmosphere and significantly reduced the Pt content. During the reaction, Pt atoms initially located at the center of the particles migrated directionally toward the edges and corners of the octahedra.24–26 More recently, the introduction of a third metal to form ternary NPs has been actively explored to further enhance the ORR activity. Zhao et al. successfully synthesized PtNiCo NPs and employed them as ORR catalysts.27 As a result, the MA and SA achieved were 14.7 and 20.2 times higher, respectively, than those of commercial Pt/C. Hu et al. synthesized PtCuCo NPs and confirmed that they exhibited improved ORR activity compared to their bimetallic counterparts, PtCu and PtCo.28 The introduction of a third metal further shortened the average distance between Pt atoms, inducing a compressive strain.29–31 A decrease in the Pt–Pt bond length promotes more frequent overlap of Pt 5d orbitals, which leads to a downshift in the d-band center relative to the Fermi level.32–34 Consequently, the bonding strength between surface Pt atoms and oxygen-containing intermediates is optimized, thereby enhancing the ORR activity.
In this study, octahedral binary and ternary skeletal NPs were synthesized via gas-phase reduction followed by acid etching. During the acid treatment step, non-noble 3d transition metals were selectively etched, exposing a robust Pt-based skeletal structure (Scheme 1). This result demonstrates that nanoscale phase segregation in binary and ternary systems can occur not only via solution-phase synthesis but also through gas-phase reactions. The synthesized ternary PtNiCo NPs exhibited significantly higher MA and SA for the ORR compared to bimetallic PtNi, PtCo, and commercial Pt/C catalysts. Accelerated durability tests (ADT), along with single-cell evaluations confirmed the superior catalytic stability and applicability of the ternary skeleton.
 | ||
| Scheme 1 Schematic illustration of the PtNiCo skeleton synthesis. Metal precursors supported on fumed silica undergo thermal treatment in a H2/CO atmosphere, forming octahedral NPs. Subsequent HF etching removes silica and selectively leaches Ni and Co, forming an octahedral skeleton. | ||
To synthesize octahedral PtNiCo NPs, metal precursors were impregnated onto fumed silica, followed by thermal annealing in a H2/CO atmosphere (PtNiCo@fumed). The resulting octahedral morphology was confirmed via transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. S1a and b, ESI†). Energy-dispersive X-ray spectroscopy (EDS) elemental mapping indicated a uniform atomic distribution within the NPs, with a Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni:Co atomic ratio of 17:60:23 (Fig. S1c–e, ESI†). For comparison, PtNi and PtCo NPs were also synthesized. The octahedral structure of PtNi NPs synthesized on fumed silica (PtNi@fumed) was verified by TEM and HAADF-STEM (Fig. S2a and b, ESI†), and EDS elemental mapping confirmed a uniform atomic distribution (Fig. S2c and d, ESI†), with a Pt:Ni atomic ratio of 5:95. In contrast, PtCo NPs synthesized on fumed silica (PtCo@fumed) exhibited a spherical morphology, as observed in TEM and HAADF-STEM images (Fig. S3a and b, ESI†). EDS analysis revealed a Pt:Co atomic ratio of 12:88. Unlike the Ni-containing samples, PtCo@fumed showed insufficient interaction with CO, hindering the formation of an octahedral structure. Furthermore, due to the difference in decomposition temperatures between Pt and Co precursors, a homogeneous alloy phase was not formed (Fig. S3c and d, ESI†).35 The NPs synthesized on silica were subsequently transferred onto a carbon support via acid treatment. This process not only facilitated the migration of NPs onto Vulcan carbon but also selectively etched non-noble metals. TEM and HAADF-STEM images confirmed the formation of octahedral PtNiCo skeletons on carbon (PtNiCo/C) (Fig. 1a and b). The average edge length of the skeletons was 9.1 ± 1.9 nm. EDS elemental mapping verified the uniform distribution of Pt, Ni, and Co within the nanocrystals (Fig. 1c–e), and the Pt:Ni:Co atomic ratio was determined to be 79:19:2, indicating significant removal of Ni and Co during acid etching. High-resolution TEM (HRTEM) analysis revealed that the (111) facet of PtNiCo exhibited a d-spacing of 0.23 nm (Fig. 1f). X-ray photoelectron spectroscopy (XPS) showed a lower fraction of oxidized Pt species in PtNiCo/C compared to commercial Pt/C (Fig. S4, ESI†). The same acid treatment process was applied to PtNi and PtCo NPs. TEM and HAADF-STEM images confirmed the formation of octahedral PtNi skeletons on carbon (PtNi/C) (Fig. 1g and Fig. S5a, ESI†), with EDS mapping revealing a Pt:Ni ratio of 70:30 (Fig. S5b and c, ESI†). Similarly, PtCo/C samples displayed a spherical morphology in TEM and HAADF-STEM images (Fig. 1h and Fig. S6a, ESI†), and EDS mapping showed a Pt:Co ratio of 71:29 (Fig. S6b and c, ESI†). These EDS results are highly consistent with the ICP-OES data (Table S1, ESI†). These observations suggest that sufficient directional diffusion of Pt atoms is essential for the formation of skeleton-structured NPs after acid treatment. To further confirm the etching of Ni and Co atoms, X-ray diffraction (XRD) patterns were acquired for both PtNiCo@fumed and PtNiCo/C (Fig. 1i). Prior to acid treatment, the main diffraction peak appeared near those of Ni and Co, indicating that the as-synthesized ternary alloy contained only a small fraction of Pt. After acid treatment, the main peak shifted significantly to a lower angle, indicating an increased Pt content. The XRD pattern displayed a face-centered cubic (fcc) crystal structure, consistent with the selected area electron diffraction (SAED)pattern of PtNiCo/C shown in the inset of Fig. 1a. A similar peak shift was also observed in the PtNi and PtCo binary alloys.
:Ni:Co atomic ratio of 17:60:23 (Fig. S1c–e, ESI†). For comparison, PtNi and PtCo NPs were also synthesized. The octahedral structure of PtNi NPs synthesized on fumed silica (PtNi@fumed) was verified by TEM and HAADF-STEM (Fig. S2a and b, ESI†), and EDS elemental mapping confirmed a uniform atomic distribution (Fig. S2c and d, ESI†), with a Pt:Ni atomic ratio of 5:95. In contrast, PtCo NPs synthesized on fumed silica (PtCo@fumed) exhibited a spherical morphology, as observed in TEM and HAADF-STEM images (Fig. S3a and b, ESI†). EDS analysis revealed a Pt:Co atomic ratio of 12:88. Unlike the Ni-containing samples, PtCo@fumed showed insufficient interaction with CO, hindering the formation of an octahedral structure. Furthermore, due to the difference in decomposition temperatures between Pt and Co precursors, a homogeneous alloy phase was not formed (Fig. S3c and d, ESI†).35 The NPs synthesized on silica were subsequently transferred onto a carbon support via acid treatment. This process not only facilitated the migration of NPs onto Vulcan carbon but also selectively etched non-noble metals. TEM and HAADF-STEM images confirmed the formation of octahedral PtNiCo skeletons on carbon (PtNiCo/C) (Fig. 1a and b). The average edge length of the skeletons was 9.1 ± 1.9 nm. EDS elemental mapping verified the uniform distribution of Pt, Ni, and Co within the nanocrystals (Fig. 1c–e), and the Pt:Ni:Co atomic ratio was determined to be 79:19:2, indicating significant removal of Ni and Co during acid etching. High-resolution TEM (HRTEM) analysis revealed that the (111) facet of PtNiCo exhibited a d-spacing of 0.23 nm (Fig. 1f). X-ray photoelectron spectroscopy (XPS) showed a lower fraction of oxidized Pt species in PtNiCo/C compared to commercial Pt/C (Fig. S4, ESI†). The same acid treatment process was applied to PtNi and PtCo NPs. TEM and HAADF-STEM images confirmed the formation of octahedral PtNi skeletons on carbon (PtNi/C) (Fig. 1g and Fig. S5a, ESI†), with EDS mapping revealing a Pt:Ni ratio of 70:30 (Fig. S5b and c, ESI†). Similarly, PtCo/C samples displayed a spherical morphology in TEM and HAADF-STEM images (Fig. 1h and Fig. S6a, ESI†), and EDS mapping showed a Pt:Co ratio of 71:29 (Fig. S6b and c, ESI†). These EDS results are highly consistent with the ICP-OES data (Table S1, ESI†). These observations suggest that sufficient directional diffusion of Pt atoms is essential for the formation of skeleton-structured NPs after acid treatment. To further confirm the etching of Ni and Co atoms, X-ray diffraction (XRD) patterns were acquired for both PtNiCo@fumed and PtNiCo/C (Fig. 1i). Prior to acid treatment, the main diffraction peak appeared near those of Ni and Co, indicating that the as-synthesized ternary alloy contained only a small fraction of Pt. After acid treatment, the main peak shifted significantly to a lower angle, indicating an increased Pt content. The XRD pattern displayed a face-centered cubic (fcc) crystal structure, consistent with the selected area electron diffraction (SAED)pattern of PtNiCo/C shown in the inset of Fig. 1a. A similar peak shift was also observed in the PtNi and PtCo binary alloys.
 | ||
| Fig. 1 Characterization of the synthesized NPs. (a) TEM image, with the inset displaying the corresponding SAED patterns. (b) STEM image and (c)–(e) EDS elemental maps of Pt, Ni and Co. (f) HRTEM image of PtNiCo/C. (g) and (h) TEM images of PtNi/C and PtCo/C. (i) XRD patterns of the synthesized NPs. | ||
The electrochemical ORR performance of as obtained PtNiCo/C was evaluated in a three-electrode electrochemical system and compared with the state-of-the-art commercial Pt/C (20 wt%). Initially, all the catalysts underwent electrochemical treatment in an Ar-purged 0.1 M HClO4 solution at a scan rate of 50 mV s−1, and CVs were obtained (Fig. 2a). The Pt–O reduction peak positions obtained from CVs, within the range of 0.7–0.9 V vs. reversible hydrogen electrode (RHE), revealed that both skeletons exhibited a positive shift compared to Pt/C. This shift indicates weaker binding strength, easier removal of adsorbed oxygen species from the catalyst surface, and improved ORR performance. The electrochemical surface area (ECSA) was determined from the hydrogen underpotential (Hupd) region in the CV curves for all catalysts (Table S2, ESI†). The measured ECSA values were 85.10, 43.82, and 43.91 m2 gPt−1 for Pt/C, PtNi/C, and PtNiCo/C, respectively.
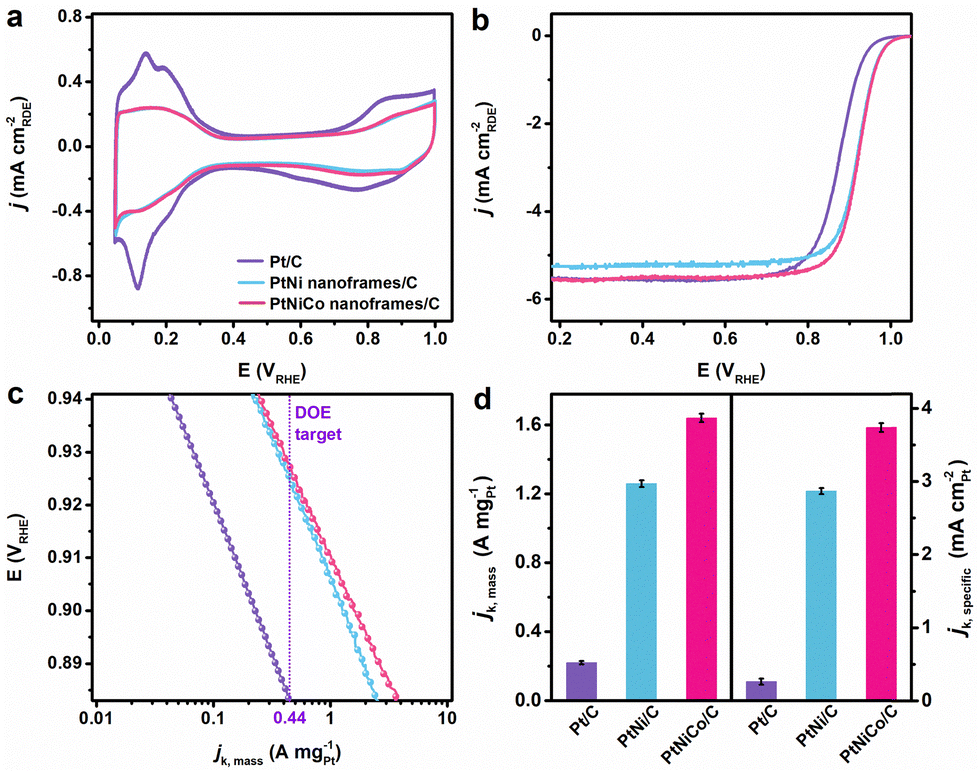 | ||
| Fig. 2 Electrochemical properties of the PtNiCo/C, PtNi/C, and Pt/C catalysts. (a) Comparison of CVs of the catalysts before ORR measurements. (b) ORR polarization curves for the corresponding catalysts in an O2-saturated 0.1 M HClO4 solution. (c) Tafel plots of mass activities given as kinetic current densities (jk,mass) normalized against the mass of total Pt loading. (d) Bar graph showing the mass and specific activities of the catalysts recorded at 0.9 V vs. RHE. | ||
ORR performances of the prepared catalysts were evaluated using polarization curves in O2-saturated 0.1 M HClO4 solutions (Fig. 2b). The ORR polarization curves demonstrated that PtNiCo/C delivered the highest catalytic activity compared to the other catalysts in this study. The kinetic current density (jk) for the ORR was derived using the Koutecky–Levich equation, and using the data Tafel plots for MAs and SAs were generated as shown in Fig. 2c and Fig. S8 (ESI†). As depicted in Fig. 2d, the ORR MAs at 0.9 V vs. RHE for Pt/C, PtNi/C, and PtNiCo/C were 0.22, 1.26, and 1.64 A mgPt−1, respectively. PtNiCo/C exhibited 1.3 and 7.5-fold increase in MA compared to PtNi/C and Pt/C, respectively. The ORR SAs (jk, specific) for these catalysts were 0.26, 2.87, and 3.73 mA cm−2, respectively, and PtNiCo/C showed 1.3 and 14.3-fold enhancements compared to PtNi/C and Pt/C, respectively. The significantly enhanced ORR activity observed in the trimetallic PtNiCo/C is likely attributed to the skeletal structure compared to the commercial Pt/C catalyst. In addition, the electronic effect of Co in PtNiCo/C improved the ORR activity compared to that of PtNi owing to the fine-tuning of the d-band center of Pt, thereby facilitating O2 adsorption, activation, and desorption.
ADT was conducted on PtNiCo/C, PtNi/C, and Pt/C catalysts to evaluate their stability. The ADT involved 10000 and 20,000 cycle tests at a scan rate of 100 mV s−1 between 0.6 and 1.0 V vs. RHE in an O2-saturated 0.1 M HClO4 solution. The stability was assessed by measuring the CV and ORR polarization curves before and after the ADT, as shown in Fig. 3a and b and Fig. S7 (ESI†). Post 20000 cycles, the ORR half-wave potential (E1/2) values for PtNiCo/C, PtNi/C, and Pt/C exhibited negative shifts of 11, 16, and 44 mV from their pristine conditions, respectively. These shifts indicate a higher stability of PtNiCo/C compared to PtNi/C and Pt/C. Furthermore, the MAs, as obtained from the Tafel plots, showed that after 20000 cycles of ADT, the PtNiCo/C, PtNi/C, and Pt/C catalysts retained 73.8%, 65.8%, and 31.9% of their original activities, respectively (Fig. 3c and Table S2, ESI†). Additionally, the SAs derived from the Tafel plots indicated that after 20000 cycles, PtNiCo/C, PtNi/C, and Pt/C retained 93.0%, 81.2%, and 57.6% of their initial activities, respectively (Fig. 3d and Table S2, ESI†). Notably, both LSV and MA measurements revealed that PtNiCo/C maintained performance comparable to that of the pristine PtNi/C even after extensive cycling. These results highlight the high ORR efficiency of PtNiCo/C. After 20000 cycles of durability testing, STEM and EDS elemental mapping were performed on individual NPs of commercial Pt/C and PtNiCo/C. In Pt/C, significant nanoparticle aggregation was observed during the ORR process (Fig. S9, ESI†). In contrast, PtNiCo/C retained its skeletal structure, with a Pt:Ni:Co atomic ratio of 76:18:6 as determined by TEM-EDS (Fig. S10, ESI†), suggesting only a minimal dissolution of transition metals, which contributes to its excellent stability.
 | ||
| Fig. 3 ORR polarization curves for (a) PtNiCo/C and (b) PtNi/C before and after 10000 and 20000 cycles in an O2-saturated 0.1 M HClO4 solution. The insets display the CVs before and after the stability test for each catalyst. Bar graph shows changes in (c) MAs and (d) SAs before and after the stability test. | ||
To evaluate the practical potential of PtNiCo/C, single-cell tests were conducted (Fig. S11, ESI†). At a cell voltage of 0.6 V (j0.6V), PtNiCo/C membrane electrode assembly (MEA) achieved a current density of 0.711 A cm−2, surpassing the performance of the Pt/C MEA (0.516 A cm−2) under H2/O2 conditions. Moreover, power density measurement revealed that PtNiCo/C exhibited a maximum power density (pMax) of 0.389 W cm−2 under H2/air and 0.701 W cm−2 under H2/O2, outperforming Pt/C, which showed 0.285 W cm−2 and 0.573 W cm−2 under the respective conditions.
The superior catalytic performance of the ternary NPs can be attributed to modifications in the d-band center and Pt–Pt bond length. Hu et al. carried out Pt L3-edge X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses of PtCuCo alloys, revealing the lowest d-band vacancy and shortest Pt–Pt distance among the tested alloys.28 The incorporation of a third metal induces stronger compressive strain on surface Pt atoms, increasing the overlap of Pt 5d orbitals and lowering the d-band center. Optimizing the oxygen adsorption energy is crucial in ORR catalysis, as the binding strength of intermediates significantly impacts reaction kinetics. In this study, we attribute the enhanced ORR performance of PtNiCo/C to the compressive strain and ligand effects introduced by Co, which together lead to an optimized d-band center and improved catalytic behavior.
In summary, we demonstrated the synthesis of binary and ternary octahedral skeletons via a gas-phase reaction followed by acid treatment. This gas-phase approach enabled a significantly shorter reaction time compared to conventional solution-based methods. Unlike typical spherical NPs, which limited catalytic activity to surface Pt atoms, the octahedral skeletons enabled more efficient utilization of noble metals by exposing surface and interior Pt atoms as active sites. The incorporation of Co as a third metal further optimized the electronic structure and oxidation state of the alloy, resulting in 7.5- and 14.3-fold enhancements in ORR MA and SA, respectively, in half-cell tests. Moreover, the PtNiCo/C catalyst outperformed commercial Pt/C in single-cell tests, exhibiting superior power density and long-term stability.
This work was financially supported by the National Research Foundation of Korea (RS-2020-NR048317, RS-2023-NR076943, RS-2024-00346153, and RS-2025-02215028)
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- Y. Yang, J. Zhou, Z. Zhao, G. Sun, S. Moniri, C. Ophus, Y. Yang, Z. Wei, Y. Yuan, C. Zhu, Y. Liu, Q. Sun, Q. Jia, H. Heinz, J. Ciston, P. Ercius, P. Sautet, Y. Huang and J. Miao, Nat. Catal., 2024, 7, 796–806 CrossRef CAS.
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- A. R. Fairhurst, J. Snyder, C. Wang, D. Strmcnik and V. R. Stamenkovic, Chem. Rev., 2025, 125, 1332–1419 CrossRef CAS PubMed.
- F. Xiao, Y.-C. Wang, Z.-P. Wu, G. Chen, F. Yang, S. Zhu, K. Siddharth, Z. Kong, A. Lu, J.-C. Li, C.-J. Zhong, Z.-Y. Zhou and M. Shao, Adv. Mater., 2021, 33, 2006292 CrossRef CAS PubMed.
- H. Cruz-Martinez, H. Rojas-Chávez, P. T. Matadamas-Ortiz, J. C. Ortiz-Herrera, E. López-Chávez, O. Solorza-Feria and D. I. Medina, Mater. Today Phys., 2021, 19, 100406 CrossRef CAS.
- F. Lin, M. Li, L. Zeng, M. Luo and S. Guo, Chem. Rev., 2023, 123, 12507–12593 CrossRef CAS PubMed.
- R. M. Kluge, R. W. Haid, A. Riss, Y. Bao, K. Seufert, T. O. Schmidt, S. A. Watzele, J. V. Barth, F. Allegretti, W. Auwärter, F. Calle-Vallejo and A. S. Bandarenka, Energy Environ. Sci., 2022, 15, 5181–5191 RSC.
- M. Zhou, C. Li and J. Fang, Chem. Rev., 2021, 121, 736–795 CrossRef CAS PubMed.
- P. P. Lopes, D. Li, H. Lv, C. Wang, D. Tripkovic, Y. Zhu, R. Schimmenti, H. Daimon, Y. Kang, J. Snyder, N. Becknell, K. L. More, D. Strmcnik, N. M. Markovic, M. Mavrikakis and V. R. Stamenkovic, Nat. Mater., 2020, 19, 1207–1214 CrossRef CAS PubMed.
- Y. He, Q. Tan, L. Lu, J. Sokolowski and G. Wu, Electrochem. Energy Rev., 2019, 2, 231–251 CrossRef CAS.
- B. Peng, Z. Liu, L. Sementa, Q. Jia, Q. Sun, C. U. Segre, E. Liu, M. Xu, Y.-H. J. Tsai, X. Yan, Z. Zhao, J. Huang, X. Pan, X. Duan, A. Fortunelli and Y. Huang, Nat. Catal., 2024, 7, 818–828 CrossRef CAS.
- Z. Zhao, Z. Liu, A. Zhang, X. Yan, W. Xue, B. Peng, H. L. Xin, X. Pan, X. Duan and Y. Huang, Nat. Nanotechnol., 2022, 17, 968–975 CrossRef CAS PubMed.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- S. Zaman, L. Huang, A. I. Douka, H. Yang, B. You and B. Y. Xia, Angew. Chem., 2021, 133, 17976–17996 CrossRef.
- J. Guan, D. Dong, N. A. Khan and Y. Zheng, Chem. Commun., 2024, 60, 1811–1825 RSC.
- Q. Chang, Y. Xu, Z. Duan, F. Xiao, F. Fu, Y. Hong, J. Kim, S.-I. Choi, D. Su and M. Shao, Nano Lett., 2017, 17, 3926–3931 CrossRef CAS PubMed.
- J. Lim, K. Shin, J. Bak, J. Roh, S.-J. Lee, G. Henkelman and E.-A. Cho, Chem. Mater., 2021, 33, 8895–8903 CrossRef CAS.
- E. Hornberger, M. Klingenhof, S. Polani, P. Paciok, A. Kormányos, R. Chattot, K. E. MacArthur, X. Wang, L. Pan, J. Drnec, S. Cherevko, M. Heggen, R. E. Dunin-Borkowski and P. Strasser, Chem. Sci., 2022, 13, 9295–9304 RSC.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- J. Hoi, J.-H. Jang, C.-W. Roh, S. Yang, J. Kim, J. Lim, S. J. Yoo and H. Lee, Appl. Catal., B, 2018, 225, 530–537 CrossRef.
- J. Zhang, H. Yang, J. Fang and S. Zou, Nano Lett., 2010, 10, 638–644 CrossRef CAS PubMed.
- A. Eichler, Surf. Sci., 2002, 498, 314–320 CrossRef CAS.
- Y. Morikawa, J. J. Mortensen, B. Hammer and J. K. Nørskov, Surf. Sci., 1997, 386, 67–72 CrossRef CAS.
- B. A. Riguetto, J. M. C. Bueno, L. Petrov and C. M. P. Marques, Spectrochim. Acta, Part A, 2003, 59, 2141–2150 CrossRef CAS PubMed.
- A. Oh, H. Baik, D. S. Choi, J. Y. Cheon, B. Kim, H. Kim, S. J. Kwon, S. H. Joo, Y. Jung and K. Lee, ACS Nano, 2015, 9, 2856–2867 CrossRef CAS PubMed.
- Z. Zhao, M. Feng, J. Zhou, Z. Liu, M. Li, Z. Fan, O. Tsen, J. Miao, X. Duan and Y. Huang, Chem. Commun., 2016, 52, 11215–11218 RSC.
- B. Hu, J. Yuan, J. Zhang, Q. Shu, D. Guan, G. Yang, W. Zhou and Z. Shao, Composites, Part B, 2021, 222, 109082 CrossRef CAS.
- T. Wang, J. Liang, Z. Zhao, S. Li, G. Lu, Z. Xia, C. Wang, J. Luo, J. Han, C. Ma, Y. Huang and Q. Li, Adv. Energy Mater., 2019, 9, 1803771 CrossRef.
- J. Liang, N. Li, Z. Zhao, L. Ma, X. Wang, S. Li, X. Liu, T. Wang, Y. Du, G. Lu, J. Han, Y. Huang, D. Su and Q. Li, Angew. Chem., Int. Ed., 2019, 58, 15471–15477 CrossRef CAS PubMed.
- R.-Y. Shao, X. Niu, X.-C. Xu, X.-H. Zhou, S. Chu, L. Tong, L. Zhang and H.-W. Liang, Nano Lett., 2024, 24, 5578–5584 CrossRef CAS PubMed.
- S.-G. Qin, J. Liu, Z.-Y. Chen, X.-H. Liu, H. Feng, Y. Feng, Z.-Q. Tian, P. Tsiakaras and P.-K. Shen, Appl. Catal., B, 2024, 349, 123832 CrossRef CAS.
- L. Huang, Y.-Q. Su, R. Qi, D. Dang, Y. Qin, S. Xi, S. Zaman, B. You, S. Ding and B. Y. Xia, Angew. Chem., 2021, 133, 25734–25741 CrossRef.
- X. Liu, Y. Wang, J. Liang, S. Li, S. Zhang, D. Su, Z. Cai, Y. Huang, L. Elbaz and Q. Li, J. Am. Chem. Soc., 2024, 146, 2033–2042 CrossRef CAS PubMed.
- R. M. Arán-Ais, F. Dionigi, T. Merzdorf, M. Gocyla, M. Heggen, R. E. Dunin-Borkowski, M. Gliech, J. Solla-Gullón, E. Herrero, J. M. Feliu and P. Strasser, Nano Lett., 2015, 15, 7473–7480 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc01349f |
| ‡ M. K. and M. K. K. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |