
In situ polymerized ether-based polymer electrolytes towards practical lithium metal batteries
Sisi
Peng
,
Jialong
Fu
,
Lu
Wei
 and
Xin
Guo
*
and
Xin
Guo
*
School of Materials Science and Engineering, State Key Laboratory of Material Processing and Die & Mould Technology, Huazhong University of Science and Technology, Wuhan 430074, P.R. China. E-mail: xguo@hust.edu.cn
First published on 11th December 2024
Abstract
Commercial lithium–ion batteries that use flammable liquid electrolytes face significant safety risks, such as fires caused by electrolyte leaks. Solid polymer electrolytes (SPEs) present a viable solution to this problem, with ether-based polymer electrolytes standing out due to their superior stability and compatibility with lithium metal. The in situ ring-opening polymerization of cyclic ether monomers not only simplifies the battery manufacturing process but also improves the solid/solid interfacial contacts between electrolytes and electrodes, thereby significantly reducing interfacial impedance. In this paper, we review the mechanisms of ring-opening polymerization for cyclic ether monomers and analyze the ionic conduction of ether-based polymer electrolytes. We also explore the in situ curing mechanisms for several representative cyclic ether monomers and assess research advancements in this area. Additionally, this paper discusses the sustainability of ether-based polymer electrolytes and provides an outlook on future research and sustainability initiatives in the field.
1 Introduction
In recent decades, electrochemical energy storage technology has rapidly advanced, driven by developments in electrical vehicles, drones, and other electronic devices. This progress has led to increased demands for higher energy density in lithium batteries.1,2 Compared to the widely used graphite anodes, lithium metal is an ideal anode material due to its exceptionally high theoretical specific capacity (3860 mA h g−1) and the lowest electrochemical potential (−3.04 V vs. the standard hydrogen electrode).3–5To achieve long-term cycling stability and ensure the service life and safety of lithium metal batteries, the performance requirements for the electrolyte must become more stringent. A high-performance electrolyte must provide sufficient ionic conductivity at room temperature for effective lithium ion (Li+) transport, while also possessing excellent mechanical properties and flexibility to inhibit lithium dendrite growth. Solid polymer electrolytes (SPEs) address these needs by offering enhanced safety, mechanical strength, and flexibility. Compared to liquid electrolytes, SPEs provide much higher mechanical strength, which helps inhibit the growth of lithium dendrites.6 However, SPEs prepared using ex situ methods often face severe interfacial contact problems. Poor interfacial contacts can lead to high interfacial impedance,7–9 ultimately contributing to potential battery failure.
These challenges can be effectively addressed through in situ curing strategies. These strategies involve preparing a homogeneous precursor solution composed of polymer monomers, lithium salts, initiators, and other components in precise ratios. This precursor solution is injected directly into the battery, where it infiltrates both the electrodes and the separator. Following the injection, the sealed cell undergoes treatments such as heating or irradiation, which cause the liquid precursor to polymerize into a quasi-solid electrolyte.
The high fluidity of the precursor solution before curing ensures excellent contacts with electrodes, while the conformal interfacial contacts are maintained after curing, thereby facilitating efficient ion transport between electrodes and electrolytes. Additionally, the in situ polymerization method ensures uniform mixing of the polymer matrix, lithium salt, and other components, which promotes consistent and rapid ion transport. This method is straightforward, produces no harmful solvent emissions, and shows significant promise for the commercialization of high-performance lithium metal solid-state batteries.
Common monomers used for in situ polymerization include cyclic ethers, carbonates, acrylates, and alkenes. Among these, cyclic ethers are particularly advantageous due to their low viscosity and ability to undergo ring-opening reactions at room temperature, making them ideal for preparing solid electrolytes via in situ polymerization. Ring-opening polymerization of cyclic ether monomers is a well-established polymerization technique widely utilized in polymer science to synthesize various polymers,10,11 thereby playing a crucial role in advancing the field. Cyclic ether polymers are important polymer types, frequently applied in the production of surfactants,12 polyurethanes,13 engineering plastics,14 and other essential materials. Another advantage of ether-based polymers is their compatibility with existing battery production processes. Therefore, the large-scale production of cyclic ether polymers requires minimal modifications to current equipment and methods, facilitating their adoption in practical applications. Additionally, ether-based polymers are distinguished by their excellent reduction stability and compatibility with lithium metal, as they typically do not react with it.15 They also feature a low glass transition temperature (Tg) and active polymer chain mobility at room temperature, which results in high ionic conductivity (Fig. 1).16
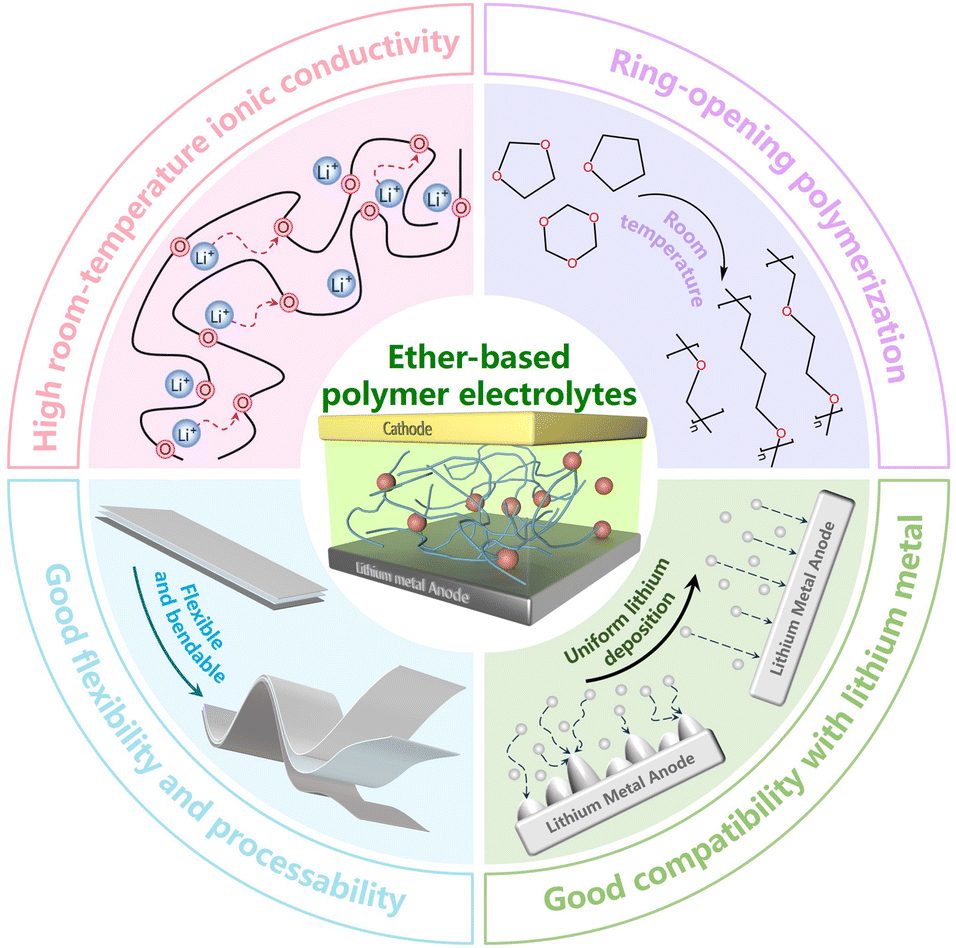 | ||
| Fig. 1 Advantages of ether-based polymer electrolytes. | ||
2 Li+ conduction in ether-based polymers
2.1 Ring-opening polymerization
Ring-opening polymerization of cyclic ether monomers serves as an effective strategy for in situ preparation of SPEs. The driving force behind this polymerization process is the ring tension inherent in cyclic ether monomers, which is mainly influenced by the size of the ring, the substituent groups attached to it and the elements constituting the ring. Generally, the more deformed the bonds within the ring structure, the greater the ring strain. This increased ring strain makes the ring-opening polymerization process easier to initiate and complete.Thermodynamically, there is a strong tendency for the ring-opening polymerization. In cyclic ether monomers, oxygen atoms contain two lone pairs of electrons, making them highly susceptible to attack by cations, which initiates the ring-opening polymerization reaction. Consequently, acids (cations) and bases (anions) can break the C–O bonds, leading to ring-opening.17Fig. 2a shows that the cationic ring-opening polymerization of cyclic ether monomers can be initiated by protonic and Lewis acids. Many lithium salts, such as lithium hexafluorophosphate (LiPF6), lithium difluoro-oxalate borate (LiDFOB), and lithium bis(fluorosulfonyl)imide (LiFSI) can be decomposed to produce Lewis acids by forming complexes with trace water in the electrolyte. The anionic groups then undergo transformation to generate anionic and cationic pairs after binding to the trace amounts of water. These pairs provide protons or cations that react with ether oxygen atoms, initiating polymerization.18,19 This process eliminates the need for additional initiators and occurs under mild conditions, even at room temperature, so it is particularly suitable for in situ curing during battery assembly.
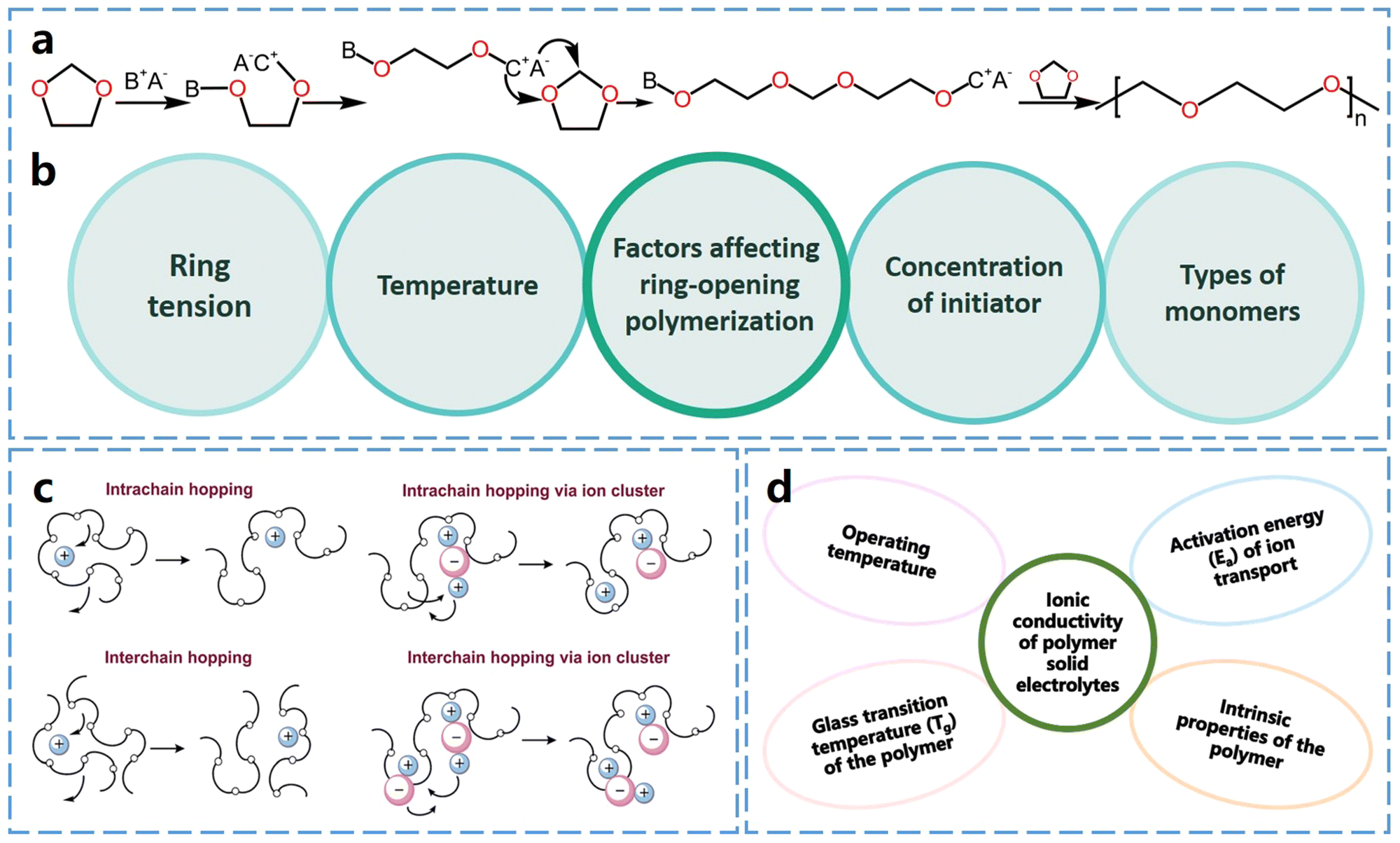 | ||
| Fig. 2 Ring-opening polymerization and Li+ conduction of ether-based polymers. (a) Schematic diagram of protonate or Lewis acid initiated cationic ring-opening polymerization of cyclic ether monomers (DOL as an example, A−B+ represents anionic and cationic pairs). (b) Factors affecting ring-opening polymerization. (c) Ion transport mechanism in the polymer electrolytes. Reproduced from ref. 20 with permission from the Royal Society of Chemistry, copyright 2015 (d) factors affecting ionic conductivity of polymer electrolytes. | ||
2.2 Li+ conduction mechanism
Ether-based polymers contain ether–oxygen atoms in the main chain, which act as Lewis base groups. The ether oxygen atoms can form complexes with Li+, influencing its dissociation and conduction. When the Gibbs free energy for the salt solvation in the polymer exceeds the salt's lattice energy, lithium salt dissociates, releasing free Li+. The Li+ ions then coordinate with ether oxygen atoms in the polymer chains and migrate via the formation and breaking of lithium–oxygen bonds.20,21 Generally, a higher concentration of ether oxygen atoms in ether-based polymer electrolytes leads to an increased concentration of charge carriers, which in turn results in higher ionic conductivity.22The ionic conductivity of polymeric solid electrolytes can be described by the Arrhenius or Vogel–Tammann–Fulcher (VTF) equations.23 The Arrhenius behavior of the ionic conductivity σ is described by eqn (1),
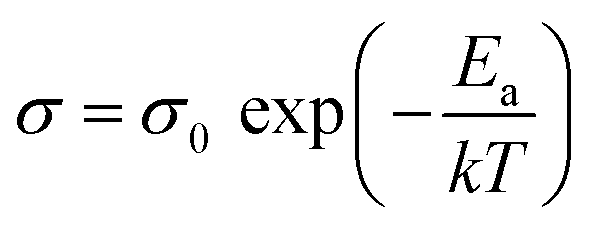 | (1) |
The ionic conductivity of polymer electrolytes is more relevant to the Vogel–Tammann–Fulcher (VTF) equation, which is described by eqn (2):
 | (2) |
3 Ether-based polymer electrolytes
Common polymer electrolytes prepared by in situ curing strategies include nitrile-based polymer electrolytes, ester-based polymer electrolytes, and ether-based polymer electrolytes, etc. Among these, ether-based polymer electrolytes have attracted significant attention due to its better chemical stability compared to nitrile-based polymer electrolytes, and they do not typically react with lithium metal. Therefore, ether-based polymer electrolytes can promote the formation of a stable SEI on the surface of the lithium metal anode. This interface effectively facilitates reversible lithium stripping and plating, while reducing side reactions between the electrolyte and lithium metal. Additionally, the in situ ring-opening polymerization of cyclic ether monomers to form solid electrolytes can significantly enhance the electrochemical stability window and cycling stability. In the following section, we introduce several commonly used ether-based polymer electrolytes prepared through in situ curing methods.3.1 Poly(1,3-dioxolane)-based electrolytes
The ring-opening polymerization of 1,3-dioxolane (DOL) has been studied for decades. Last century, Okada et al.29 studied the polymerization reaction of DOL using an organo-aluminium compound, acetyl chloride-metal halide, and acetic anhydride-perchloric acid as initiators. They obtained poly(1,3-dioxolane) (P-DOL) as a white crystalline solid with a melting point of 50–55 °C and discussed the location of bond breakage in the cationic polymerization of DOL. More recently, Guo et al.19 used LiPF6 to induce the cationic ring-opening polymerization of the cyclic ether monomer DOL at ambient temperature, converting the conventional mixed ether liquid electrolyte of glycol dimethyl ether (DME) and DOL into quasi-solid state (Fig. 3a), with the DOL polymerization conversion rate reaching 91%. The resulted gel polymer electrolytes demonstrated enhanced stability against lithium metal. The Li‖Li battery assembled with this gel polymer electrolytes cycled stably for over 400 hours at a high current density of 1.0 mA cm−2. Archer et al.30 used a low concentration of Al(OTf)3 to initiate the ring-opening polymerization of DOL to form SPEs (Fig. 3b). These SPEs were able to cycle stably in Li–S, Li–LiNi0.6Mn0.2Co0.2O2 (NCM), and Li–LiFePO4 (LFP) batteries, demonstrating the potential of P-DOL as SPEs. DOL's higher ring tension, caused by the presence of two oxygen atoms in its five-membered ring, leads to greater ring-opening activity and higher conversion rates.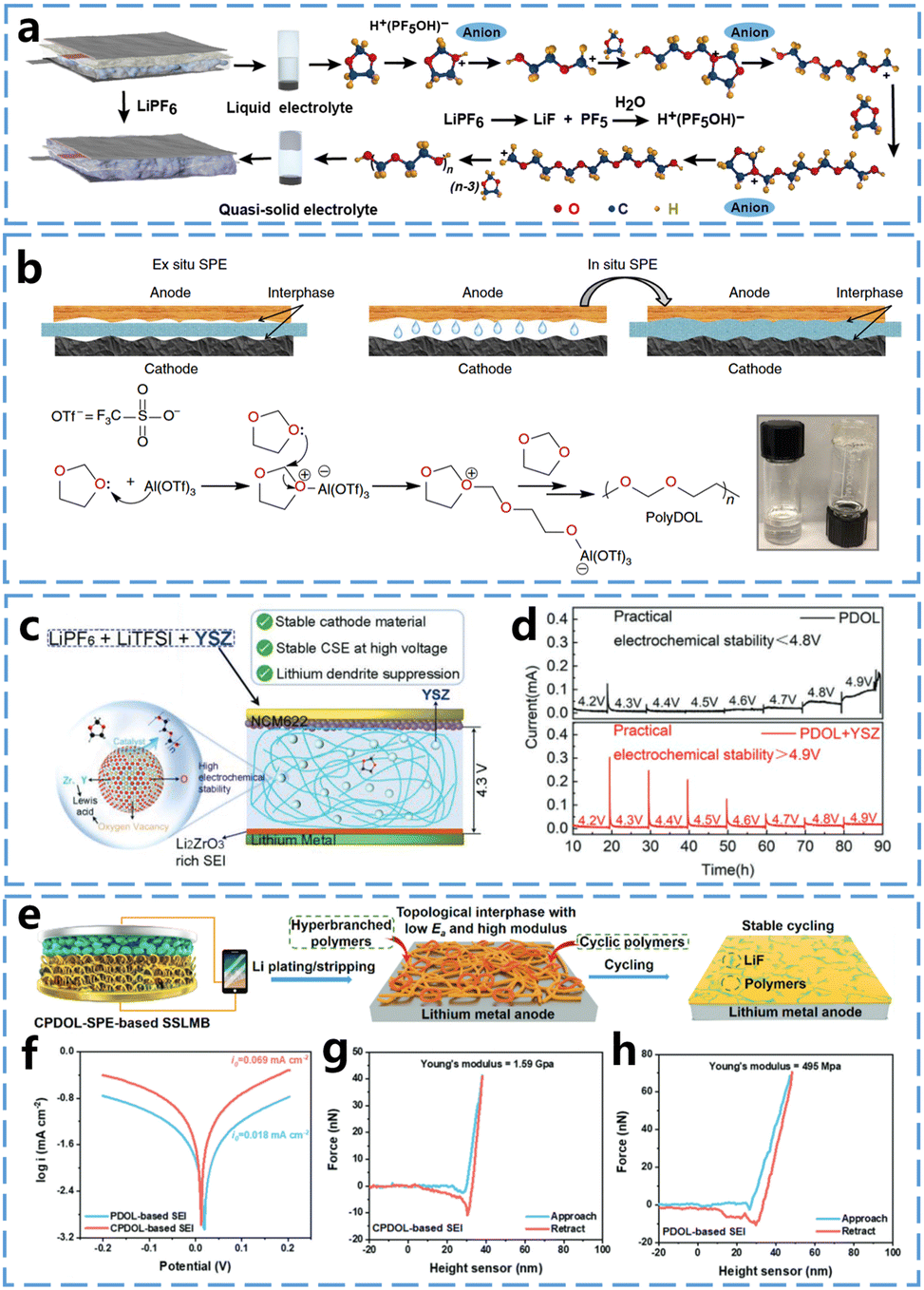 | ||
| Fig. 3 In situ curing mechanisms of DOL and properties of P-DOL electrolytes. (a) Reaction mechanism of LiPF6 initiating DOL polymerization. Reproduced from ref. 19 with permission from American Association for the Advancement of Science, copyright 2018. (b) Schematic representation of solid-state battery preparation by in situ curing and Al(OTf)3 initiated DOL ring-opening polymerization. Reproduced from ref. 30 with permission from Springer Nature, copyright 2019. (c) Schematic design principle of YSZ to enhance the performance of P-DOL based solid-state batteries. (d) Electrochemical float test of P-DOL and P-DOL + YSZ nanoparticle-assembled NCM622 cathode battery. Reproduced from ref. 31 with permission from John Wiley and Sons, copyright 2022. (e) Schematic of in situ generation of topologically structured SEI in solid-state lithium metal battery. (f) Tafel plots of CPDOL-SPEs and P-DOL-SPEs after 10 cycles and the corresponding exchange current densities. (g) Force–displacement curves of SEI nano-indentation experiments based on topological CPDOL and h P-DOL. Reproduced from ref. 32 with permission from John Wiley and Sons, copyright 2023. | ||
Although the use of lithium salts as initiators has enabled DOL to achieve high conversion rates, the presence of residual monomers continues to limit the battery performance. Therefore, further enhancing the conversion efficiency of DOL is essential for optimizing the battery performance. He et al.31 found that yttria stabilized zirconia (YSZ) nanoparticles containing a large number of Lewis acid sites that can catalyze the polymerization of DOL, as shown in Fig. 3c. These YSZ nanoparticles can work in synergy with LiPF6, and compared to the direct use of LiPF6 alone, the combination of YSZ with LiPF6, along with the presence of numerous Lewis acid sites on the surface of the YSZ nanoparticles (including Zr4+, Y3+, and oxygen vacancies), significantly enhances the conversion rate of DOL monomers to solid electrolytes. The conversion rate increased from 81.8% to 98.5% when using LiPF6 and YSZ particles compared to LiPF6 alone, demonstrating the effectiveness of YSZ nanoparticles in improving monomer DOL conversion. This increased conversion rate also significantly improved the high-voltage stability of the polymer electrolyte. The leakage current during constant voltage charging was measured by electrochemical float test to assess the oxidative stability of the solid electrolyte, as shown in Fig. 3d. The leakage current of P-DOL began to increase at a voltage of 4.6 V and rose dramatically at 4.9 V. In contrast, the leakage current of the solid electrolyte using the YSZ nanoparticles remained stable between 4.2 V and 4.9 V, with only a weak leakage current of 16 μA at 4.9 V.
P-DOL based electrolyte also facilitates the formation of a stable SEI layer on the surface of lithium metal, promoting the long-term stable operation of the battery. Mai et al.,32 used DOL to synthesize a topologically cross-linked P-DOL based solid polymer electrolytes (CPDOL-SPEs). CPDOL-SPEs undergo mild redox reactions with the lithium metal anode, and the cyclic hyperbranched polymer can in situ construct a topological interfacial layer on the lithium metal surface (Fig. 3e). This topological interfacial layer regulates the aggregation of TFSI− anions through supramolecular hydrogen-bonding interactions between –NH and –F, which facilitates the formation of LiF-rich SEI layers. Consequently, this interfacial layer exhibits both high mechanochemical stability and rapid Li+ transport. The SEI layer demonstrates low charge transfer impedance, inhibits lithium dendrite penetration and SEI cracking, and effectively suppresses further side reactions between CPDOL-SPEs and the lithium metal anode. According to the Tafel curves in Fig. 3f, the calculated exchange current density of the topological CPDOL-based SEI is i0 = 0.069 mA cm−2, which is much higher than the P-DOL-based SEI's i0 = 0.018 mA cm−2. This result indicates that the fast diffusion of Li+ in the CPDOL-based SEI promotes the uniform deposition of Li+ and inhibits the lithium dendrite growth. As shown in Fig. 3g and h, the CPDOL-based SEI exhibits a higher Young's modulus of 1.59 GPa, compared to the P-DOL-based SEI's 0.49 GPa. Additionally, the force-displacement curves during loading and unloading of the CPDOL-based SEI are more reversible than those of the P-DOL-based SEI, indicating that the mechanical properties of the CPDOL-based SEI are closer to elastic deformation.
3.2 Poly(tetrahydrofuran)-based electrolytes
Tetrahydrofuran (THF) is a polar ether solvent with a five-membered cyclic ether structure containing one oxygen atom. Cui et al.33 utilized boron trifluoride ethyl ether as an initiator for the cationic ring-opening polymerization of THF, to obtain a solid-state polymer electrolyte for lithium batteries (Fig. 4b). The boron trifluoride facilitated the formation of a stable SEI containing lithium fluoride (LiF) and B–O, enhancing long-cycle performance in LiFePO4‖Li metal batteries assembled with the polymer electrolyte.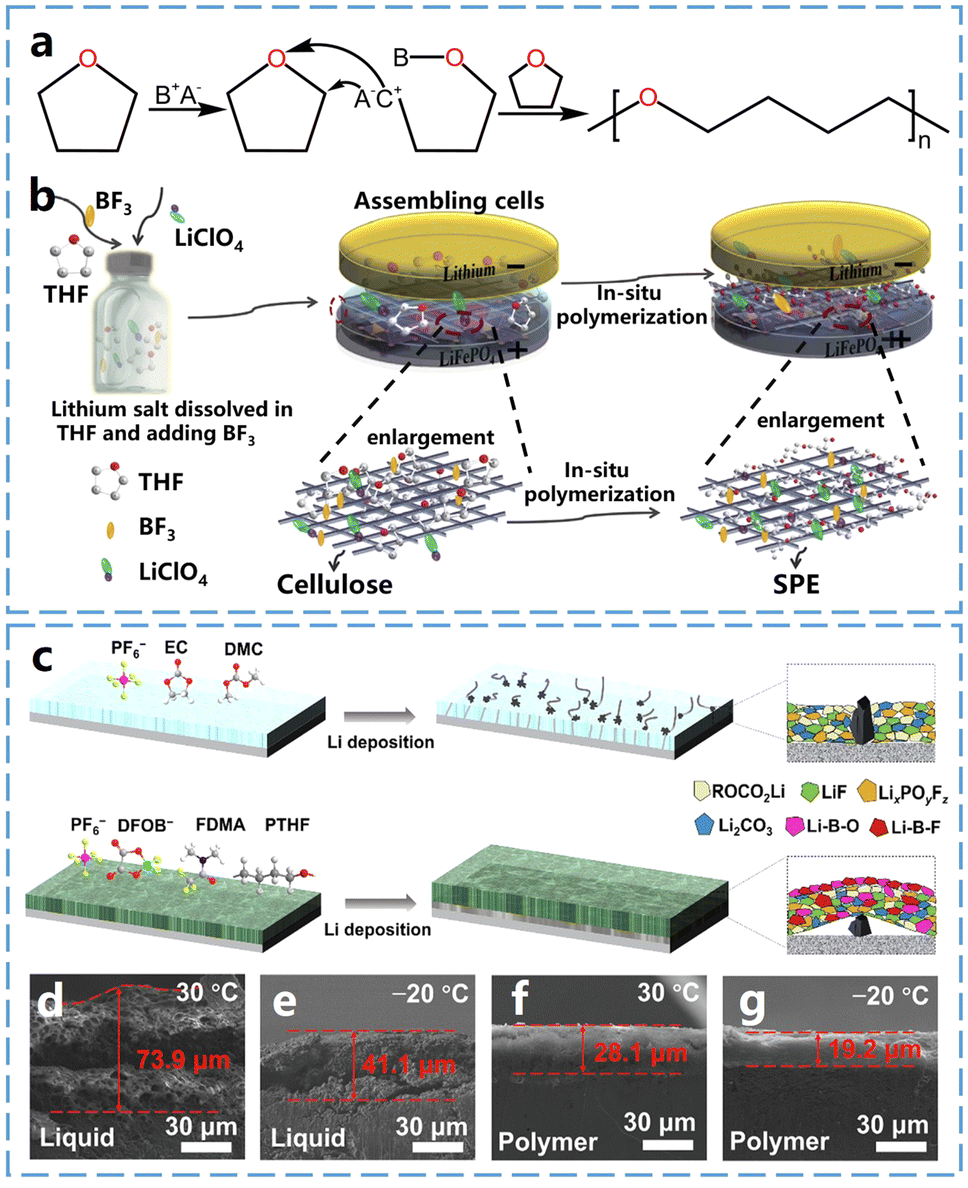 | ||
| Fig. 4 Ring-opening polymerization of THF and properties of P-THF electrolytes. (a) Schematic diagram of THF open-loop polymerization. (b) Schematic of the in situ polymerization process of P-THF electrolyte. Reproduced from ref. 33 with permission from Elsevier, copyright 2019. (c) Schematic representation of the formation process and SEI layer of carbonate-based and P-THF quasi-solid electrolytes. (d) Cross-sectional SEM images of lithium anode with liquid electrolyte at 30 °C, and (e) −20 °C after cycling. (f) Cross-sectional SEM images of lithium anode with P-THF electrolyte at 30 °C and (g) −20 °C after cycling. Reproduced from ref. 34 with permission from Elsevier, copyright 2024. | ||
Poly(tetrahydrofuran) (P-THF) retains the low melting point properties of its monomer, and therefore it is expected to be used in low-temperature solid electrolytes. For instance, Guo et al.34 prepared a quasi-solid polymer electrolyte by ring-opening polymerization of THF, using a dual-salt system composed of 1 M LiDFOB and 1 M LiPF6 as initiators. The anionic group DFOB− not only initiates the ring-opening polymerization of THF but also promotes the formation of a high ionically conductive and stable interface on the lithium metal anode surface (Fig. 4c). As shown in Fig. 4d and e, the Li thicknesses deposited from the carbonate-based liquid electrolytes were 73.9 μm and 41.1 μm at 30 °C and −20 °C, respectively. It is evident that lithium dendrites with highly porous structures were formed on the surface of lithium metal anodes when liquid electrolytes were used, indicating that the lithium metal anode experienced inhomogeneous Li–metal deposition during battery charging and discharging processes. In contrast, as shown in Fig. 4f and g, the thicknesses of the Li plated on the SPEs were approximately 28.1 μm and 19.2 μm at 30 °C and −20 °C, respectively, which are lower and denser compared to those of the liquid electrolytes. This dendrite-free Li deposition layer with a blocky aggregation morphology effectively reduces harmful parasitic reactions between metallic Li and the electrolyte, significantly improving the stability of the lithium metal anode.
3.3 Poly(1,3,5-trioxane)-based electrolytes
1,3,5-Trioxane (TXE) features three oxygen atoms alternately distributed in its six-membered ring, resulting in very high ring-opening activity. During TXE polymerization, polymers with high crystallinity and molecular weight are typically formed. These polymers have high strength but relatively low ionic conductivity.To address the issue of high crystallinity of poly(1,3,5-trioxane) (P-TXE) at room temperature, Cui et al.35 introduced succinonitrile (SN) as a polymerization retarder, which successfully reduced the polymerization rate of TXE. This reduction in polymerization rate affects the molecular weight of the resulted polymer, thereby decreasing its crystallinity. As shown in Fig. 5a, both TXE and SN are solids at room temperature. Due to their strong interactions, TXE can form a deep eutectic solution when mixed with SN. This method simplifies the in situ curing process and enhances the ionic conductivity of the resulted SPEs. In addition, this SPE design can also produce a protective layer on both the LiCoO2 cathode and the Li metal anode. In contrast, SPEs with TXE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SN = 5:3 mass ratio (PSL53) did not exhibit such a behavior. DFT calculations indicated that among the three electrolyte components (LiDFOB, SN, and P-TXE), LiDFOB has the lowest LUMO energy, making it more likely to reduce on Li metal and generate BF3. This BF3 triggers the polymerization of TXE on the Li metal, forming a passivated protective layer that preferentially adheres to lithium (Fig. 5b). Consequently, this thin P-TXE layer effectively prevents side reactions between Li metal and SN, improving the interfacial compatibility between PSL53 and Li metal.
:SN = 5:3 mass ratio (PSL53) did not exhibit such a behavior. DFT calculations indicated that among the three electrolyte components (LiDFOB, SN, and P-TXE), LiDFOB has the lowest LUMO energy, making it more likely to reduce on Li metal and generate BF3. This BF3 triggers the polymerization of TXE on the Li metal, forming a passivated protective layer that preferentially adheres to lithium (Fig. 5b). Consequently, this thin P-TXE layer effectively prevents side reactions between Li metal and SN, improving the interfacial compatibility between PSL53 and Li metal.
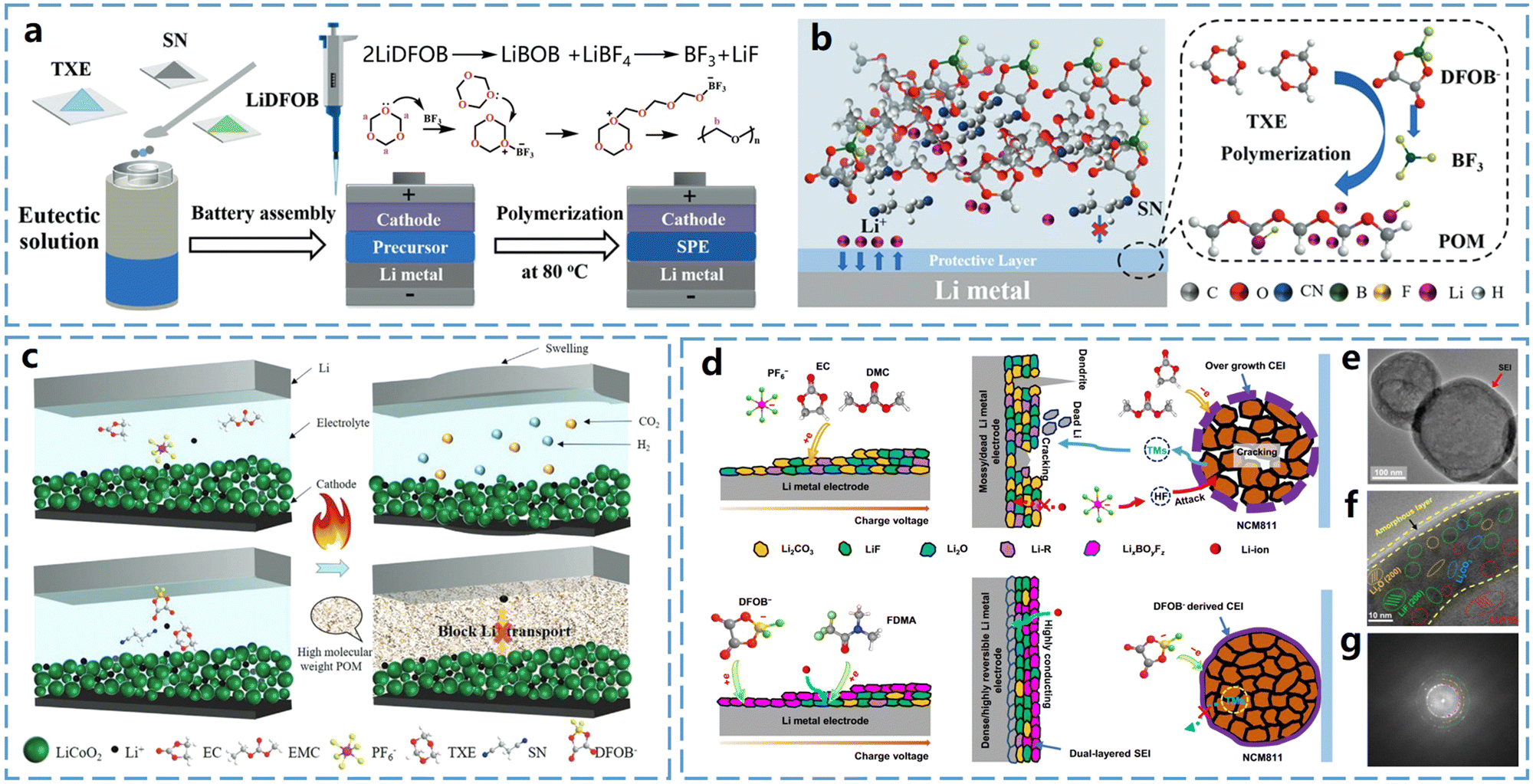 | ||
| Fig. 5 Ring-opening polymerization of TXE monomers and application of P-TXE electrolytes in batteries. (a) Schematic diagram of the in situ generation process of P-TXE-based SPEs. (b) Mechanism of formation of protective layer on lithium metal surface by P-TXE-based SPEs. Reproduced from ref. 35 with permission from John Wiley and Sons, copyright 2020. (c) Schematic of thermal failure of carbonate-based electrolyte and thermal shutdown function using DEE electrolyte. Reproduced from ref. 36 with permission from John Wiley and Sons, copyright 2023. (d) Schematic representation of the solid electrolyte interface (SEI) formed on Li metal electrode using non-aqueous carbonate-based electrolyte and the degradation process that occurs in Li‖NCM811 cell, as well as schematic representation of the solid electrolyte interface (SEI) formed by P-TXE-based polymer electrolyte on Li metal electrode and the process of inhibiting degradation in Li‖NCM811 cell. (e) and (f) Cryo-TEM images of lithium deposited with polymer electrolyte in the cell at different scales. (g) Schematic of the SEI structure observed on the Li metal surface. Reproduced from ref. 37 with permission from Springer Nature, copyright 2023. | ||
Utilizing the rapid polymerization of TXE and its high crystallinity, Cui et al.36 prepared a thermally stable polymer electrolyte. The electrolyte consisted of TXE, SN, and LiDFOB, where the deep eutectic solution formed by TXE and SN hindered the ring-opening polymerization of TXE at low temperatures. This allowed the electrolyte to remain liquid at room temperature, enabling fast ionic conduction and stable battery operation. However, during thermal runaway, as shown in Fig. 5c, TXE rapidly polymerizes at high temperatures to produce a crystalline polymer electrolyte that blocks the ionic conduction and provides thermal protection.
P-TXE can also be used for low-temperature solid-state lithium–metal batteries. Guo et al.37 prepared an electrolyte through in situ polymerization using TXE monomer (Fig. 5d). In this system, LiDFOB exhibits the lowest LUMO energy, making it the first to be reduced and thereby dominating the formation of the outer SEI layer. 2,2,2-Trifluoro-N,N-dimethylacetamide (FDMA), with the second lowest LUMO energy, further accepts electrons from the anode, contributing to the development of a LiF-rich, Li2CO3-free inner SEI beneath the LixBOyFz layer. Using this strategy, the polymer-based electrolyte formed a bilayer solid electrolyte interface (SEI) on the Li metal electrode, which stabilized the LiNi0.8Co0.1Mn0.1O2 anode, enhanced the interfacial charge transfer at low temperatures, and suppressed the growth of lithium dendrites. Because of the formation of an effective SEI layer, this electrolyte enabled the stable operation of Li‖LiNi0.8Co0.1Mn0.1O2 coin cells and pouch batteries even at −30 °C. To further reveal the bilayer structure, cryogenic transmission electron microscopy (Cryo-TEM) was used to investigate the nanostructures of SEI formed at low temperatures. The results revealed a continuous and uniform SEI on the deposited Li surface (Fig. 5e). At the atomic scale, a bilayer SEI with an inorganic inner phase and an amorphous outer layer was observed in the polymer system (Fig. 5f). The inner layer contains a small amount of Li2CO3 and Li2O, along with a significant amount of inorganic species like LiF. Fig. 5g illustrates the SEI bilayer structure, where this bilayer SEI inhibits lithium–electrolyte interactions, minimizes lithium loss, and enhances the cycling performance of lithium metal electrodes.
3.4 Poly(1,3-dioxane)-based electrolytes
1,3-Dioxane (DOX) is a six-membered cyclic ether with a structure similar to that of DOL. However, the polymerization product of six-membered DOX has a alkyl chain longer than that of the five-membered ring structure in DOL. The ring tension of DOX is lower than that of DOL, resulting in a reduced driving force for ring-opening.Zheng et al.38 firstly reported the in situ polymerization of 1,3-dioxane (DOX) monomer as a novel polymer electrolyte (PE) (Fig. 6a). Poly(1,3-dioxane) (P-DOX) exhibits higher oxidative stability and better lithium compatibility due to the increased number of carbon atoms in the polymer backbone. Furthermore, as shown in Fig. 6b, P-DOX has a alkyl chain longer than that of P-DOL, which reduces its highest occupied molecular orbital (HOMO) energy level, thereby enhancing its oxidative stability. For instance, as shown in the electrochemical floating test results in Fig. 6c, a sharp increase in the leakage current of the L-DOX liquid electrolyte was observed when the voltage was increased to 4.1 V, indicating oxidation of the liquid electrolyte at this point. Furthermore, a leakage current of 22 mA was detected for P-DOL PE at 4.0 V, and the leakage current dramatically increased to more than 200 mA at 4.6 V, suggesting that P-DOL PE is unstable at high voltages. In comparison, the leakage current of P-DOX PE remains very low even at a high voltage of 4.6 V. This result demonstrates that the oxidative stability of in situ PEs can be effectively improved by modulating the molecular structure of cyclic ether monomers. Additionally, the extension of alkyl chains reduces the solvation ability of P-DOX, promoting the formation of an anion-derived, inorganic-rich SEI, which significantly enhances the interfacial compatibility between lithium metal and P-DOX PEs.
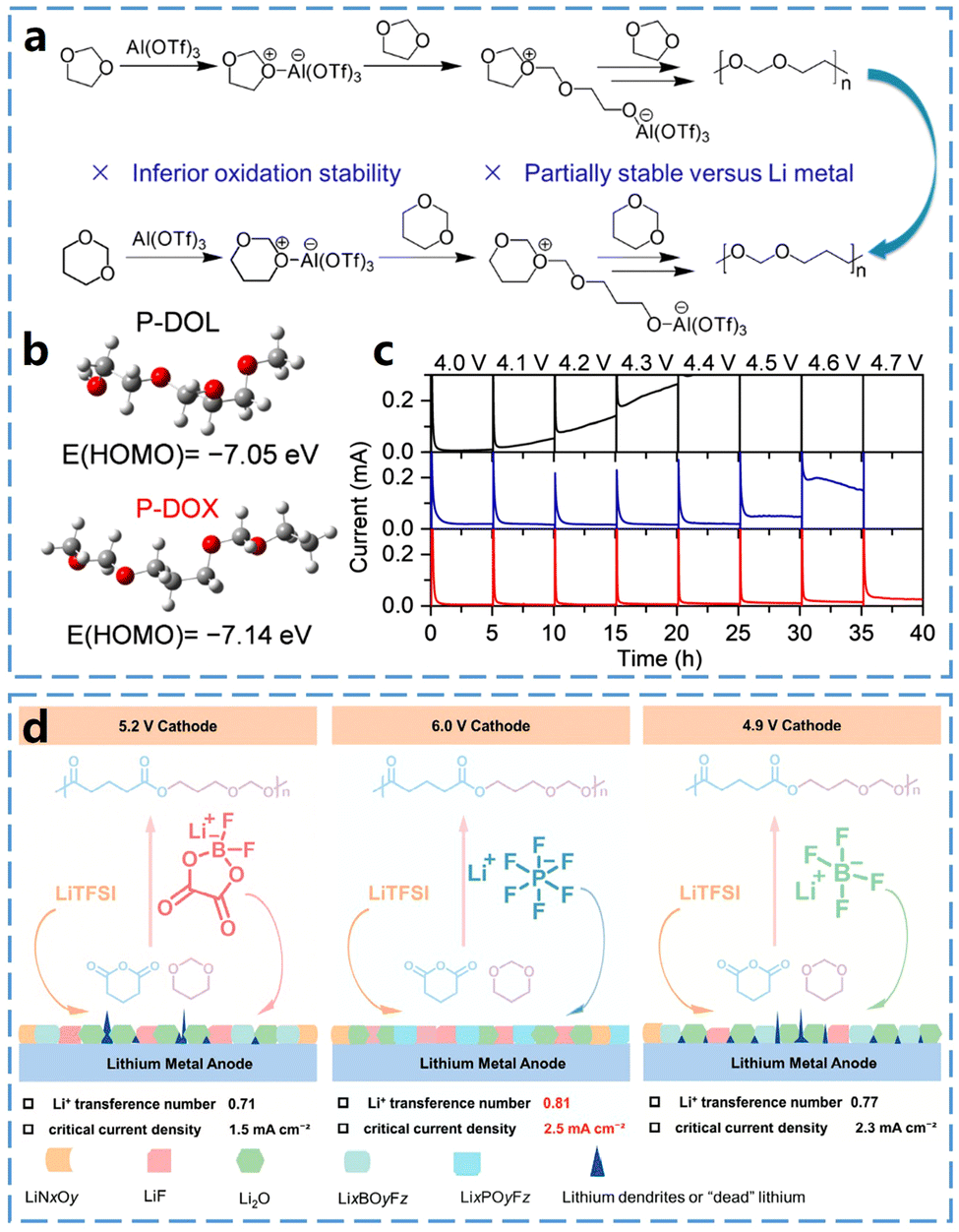 | ||
| Fig. 6 In situ curing mechanisms of DOX and properties of P-DOX electrolytes. (a) Schematic of the DOL and DOX polymerization processes initiated by Al(OTf)3. (b) HOMO energies of P-DOL and P-DOX polymer chains. (c) Electrochemical float test of electrolyte in Li‖NCM111 battery. Reproduced from ref. 38 with permission from the Royal Society of Chemistry, copyright 2023. (d) Schematic illustration of the fabrication of PEA-PEs and the effect of salts on the compositions of SEI layers. Reproduced from ref. 39 with permission from John Wiley and Sons, copyright 2024. | ||
The enhanced electrochemical stability of P-DOX highlights its potential as a promising cyclic ether monomer, and its copolymerization with other polymer monomers may be an effective way to obtain high-performance electrolytes. For instance, Zhang et al.39 developed a novel polyester acetal (PEA) electrolyte by in situ co-polymerization of valeric anhydride and DOX (Fig. 6d). The use of three lithium salts as initiators ensured the polymerization of the cyclic ether monomers, while their decomposition products contributed to the formation of a better SEI layer and prevented the growth of lithium dendrites. This novel PEA electrolyte exhibited an ionic conductivity of 0.43 mS cm−1 at 30 °C, an electrochemical stability window of 6.0 V, and a Li+ transfer number of 0.81, along with long-lasting stability over extended cycling periods with various high-voltage cathodes.
3.5 Poly(multicyclic ether)-based electrolytes
Multicyclic ether monomers contain multiple epoxy ether groups. Due to the presence of multiple ring-opening sites on these monomers, multicyclic ethers can form ether-based polymer electrolytes with more diverse structures. Moreover, cross-linking can occur between different polymer backbones due to the presence of excess rings. This typically results in enhanced mechanical properties and broader electrochemical windows for multicyclic ether-based polymer electrolytes. For example, Deng et al.40 used DOL and trimethylolpropane triglycidyl ether (TTE) in a one-step cross-linked polymerization within a lithium nitrate-releasing mesoporous polymer (LP) matrix to generate a poly(DOL-TTE)-LP cross-linked solid polymer electrolyte (Fig. 7a). In this case, both poly(DOL-TTE) and poly(DOL-TTE)-LP exhibited mechanical strengths far exceeding that of PEO (Fig. 7b). Similarly, as shown in Fig. 7c, Ma et al.41 demonstrated this by using LiDFOB as an initiator to induce the ring-opening polymerization of poly(ethylene glycol) diglycidyl ether (PEGDE). They further complexed the resulted polymer with a nanocellulose framework, producing the composite polymer electrolytes, P-PEGDE-NC. As illustrated in Fig. 7d, the combined effect of the cross-linked electrolyte and the nanocellulose framework significantly improves the mechanical strength of the polymeric solid electrolyte. At a thickness of 10 μm, the mechanical strength of the P-PEGDE-NC electrolyte (11.31 MPa) and the single P-PEGDE (3.17 MPa) are significantly higher than that of PEO (0.92 MPa).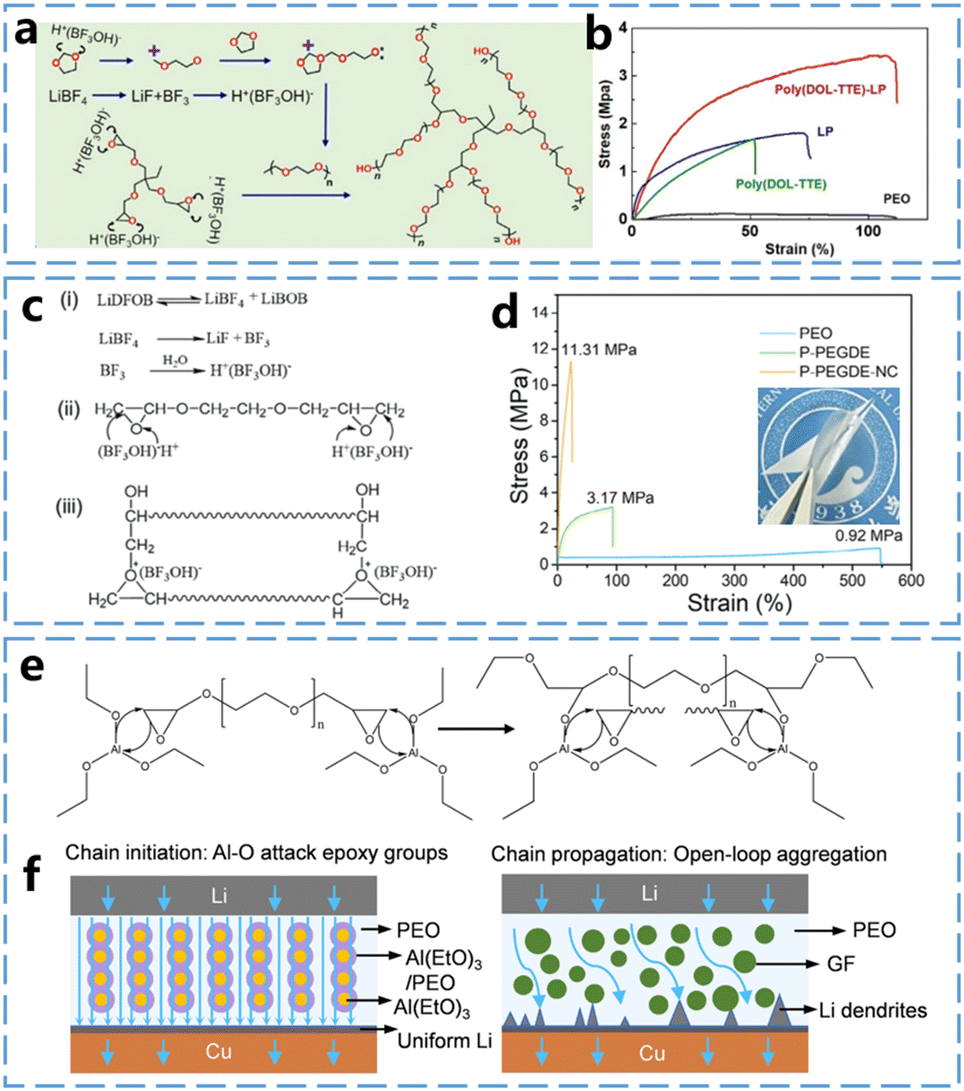 | ||
| Fig. 7 In situ curing mechanisms of multicyclic ether and properties of Poly(multicyclic ether)-based electrolyte (part 1) (a) Cross-linked polymerization of DOL and TTE monomers assisted by LiBF4 initiator. (b) Stress–strain curves of poly(DOL-TTE)-LP, LP, poly(DOL-DTE), and PEO. Reproduced from ref. 40 with permission from Elsevier, copyright 2022. (c) Ring-opening polymerization of PEGDE initiated by LiDFOB. (d) Stress–strain curves of PEO, P-PEGDE and P-PEGDE-NC electrolytes. Reproduced from ref. 41 with permission from John Wiley and Sons, copyright 2024. (e) Schematic of AGPE reaction mechanism. (f) Li+ transport and deposition of AGPE and FGPE in Li–Cu battery. Reproduced from ref. 42 with permission from Elsevier, copyright 2024. | ||
Good mechanical properties contribute to the stable operation of the battery. Multicyclic ether-based polymer electrolytes exhibit unique advantages in the ion transport process. Wang et al.42 developed a composite polymer electrolyte using PEGDE (Fig. 7e), where an Al(EtO)3 nanowire framework was employed to fill the PEGDE precursor. Notably, Al(EtO)3 not only catalyzed the ring-opening polymerization of PEGDE but also provided fast and orderly Li+ transport channels between the Al(EtO)3 nanowire framework and the PEO matrix. As illustrated in Fig. 7f, Li+ can transport through the PEO matrix and along the surface of the PEO matrix and the Al(EtO)3 nanowires in the composite polymer electrolyte AGPE. However, in the composite polymer electrolyte FGPE, which uses glass fibers as the filler frame, Li+ can transport only through the PEO matrix. Consequently, the Al(EtO)3 nanowire framework provides a new ionic transport pathway and enhances the ionic conductivity. This is reflected by the higher ionic conductivity of AGPE (6.8 × 10−4 S cm−1) compared to FGPE (1.6 × 10−4 S cm−1) at 30 °C.
The combination of good mechanical properties and high ionic conductivity indicates that multicyclic ether-based polymer electrolytes are excellent electrolyte materials. However, the end groups of cross-linked multicyclic ethers often contain unstable hydroxyl groups, which significantly reduce the electrochemical window of the cell, leading to lower oxidative stability and poor thermal stability. To address this problem, Wang et al.43 developed a cross-linked poly(ethylene glycol)-based resin (c-PEGR) by ring-opening copolymerization of PEGDE and poly(ether amine) (PEA) with amino groups (Fig. 8a). This c-PEGR confines the less oxidatively stable hydroxyl groups within the polymer backbone, thereby reducing reactivity and improving its electrochemical window. Fig. 8b illustrates a quasi-static linear scanning voltammetry (QS-LSV) method for accurately measuring the oxidation potential and electrochemical stability window of low-conductivity materials such as polymers. The results indicate that the oxidation potential of c-PEGR was increased to 4.36 V. The 3D cross-linked backbone of c-PEGR significantly enhances the structural stability of the gel electrolyte and effectively confines the hydroxyl groups, preventing them from undergoing redox reactions with the electrode surface. This confinement greatly reduces the reactivity of c-PEGR compared to poly(ethylene glycol) (PEG), which has free-moving hydroxyl groups, thereby resulting in good lithium compatibility. As shown in Fig. 8c, the cell utilizing the c-PEGR gel electrolyte maintains a consistent or even slightly reduced overpotential at a high current density of 4.5 mA cm−2.
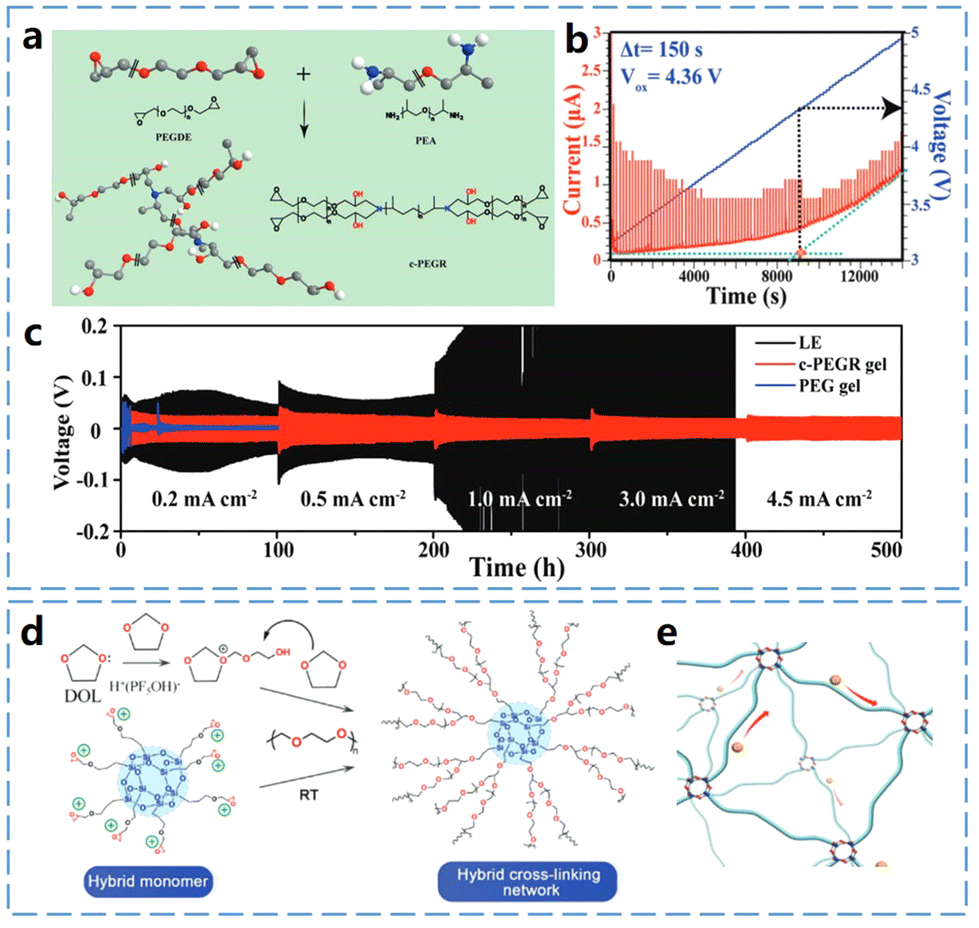 | ||
| Fig. 8 In situ curing of multicyclic ether and properties of Poly(multicyclic ether)-based electrolyte (part 2) (a) schematic of the synthesis process of c-PEGR. (b) Oxidation potential of c-PEGR gels was measured by setting QS-LSV to Δt for 150 s. (c) Voltage distribution of lithium symmetric cells at different current densities. Reproduced from ref. 43 with permission from John Wiley and Sons, copyright 2021. (d) Schematic diagram of the HCPE polymerization process. (e) Schematic representation of ion transport in DOL long-chain cross-linked and PS-hybridized cross-linked polymer backbones. Reproduced from ref. 44 with permission from John Wiley and Sons, copyright 2023. | ||
In response to the same problem, Tian et al.44 proposed a strategy for organic/inorganic hybrid cross-linked polymer electrolytes (HCPE). As shown in Fig. 8d, these electrolytes were obtained by cross-linking P-DOL with glycidyl ether oxypropyl cage polyhedral silsesquioxane (PS), which served as both the cross-linker and hybrid center. In this design, the hydrophobic PS cages function as hubs within the hydrophilic P-DOL chain network. These cages cross-link with the linear P-DOL chains, creating a spatial network structure. This network can accommodate free spaces for lithium salts and promote polymer chain motion, thereby facilitating the decomposition of lithium ions and enhancing ion transport. As shown in Fig. 8e, the ion transport pathway within the P-DOL-PS network structure demonstrates that HCPE has strong interactions with Li+. These interactions allow HCPE to enter the solvated shell of Li+, which is crucial for enhancing the thermal stability, ionic conductivity, and overall electrochemical properties of the HCPE.
In conclusion, the performance of ether polymer electrolytes relies on the dissociation of Li+ ions by ether oxygen atoms, and the conduction of Li+ ions through the movement of polymer chain segments, making it highly dependent on the proportion of ether oxygen atoms. Additionally, factors such as polymer molecular weight, the presence of solvents, and the type and concentration of additives can significantly influence battery performances. The compositions and properties of the ether polymer electrolytes discussed in this section are summarized in Table 1.
| Compositions of electrolytes | Conversion rate (%) | Molecular weight | Ionic conductivity (mS cm−1) | Li+ transference number | Oxidation potential (V) | Ref. |
|---|---|---|---|---|---|---|
| 1 M LiTFSI, 2 M LiPF6 in DOL:DME = 1:1(vol) |
91.0% | 5000 (Mn) | 3.8 | — | 4.6 | 19 |
| 2 M LiTFSI, 0.5 mmol Al(OTf)3 in DOL | 86% | 15000 (Mn) |
1.1 | — | 5.0 | 30 |
| 1 wt% LiPF6, 0.2 wt%YSZ nanoparticles and 0.1 M LiTFSI (relative to DOL monomer) | 98.5% | 23588 (Mw) |
0.28 | 0.65 | 5.2 | 31 |
| 0.4 g LiTFSI in 0.5 g cross-linked P-DOL | — | 110000 (Mw) |
0.101 | 0.46 | 5.1 | 32 |
| 0.6 M LiClO4 in 1 mL THF 0.6 M boron trifluoride diethyl ether | — | 6530 (Mn) | 0.23 (60 °C) | 0.36 | 4.5 | 33 |
| 1 M LiPF6, 1 M LiDFOB, in THF FDMA (at volume ratio of 4:1), 10 wt% FEC, 1 wt%EO |
— | 18716 (Mw) |
2.6 | 0.58 | 5.2 | 34 |
| 2.5 g TXE 1.5 g SN 0.365 g LiDFOB | — | 5083 (Mn) 5388 (Mw) | 0.114 | — | 5.5 | 35 |
| 0.365 g LiDFOB, 2 g SN, 2 g TXE | — | 725500 (Mn) (after shutdown) |
4.1 (before shutdown) 0.0002 (after shutdown) | 0.4 | 5.6 | 36 |
| TXE:FDMA:FEC = 5:3:1 wt%, 1 M LiDFOB |
— | — | 2.5 | 0.8 | 5.6 | 37 |
| Al(OTf)3 20 mmol in LiFSI-DOX (molar ratio of 1:8) |
— | 9353 (Mw) | 0.17 | 0.75 | 4.7 | 38 |
| 10 mmol glutaric anhydride (GA), 10 mmol DOX, 25 wt% LiTFSI, 0.4 mmol LiPF6 | — | 11000 (Mn) |
0.43 | 0.81 | 6.0 | 39 |
| 2.0 wt% TTE, 1.0 M LiTFSI, and 0.2 M LiBF4 in DOL | — | — | 0.3 | 0.35 | 4.9 | 40 |
| 1 wt% LiDFOB and 16 wt% LiTFSI in PEGDE TPP 2 wt%. Zn(TFSI)2 3 wt% | — | — | 0.078 | 0.62 | 4.78 | 41 |
| 12 mg precursor (PEGDE:LiTFSI:LiDFOB = 100:12.8:1 wt%) applying onto Al(EtO)3 thin slice |
90.06% | — | 0.68 | 0.59 | 5.3 | 42 |
| Applying PEGDE:PEA = 7:20 to form c-PEGR. c-PEGR was immersed in 1 m LiPF6 in DMC:FEC 1:1 vol% |
— | — | 0.7 | 0.47 | 4.36 | 43 |
| 1 M LiTFSI, 2 M LiPF6 and 5% wt% PS dissolved in DOL/DME (1:1, vol) |
— | — | 2.22 | 0.88 | 5.2 | 44 |
4 Recyclability of ether-based polymer electrolytes
As lithium batteries become the most widely used energy storage technology, repurposing used lithium batteries as raw materials for manufacturing is essential for promoting resource conservation and environmental sustainability. Achieving sustainability for end-of-life lithium batteries relies on developing recycling processes that maximize material recovery and minimize both waste generation and energy consumption. Although an increasing number of researchers are focusing on lithium battery recycling, the use of solid-state electrolytes in lithium batteries introduces distinct challenges primarily due to their structural differences from conventional liquid electrolyte batteries.45 Solid-state electrolytes, especially SPEs produced through in situ curing, are often strongly bonded to the electrodes to maintain ionic conduction at the interfaces, which complicates the separation of components during recycling. The easily decomposable nature of ether-based polymer electrolytes simplifies the battery recycling by enabling the efficient separation of the electrolyte from the electrode material. By utilizing this property, the electrolyte, electrode material, current collector, and other battery components can be easily separated and reused, while the lithium salt in the electrolyte can be recovered through polymer decomposition. This approach significantly reduces battery waste and enhances resource utilization efficiency.Studying the thermal decomposition behavior of ether-based polymer electrolytes is essential for enhancing the efficiency of battery recycling processes. Conventional recycling methods typically involve a combination of mechanical separation, pyrometallurgical, and hydrometallurgical techniques to separate battery components.46 These recovered components can then be reused in the production of new batteries, thereby supporting the sustainable use of lithium battery materials. However, the entire recycling process is energy-intensive and produces considerable liquid waste, leading to resource loss and potential environmental harm. Therefore, developing a simple and efficient recycling method that minimizes waste generation and maximizes resource recovery is a critical priority in this field. Zhou et al.47 observed the reversible decomposition of P-DOL-SPEs into formaldehyde and small-molecule epoxides at 110 °C. The thermal decomposition of P-DOL-SPEs in batteries leads to significant volume expansion, as shown in Fig. 9a and b, where a sealed aluminum-plastic film bag containing P-DOL-SPEs produced a substantial amount of gas at 110 °C after only 20 minutes. Thermal analysis of P-DOL-SPEs was conducted by the use of thermogravimetric analysis (TGA), derivative thermogravimetric (DTG) and differential scanning calorimetry (DSC). As shown in Fig. 9c, the TGA and DTG results of the P-DOL electrolyte show a stable plateau below 97 °C, indicating that the ether-based polymer electrolyte remains stable under this temperature. A sharp weight loss of 76.8% between 97 °C and 127 °C is attributed to the degradation of P-DOL. Moreover, the results show the fastest weight loss at 113 °C, indicating the onset of polymer decomposition and gas production. Additionally, as shown in Fig. 9d, the P-DOL electrolyte exhibits three main endothermic peaks during the heating process: −64 °C, 60 °C, and 123 °C. These peaks correspond to the glass transition temperature (Tg), melting point (Tm), and thermal decomposition temperature, respectively, these results are consistent with previous tests. The thermal stability studies of ether-based polymer electrolytes have significantly contributed to advancing sustainability efforts in this field.
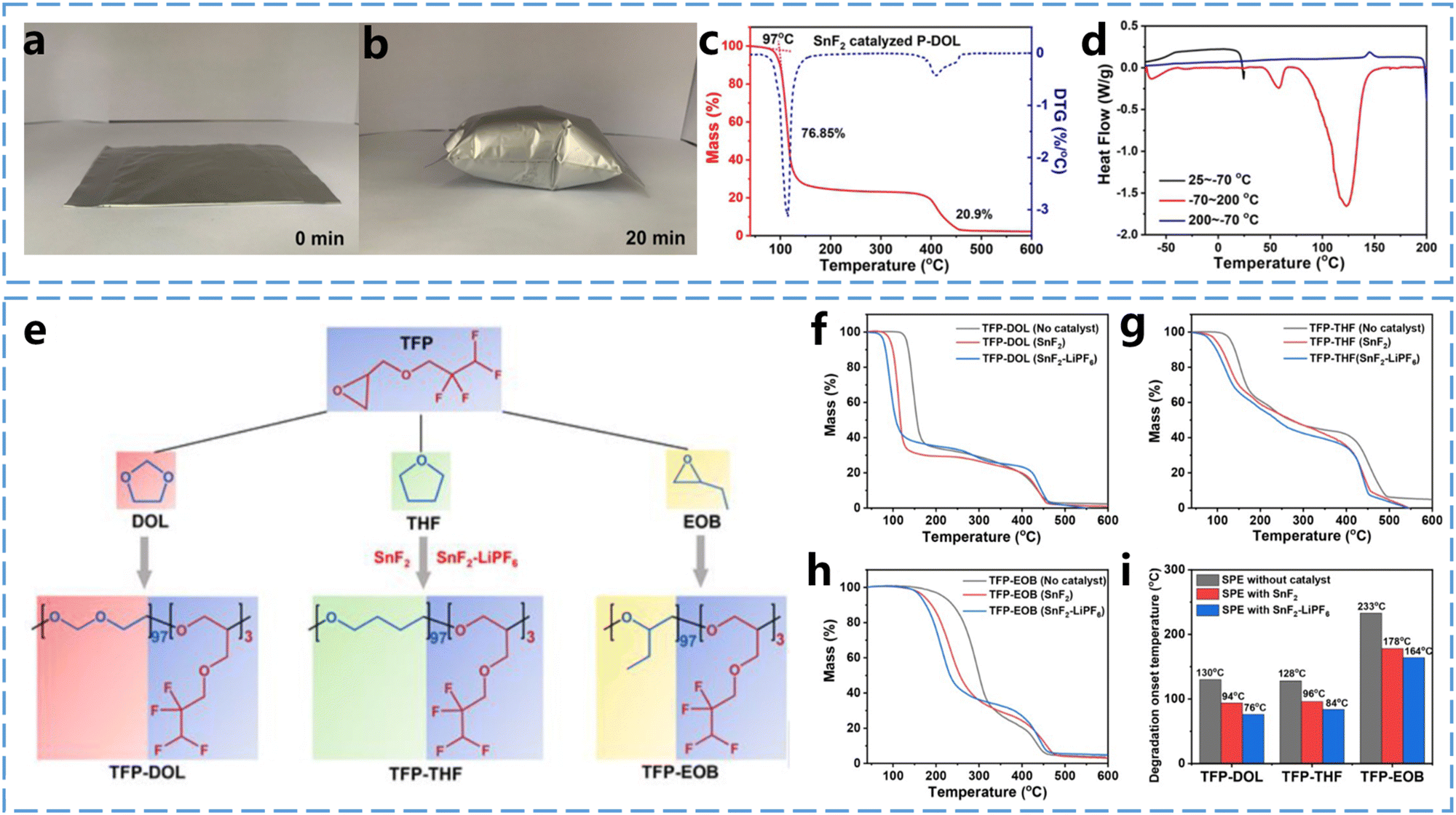 | ||
| Fig. 9 Thermal stability of ether-based polymer electrolytes. (a) and (b) Variation of P-DOL SPEs in aluminum-plastic film sealed bag at 110 °C with time. (c) TGA and DTG curves of P-DOL-SPEs. (d) DSC curve of P-DOL-SPEs. Reproduced from ref. 47 with permission from John Wiley and Sons, copyright 2022. (e) SnF2 and SnF2–LiPF6 catalyzed synthetic routes for the three copolymers TFP-DOL, TFP-THF and TFP-EOB. (f) TGA profiles of TFP-DOL. (g) TFP-THF and (h) TFP-EOB SPEs in the presence and absence of catalysts containing 1 wt% SnF2, SnF2 (1 wt%)-LiPF6 catalyst. (i) Comprehensive comparison of thermal degradation temperatures of the three SPEs without and with catalyst. Reproduced from ref. 48 with permission from John Wiley and Sons, copyright 2022. | ||
To recover lithium salts from mixed electrolytes, Zhou et al.48 synthesized three copolymers by cationic ring-opening polymerization of epoxides using two Lewis acid catalysts, SnF2 or SnF2–LiPF6 (1:1 molar ratio) (Fig. 9e). They compared the thermal decomposition behaviors of polymers obtained by copolymerizing 2,2,3,3-tetrafluoropropyl ether (TFP) with three fluorine-free epoxides—DOL, THF, and EOB (1,2-epoxybutane). The TGA curves in Fig. 9f–i reveal that after the removal of the catalyst, the decomposition temperatures of these copolymers increased by 30–55 °C. It was found that the strong interactions between LiTFSI and the long polymer chains in the SPEs in PEO were intrinsic to the dissociation of LiTFSI and the high Li+ conductivities, but these interactions also impeded the recovery of LiTFSI. As a result, PEOs catalyzed by SnF2–2LiPF6 (1:2 molar ratio) were decomposed at 70 °C lower than catalyst-free PEO–LiTFSI. By analyzing the thermal decomposition behavior of these homemade ether-based SPEs and commercial PEOs containing initiators, it was found that the long polymer chains were broken, leading to the disappearance of their originally strong interaction with LiTFSI. This breakdown may explain the thermal decomposition of the polymer. Finally, the recovery rate of the ether-based SPEs was as high as 80%, which exceeded the 70% recovery rate of PEO–LiTFSI.
Many studies have explored the recovery of rechargeable batteries,49–52 however, these studies have predominantly focused on the recovery of electrode materials, current collectors in the batteries, and lithium salts in electrolytes.53–55 There are relatively little reports on the degradation of ether-based polymer electrolytes. Ether-based polymer electrolytes exhibit certain degradable properties, suggesting that under the right conditions it may be possible to realize the degradation of the polymer chains and recover the monomers. For instance, Wang et al.56 explored the degradation process of P-THF to produce THF monomers. They investigated various catalysts, including different Lewis acids and proton acids, in the degradation of P-THF. The results demonstrated that heteropoly-acids exhibited higher catalytic activity and stability compared to other catalysts. The optimum conditions for the degradation of P-THF using heteropolyacids, such as phosphotungstic acid, as catalysts were a mass ratio of phosphotungstic acid to P-THF of 1:10, at 130 °C for 15 minutes. Under these conditions, the yield of THF exceeded 95%. Meanwhile, their study demonstrated that heteropolyacid catalysts maintained good stability throughout the reaction process. For example, when the phosphotungstic acid catalyst was reused 10 times, the yield of THF remained as high as 90%. Besides, they explored the degradation of P-THF using phosphotungstic acid as a catalyst via nuclear magnetic resonance (NMR) and Fourier transform infrared (FTIR) methods, proposing a mechanism for ether bond (C–O–C) cleavage. Their study provides a valuable reference for the decomposition and recycling of ether-based polymer electrolytes. Common ether-based polymers such as P-DOL and P-TXE can also be degraded by using catalysts to recover monomers.
5 Perspectives and conclusions
In summary, the in situ ring-opening polymerization of cyclic ether monomers not only simplifies battery fabrication but also improves the solid/solid interfacial contacts between electrolytes and electrodes, thereby significantly reducing interfacial impedance. Cyclic ether monomers that can undergo ring-opening polymerization at room temperature are particularly beneficial for the in situ preparation of solid polymer electrolytes, which is essential for producing solid-state batteries that meet high safety standards.In this paper, we reviewe the mechanisms of ring-opening polymerization for cyclic ether monomers and analyze the ionic conduction behavior in ether-based polymer electrolytes. We also detail the in situ curing mechanisms for several representative cyclic ether monomers and exam research progress in this area. Despite these advancements, ether-based polymer electrolytes still face certain performance limitations that impede their practical applications. To address these challenges, we propose six key areas for future research: (Fig. 10)
 | ||
| Fig. 10 Perspectives and outlooks of in situ polymerized ether-based polymer electrolytes. | ||
(1) Ionic conduction in ether-based polymer electrolytes
Research on the ionic conduction behavior of ether-based polymer electrolytes remains limited, and the mechanisms underlying the interactions between ions and polymer chains during the ionic conduction are still not fully understood. Moreover, the ionic conductivity of ether-based polymer electrolytes depends on the mobility of chain segments in the amorphous region. Increasing the amorphous proportion can effectively enhance the ionic conductivity. However, this often leads to a rapid degradation of mechanical properties. Therefore, it is crucial to develop methods that can boost ionic conductivity while preserving mechanical integrity.
(2) Regulation of ring-opening polymerization reactions of cyclic ether monomers
Residual monomers and low molecular weight polymers in ether-based polymer electrolytes can lead to continuous oxidative decomposition of the electrolytes, thereby reducing the electrochemical window and mechanical stability of the battery. Therefore, it is extremely important to regulate conditions such as temperature, initiator type, and concentration of the cyclic ether monomers during ring-opening polymerization to achieve a desirable molecular weight. Additionally, the development of novel cyclic ether monomers and modification of the polymer backbone structure through copolymerization may be effective strategies to enhance the performance of SPEs.
(3) Enhancing the long-term stability of ether-based polymer electrolytes under extreme conditions
Ether-based polymer electrolytes face significant challenges in maintaining stability over extended periods, particularly under high temperatures, high current densities, and prolonged cycling. Ether-based polymer electrolytes exhibit thermal instability, and their prolonged degradation at high temperatures can lead to severe outgassing, which adversely impacts battery performances.47 Consequently, solid-state batteries using ether-based polymer electrolytes are suitable only for a narrow operational temperature range. To improve the high-temperature stability of ether polymer electrolytes, further research is required. Furthermore, the elevated internal polarization of the cell under high current density conditions induces electrolyte degradation and severe side reactions during prolonged charge–discharge cycles. Although the ion transport within the bulk phase of the electrolyte remains relatively stable, these reactions can result in the formation of an excessively thick SEI layer or the accumulation of unwanted by-products.57 These factors impede ion transport pathways, increase internal impedance, and ultimately hinder the ionic conduction between the electrolyte and the electrode. Additionally, lithium dendrite growth on the anode can cause direct contact between the positive and negative electrodes, which disrupts the ion transport within the electrolyte and results in battery failure, presenting a serious safety risk. To make ether polymer solid electrolytes viable for broader practical applications, addressing these stability challenges under extreme conditions is essential. Developing improved electrolyte formulations and advanced SEI management strategies are crucial to enhance high-temperature performance, preventing dendrite formation, and ensuring safe, longe-lasting solid-state batteries.
(4) Broaden the electrochemical window of ether-based polymer electrolytes
The electrochemical window of the electrolyte plays a crucial role in determining the battery's energy density and ensuring its stable operation. For example, utilizing high-voltage, high-capacity cathodes, such as LiCoO2 (LCO),58 LiNixMnyCozO2,59,60 LiNi0.8Co0.15Al0.05O2 (NCA),61 in combination with lithium metal anode is an effective approach to enhance the energy density.62 However, ether polymer chains are susceptible to oxidative degradation at high voltages, which compromises battery stability. Decomposition of ether-based polymer solid electrolytes under high-voltages can damage the SEI layer, thereby promoting lithium dendrite growth and accelerating cathode degradation, which may shorten cycle lifespan and compromise battery safety. A critical safety concern is that lithium dendrite growth may cause short circuits and lead to thermal runaway, presenting significant risks to battery operation. Previous studies have shown that strategies such as polymer blending,63 cross-linking, incorporation of inorganic fillers,31 and adjustment of polymer end groups64 can improve stability. Nevertheless, the improvements achieved through these methods are still limited, indicating the need for new strategies to meet practical requirements. Moreover, ether-based polymer electrolytes exhibit good stability with lithium metal.65,66 However, the underlying mechanism remains insufficiently understood.
(5) Application of new characterization methods and theoretical calculations in ether-based polymer electrolytes
Advances in fields such as instrumentation science and computational materials science have introduced novel characterization methods to materials science. However, these methods remain underutilized in the study of ether-based polymer electrolytes. Current research remains insufficient to fully address key issues such as the ring-opening polymerization process, ionic conduction behavior in ether-based polymer electrolytes, and Li+ deposition behavior on the anode during long-term cycling. Advanced characterization techniques are needed to gain deeper insights into these challenges. Recent developments in in situ characterization methods, such as in situ scanning electron microscopy and in situ Raman spectroscopy, have introduced valuable new approaches for these investigations.67 For example, understanding the failure mechanism of lithium metal anodes in lithium metal batteries remains a major scientific challenge. While numerous theoretical models of lithium metal failure have been proposed,68,69 however, limited characterization techniques have constrained further exploration. The development of in situ characterization techniques has facilitated significant progress in this area. Recently, Zhao et al.70 studied the variation in anion concentration at the lithium metal/electrolyte interface using quantitative in situ Raman spectroscopy, providing Raman spectroscopic evidence for the quantitative changes in anion concentration at the lithium metal anode interface during lithium deposition. Their results indicate that the formation of an anion depletion layer at the electrode/electrolyte interface contributes significantly to the generation of an inhomogeneous electric field, promoting dendrite growth. Additionally, computational materials science offers promising approaches for developing cyclic ether monomers, substantially advancing research and development in ether-based polymer electrolytes.
(6) Recovery of solid electrolyte components
The recycling of ether-based polymer electrolytes has not been extensively studied. However, the recycling of electrolytes is crucial for the efficient utilization of resources. Therefore, developing efficient catalysts to promote the decomposition of ether polymers, enabling the separation of battery components and the recycling of battery materials, is an issue that urgently needs to be addressed.
Author contributions
Jialong Fu, Lu Wei and Xin Guo designed the project, contributed significant conceptual ideas, and revised the manuscript. Sisi Peng reviewed pertinent information and wrote the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to express their appreciation to the Natural Science Foundation of Hubei Province, China (Grant No. 2022CFA031).Notes and references
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 1–16 CrossRef.
- J. B. Goodenough, Energy Environ. Sci., 2013, 7, 14–18 RSC.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- J. Holoubek, H. Liu, Z. Wu, Y. Yin, X. Xing, G. Cai, S. Yu, H. Zhou, T. A. Pascal, Z. Chen and P. Liu, Nat. Energy, 2021, 6, 303–313 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- D. Cao, X. Sun, Q. Li, A. Natan, P. Xiang and H. Zhu, Matter, 2020, 3, 57–94 CrossRef.
- Z. Xiao, L. Jiang, L. Song, T. Zhao, M. Xiao, Q. Yan and L. Li, J. Energy Storage, 2024, 96, 112696 CrossRef.
- D. Zhang, S. Li, Q. Xiong, Z. Huang, H. Hong, S. Yang, J. Zhu and C. Zhi, MetalMat, 2024, 1, e13 CrossRef.
- S. Zou, Y. Yang, J. Wang, X. Zhou, X. Wan, M. Zhu and J. Liu, Energy Environ. Sci., 2024, 17, 4426–4460 RSC.
- P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 457–468 CrossRef CAS.
- A.-L. Brocas, C. Mantzaridis, D. Tunc and S. Carlotti, Prog. Polym. Sci., 2013, 38, 845–873 CrossRef CAS.
- R. Dong and J. Hao, Chem. Rev., 2010, 110, 4978–5022 CrossRef CAS.
- V. Ali, Neelkamal, F. Z. Haque, M. Zulfequar and M. Husain, J. Appl. Polym. Sci., 2007, 103, 2337–2342 CrossRef CAS.
- Y.-T. Feng, H. Zhan, H.-Y. Mi, M. F. Antwi-Afari, Y. Chen, L. Gu, B. Dong, C. Liu and C. Shen, J. Appl. Polym. Sci., 2023, 140, e54336 CrossRef CAS.
- Z. Chen, J. Xian, X. Pan, F. Ren, Y. Li, Y. Tan, Y. Bai and J. Wu, J. Mater. Chem. A, 2023, 11, 26794–26803 RSC.
- H. Zhang, J. Zhang, J. Ma, G. Xu, T. Dong and G. Cui, Electrochem. Energy Rev., 2019, 2, 128–148 CrossRef CAS.
- S. Wang, L. Zhang, Q. Zeng, J. Guan, H. Gao, L. Zhang, J. Zhong, W.-Y. Lai and Q. Wang, Adv. Energy Mater., 2024, 14, 2302876 CrossRef CAS.
- A. Hu, Z. Liao, J. Huang, Y. Zhang, Q. Yang, Z. Zhang, L. Yang and S. Hirano, Chem. Eng. J., 2022, 448, 137661 CrossRef CAS.
- F.-Q. Liu, W.-P. Wang, Y.-X. Yin, S.-F. Zhang, J.-L. Shi, L. Wang, X.-D. Zhang, Y. Zheng, J.-J. Zhou, L. Li and Y.-G. Guo, Sci. Adv., 2018, 4, eaat5383 CrossRef CAS PubMed.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- Y. Zhao, L. Wang, Y. Zhou, Z. Liang, N. Tavajohi, B. Li and T. Li, Adv. Sci., 2021, 8, 2003675 CrossRef CAS PubMed.
- Z. Li, J. Fu, X. Zhou, S. Gui, L. Wei, H. Yang, H. Li and X. Guo, Adv. Sci., 2023, 10, 2201718 CrossRef CAS PubMed.
- Y. An, X. Han, Y. Liu, A. Azhar, J. Na, A. K. Nanjundan, S. Wang, J. Yu and Y. Yamauchi, Small, 2022, 18, 2103617 CrossRef CAS.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- D. J. Brooks, B. V. Merinov, W. A. I. Goddard, B. Kozinsky and J. Mailoa, Macromolecules, 2018, 51, 8987–8995 CrossRef CAS.
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC.
- S. B. Aziz, T. J. Woo, M. F. Z. Kadir and H. M. Ahmed, J. Sci.: Adv. Mater. Devices, 2018, 3, 1–17 Search PubMed.
- R. Chen, W. Qu, X. Guo, L. Li and F. Wu, Mater. Horiz., 2016, 3, 487–516 RSC.
- M. Okada, Y. Yamashita and Y. Ishii, Die Makromol. Chem., 1964, 80, 196–207 CrossRef CAS.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef CAS.
- H. Yang, B. Zhang, M. Jing, X. Shen, L. Wang, H. Xu, X. Yan and X. He, Adv. Energy Mater., 2022, 12, 2201762 CrossRef CAS.
- H. Xu, J. Zhang, H. Zhang, J. Long, L. Xu and L. Mai, Adv. Energy Mater., 2023, 13, 2204411 CrossRef CAS.
- S. Huang, Z. Cui, L. Qiao, G. Xu, J. Zhang, K. Tang, X. Liu, Q. Wang, X. Zhou, B. Zhang and G. Cui, Electrochim. Acta, 2019, 299, 820–827 CrossRef CAS.
- Z. Li, Z. Li, R. Yu and X. Guo, J. Energy Chem., 2024, 96, 456–463 CrossRef CAS.
- H. Wu, B. Tang, X. Du, J. Zhang, X. Yu, Y. Wang, J. Ma, Q. Zhou, J. Zhao, S. Dong, G. Xu, J. Zhang, H. Xu, G. Cui and L. Chen, Adv. Sci., 2020, 7, 2003370 CrossRef CAS.
- J. Zhang, H. Wu, X. Du, H. Zhang, L. Huang, F. Sun, T. Liu, S. Tian, L. Zhou, S. Hu, Z. Yuan, B. Zhang, J. Zhang and G. Cui, Adv. Energy Mater., 2023, 13, 2202529 CrossRef CAS.
- Z. Li, R. Yu, S. Weng, Q. Zhang, X. Wang and X. Guo, Nat. Commun., 2023, 14, 482 CrossRef PubMed.
- Y. Liu, H. Zou, Z. Huang, Q. Wen, J. Lai, Y. Zhang, J. Li, K. Ding, J. Wang, Y.-Q. Lan and Q. Zheng, Energy Environ. Sci., 2023, 16, 6110–6119 RSC.
- J. Guo, X. Liu, Z. Shen, Y. Lv, X. Zhang, C. Zhang and X. Zhang, Adv. Funct. Mater., 2024, 2405951 CrossRef CAS.
- S. Wen, C. Luo, Q. Wang, Z. Wei, Y. Zeng, Y. Jiang, G. Zhang, H. Xu, J. Wang, C. Wang, J. Chang and Y. Deng, Energy Storage Mater., 2022, 47, 453–461 CrossRef.
- M. Zhang, H. Wang, A. Shao, Z. Wang, X. Tang, S. Li, J. Liu and Y. Ma, Adv. Energy Mater., 2024, 14, 2303932 CrossRef CAS.
- C. Ou, S. Ye, Z. Li, X. Zheng, F. Tian, D. Lei and C. Wang, Energy Storage Mater., 2024, 67, 103277 CrossRef.
- Z. Fang, Y. Luo, H. Liu, Z. Hong, H. Wu, F. Zhao, P. Liu, Q. Li, S. Fan, W. Duan and J. Wang, Adv. Sci., 2021, 8, 2100736 CrossRef CAS.
- K. Mu, D. Wang, W. Dong, Q. Liu, Z. Song, W. Xu, P. Yao, Y. Chen, B. Yang, C. Li, L. Tian, C. Zhu and J. Xu, Adv. Mater., 2023, 35, 2304686 CrossRef CAS PubMed.
- X. Wu, G. Ji, J. Wang, G. Zhou and Z. Liang, Adv. Mater., 2023, 35, 2301540 CrossRef CAS PubMed.
- Z. J. Baum, R. E. Bird, X. Yu and J. Ma, ACS Energy Lett., 2022, 7, 712–719 CrossRef CAS.
- W. Li, J. Gao, H. Tian, X. Li, S. He, J. Li, W. Wang, L. Li, H. Li, J. Qiu and W. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202114805 CrossRef CAS PubMed.
- W. Li, L. Ma, S. Liu, X. Li, J. Gao, S. Hao and W. Zhou, Angew. Chem., 2022, 134, e202209169 CrossRef.
- D. Jeong, D.-S. Kwon, G. Won, S. Kim, J. Bang and J. Shim, ChemSusChem, 2024, e202401010 CrossRef CAS.
- M. Ahuis, S. Doose, D. Vogt, P. Michalowski, S. Zellmer and A. Kwade, Nat. Energy, 2024, 9, 373–385 CrossRef CAS.
- Y. Cao, J. Li, H. Ji, X. Wei, G. Zhou and H.-M. Cheng, Energy Storage Mater., 2024, 70, 103475 CrossRef.
- J. J. Roy, D. M. Phuong, V. Verma, R. Chaudhary, M. Carboni, D. Meyer, B. Cao and M. Srinivasan, Carbon Energy, 2024, 6, e492 CrossRef CAS.
- Y. Liu, X. Zhang, W. Ma, H. Duan, Q. Zhao and Z. Liang, J. Energy Storage, 2024, 99, 113407 CrossRef.
- X. Wu, Y. Liu, J. Wang, Y. Tan, Z. Liang and G. Zhou, Adv. Mater., 2024, 36, 2403818 CrossRef CAS.
- C. Qi, T. Yao, W. Zhai, M. Zhang, L. Song and J. He, Energy Storage Mater., 2024, 71, 103623 CrossRef.
- Y. Wang, Y. Hou and H. Song, Polym. Degrad. Stab., 2017, 144, 17–23 CrossRef CAS.
- J.-F. Ding, R. Xu, C. Yan, B.-Q. Li, H. Yuan and J.-Q. Huang, J. Energy Chem., 2021, 59, 306–319 CrossRef CAS.
- J. Liang, Y. Sun, Y. Zhao, Q. Sun, J. Luo, F. Zhao, X. Lin, X. Li, R. Li, L. Zhang, S. Lu, H. Huang and X. Sun, J. Mater. Chem. A, 2020, 8, 2769–2776 RSC.
- Z. Wang, X. Che, D. Wang, Y. Wang, X. He, Y. Zhu and B. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202404109 CrossRef CAS PubMed.
- H. Sun, X. Xie, Q. Huang, Z. Wang, K. Chen, X. Li, J. Gao, Y. Li, H. Li, J. Qiu and W. Zhou, Angew. Chem., Int. Ed., 2021, 60, 18335–18343 CrossRef CAS PubMed.
- X. Liu, A. Mariani, T. Diemant, M. E. Di Pietro, X. Dong, A. Mele and S. Passerini, Adv. Mater., 2024, 36, 2309062 CrossRef CAS PubMed.
- S. Randau, D. A. Weber, O. Koetz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffee, T. Adermann, J. Kulisch, W. G. Zeier, F. H. Richter and J. Janek, Nat. Energy, 2020, 5, 259–270 CrossRef CAS.
- Y. Liu, Y. Zhao, W. Lu, L. Sun, L. Lin, M. Zheng, X. Sun and H. Xie, Nano Energy, 2021, 88, 106205 CrossRef CAS.
- X. Yang, M. Jiang, X. Gao, D. Bao, Q. Sun, N. Holmes, H. Duan, S. Mukherjee, K. Adair, C. Zhao, J. Liang, W. Li, J. Li, Y. Liu, H. Huang, L. Zhang, S. Lu, Q. Lu, R. Li, C. V. Singh and X. Sun, Energy Environ. Sci., 2020, 13, 1318–1325 RSC.
- S. Xiao, L. Ren, W. Liu, L. Zhang and Q. Wang, Energy Storage Mater., 2023, 63, 102970 CrossRef.
- J. Xiang and Y.-C. Lu, ACS Nano, 2024, 18, 10726–10737 CrossRef CAS PubMed.
- J. Pu, C. Zhong, J. Liu, Z. Wang and D. Chao, Energy Environ. Sci., 2021, 14, 3872–3911 RSC.
- P. Li, Z. Fang, X. Dong, C. Wang and Y. Xia, Natl. Sci. Rev., 2022, 9, nwac031 CrossRef CAS.
- W. Fan, H. Wang and J. Wu, eScience, 2024, 4, 100248 CrossRef.
- J. Liu, H. Hua, J. Lin, Y. Deng, N. Pei, P. Zhang, J.-C. Dong, J.-F. Li and J. Zhao, Energy Environ. Sci., 2024, 17, 5993–6002 RSC.
| This journal is © The Royal Society of Chemistry 2025 |