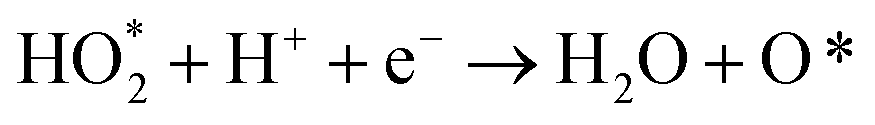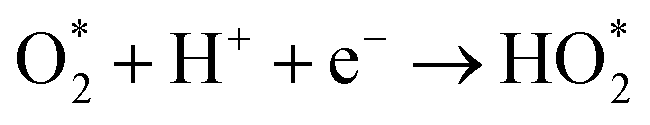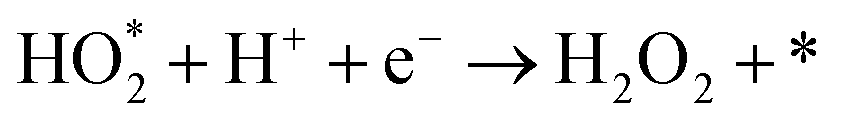DOI:
10.1039/D4TA01475H
(Paper)
J. Mater. Chem. A, 2024,
12, 11277-11285
S-doped TiN supported N, P, S-tridoped TiO2 with hetero-phase junctions for fuel cell startup/shutdown durability†
Received
3rd March 2024
, Accepted 17th April 2024
First published on 17th April 2024
Abstract
TiO2 layers codoped with N and P formed on S-doped TiN are recently developed platinum-group metal (PGM)-free catalysts (N, P-TiO2/S-TiN) for the oxygen reduction reaction (ORR) in acidic media. Conventional Pt-based catalysts lack durability as the necessary carbon supports are oxidized at high potentials (>1.0 V) during startup and shutdown. N, P-TiO2/S-TiN catalysts do not require carbon supports and are expected to tolerate oxidation at high potentials. However, N, P-TiO2/S-TiN loses ORR activity during startup or shutdown at potentials >1.0 V due to removal of N- and P-atoms. We therefore report a new pathway to enhance durability and ORR activity of N, P-TiO2/S-TiN. By annealing N, P-TiO2/S-TiN with NH4F under N2, ORR active anatase/rutile TiO2 hetero-phase junctions are produced in bulk with remaining TiN supports, and the surface O-atoms in the TiO2 lattice are substituted by S2−. S-doped TiN-supported N, P, S-tridoped TiO2 exhibits the highest ORR activity among reported oxide/oxynitride catalysts, although the ORR activity of the N, P, S-TiO2/S-TiN catalyst remains lower than that of state-of-the-art carbon-based catalysts. The N, P, S-TiO2/S-TiN catalyst exhibits superior durability among PGM-free catalysts as the anionic dopants are not removed after 5000 potential cycles (1.0–1.5 V), leading to a 0.02 V reduction in half-wave potential.
1. Introduction
The global transport sector currently accounts for one-quarter of energy consumption overall, and is responsible for approximately 40% of carbon emissions from end-use sectors.1 In 2021, road vehicles emitted 5.9 gigatons of CO2, representing three-quarters of all CO2 emissions from the transport sector.1 By the end of 2020, the total number of vehicles globally reached 1.5 billion, including 1.1 billion passenger vehicles and 0.4 billion buses or trucks.2 Government regulations to limit emissions from passenger vehicles are becoming increasingly stringent, and some countries and regions plan to phase out passenger vehicles powered by the conventional internal combustion engine (ICE) between 2025 and 2040.3
Polymer electrolyte fuel cells (PEFCs) have emerged as a promising alternative to ICEs in light-duty passenger vehicles for journeys over 300 miles,4 as well as in mid- to heavy-duty vehicles.5 Although PEFC-powered passenger vehicles have been available since 2014, they still constitute a small percentage of the current market share. At present, carbon-supported platinum (Pt/C) and platinum-cobalt (PtCo/C) catalysts are the most common means to catalyze the hydrogen oxidation reaction (HOR) at the anode and the oxygen reduction reaction (ORR) at the cathode of PEFC catalyst layers, respectively.5,6 Cathode platinum loading is typically four-times higher than that of the corresponding anode due to the slower kinetics of ORR compared with HOR.7 The high platinum loading requirement is assumed to be the greatest cost barrier to the widespread adoption of fuel cell vehicles (FCVs).8 Therefore, efforts have been made over the last two decades to reduce the amount of platinum required in PEFCs by an order of magnitude, to ∼20 g per 128 kW class passenger vehicle.4,6 However, a further reduction to ∼6 g per vehicle is necessary to make PEFC-powered passenger vehicles affordable and allow them to become widespread.7
In addition to the high costs, carbon-supported platinum-based catalysts currently lack stability, with the instability of platinum/platinum-alloy catalysts and corrosion of carbon supports being identified as major challenges in improving the durability of PEFC stacks.8 In an ideal PEFC, the anode and cathode reactions would be described as follows (eqn (1) and (2)):
| | | HOR at anodes: H2 → 2H+ + 2e− | (1) |
| | | ORR at cathodes: O2 + 4H+ + 4e− → 2H2O | (2) |
During the startup or shutdown of PEFCs, the anode is contaminated with oxygen molecules originating either from the cathode or the air. Later, the contaminating oxygen molecules are reduced to water as the anode Pt/C catalyzes the ORR (eqn (2)), giving rise to a counter cathode potential of up to ∼1.5 V via a reverse current decay mechanism.9 Carbon blacks in a PtCo/C cathode are oxidized viaeqn (3) at such high potentials.
| | | C + 2H2O → CO2 + 4H+ + 4e− | (3) |
The PtCo nanoparticles on the carbon supports can no longer be used once the carbon supports are corroded. To avoid this issue, carbon supports are usually protected by system-level measures, such as reducing the cathode air flow rate during shutdown to minimize O2 diffusion to the anode and injecting a small amount of H2 to the anode during FCV off-time to react with O2 contaminants.10 However, as these measures further increase the cost of FCVs, considerable efforts have been made over the last two decades to develop carbon-free materials as supports for platinum or platinum alloys.11–14
The high loading of expensive and scarce platinum catalysts and the low durability of carbon supports are the major motivations to seek cathode catalysts that do not use platinum-group metals (PGMs) or carbon supports. Researchers in the PGM-free catalyst community have developed so-called M/N/C catalysts, which are graphitic carbons with abundant defects doped with N and one or two metals (M); typically Fe,15–20 Co,21 or Mn.22 The highest activity reported to date has been seen with Fe/N/C catalysts, although durability remains an issue, as explored in the literature. The formation of carbon dioxides by the oxidation of carbon species has been experimentally verified, even at potentials below 1.0 V,16,18,21 and is regarded as one of the pivotal causes of the degradation of M/N/C catalysts. To minimize carbon oxidation, a recent standardized protocol for evaluating the activity of M/N/C catalysts has limited the upper potential to 0.925 V, which is lower than that for automotive carbon-supported platinum-cobalt catalysts (0.95 or 1.0 V).20 The equilibrium potential in eqn (3) with respect to a standard hydrogen electrode is 0.207 V, and the reaction rate in eqn (3) is accelerated at high potentials. Thus, even when M/N/C catalysts are used, the system-level measures for protecting the carbon blacks of PtCo/C catalysts, as mentioned above, are also necessary.
Oxide/oxynitride-type catalysts containing group IV or V metals have been less well studied in the field of PGM-free catalysts owing to their low conductivity when compared with M/N/C catalysts; a property which also acts as a barrier for activity evaluation.23 Zirconium oxynitride (ZrOxNy) nanoparticles have shown the best activity in this type of catalyst after being supported on conductive multiwall carbon nanotubes (MWCNTs). However, as MWCNTs were found not to be sufficiently conductive, carbon blacks were added to ZrOxNy-MWCNT catalysts to enhance the conductivity in the catalyst layer.24 Nevertheless, the resulting durability was still insufficient, even below 1.0 V.24
We have thus shifted our attention toward nitrogen-doped titanium dioxide (TiO2) layer catalysts formed on conductive titanium nitride (TiN) particles without employing any carbon supports.25–29 Various control experiments confirmed that both the activity and conductivity of the catalyst originated from TiN, not from the carbon residues of the urea precursor.25,26 Recently, P- and N- atoms were codoped into the surface TiO2 layer on TiN.27,28 This led to enhanced activity and durability due to the augmented surface phosphorous doping level, and the catalyst exhibited no degradation after repeated load cycling tests at potentials of 0.6 and 1.0 V.28 Since the catalyst was free from carbon supports, there was no corrosion of carbon support as shown in eqn (3); however, after startup/shutdown tests in which the potential was cycled between 1.0 and 1.5 V, the activity deteriorated significantly due to removal of surface P- and N-atoms.27,28 In particular, a large number of N-atoms substituting for O-atoms in the TiO2 lattice were removed after 5000 cycles.28 Thus, high-cost measures to maintain the potential below 1.0 V are still necessary for the utilization of P- and N-codoped TiO2 catalysts (hereafter denoted as N, P-TiO2/S-TiN as bulk TiN contains sulfur species from the precursor, which is described later), despite the absence of carbon supports.
Here, we report a new strategy for the dramatic enhancement of both ORR activity and startup/shutdown durability above 1.0 V by introducing (1) hetero-phase junctions from two different TiO2 phases (anatase and rutile in bulk) and (2) a new dopant (S-atoms which substituted for O-atoms in the surface TiO2). This approach resulted in higher ORR activity and durability than reported for other oxide/oxynitride catalysts. In particular, durability during startup/shutdown cycles ranks the highest among the PGM-free catalysts, which opens the door to significantly reducing the production and operating costs of FCVs.
2. Experimental methods
2.1. Catalyst synthesis
The N, P-TiO2/S-TiN catalysts were synthesized using a recently reported solution phase combustion route,28 and the details of catalyst synthesis and characterization methods are described in S1; ESI.† Briefly, powders of titanium oxysulfate, urea, and hypophosphorous acid solution were mixed in 1.0 mol dm−3 hydrochloric acid solution at room temperature. The mass ratio of urea to titanium oxysulfate-derived TiO2 was set at 100. The atomic ratio of phosphorous to titanium was set at 0.2, which is the optimized value for the activity.28 The dispersion was then heated with continuous mixing, followed by drying. The dried powders were pyrolyzed at 1173 K for 2 h under N2 gas. Then the obtained catalysts and ammonium fluoride (NH4F) powders were ground in an agate mortar and annealed under N2 gas at various temperatures and for different durations to yield TiO2 catalysts doped with N-, P- and S-atoms; hereafter denoted as N, P, S-TiO2/S-TiN.
2.2. Characterization
The bulk and outermost surface structures of catalysts were investigated by analyzing X-ray diffraction (XRD) patterns and ultraviolet (UV)-Raman spectra, respectively. The chemical states of the catalysts were determined by X-ray photoelectron spectroscopy (XPS). The peak shifts due to surface charge were corrected using the binding energy of C 1s (284.8 eV), originating from the hydrocarbon contaminants in the spectrometer or air. The morphology of the N, P, S-TiO2/S-TiN catalysts was investigated by transmission electron microscopy (TEM) images. Elemental compositions were evaluated using energy dispersive X-ray spectroscopy (EDS).
2.3. ORR activity, selectivity, and durability measurements
Rotating disk electrode (RDE) and rotating ring disk electrode (RRDE) voltammograms were obtained to evaluate the ORR activity and selectivity, respectively, of the catalysts. The mass fraction of Nafion in the catalyst layer was set at 0.05 and the catalyst loading was set at 0.86 mg cm−2 unless otherwise noted. A conventional three-electrode cell was used for the room-temperature electrochemical measurements performed in 0.1 mol dm−3 H2SO4. After sequentially bubbling O2 and N2 for 1800 s, RDE and RRDE voltammograms were recorded by applying a disk potential (E) of 0.05–1.2 V versus the reversible hydrogen electrode (RHE) at a scan rate of 5 mV s−1 and a rotation speed of 1500 rpm. The ring potential was maintained at 1.2 V to obtain RRDE voltammograms used to calculate the hydrogen peroxide yield, XH2O2. The ORR was measured by j = jO − jN, the difference between the current per unit geometrical area, S, of the disk electrode obtained in N2 (jN = INS−1) and in O2 (jO = IOS−1).
3. Results and discussion
3.1. Catalyst synthesis and crystal structure, morphology
Fig. 1(a) schematically depicts the synthesis of the N, P, S-TiO2/S-TiN catalysts (see also S1; ESI†) and the roles of dopants and supports. First, N, P-TiO2 layer catalysts were formed on S-TiN by the pyrolysis of precursor powders prepared from titanium oxysulfate, urea, and hypophosphorous acid using the previously reported solution phase combustion route.28 The N, P-TiO2/S-TiN catalysts were then mixed with NH4F and annealed under N2 gas. The surface N, P-TiO2/S-TiN was etched during NH4F-annealing due to the decomposed HF from NH4F. Sulfur species inside the bulk TiN appeared on the surface as substitutional dopants for TiO2. Furthermore, the bulk TiN was oxidized to display hetero-phase junctions with two TiO2 polymorphs: anatase and rutile.
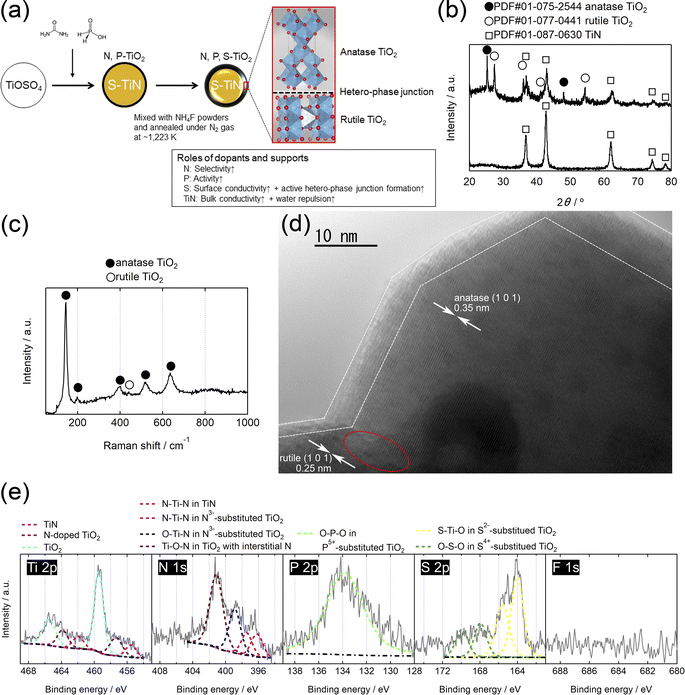 |
| | Fig. 1 (a) Schematic diagram for the synthesis of N, P, S-TiO2/S-TiN and the roles of dopants and S-TiN supports, (b) X-ray diffraction (XRD) patterns of (top) N, P, S-TiO2/S-TiN after NH4F-annealing at 1223 K and (bottom) N, P-TiO2/S-TiN before NH4F-annealing, (c) Ultraviolet (UV)-Raman spectrum, (d) transmission electron microscopy (TEM) image, and (e) X-ray photoelectron (XP) spectra of N, P, S-TiO2/S-TiN. | |
The effect of the NH4F-annealing on the bulk crystal structures of the catalysts was evaluated using XRD patterns, as shown in Fig. 1(b). Similar to previous studies on N, P-TiO2/S-TiN (Fig. S1, ESI†),27,28 a single TiN phase is observed before NH4F-annealing. Both anatase and rutile TiO2 phases appear after the NH4F-annealing at 1223 K under N2 gas. The process is as follows: NH4F decomposes at ∼398 K to produce NH3 and HF gases.30 Subsequently, part of the TiN reacts with HF to form TiF3 at elevated temperatures in the range 473–573 K.31 TiF3 is not a stable chemical, and the results shown in Fig. 1(b) indicate that it reacts with trace O2 molecules contaminating the N2 gas during annealing to form anatase/rutile TiO2. Indeed, the single TiN phase persists even when the gas flowing during annealing was changed to 10% v/v H2/Ar (Fig. S3, ESI†). Thus, the N2 atmosphere is necessary to produce a mixture of anatase/rutile TiO2 and TiN phases during the annealing with NH4F. The crystal structure of N, P, S-TiO2/S-TiN on the top surface is different from the bulk, as shown in Fig. 1(c). As the ORR is a surface reaction, it was evaluated using ultraviolet (UV)-Raman spectroscopy, which is highly sensitive to surfaces.32 Five clear peaks are observed at ∼145, 198, 401, 521, and 636 cm−1, which are assigned to the Eg, Eg, B1g, B1g, and Eg vibration modes of anatase TiO2, respectively.32,33 A weak peak at ∼441 cm−1 is an Eg vibration mode of rutile TiO2.33 The absence of peaks from TiN indicates that the surface of TiN was oxidized to form TiO2 layers. The morphology of these different phases in N, P, S-TiO2/S-TiN was characterized using TEM. Clear lattice fringes with interplanar distances of 0.35 and 0.25 nm are observed at the inner cores in Fig. 1(d) and S4 (ESI),† which correspond well to the (1 0 1) plane of anatase and rutile TiO2, respectively. These two phases are directly connected to each other via a hetero-phase junction, as indicated by the circle. In addition, the outer surfaces indicated by the dashed lines are disordered to show no clear fringes. Such disordered surfaces are known to promote ORR activity of N, P-TiO2/S-TiN28 and N-TiO2/S-TiN.34 Other plane combinations of anatase (1 0 1)-rutile (1 1 0) and anatase (1 0 1)-TiN (2 0 0) are displayed in Fig. S4 (ESI),† and these different phases are also directly connected to each other via the hetero-phase junction. These results indicate that the bulk of the N, P, S-TiO2/S-TiN is composed of anatase/rutile TiO2 and TiN and contains hetero-phase junctions between anatase and rutile or anatase and TiN. On the other hand, the outermost surface consists mostly of TiO2.
3.2. Chemical states
The oxidized outermost surface is also verified by XPS, as shown in Fig. 1(e). The Ti 2p level splits into Ti 2p3/2 and 2p1/2 sublevels to display doublets at three different binding energies. In the Ti 2p3/2 region, the highest peak at ∼459 eV is assigned to Ti-atoms in the TiO2 lattice while weak shoulders at ∼457 and ∼455 eV are assigned to those in N-doped TiO2 and TiN, respectively.35,36 Thus, the surface is mostly TiO2 phases, which is consistent with the results of UV-Raman analyses. Four different chemical states are observed from the doped N-atoms. The highest peak at ∼401 eV is assigned to interstitial N-atoms in TiO2, while two peaks at ∼399 and ∼397 eV are substitutional N-atoms in TiO2.37–39 The peak at a low binding energy of ∼396 eV is assigned to N-atoms in TiN.35,36 Therefore, most of the N-atoms on the surface are embedded in the TiO2 lattice. In the P 2p region, a peak is observed at ∼134 eV, which is assigned to cationic pentavalent phosphorous atoms (P5+) which substituted for Ti-atoms in TiO2.40 No peaks are observed at ∼129 eV, indicating the absence of anionic phosphorous atoms (P3−). These chemical states were also observed in our previous works on N, P-TiO2/S-TiN without the NH4F-annealing used in this study (Fig. S2, ESI†),27,28 although an unexpected difference is observed in the S 2p spectrum. The clear doublets at ∼164 and ∼166 eV are the S 2p3/2 and 2p1/2 peaks, respectively, of divalent anionic sulfur atoms (S2−) substituted for O-atoms in the TiO2 lattice, while the smaller peaks at ∼168 and ∼170 eV with higher binding energy are the S 2p3/2 and 2p1/2 peaks, respectively, which are tetravalent cationic sulfur atoms (S4+) substituted for Ti-atoms in TiO2.41,42 The binding energy of anionic sulfur species doped into TiN is lower than that observed in Fig. 1(e), ∼163 eV,43 ruling out the presence of such species on the surface of N, P, S-TiO2/S-TiN. Unlike N-doped TiO2, no clear peak shifts in the Ti 2p regions have been reported for TiO2 doped with S2− and S4+.41 In XPS, no peak was detected from the S 2p region in the previous N, P-TiO2/S-TiN catalysts without NH4F-annealing (Fig. S2, ESI†)27,28 or in the current N, P-TiO2/S-TiN before NH4F-annealing (Fig. S5, ESI†). However, sulfur species were detected on EDS in the N, P-TiO2/S-TiN before NH4F-annealing, with an atomic ratio of sulfur to titanium of 0.075 ± 0.007. These results indicate that sulfur species from titanium oxysulfate precursors are absent on the top surface before NH4F-annealing but present in the lower layers, since XPS is much more surface sensitive than EDS. As mentioned above, NH4F should decompose to form NH3 and HF gases during annealing30 and the latter product, HF, is a well-known etchant. During annealing, the sulfur-free surface of N, P-TiO2/S-TiN was etched with HF to expose S-atoms from inside N, P-TiO2/S-TiN to the surface. Indeed, the TiN content inside N, P-TiO2/S-TiN was decreased with increasing mass ratio of NH4F to N, P-TiO2/S-TiN (Fig. S6, ESI†). The TiN is necessary to give the catalyst conductivity and to improve the ORR activity (Fig. S6, ESI†); however, after NH4F-annealing, the surface is an N, P, S-tridoped TiO2 phase. NH3, another product of NH4F-decomposition, further decomposes to N2 and H2 at high temperatures, which would be expected to produce a reducing atmosphere, and most of the surface S-atoms are in the reduced state, S2−, rather than the oxidated state, S4+. The EDS analyses on N, P, S-TiO2/S-TiN revealed that an atomic ratio of fluorine to titanium is at 0.000 ± 0.000. These results and F 1s spectrum shown in Fig. 1(e) indicate that there are no fluorine species in the bulk or surface of N, P, S-TiO2/S-TiN. Another possible role of the subsurface TiN is to “support” facilitating ORR process. Seifitokaldani et al. reported that TiN (2 0 0) facet showed a high activity for both (i) converting OH intermediate to water molecules and (ii) removing the produced water molecules during ORR by using a density functional theory (DFT) calculation.44 These two steps are necessary for the ORR after the first dissociative adsorption of O2 molecules which proceeded on the surface N, P, S-TiO2. Particularly, the step (ii) could proceed faster on TiN (2 0 0) facet than the N, P, S-TiO2 as TiO2 is hydrophilic in general. Therefore, TiN could support ORR by both giving the catalyst conductivity and helping the process, particularly by removing the produced water molecules.
3.3. ORR activity and selectivity
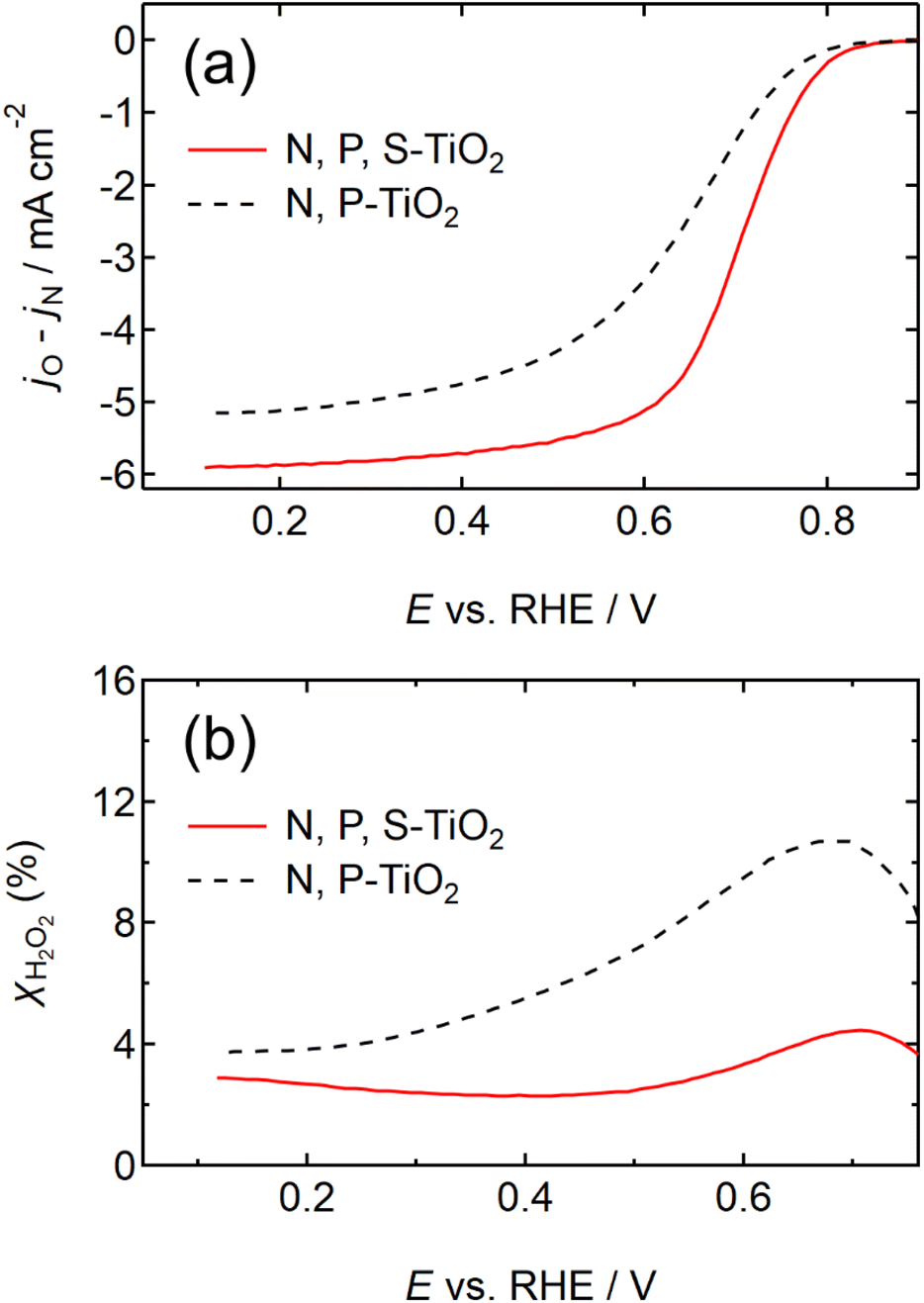
The NH4F-annealing conditions such as temperature, duration, N2-gas flow rate, and mass ratio of NH4F to N, P-TiO2/S-TiN were systematically optimized to maximize the ORR activity (Fig. S6 and S7, ESI†). The RDE voltammograms of the optimized N, P, S-TiO2/S-TiN and previously reported N, P-TiO2/S-TiN28 are shown in Fig. 2(a). The j from N, P, S-TiO2/S-TiN outperforms N, P-TiO2/S-TiN at any E. The half-wave potential (i.e., E where half of the maximum j is obtained) has been used to measure the ORR activity of catalysts, and hereafter is denoted as E1/2. The E1/2 of N, P, S-TiO2/S-TiN reached 0.72 V, which is 0.08 V higher than that of N, P-TiO2/S-TiN, and higher than that of any other carbon-support-free, or even carbon-supported, oxide/oxynitride catalysts reported to date, including ZrOxNy-MWCNT24 (Table S1, ESI†). If ORR proceeds via a two-electron reaction, hydrogen peroxide is produced (eqn (4)).
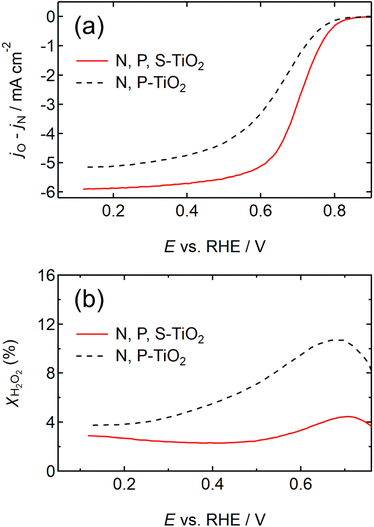 |
| | Fig. 2 (a) Rotating disk electrode (RDE) voltammograms and (b) hydrogen peroxide yield versus potential (XH2O2– E) curves of N, P, S-TiO2/S-TiN catalyst after NH4F-annealing at 1223 K and N, P-TiO2/S-TiN catalyst. The N, P-TiO2/S-TiN catalyst's curves were reproduced with permission.28 Copyright 2020, American Chemical Society. The scans were performed in N2 and O2 atmospheres at a rotation speed of 1500 revolutions per minute (rpm) and a cathodic scan rate of −5 mV s−1 in 0.1 mol dm−3 H2SO4 solution. | |
The produced H2O2 molecules decompose membranes and a perfluorosulfonate ionomer in catalyst layers to deteriorate the performance of PEFCs.45 The H2O2 yield measured by RRDE voltammograms, XH2O2, is plotted as a function of E in Fig. 2(b). Less than half of XH2O2 is obtained from N, P, S-TiO2/S-TiN when compared with N, P-TiO2/S-TiN at E ≥ 0.6 V, which is an operating potential range for FCVs.46 Therefore, N, P, S-TiO2/S-TiN displays higher ORR activity and selectivity toward the four-electron reaction than those of previously reported N, P-TiO2/S-TiN. The N, P, S-TiO2/S-TiN was synthesized by annealing N, P-TiO2/S-TiN and NH4F under N2 gas. The NH4F-annealing produced etchant HF to (1) convert a single TiN phase of N, P-TiO2 to a mixture of anatase/rutile TiO2 and TiN phases of N, P, S-TiO2/S-TiN and (2) expose S-atoms from the inside of N, P-TiO2/S-TiN to the surface of N, P, S-TiO2/S-TiN, where they mostly substituted for O-atoms in the TiO2 lattice. In previous reports of carbon-support-free N, P-TiO2/S-TiN or N-TiO2/S-TiN synthesized via the solution phase combustion route used in this study,27,28 all XRD patterns displayed a single TiN phase, while the surface was rutile TiO2 phase according to Raman analyses with a visible laser (Fig. S1, ESI†). When the flowing gas was N2 during the NH4F-annealing, anatase/rutile TiO2 phases appeared in N, P, S-TiO2/S-TiN due to oxygen contaminants in N2 flowing in the tube furnace. However, when the flow gas was changed to 10% v/v H2/Ar instead of N2 with an identical gas flow rate, the oxygen contaminants were reduced by H2, leaving a single TiN phase (Fig. S3, ESI†). As a result, much lower activity was observed when the flow gas was 10% v/v H2/Ar gas than when N2 was used (Fig. S3, ESI†). These results indicate that a new active site could be formed in the anatase/rutile TiO2 phases of N, P, S-TiO2/S-TiN, which is in contrast to previously reported N, P-TiO2/S-TiN and N-TiO2/S-TiN catalysts. Anatase/rutile hetero-phase junctions have recently been used to enhance the electrocatalytic activity of TiO2 for the oxygen evolution reaction (OER)47 and the selective nitrate reduction to ammonia.48 Since the conduction band minimum of rutile is higher than that of anatase, electrons are transferred from rutile to anatase and rutile becomes positively charged.47,48 The positively charged site acts as a Lewis acid, attracting and dissociating the O-atoms of the O2 molecule,49 which is the first step for the ORR. Furthermore, the d-band center of Ti in rutile relative to the Fermi level is upshifted by the electron transfer to anatase. In general, the upshift/downshift of d-band center of transition metals strengthens/weakens the interaction between the metal and intermediates formed during the catalytic reactions.50–54 When the interaction between reaction intermediates and the catalyst is too strong, the desorption of the intermediates or the next reaction step becomes the rate determining step to delay the overall reaction. On the other hand, too weak interaction also delays the reaction. An optimum adsorption strength of intermediates for various catalytic reactions on several PGM and PGM-free catalysts has been suggested; hydrogen evolution reaction (HER) on P-doped TiO2 supported Ru cluster catalyst,50 ORR on various binary/ternary oxide supported Pt catalysts,51 ORR on activated N-doped porous carbon supported Pt skin shell covered on Pt3Co bimetallic core catalyst,52 ORR and OER on Co3O4 shell covered on Co core catalyst53 and ORR on Zn–N–C catalyst.54 Four-electron (eqn (2)) and two-electron ORR (eqn (4)) in acidic media have been assumed to proceed via several elementary steps described by eqn (2a)–(2e) and (4a)–(4c), respectively on conventional catalysts55,56 and oxynitride catalysts.57
Four electron ORR
| | 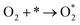 | (2a) |
| | 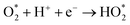 | (2b) |
| | 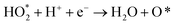 | (2c) |
| | | HO* + H+ + e− → H2O + * | (2e) |
Two electron ORR
| | 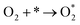 | (4a) |
| | 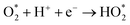 | (4b) |
| | 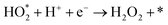 | (4c) |
where * is the active site. After the adsorption of O
2 molecules on the positively charged rutile surface of N, P, S-TiO
2/S-TiN, a peroxy intermediate HO
2 is produced in both four electron and two electron ORR processes (
eqn (2b) and
(4b)). When the interaction between HO
2 and * is too weak or too strong, it inhibits the breaking of O–O bonding in the HO
2 to delay ORR process to favor two electron reactions. Cheng group recently reported that positively charged Zn atoms in their Zn–N–C catalyst facilitate the adsorption of O
2 and reaction intermediates by the upshift of the d-band center of Zn to promote electron transfer and enhance the intrinsic ORR activity in alkaline media.
54 Although the ORR pathway is different between the alkaline media and acidic counterparts used in this study, the HO
2 intermediate is also produced during ORR in alkaline media. Therefore, the mechanism similar to the positively charged Zn atoms in Zn–N–C catalysts reported by Cheng group is expected for positively charged rutile TiO
2. As the rutile at hetero-phase junctions interacts with HO
2 strongly owing to the upshift of d-band center of Ti as electrons are transferred from the rutile to anatase, it could facilitate adsorption of HO
2 to enhance ORR activity and selectivity towards four electron reaction (
eqn (2b) and
(2c)). In contrast, anatase directly connected to rutile
via the hetero-phase junction is expected to interact with HO
2 weaker than the rutile as the d-band center of Ti in anatase downshifted owing to the transferred electrons from rutile. If the interaction between rutile and HO
2 is too strong, ORR can proceed on anatase in the vicinity of rutile due to the weaker adsorption strength of anatase than rutile. Therefore, one possible mechanism for the enhancement of ORR activity on N, P, S-TiO
2/S-TiN could be hetero-phase junction formation. Another new product by NH
4F-annealing, substitutional S
2− doping into surface TiO
2, could enhance the electrical conductivity of surface TiO
2, which has been used in S
2−-doped TiO
2 for sodium ion battery anodes
41 and N
3−, S
2− codoped TiO
2 for high-rate lithium ion battery anodes.
42 Indeed, N, P-TiO
2/TiN synthesized from sulfur-free titanium source shows higher Tafel slope and lower ORR activity than N, P, S-TiO
2/S-TiN even after the NH
4F-annealing under the conditions identical to those of N, P, S-TiO
2/S-TiN (Fig. S8, ESI
†). Another role of sulfur is to inhibit the complete phase transition of anatase to rutile at the high annealing temperature to leave anatase/rutile hetero-phase junctions, which are active toward ORR. Sulfur-doping is known to increase the anatase content in TiO
2 synthesized
via both a low temperature hydrothermal route
58 and a high temperature annealing route.
59 Similar to these previous reports, anatase content in N, P, S-TiO
2/S-TiN is higher than rutile counterpart even after the high temperature NH
4F-annealing at 1223 K as shown in
Fig. 1(b). Therefore, substitutional S
2− doping indirectly tuned the strength of ORR intermediates with TiO
2 as mentioned above by forming the anatase/rutile hetero-phase junctions to keep the anatase phase after the NH
4F-annealing. Precise control of the adsorption strength of ORR intermediates by for example, controlling the amount of oxygen vacancy
53 is our next research topic. The grain size of TiO
2 is also reported to be decreased by sulfur-doping to increase the surface area.
58
Based on the results obtained in this study and previous studies on N, P-TiO2/S-TiN,27,28 the roles of three dopants, N, P and S are proposed as follows. Substitutional N3− and P5+ dopants have promoted ORR selectivity and activity, respectively of N, P-TiO2/S-TiN.28 After NH4F annealing used in this study, the surface TiO2 is doped with S2− anions. The S2− substituted for O-atoms in TiO2 to increase both the surface conductivity and anatase content, which is the origin of the significantly enhanced ORR activity and selectivity towards four electron reaction via forming the anatase/rutile hetero-phase junctions. S-doped TiN “supports” both the bulk conductivity60 and facilitating ORR via removing water from the active sites continuously44 even after the durability tests, which are shown in the next subsection.
3.4. Durability against fuel cell startup and shutdown protocol
As N, P, S-TiO2/S-TiN catalysts are free from carbon supports, they are expected to be tolerant to the high potentials faced by FCVs during startup and shutdown. The durability of N, P, S-TiO2/S-TiN, which showed the highest activity level, was investigated using the common protocol for FCVs proposed by the Fuel Cell Commercialization Conference in Japan (FCCJ) in 2011.46 The E was cycled between 1.0 and 1.5 V at 0.5 V s−1 to simulate the startup/shutdown of FCVs.46 The RDE voltammograms before and after 5000 cycles are shown in Fig. 3. To compare the startup/shutdown durability of N, P, S-TiO2/S-TiN catalyst with other limited number of non-PGM catalysts reported to date, the cycle number was selected at 5000 as most works performed below 5000 of startup/shutdown cycles (Table S2, ESI†). Excellent durability within the stringent protocol is observed from N, P, S-TiO2/S-TiN, showing only a 0.02 V decrease in E1/2 after 5000 cycles, which is one-quarter of that shown by N, P-TiO2/S-TiN under identical conditions (0.08 V).28 As mentioned in the introduction, carbon oxidation leads to a severe loss of activity in both PtCo/C and PGM-free catalysts. Most studies into PGM-free catalysts investigated durability with a protocol based on <1.0 V (typically between 0.6 and 1.0 V). A limited number of publications have investigated durability to potentials >1.0 V, with a comparison of changes in E1/2 during potential cycling provided in Table S2 (ESI†). State-of-the-art Fe/N/C catalysts display significant E1/2 losses, and often show no limiting current plateaus. The durability of N, P, S-TiO2/S-TiN to the startup/shutdown protocol ranks the highest among PGM-free catalysts. While the initial activity of N, P, S-TiO2/S-TiN (E1/2 of 0.72 V) is still lower than that of state-of-the-art M/N/C catalysts,19,21,22 its durability against startup/shutdown cycles remains the highest. A similar durability was also observed for two other N, P, S-TiO2/S-TiN catalysts synthesized under different conditions (Fig. S9, ESI†). The extent of titanium ions dissolved from all three N, P, S-TiO2/S-TiN catalysts in the electrolyte solution after 5000 cycles, as shown in Fig. 3 and S9 (ESI),† was below the detection limit of the inductively coupled plasma (ICP) spectrometer, indicating excellent stability of N, P, S-TiO2/S-TiN in bulk. The bulk stability suggests the excellent durability of S-TiN supports. Although the surface conductivity of N, P, S-TiO2/S-TiN is significantly enhanced by S2−-doping into TiO2 as discussed in the previous subsection, S-TiN supports should be the origin of bulk conductivity as a well-known metallic nitride, TiN displays 17 orders of magnitude higher electrical conductivity than that of the semiconductor TiO2.60 The stable activity shown in Fig. 3 and S9† and the ICP analyses on the electrolyte solution after the tests indicate that S-TiN did not dissolve to keep the conductivity during the 5000 startup/shutdown cycles.
 |
| | Fig. 3 RDE voltammograms of N, P, S-TiO2/S-TiN after NH4F-annealing at 1133 K before (solid curve) and after (dashed curve) 5000 potential cycles between 1.0 and 1.5 V vs. reversible hydrogen electrode (RHE) at 0.5 V s−1 in 0.1 mol dm−3 H2SO4 solution. Before and after 5000 cycles, the scans were performed in N2 and O2 atmospheres at a rotation speed of 1500 rpm and a cathodic scan rate of −5 mV s−1 for evaluating the activity. For reference, RDE voltammograms of N, P-TiO2/S-TiN before (dash dotted curve) and after (dash double dotted curve) 5000 cycles are also shown with permission.28 Copyright 2020, American Chemical Society. | |
The surface stability was next examined by XPS, as shown in Fig. 4. The Ti 2p, N 1s and S2− peaks in the S 2p spectra remained unchanged after 5000 cycles, indicating that these surface chemical states are highly stable. Due to the use of Nafion in the catalyst layer, the stability of S4+ cannot be evaluated for the N, P, S-TiO2/S-TiN catalyst after 5000 cycles because the contribution of the sulfonate group of Nafion and N, P, S-TiO2/S-TiN cannot be separated. The P 2p spectrum changed to indicate loss of P5+ after 5000 cycles. These results suggest that at least the Ti-atoms and anionic dopants which substituted for O-atoms in the TiO2 lattice are stable. In previous studies on N, P-TiO2/S-TiN, the Ti 2p, N 1s and P 2p spectra all changed significantly during 5000 cycles from 1.0 to 1.5 V, with the loss of N- and P-atoms from the surface of the TiO2 lattice.27,28 Chen et al. reported that oxidative annealing of the N-doped TiO2 in air stabilized N-atoms in the TiO2 lattice.61 Although air-annealing reduced the amount of nitrogen in TiO2, the surviving N-atoms were stable and were not removed during the photocatalytic oxidation of ethylene under highly oxidizing conditions. As a result, the Ti 2p and N 1s spectra of the N-doped TiO2 did not change significantly during the photocatalytic oxidation of ethylene, and no significant change in photocatalytic performance was observed.61 In the work of Chen et al.,61 the oxidant that removes the N-atoms from the TiO2 lattice without the air-annealing was photogenerated holes (2N3− + 6h+ → N2) produced by visible light irradiation from a 500 W Xe-arc ramp.62 This differs from the oxidant used in this work; i.e., high potential sites in acidic media. However, one reason for the stabilization of N-atoms in the TiO2 lattice of N, P, S-TiO2/S-TiN is that NH4F-annealing oxidatively forms the TiO2 phases from TiN, during which N-atoms are embedded in the TiO2 lattice. The results from Chen et al.61 and this study suggest that to avoid oxidation of the doped TiO2-based materials during the oxidative catalytic conditions, an oxidative annealing of the material before use will be an effective pathway. Indeed, these phases are sufficiently stable that the XRD patterns remain unchanged after ORR (Fig. S10, ESI†). This is similar to the phase stability of oxynitride photocatalysts during photocatalytic reactions.62 Furthermore, the morphology of N, P, S-TiO2/S-TiN did not change significantly during the 5000 cycles (Fig. S11, ESI†) which indicates the structural stability.
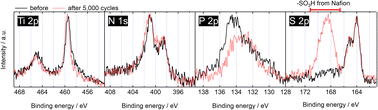 |
| | Fig. 4 XP spectra of N, P, S-TiO2/S-TiN catalyst before (solid curves) and after (dashed curves) 5000 potential cycles between 1.0 and 1.5 V vs. RHE at 0.5 V s−1 in 0.1 mol dm−3 H2SO4 solution. The N, P, S-TiO2/S-TiN catalyst after 5000 cycles contains Nafion ionomer used in the catalyst layer. | |
4. Conclusions
N, P-TiO2 layer catalysts formed on S-doped TiN were annealed with NH4F under N2 gas to dramatically improve their ORR activity and durability against startup/shutdown cycles for FCVs. NH4F-annealing oxidized the N, P-TiO2/S-TiN, resulting in the formation of S-doped TiN-supported N, P, S-tridoped TiO2 catalysts. The anatase and rutile TiO2 appeared in the bulk, with the remaining TiN during NH4F-annealing forming hetero-phase junctions that produced positively charged ORR active sites. The NH4F-annealing exposed sulfur species from inside the core to the surface, forming anionic S2−-substituted TiO2 with enhanced ORR activity. The E1/2 after NH4F annealing improved by 0.08 V, reaching 0.72 V, which is higher than that of any other oxide/oxynitride catalyst reported to date. Although the improved E1/2 is still lower than that of state-of-the-art M/N/C catalysts, the N, P, S-TiO2/S-TiN exhibited excellent durability against startup/shutdown cycles. After 5000 cycles at high potential between 1.0 and 1.5 V, the decrease in E1/2 was only 0.02 V, which is among the highest durability of any PGM-free catalysts. The anionic dopants and Ti-atoms on the surface are stable, are not removed even after 5000 cycles, and remain active. Simple NH4F-annealing with the formation of hetero-phase junctions as well as S2− substitution of O-atoms in TiO2 lattice has been shown to be a new route to improve both ORR activity and startup/shutdown durability and reduce the cost of FCVs. In the proposed mechanism for the enhanced activity and durability, ORR starts from the O2 adsorption on positively charged rutile TiO2 followed by smooth adsorption/desorption of reaction intermediates at the hetero-phase junction formed by the S2−-substitution. Validation of the proposed mechanism, precise control of S2− doping level and stabilization of P5+-dopants could further enhance the ORR activity, and these are our next research topics.
Author contributions
Mitsuharu Chisaka: conceptualization, investigation, data curation, formal analyses, writing – original draft, writing – review & editing, funding acquisition. Jubair A. Shamim: investigation, validation, writing – review & editing. Wei-Lun Hsu: validation, writing – review & editing. Hirofumi Daiguji: investigation, validation, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge Yusei Tsushima at Hirosaki University, Keishi Yamashita at Horiba Techno Service Co., and Daisuke Nishiya at Public Nuisance & Medical Research Institute for acquiring TEM images–ED spectra, Raman spectra and ICP spectra, respectively. This work was partially supported by a Grant-in-Aid for Scientific Research, Grant Number JP23H01347 from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) in Japan, and a research grant from Takahashi Industrial and Economic Research Foundation in Japan. The XP spectra were acquired at the University of Tokyo with the support by the Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM) of the MEXT, Proposal Number JPMXP1223UT0203.
Notes and references
-
World Energy Outlook 2022, International Energy Agency, 2022, p. , p. 146, https://iea.blob.core.windows.net/assets/830fe099-5530-48f2-a7c1-11f35d510983/WorldEnergyOutlook2022.pdf, accessed: Feb. 27, 2024 Search PubMed.
-
Japan Automobile Manufacturers Association, Inc, Active Matrix DB, https://www.jama.or.jp/statistics/facts/world/index.html, accessed: Feb. 27, 2024 Search PubMed.
- W. Jia, Z. Jiang, Q. Wang, B. Xu and M. Xiao, Transp. Policy, 2023, 135, 21 CrossRef.
- O. Gröger, H. A. Gasteiger and J. P. Suchsland, J. Electrochem. Soc., 2015, 162, A2605 CrossRef.
- D. A. Cullen, K. C. Neyerlin, R. K. Ahluwalia, R. Mukundan, K. L. More, R. L. Borup, A. Z. Weber, D. J. Myers and A. Kusoglu, Nat. Energy, 2021, 6, 462 CrossRef CAS.
-
T. Nakamichi, Nikkei Electronics, 2022, vol. 6, p. 73 Search PubMed.
- A. Kongkanand and M. F. Mathias, J. Phys. Chem. Lett., 2016, 7, 1127 CrossRef CAS.
- M. M. Whiston, I. L. Azevedo, S. Litster, K. S. Whitefoot, C. Samaras and J. F. Whitacre, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4899 CrossRef CAS PubMed.
- C. A. Reiser, L. Bregoli, T. W. Patterson, J. S. Yi, J. D. Yang, M. L. Perry and T. D. Jarvi, Electrochem. Solid-State Lett., 2005, 8, A273 CrossRef CAS.
- U. Eberle, B. Müller and R. von Helmolt, Energy Environ. Sci., 2012, 5, 8780 RSC.
- T. Ioroi, Z. Siroma, N. Fujiwara, S. Yamazaki and K. Yasuda, Electrochem. Commun., 2005, 7, 183 CrossRef CAS.
- J. Parrondo, T. Han, E. Niangar, C. Wang, N. Dale, K. Adjemian and V. Ramani, Proc. Natl. Acad. Sci., 2014, 111, 45 CrossRef CAS.
- I. Jiménez-Morales, F. Haidar, S. Cavaliere, D. Jones and J. Rozière, ACS Catal., 2020, 10, 10399 CrossRef.
- M. Chisaka, W. Nagano, B. Delgertsetseg and T. Takeguchi, Chem. Commun., 2021, 57, 12772 RSC.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef.
- C. H. Choi, C. Baldizzone, J. P. Grote, A. K. Schuppert, F. Jaouen and K. J. J. Mayrhofer, Angew Chem. Int. Ed. Engl., 2015, 54, 12753 CrossRef CAS.
- K. Strickland, E. Miner, Q. Jia, U. Tylus, N. Ramaswamy, W. Liang, M. T. Sougrati, F. Jaouen and S. Mukerjee, Nat. Commun., 2015, 6, 7343 CrossRef CAS.
- Y. Shao, J. P. Dodelet, G. Wu and P. Zelenay, Adv. Mater., 2019, 31, 1807615 CrossRef.
- X. Zhao, X. Yang, M. Wang, S. Hwang, S. Karakalos, M. Chen, Z. Qiao, L. Wang, B. Liu, Q. Ma, D. A. Cullen, D. Su, H. Yang, H. Y. Zang, Z. Feng and G. Wu, Appl. Catal., B, 2020, 279, 119400 CrossRef CAS.
- H. Zhang, L. Osmieri, J. H. Park, H. T. Chung, D. A. Cullen, K. C. Neyerlin, D. J. Myers and P. Zelenay, Nat. Catal., 2022, 5, 455 CrossRef CAS.
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. Nie, J. Liu, D. J. Myers, P. Zelenay, G. Wang, G. Wu, V. Ramani and Y. Shao, Nat. Catal., 2020, 3, 1044 CrossRef CAS.
- M. Chen, X. Li, F. Yang, B. Li, T. Stracensky, S. Karakalos, S. Mukerjee, Q. Jia, D. Su, G. Wang, G. Wu and H. Xu, ACS Catal., 2020, 10, 10523 CrossRef CAS.
-
M. Chisaka, in Electrocatalysts for Low Temperature Fuel Cells: Fundamentals and Recent Trends, ed. T. Maiyalagan and V. S. Saji, Wiley-VCH, Weinheim, Germany, 2017, ch. 14 Search PubMed.
- M. Chisaka, A. Ishihara, H. Morioka, T. Nagai, S. Yin, Y. Ohgi, K. Matsuzawa, S. Mitsushima and K. Ota, ACS Omega, 2017, 2, 678 CrossRef CAS.
- M. Chisaka, Y. Ando, Y. Yamamoto and N. Itagaki, Electrochim. Acta, 2016, 214, 165 CrossRef CAS.
- M. Chisaka, Y. Yamamoto, N. Itagaki and Y. Hattori, ACS Appl. Energy Mater., 2018, 1, 211 CrossRef CAS.
- M. Chisaka and H. Morioka, Catal. Sci. Technol., 2019, 9, 611 RSC.
- M. Chisaka, R. Xiang, S. Maruyama and H. Daiguji, ACS Appl. Energy Mater., 2020, 3, 9866 CrossRef CAS.
- M. Chisaka, R. Xiang, S. Maruyama and H. Daiguji, Energy Fuels, 2022, 36, 539 CrossRef CAS.
- J. S. Sanghera, P. Hart, M. G. Sachon, K. J. Ewing and I. Aggarwal, J. Am. Ceram. Soc., 1990, 73, 1339 CrossRef CAS.
- Y. Lee, C. Huffman and S. M. George, Chem. Mater., 2016, 28, 7657 CrossRef CAS.
- J. Zhang, M. Li, Z. Feng, J. Chen and C. Li, J. Phys. Chem. B, 2006, 110, 927 CrossRef CAS PubMed.
- U. Balachandran and N. G. Eror, J. Solid State Chem., 1982, 42, 276 CrossRef CAS.
- M. Chisaka, Phys. Chem. Chem. Phys., 2018, 20, 15613 RSC.
- K. S. Robinson and P. M. A. Sherwood, Surf. Interface Anal., 1984, 6, 261 CrossRef CAS.
- N. C. Saha and H. G. Tompkins, J. Appl. Phys., 1992, 72, 3072 CrossRef CAS.
- C. D. Valentin, G. Pcchioni, A. Selloni, S. Livraghi and E. Giamello, J. Phys. Chem. B, 2005, 109, 11414 CrossRef PubMed.
- J. Wang, W. Zhu, Y. Zhang and S. Liu, J. Phys. Chem. C, 2007, 111, 1010 CrossRef CAS.
- G. Wang, Z. Jiang, H. Shi, T. Xiao and Z. Yan, J. Mater. Chem., 2010, 20, 5301 RSC.
- S. P. Madhusudanan, B. Gangaja, A. G. Shyla, A. S. Nair, S. V. Nair and D. Santhanagopalan, ACS Sustainable Chem. Eng., 2017, 5, 2393 CrossRef CAS.
- J. Ni, S. Fu, C. Wu, J. Maier, Y. Yu and L. Li, Adv. Mater., 2016, 28, 2259 CrossRef CAS PubMed.
- W. Jiao, N. Li, L. Wang, F. Li, G. Liu and H. M. Cheng, Chem. Commun., 2013, 49, 3461 RSC.
- C. Li, J. Shi, L. Zhu, Y. Zhao, J. Lu and L. Xu, Nano Res., 2018, 11, 4302 CrossRef CAS.
- A. Seifitokaldani, O. Savadogo and M. Perrier, Electrochim. Acta, 2014, 141, 25 CrossRef CAS.
- T. Kinumoto, M. Inaba, Y. Nakayama, K. Ogata, R. Umebayashi, A. Tasaka, Y. Iriyama, T. Abe and Z. Ogumi, J. Power Sources, 2006, 158, 1222 CrossRef CAS.
- A. Ohma, K. Shinohara, A. Iiyama, T. Yoshida and A. Daimaru, ECS Trans., 2011, 41, 775 CrossRef CAS.
- Y. Hu, T. Ding, K. Zhang, B. Li, B. Zhu and K. Tang, ChemNanoMat, 2018, 4, 1133 CrossRef CAS.
- Z. Wei, X. Niu, H. Yin, S. Yu and J. Li, Appl. Catal., A, 2022, 636, 118596 CrossRef CAS.
- C. Jia, G. Zhang, W. Zhong and J. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 10315 CrossRef CAS.
- S. Zhou, H. Jang, Q. Qin, L. Hou, M. G. Kim, S. Liu, X. Liu and J. Cho, Angew. Chem., Int. Ed., 2022, 61, e202212196 CrossRef CAS.
- F. Ando, T. Gunji, T. Tanabe, I. Fukano, H. D. Abruña, J. Wu, T. Ohsaka and F. Matsumoto, ACS Catal., 2021, 11, 9317 CrossRef CAS.
- Z. Wang, S. Chen, W. Wu, R. Chen, Y. Zhu, H. Jiang, L. Yu and N. Cheng, Adv. Mater., 2023, 35, 2301310 CrossRef CAS PubMed.
- W. Wu, R. Chen, S. Chen, Z. Wang and N. Cheng, Small, 2023, 19, 2300621 CrossRef CAS PubMed.
- Y. Tan, Z. Zhang, S. Chen, W. Wu, L. Yu, R. Chen, F. Guo, Z. Wang and N. Cheng, Adv. Funct. Mater., 2024, 34, 2311337 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886 CrossRef.
- S. Siahrostami, S. J. Villegas, A. H. B. Mostaghimi, S. Back, A. B. Farimani, H. Wang, K. A. Persson and J. Montoya, ACS Catal., 2020, 10, 7495 CrossRef CAS.
- M. Chisaka, T. Abe, R. Xiang, S. Maruyama and H. Daiguji, Phys. Chem. Chem. Phys., 2022, 24, 29328 RSC.
- H. Tian, J. Ma, K. Li and J. Li, Ceram. Int., 2009, 35, 1289 CrossRef CAS.
- N. Sharotri and D. Sud, New J. Chem., 2015, 39, 2217 RSC.
-
F. Cardarelli, in Materials Handbook: A Concise Desktop Reference, ed. F. Cardarelli, Springer-Verlag, London, UK, 2nd edn, 2008, ch. 10 Search PubMed.
- X. Chen, X. Wang, Y. Hou, J. Huang, L. Wu and X. Fu, J. Catal., 2008, 255, 59 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, XRD patterns, XP spectra, RDE voltammograms and TEM images. See DOI: https://doi.org/10.1039/d4ta01475h |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Jubair A.
Shamim
b,
Wei-Lun
Hsu
*a,
Jubair A.
Shamim
b,
Wei-Lun
Hsu