
Selective grafting of phosphorus onto Ti3C2Tx MXene enables a two-proton process and enhanced charge storage†
Hao
Li‡
 a,
Ke
Fan‡
a,
Pei
Xiong
a,
Hanmo
Zhou
a,
Zezhou
Lin
a,
Keyu
Tao
b,
Tiancheng
Liu
a,
Xuyun
Guo
a,
Ye
Zhu
a,
Lyuchao
Zhuang
a,
Wei
Han
c,
Chen
Yang
a,
Yan
Liu
d,
Molly
Meng-Jung Li
*a,
Mingwang
Fu
e,
John
Wang
f and
Haitao
Huang
*a
a,
Ke
Fan‡
a,
Pei
Xiong
a,
Hanmo
Zhou
a,
Zezhou
Lin
a,
Keyu
Tao
b,
Tiancheng
Liu
a,
Xuyun
Guo
a,
Ye
Zhu
a,
Lyuchao
Zhuang
a,
Wei
Han
c,
Chen
Yang
a,
Yan
Liu
d,
Molly
Meng-Jung Li
*a,
Mingwang
Fu
e,
John
Wang
f and
Haitao
Huang
*a
aDepartment of Applied Physics and Research Institute for Smart Energy, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong 999077, P. R. China. E-mail: molly.li@polyu.edu.hk; aphhuang@polyu.edu.hk
bCollege of Chemistry and Chemical Engineering, Chongqing University, Shapingba, Chongqing 401331, P. R. China
cHubei Yangtze Memory Laboratories, Wuhan, Hubei 430205, P. R. China
dSchool of Chemical Engineering and Technology, Sun Yat-sen University, Tangjiawan, Zhuhai 519082, P. R. China
eDepartment of Mechanical Engineering, Research Institute for Advanced Manufacturing, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong 999077, P. R. China
fDepartment of Materials Science and Engineering, National University of Singapore, 117574, Singapore
First published on 28th December 2023
Abstract
Ti3C2Tx MXene shows great promise as a supercapacitor electrode material owing to its high conductivity and pseudocapacitive nature. Phosphorus doping is an efficient strategy to boost its capacitance due to the synergistic effect of the P–O and P–C species formed. However, the contribution to enhanced capacitance from specific phosphorus doped species in P-doped Ti3C2Tx remains largely unexplored. Herein, phosphorus atoms are selectively grafted onto Ti3C2Tx MXene, introducing only P–O doped species and how this doping configuration contributes to capacitance is unraveled. The results show that 2.1 at% P-doped Ti3C2Tx delivers a capacitance enhancement of 35% (437 F g−1 at 2 mV s−1) in comparison with pristine MXene and outstanding cycling stability. Multiple in situ and ex situ characterization studies along with DFT calculations collectively reveal that the formed P–O bonds are new active sites for a two-proton bonding-debonding process, leading to enhanced charge storage and capacitive performance in MXene. However, higher surface phosphorus doping would destroy crystal integrity of MXene and leads to performance deterioration.
1 Introduction
Two-dimensional (2D) materials, such as graphene, transition metal chalcogenides (TMDs) and MXenes are promising electrode materials for high power electrochemical energy storage devices (including batteries and supercapacitors) due to the merits of high specific area, low ion diffusion barrier and short ion transport path.1–3 Their electrochemical performance can be further boosted by heteroatom incorporation that adjusts the crystal structure and electron states of materials.2,4 Doping elements, amounts and sites synergistically determine the performance change of doped materials. For instance, surface O doping can endow graphene with extra pseudocapacitance but decreased charge mobility, while incorporation of nitrogen into the lattice of graphene not only offers new active sites for pseudocapacitive reactions but also improves its charge carrier concentration.5,6 MXenes have been investigated as pseudocapacitive electrode materials since they were discovered in 2011, owing to their metallic conductivity (up to 2.4 × 106 S m−1), hydrophilic surface, outstanding capacitance and cycling stability in acidic electrolyte.1,7–10 But heteroatom doping to boost charge storage in MXenes is more complicated than that in graphene and TMDs due to their special chemical composition and crystal structure.11MXenes have a general formula of MnXn+1Tx, where M represents an early transition metal (Ti, Cr, V, Mo, Nb, Zr, Hf, Ta, Sc, W, and Y), X is C and/or N, and T refers to functional groups linked with surface metal atoms of the material, e.g., –O, –OH, –F, and –Cl.12,13 A typical synthesis method for MXenes is selectively etching off the A-layer of hexagonal stratified Mn+1AXn (MAX) precursors in hydrofluoric acid or other etchants.12,14,15 The unique crystal structure endows MXenes with good conductive behavior and redox-active surface, which make them promising for supercapacitor electrode materials.16,17 Their supercapacitive performance can be further boosted via functional group modification, interlayer path design and heteroatom doping.18–22 When heteroatoms are incorporated into MXenes, M, X, and T are three kinds of available doping sites. Therefore, the doping of different elements may give rise to different doping configurations that contribute differently to performance variation of MXenes. Ti3C2Tx is the most studied MXene for supercapacitor electrode materials and delivers the highest specific capacitance in acidic electrolyte among all as-made MXenes, which store energy via proton bonding/debonding with surface oxygen terminals.23,24 Phosphorus has been doped into Ti3C2Tx, MXene to boost its supercapacitive performance.25–27 The doped atoms either fill surface titanium vacancies to form P–C bonds or are grafted onto the surface oxygen terminations with the formation of P–O species.25–28 Ti–P bonds through substituting T sites within Ti3C2Tx are rarely reported due to high formation energy.25 Note that P–C bonds and P–O bonds simultaneously exist in P-doped MXene in previous studies and the ameliorative capacitance is largely attributed to a “synergistic effect” of P–C and P–O species,25–28 while the underlying mechanism that how each specific phosphorus doping configuration cannot be differentiated. Specifically, P–O bonds are the outmost functional groups on the MXene surface, which directly influence the interaction between MXene and electrolyte ions. Therefore, its role in boosting capacitance needs to be critically evaluated.
Herein, phosphorus atoms are selectively grafted onto the Ti3C2Tx surface through oxygen bridging by heating the MXene aerogel in a mixed atmosphere of argon and phosphine that is decomposed from sodium hypophosphite (Fig. 1a), leading to the sole formation of P–O doped species. The underlying mechanism of how the P–O bonds boost charge storage capability of MXene is unraveled by combining X-ray diffraction (XRD), X-ray absorption near edge structure spectra (XANES), X-ray photoelectron spectroscopy (XPS), and in situ Raman spectroscopy. The results show that the selectively phosphorus-doped Ti3C2Tx MXene (PMX) with 2.1 wt% P shows a high gravimetric capacitance of 437 F g−1 at 2 mV s−1 in 1 M H2SO4 electrolyte, much superior to that of pristine MXene (324 F g−1). The formed surface P–O (Ti–O–P) groups act as new active sites for more proton bonding/debonding and thereby result in greater valence state variation of titanium during the charge/discharge process (Fig. 1b–d). It is also revealed that moderate surface phosphorus doping can maintain the MXene crystal structure intact and boost the capacitance, but further incorporation of the hybrid element would injure the crystal structure and even induce amorphization, adverse to its energy storage ability.
 | ||
| Fig. 1 Schematic illustration for (a) phosphorus doping strategy, (b) proton interaction with the surface of pristine Ti3C2Tx, and (c and d) proton interaction with the surface of PMX. | ||
2 Results and discussion
Multilayered Ti3C2Tx MXene was first prepared by selectively etching the aluminum layer of the Ti3AlC2 precursor in HCl and LiF mixed liquid, followed by fierce oscillation for its delamination to obtain few-layered MXene. The resultant MXene aqueous solution shows the Tyndall effect when a ray transmits through it (Fig. S1a†), indicating its colloidal nature. The diameter of MXene sheets is 0.50–8 μm, as confirmed by the scanning electron microscope (SEM) image (Fig. S1b†). The atomic force microscope (AFM) result shows that the MXene sheets have a thickness of 5.56 nm (Fig. S1c†), corresponding to a stacking of 5–6 layers. P–C doped species forms at titanium vacancies, and the long-time heating aggravates the formation of the vacancies and the doped species.26 Thus, selective doping with only P–O doped bonds requires Ti3C2Tx MXene with few defects and short doping time. The defects of Ti3C2Tx are suppressed by etching the Ti3AlC2 precursor with argon bubbling in this work.10 The gas-state doping strategy (Fig. 1a) was applied here to avoid the subsequent cumbersome washing procedures for removing residual doping sources. To increase doping efficiency and shorten doping time, the MXene sheets were assembled into a porous MXene aerogel (Fig. 2a and S1d†) by flash freezing the MXene solution and subsequent freeze-drying.29 Sodium hypophosphite was heated at 350 °C to produce phosphine, which was brought by flowing Ar to the downstream MXene aerogel (at 400 °C) to fulfill the phosphorus doping within 2 hours. The mass ratio between MXene and NaH2PO2 was varied from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:2 to tune the phosphorus doping amount, and the obtained samples were named PMX1 and PMX2, respectively. Meanwhile, a control sample (CMX) was also fabricated by heating pristine Ti3C2Tx MXene aerogel (MX) in pure argon under the same heating conditions as those used in the doping process.
:1 to 1:2 to tune the phosphorus doping amount, and the obtained samples were named PMX1 and PMX2, respectively. Meanwhile, a control sample (CMX) was also fabricated by heating pristine Ti3C2Tx MXene aerogel (MX) in pure argon under the same heating conditions as those used in the doping process.
 | ||
| Fig. 2 SEM images of (a) MX, (b) PMX1, (c) PMX2 and (d) CMX. (e) XRD patterns of the samples. (f) Enlarged (002) peak in (e). | ||
The SEM results show that both PMX1 and PMX2 retain a macroporous structure similar to that of MX (Fig. 2b and c) after phosphorus incorporation, implying a robust pore structure in the MXene aerogel. A similar porous structure is also observed in CMX, but with some white spots (enclosed by the pink rectangles in Fig. 2d), which could be due to the oxidation of Ti3C2Tx. The existence of titanium dioxide in CMX is confirmed by the XRD results, while no distinct oxide peaks are detected in both PMX1 and PMX2, as shown in Fig. 2e. This result implies that phosphine not only serves as the doping source but also restricts the thermal degradation of Ti3C2Tx. The position and full width at half maximum (FWHM) of the (002) XRD peak of MXene can reveal the changes of interlayer distance (d) and crystallinity, respectively, in Ti3C2Tx MXene.30 Enlarged (002) peaks of the samples are shown in Fig. 2f. MX shows an unsymmetrical (002) peak at a 2θ value of 6.02° and a right shoulder, indicating its main interlayer space of 14.67 Å and smaller interlayer distance for some of the MXene sheets. The (002) peak of PMX1 remains at 6.02° but its FWHM becomes narrower than that of MX, which implies increased uniformity of interlayer distance. Noticeably, the (002) peak of PMX2 left shifts to 5.91° (d = 14.94 Å) with smaller FWHM than MX, manifesting the more spacious and uniform interlayer path of MXene sheets due to phosphorus doping. This broadened but uniform interlayer path is expected to boost ion diffusion to active sites.19 However, CMX without phosphorus doping shows a much broader (002) peak than the other samples, implying inferior MXene crystallinity and a non-uniform interlayer path. The peak shoulder at 2θ ≈ 8.52° in CMX is indicative of the presence of some MXene sheets with a narrower interlayer path, which could originate from interlayer shrinkage of MXene during its heating process.31 Overall, the XRD results demonstrate that the phosphorus incorporation not only boosts the interlayer space of MXene but also retards its oxidation during the heating process.
Nitrogen gas adsorption/desorption at 77 K was conducted to analyze specific surface area and pore size distribution of the samples (Fig. S2a and b†). The Brunauer–Emmett–Teller (BET) surface area of MX is 41.03 m3 g−1, while those of PMX1, PMX2 and CMX are increased to 43.88, 41.83 and 47.63 m3 g−1, respectively (Fig. S2a†). These results indicate that the heating procedure (400 °C) causes the enhancement of specific surface area of the MXene aerogel, while P-doped MXenes (PMX1 and PMX2) exhibit a lower increment than that with only heating (CMX). The lower increment of specific surface area in P-doped MXenes can be attributed to their suppressed oxidation, while the larger increment of specific surface area in CMX could be due to its oxidation induced breakage of MXene sheets. Fig. S2b† indicates that MX, PMX1, PMX2 and CMX all possess a similar hierarchical pore size distribution (1.50–150 nm). But P-doped samples, especially PMX2, exhibit a higher differential pore volume at a pore width of ∼1.50 nm (inset of Fig. S2b†), probably resulting from the phosphorus incorporation.
To further investigate the crystal structure change of MX after phosphorus doping, high resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) characterization studies were carried out (Fig. 3). HRTEM images show that the phosphorus doping in PMX1 and PMX2 has not led to an obvious variation in interplanar distance of (010) planes in Ti3C2Tx MXene (Fig. 3a–c), which is consistent with the XRD results of MX, PMX1 and PMX2 showing almost identical (010) peak locations (Fig. 2e). However, CMX possesses a larger (010) interplanar distance (0.26 nm), consistent with its left shifted (010) diffraction peak compared with MX (Fig. 2e). The SAED pattern (Fig. 3a) of MX demonstrates the hexagonal symmetry of Ti3C2Tx MXene inherited from the Ti3AlC2 parent phase.7 Its innermost circle of diffraction points corresponds to (010) planes and the second circle of diffraction points originates from (110) planes.32 PMX1 and PMX2 with different doping contents exhibit identical crystal symmetry to pristine MX, as shown in Fig. 3b and c. However, if the doping amount is further enhanced via increasing the mass ratio of NaH2PO2:MXene to 8 and 20 (with the corresponding samples named PMX8 and PMX20, respectively), a shortened interplanar distance of (010) planes is observed in PMX8 (Fig. S3a and b†) and amorphization is observed in PMX20 (Fig. S3c and d†), accompanied by the disappearance of the six-fold symmetry of (010) planes (Fig. S3e†). These results conclude that a large doping amount of phosphorus would break the crystal periodicity and symmetry of Ti3C2Tx MXene. By contrast, CMX exhibits diffraction rings showing its polycrystalline nature and some newborn diffraction points corresponding to the TiO2 anatase phase (Fig. 3d), collectively verifying the fragmentation and oxidation of MXene sheets in the sample, which is also consistent with the foregoing XRD and BET results.
 | ||
| Fig. 3 HRTEM images and the corresponding SAED patterns of (a) MX, (b) PMX1, (c) PMX2 and (d) CMX, respectively. (e) EELS elemental mapping results (Ti, O, and P) of PMX2. | ||
Electron energy loss spectroscopy (EELS) was conducted to analyze the elemental distribution and change of the chemical environment with phosphorus incorporation in P-doped MXene. For simplicity, only MX, PMX2 and CMX are selected for EELS analysis. Both O K-edge and Ti L2.3-edge spectra of the samples were normalized to their corresponding Ti L2 peak intensities, as shown in Fig. S4a and b.† PMX2 exhibits the highest O K-edge intensity after normalization (Fig. S4b†), implying its richest oxygen composition. This also denotes that the doped phosphorus could be favorable for maintaining more surface oxygen on Ti3C2Tx. In addition, an asymmetric O K-edge is detected in CMX, which can be attributed to the titanium dioxide (anatase) formed in the sample.33 Note that the first peak in O K-edge spectra is ascribed to the hybridization between Ti 3d orbitals and surface oxygen terminations.34 To compare the chemical environment of oxygen in the samples, their O K-edge EELS spectra are normalized to the first peak (Fig. S4c†). The almost identical shape of the first O K-edge peaks for MX and PMX2 indicates that the oxygen maintains bonding with the titanium after the phosphorus incorporation. But the difference in peak 2 of O K-edge spectra between pristine MX and PMX2 confirms the change in the local environment of oxygen atoms in PMX2,33 which could be attributed to the introduced hybrid phosphorus element. The detailed change in chemical valence and the environment will be discussed in the following XPS and extended X-ray absorption fine structure (EXAFS) results. The EELS element mapping of PMX2 (Fig. 3e) shows that the doped phosphorus is uniformly dispersed at a nanometer scale, and the energy-dispersive X-ray spectroscopy (EDS) elemental mapping results (Fig. S5†) also demonstrate a uniform distribution of phosphorus over a larger area.
The composition of samples was investigated using XPS survey spectra and is shown in Fig. 4a. PMX1 and PMX2 possess 1.4 and 2.1 at% phosphorus, respectively, implying successful phosphorus incorporation and the doping amount increases with an increasing amount of NaH2PO2 added. It is also noted that PMX1 and PMX2 have 20.4 and 25.1 at% oxygen contents, respectively, which are higher than those of MX and CMX (17.4 and 19.3 at%, respectively). This indicates that the doped phosphorus is favorable for keeping surface oxygen on Ti3C2Tx, consistent with the aforementioned EELS results (Fig. S4b†). In the meantime, the lower contents of –F and –Cl in PMX2 than those in MX and CMX indicate that halogen terminations may be more easily eliminated in a reducing atmosphere (PH3). All XPS spectra were calibrated through the C–Ti–Tx bond (282 eV) in C 1s for reliable analysis,35 as shown in Fig. S6.† P 2p high resolution XPS spectra (Fig. 4b) demonstrate that the formed P species contains a P–O bond in both PMX1 and PMX2, corresponding to a binding energy of 133.5 eV.36 Because of the absence of the Ti–P bond (129 eV) in P 2p,36 the doped phosphorus is only grafted onto Ti3C2Tx MXene via surface oxygen bridging, forming a Ti–O–P bond. This result demonstrates that the applied doping strategy successfully achieves selective phosphorus doping with the only formation of P–O doped species. The density functional theory (DFT) calculation results (Fig. S7†) further verify that the Ti–O–P bond can stably exist on the Ti3C2Tx surface. And it is predicted that the adjacent phosphorus atoms grafted on the surface tend to interact with each other and bond together to form clusters (Fig. S8†), which could adversely affect the crystal structure of MXene due to lattice stress. This calculation result also explains why high surface P-doping damages the crystallinity of MXene and even leads to its amorphization, as observed in PMX8 and PMX20. The incorporated phosphorus in PMX8 still exists with the formation of P–O (Ti–O–P) species (133.6 eV), while P 2p of PMX20 is deconvoluted into P–O and phosphorus oxides (134.9 eV),25,37 as shown in Fig. S9.† The newly formed phosphorus oxides in PMX20 could be due to the oxidation of the phosphorus clusters in air. O 1s of MX and CMX (Fig. 4c) can be deconvoluted into Ti–O, C–Ti–O, C–Ti–OH, H2O and organics (OR), which correspond to binding energies of 529.8, 531, 532.2, 533.2, and 534.9 eV, respectively.35,38 However, for PMX1 and PMX2, some of their surface C–Ti–O was grafted with phosphorus, leading to the formation of a Ti–O–P bond at a higher binding energy (531.6 eV),39 and the amount of Ti–O–P increases with an increasing phosphorus doping amount, as shown in the middle two spectra of Fig. 4c. The comparison of Ti 2p of different samples is shown in Fig. 4d. The binding energies for the Ti 2p3 peak of PMX1 and PMX2, and CMX exhibit blue shifts of 0.27, 0.35, and 0.16 eV, respectively, compared with that of MX, indicating their elevated titanium valence states, which is similar to the case of nitrogen doped Ti3C2Tx MXene.30 Titanium with higher valence states due to the phosphorus doping in PMX1 and PMX2 is expected to contribute more charge transfer during the charge/discharge process.30,40
 | ||
| Fig. 4 (a) Composition analysis of MX, PMX1, PMX2 and CMX from XPS survey spectra. (b) High resolution P 2p XPS spectra of PMX1 and PMX2. High resolution (c) O 1s and (d) Ti 2p XPS spectra of MX, PMX1, PMX2 and CMX. (e) Raman spectra of the samples. (f) Ti K-edge of EXAFS spectra and (g) WT-EXAFS profiles for MX and PMX2. | ||
Surface sensitive Raman spectra of MX, PMX1, PMX2 and CMX are shown in Fig. 4e. The Eg band at 230–470 cm−1 represents the in-plane vibration of surface functional groups linked with outer titanium atoms in Ti3C2Tx and the A1g band at around 720 cm−1 is attributed to out-of-plane vibrations of carbon.41 The wavenumbers of the Eg band in PMX1 and PMX2 are similar (370 cm−1) but lower than those (∼382 cm−1) of MX and CMX, suggesting that the incorporation of phosphorus engenders attenuated bond strength between functional groups and surface titanium atoms in MXene. And a larger wavenumber of the A1g band (∼720 cm−1) is observed in P-doped Ti3C2Tx, which indicates the enhanced Ti–C bond strength after P-doping. The change of the A1g band with applied potential can be used to investigate the charge storage behavior of Ti3C2Tx,41 which will be discussed in the following sections for differentiating the energy storage behavior in P-doped Ti3C2Tx MXene from that in pristine MXene.
The effect of incorporated P on the coordination environment of Ti in Ti3C2Tx is investigated via EXAFS. Fig. 4f compares the Fourier-transform (FT) Ti K-edge EXAFS results of MX and PMX2. FT-EXAFS of both samples display two peaks in the range of 1.0–2.0 Å and 2.0–3.0 Å, corresponding to the Ti–O/F/C and Ti–Ti contribution, respectively.42 However, PMX2 exhibits a much higher 2nd-shell peak than MX, which is assigned to an extra Ti–P coordination at 3.48 Å in PMX according to the EXAFS fitting results (Fig. S10 and Table S1†) and DFT models (Fig. S7b†). The EXAFS Ti K-edge wavelet transform (WT) results (Fig. 4g) consistently show that PMX2 exhibits increased intensity at both larger K and R spaces of Ti–Ti coordination compared to MX, as circled by a pink line, which corresponds to Ti–P coordination. Therefore, these results collectively further confirm Ti–O–P coordination in P-doped MXene.
The electrochemical performances (ECs) of the as-prepared samples were studied in 1 M H2SO4 with three-electrode configuration, in which Ag/AgCl (sat. KCl) and a graphite bar are reference and counter electrodes, respectively. Fig. 5a shows cyclic voltammetry (CV) profiles of MX, PMX1, PMX2 and CMX at a scan rate of 20 mV s−1. Redox peaks are observed in all of them, implying their pseudocapacitive energy storage mechanism.40,43 PMX1 and PMX2 exhibit larger integral areas than MX and CMX, indicative of their higher capacitances. In addition, PMX2 with richer phosphorus doping exhibits better energy storage ability than PMX1. A similar result is also shown in galvanostatic charge/discharge (GCD) curves (Fig. 5b). These enhanced capacitances in PMX1 and PMX2 should be attributable to the incorporated phosphorus. The capacitances of the samples at various scan rates (2–200 mV s−1) and current densities (1–20 A g−1) are compared and summarized in Fig. 5c and d. PMX2 delivers the highest capacitances of 437 F g−1 and 435 F g−1 at 2 mV s−1 and 1 A g−1 respectively, which are comparable with and even higher than those of other heteroatom doped (N, S, V etc.) Ti3C2Tx MXenes, as demonstrated in Table S2.† When the scan rate increases from 2 to 200 mVs−1, MX, PMX1, PMX2 and CMX exhibit 34.3%, 35.4%, 34.3% and 37.7% capacitance decay, respectively, showing similar rate performance. To investigate the effect of phosphorus doping amount on ECs, PMX8 (2.8 at% P) and PMX20 (3.5 at% P) were also studied, and the corresponding results are shown in Fig. S11.† Unfortunately, a further increase in phosphorus doping does not contribute to better energy storage ability but results in performance deterioration, which could be attributed to their degraded crystallinity (Fig. S3†).
 | ||
| Fig. 5 (a) CV curves of MX, PMX1, PMX2 and CMX at a scan rate of 20 mV s−1 and (b) GCD curves at a current density of 1 A g−1 for the four samples. Specific capacitances of the samples at (c) scan rates from 2 to 200 mV s−1 and (d) current densities of 1–20 A g−1. (e) EIS spectra of the samples at −0.20 V vs. Ag/AgCl. Insets show the enlarged EIS spectra at intercept with the real axis and a fitted equivalent circuit. (f) Capacitive contributions of the four samples at 2 mV s−1. (g) Cyclic test of PMX2 at 10 A g−1. The inset shows its GCD curves at different cycles. | ||
The electrochemical impedance spectroscopy (EIS) results were used to evaluate redox kinetics and electrical conductivity of different samples. The Nyquist plots of MX PMX1, PMX2 and CMX at −0.20 V vs. Ag/AgCl are shown in Fig. 5e. The slope of the linear part of the plots reflects ion diffusion resistance in electrode materials. The similar slope of the four samples implies that the surface grafted phosphorus atoms on Ti3C2Tx do not block electrolyte ions from reaching active sites. The semicircle of the Nyquist plot (upper inset of Fig. 5e) in the high frequency range denotes the charge transfer resistance (Rct) at the electrode/electrolyte interface.44 Its intercept with the real axis in the plot represents equivalent series resistance (Rs), which is composed of electrical resistance of the electrode material, interface resistance and ionic resistance of electrolyte. To quantitatively compare the values of Rct and Rs, an equivalent circuit (lower inset in Fig. 5e) was constructed for the fitting. The higher Rs (Table S3†) of PMX1 and PMX2 than MX and CMX manifests that the phosphorus incorporation causes decreased electrical conductivity of MXene. This decrement in conductivity is also observed in PMX8 and PMX20 with higher phosphorus content (Fig. S11c†). The phosphorus doping induced lattice distortion and amorphization are regarded as responsible for the decrement in conductivity. In addition, the changed surface functionalization also leads to a decrement of density of states at the Femi level,34 which is another reason for the decreased electrical conductivity in phosphorus-doped MXenes. The increased electrical resistance of the doped samples causes the shift of cathodic redox peaks to higher potential in CV curves compared with pristine MX, as shown in Fig. 5a. It is also observed from Table S3† that phosphorus doping contributes to smaller Rct, indicative of increased active sites in PMX1 and PMX2.
The redox reactions of Ti3C2Tx MXene are governed by reversible proton bonding-debonding on surface –O terminations.45,46 The charge storage in the sample can be divided into diffusion-controlled and capacitive parts,
| i(V) = k1ν + k2ν0.5 | (1) |
000 cycles, suggesting its robust structure and good prospects for supercapacitor electrodes.
Though the boosting effect of phosphorus on EC performance of Ti3C2Tx MXene has been confirmed in the previous discussion, the underlying mechanism for the enhancement is still unclear. The ex situ XRD results (Fig. S13†) first reveal that MX, PMX2 and CMX show different interlayer distances at −0.35 and 0.30 V vs. Ag/AgCl, indicating their intercalation mechanism in pseudo-capacitive energy storage as reported in the literature.47 When the applied potential decreases from 0.30 to −0.35 V, all the samples show a decreased interlayer spacing since the intercalated protons increase the electrostatic attraction between MXene layers.8,47
The redox reaction of Ti3C2Tx MXene involves the change of titanium valence states,23 which was investigated through in situ Raman,48ex situ XPS18 and in situ XANES.23 The Raman spectra of MX and PMX2 at potentials from −0.25 to 0.30 V vs. Ag/AgCl are shown in Fig. 6a and b, measured using a homemade device (Fig. S14†). The Eg band at 230–470 cm−1 has not shown an evident shift with the applied potentials, while both Eg (500–690 cm−1) and A1g (∼720 cm−1) bands exhibit a reversible peak shift with the applied potentials. The shift of these two bands in pristine MX is due to the bonding/debonding of protons with surface –O functional groups.41 The similar change observed in PMX2 verifies its similar electrochemical behavior. Noting that the peak shift of the A1g (∼720 cm−1) band is associated with the change of titanium valence states,48 we compare the peak positions of the A1g bands of the two samples at potentials from −0.25 to 0.30 V vs. Ag/AgCl in Fig. 5c. The variation range of the peak position of the A1g band in PMX2 is 718–736 cm−1, which is much broader than that of MX (717–726 cm−1), indicative of a greater change of titanium valence states occurring in PMX2. To further affirm the more pronounced variation of the titanium valence state in PMX2, XANES and XPS spectra of MX and PMX2 at −0.35 and 0.30 V vs. Ag/AgCl were also collected and compared. The change in white-line peak intensity in Ti K-edge XANES can reflect its variation in the valence state.49 The white-line peak of the XANES spectrum of PMX2 exhibits an intensity difference higher than that of MX (0.037 vs. 0.020) between −0.35 and 0.30 V vs. Ag/AgCl potentials (Fig. 6d and e), validating the larger variation of the titanium valence state in PMX2. Consistently, the ex situ XPS results (Fig. S15†) also demonstrate an identical trend. S 2p XPS spectra of residual sulfate radicals (SO42−) in all samples were used for calibration because of its nonparticipation in the redox action (Fig. S16†). The shift of the right edge in normalized Ti 2p3/2 peaks was used to evaluate the change of the titanium valence state (Fig. S15†). The edge shifts in MX and PMX2 are confirmed as 0.13 and 0.07 eV, respectively, indicating more electron gain and loss in PMX2 during the charge/discharge process. In addition, the P 2p XPS spectra of PXM2 at −0.35 and 0.30 V vs. Ag/AgCl were also studied (Fig. 6f). A 0.15 eV difference in binding energies between the two spectra elucidates that the surface grafted phosphorus atoms are involved in the energy storage process. It has been reported that one oxygen termination on Ti3C2Tx MXene bonds with one proton, contributing to pseudocapacitance,24,47,50 while in the current case, as shown by DFT calculations (Fig. S17†), one surface grafted phosphorus can bond with two protons, more than pure oxygen termination does, resulting in more charge storage. In addition, the differential charge density distribution results (Fig. S18†) further verify that this two-proton bonding process also induces charge transfer in titanium of Ti3C2Tx MXene to protons.
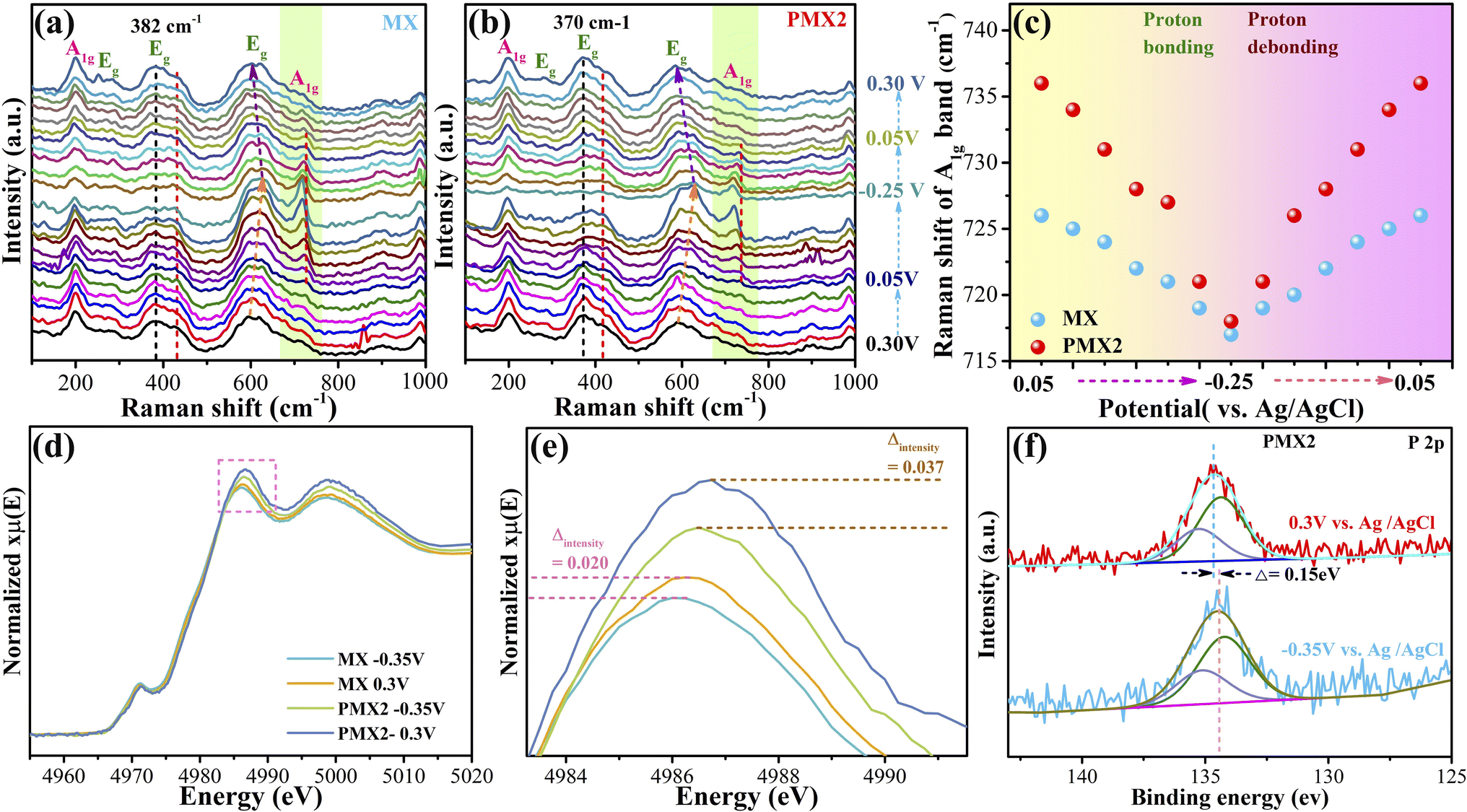 | ||
| Fig. 6 In situ Raman spectra of (a) MX and (b) PMX2 in the potential range of −0.25–0.30 V versus the Ag/AgCl reference electrode. (c) Raman shift of the A1g band of MX and PMX2 with applied potential. (d) Ti K-edge XANES spectra of MX and PMX2 at −0.35 and 0.30 V versus Ag/AgCl, and (e) enlarged profile of the rectangular area in (d). (f) P 2p XPS spectrum of PMX2 at −0.35 and 0.30 V versus Ag/AgCl. | ||
Based on the above results and discussion, it can be summarized that the surface grafted phosphorus serves as a new active site to enable a two-proton bonding/debonding process, which can boost a greater change of the titanium valence sate in Ti3C2Tx and lead to more charge transfer during its charge/discharge process, contributing to enhanced specific capacitance.
Finally, PMX2 was selected to assemble a symmetric supercapacitor to evaluate the practicability of phosphorus doped Ti3C2Tx MXene. The resultant device exhibits a voltage window of 1 V and delivers a maximum capacitance of 97 F g−1 based on the total mass of two electrodes at 2 mV s−1, superior to the device assembled with MX, as shown in Fig. S19a and b.† The capacitance values of the PMX2-based device are always higher than those of the MX-based supercapacitor at various scan rates (Fig. S19b†), although the latter shows better rate performance. The PMX2-based supercapacitor delivers 85.6% of the initial capacitance after 10000 GCD cycles at 5 A g−1 (Fig. S19c†), justifying the remarkable stability of the PMX2 electrode. In addition, the Ragone plots (Fig. S19d†) show that the PMX2-based device exhibits a maximum energy density of 13.41 W h kg−1 at a power density of 97 W kg−1 and retains 5.99 W h kg−1 at 4310 W kg−1, which not only excels those of MX-based devices, but also outperforms KOH treated (washing + annealing) Ti3C2Tx MXene (7.25 W h kg−1 at 250 W kg−1),18 a porous Ti3C2Tx MXene film (9.2 W h kg−1 at 100 W kg−1),51 and N, S co-doped Ti3C2Tx MXene (3.19 W h kg−1 at 349 W kg−1),52 proving the competent role played by the grafted phosphorus on Ti3C2Tx MXene in boosting its supercapacitive performance.
3 Conclusions
In summary, we demonstrate a phosphorus doping strategy that achieves the only formation of P–O doped species in Ti3C2Tx MXene by grafting phosphorus atoms onto its surface through oxygen bridging. The underlying mechanism that how this type of species boosts capacitive performance of Ti3C2Tx MXene is also revealed. The resultant PMX2 exhibits enhanced electrochemical performance compared to pristine MXene and delivers a maximum capacitance of 437 F g−1 at 2 mV s−1 in 1 M H2SO4 aqueous electrolyte along with excellent stability (94.3% capacitance retention after 20000 cycles). The phosphorus atoms of P–O bonds on Ti3C2Tx MXene serve as new active sites to enable a two-proton bonding-debonding process, contributing to improved charge transfer and energy storage ability in P-doped Ti3C2Tx MXene. However, too much surface P-doping is detrimental due to the damage to the crystal structure of MXene. It is believed that our work paves the way for doping MXene with other heteroatoms to achieve even better performance modulation.
4 Methods and experimental section
4.1 Preparation of Ti3C2Tx MXene aerogel
Ti3C2Tx MXene aerogel was prepared by a two-step method. Ti3C2Tx colloidal solution was first synthesized by the minimal intensive layer delamination (MILD) method.53 Two grams of 400 mesh Ti3AlC2 precursor were slowly added into 40 ml 9 M HCl + 3.2 g LiF etchant, followed by water bath heating at 40 °C and continuously stirring under Ar bubbling for 24 h. The resultant product was washed via repetitive 3500 rpm × 5 min centrifugation procedures until the pH of the supernatant reached 6. After the supernatant was decanted, extra deionized (DI) water was poured into the centrifugation tube, which was then loaded onto an orbital shaker for delaminating MXene. Finally, the black supernatant in the tube was collected after centrifugation at 3500 rpm for 60 min for the next fabrication step. Note that shaking for one time is not enough to delaminate all multilayered MXene, so here multiple shakings and collections were conducted to increase the yield. In our experiment, around 600 ml 2.9 g ml−1 MXene solution was eventually obtained.The Ti3C2Tx MXene colloidal solution was frozen in liquid nitrogen for 10 min to assemble the MXene aerogel framework, followed by vacuum drying at −65 °C in a freeze-drying machine for five days (pressure <1 × 10−3 atm) in the second step. The water between MXene sheets was vaporized during this procedure, generating the final porous MXene aerogel (Fig. S1b†).
4.2 Synthesis of phosphorus grafted Ti3C2Tx
Grafting phosphorus atoms onto Ti3C2Tx was conducted in a tube furnace with two temperature regions. A certain amount of NaH2PO2 and 50 mg as-prepared Ti3C2Tx aerogel were placed in the upstream and downwind temperature regions and heated at 350 and 400 °C, respectively, under a continuous 50 sccm argon flow. In detail, the phosphine produced in the first region flows to downstream heated MXene to achieve surface phosphorus graft. The mass ratio between NaH2PO2 and MXene was controlled as 1, 2, 8 and 20, with the corresponding synthesized samples named PMX1, PMX2, PMX8 and PMX20, respectively.4.3 Materials characterization
The morphologies and microstructure of the synthesized samples were characterized using a scanning electron microscope (SEM, JEOL Model JSM-6490) equipped with an energy dispersive X-ray spectrometry (EDS) system and scanning transmission electron microscopy (STEM). X-ray diffraction (XRD) patterns of the samples were recorded using an X-ray diffractometer (Rigaku SmartLab) with a monochromatic Cu Kα X-ray source (λ = 0.15406 nm). Specific area and pore size distribution of the samples were characterized through a MICROMERITICS surface area and porosity analyzer (ASAP2020). The chemical composition and elemental valence states of the samples were investigated on a Nexsa G2 XPS system equipped with monochromatic and focused 12 kV aluminum Kα X-rays. Raman spectra including in situ Raman spectra were measured using a WITEC confocal Raman system (Alpha300 R).4.4 Electrochemical measurements
The slurry for working electrodes was made through mixing the MXene sample, carbon black and PVDF with a mass ratio of 8:1:1 in 600 μL N-methylpyrrolidone (NMP) with continuous grinding for 30 min. The resultant slurry was then coated on 2 × 3 cm2 graphite paper, followed by vacuum drying at 80 °C for 6 h to obtain working electrodes (∼2.2 mg cm−2 active material loading). Supercapacitive performance of working electrodes was investigated through a three-electrode configuration with Ag/AgCl (sat. KCl), graphite bar and 1 M H2SO4 as the reference electrode, the counter electrode and electrolyte, respectively, conducted on a SOLARTRON electrochemical workstation. For assembling symmetric supercapacitors, two Φ14 mm working electrodes were separated by a Nafion 117 film and a glass fiber membrane and then assembled in a Swagelok cell, followed by injecting 200 μL 1 M H2SO4 electrolyte. The energy storage ability of symmetric supercapacitors was also studied through the SOLARTRON workstation. The cycling stability of both working electrodes and symmetric supercapacitors was all tested on an Arbin battery testing system.
The specific capacitance values C (F g−1) of working electrodes are calculated based on CV curves through the following formula (2),
| C = ∫idV/Vmν | (2) |
| C = iΔt/Vm | (3) |
The specific capacitance of the assembled device is also calculated based on formula (2) through CV curves, where m refers to the total mass of two electrodes (g).
The energy density E (W h kg−1) and power density P (W kg−1) of the assembled symmetric supercapacitors are calculated viaeqn (4) and (5), respectively.
| E = 0.5CV2/3.6 | (4) |
| P = 3600 × Eν/V | (5) |
Author contributions
Hao Li conceptualization, data curation, investigation, formal analysis, writing – original draft. Ke Fan theoretical calculation, writing. Pei Xiong data curation, writing. Hanmo Zhou investigation, data curation. Zezhou Lin visualization, Keyu Tao validation, conceptualization. Tiancheng Liu resources. Xuyun Guo validation, formal analysis. Ye Zhu resources. Lyuchao Zhuang investigation. Wei Han visualization. Chen Yang data curation. Yan Liu resources. Molly Meng-Jung Li data curation, investigation, supervision. Mingwang Fu validation, formal analysis. John Wang validation, formal analysis. Haitao Huang project administration, conceptualization, data curation, writing – review & editing, funding acquisition, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Hong Kong Polytechnic University (1-CD4H, 1-CD8W, and G-SAC1).References
- M. Hu, H. Zhang, T. Hu, B. Fan, X. Wang and Z. Li, Chem. Soc. Rev., 2020, 49, 6666–6693 RSC.
- H. Liu, W. Lei, Z. Tong, X. Li, Z. Wu, Q. Jia, S. Zhang and H. Zhang, Adv. Mater. Interfaces, 2020, 7, 2000494 CrossRef CAS.
- H. Cui, Y. Guo, W. Ma and Z. Zhou, ChemSusChem, 2020, 13, 1155–1171 CrossRef CAS PubMed.
- R. Wang, M. Li, K. Sun, Y. Zhang, J. Li and W. Bao, Small, 2022, 18, e2201740 CrossRef PubMed.
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 RSC.
- Z. Li, S. Gadipelli, Y. Yang, G. He, J. Guo, J. Li, Y. Lu, C. A. Howard, D. J. L. Brett, I. P. Parkin, F. Li and Z. Guo, Energy Storage Mater., 2019, 17, 12–21 CrossRef.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- M. Ghidiu, M. R. Lukatskaya, M. Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed.
- A. Shayesteh Zeraati, S. A. Mirkhani, P. Sun, M. Naguib, P. V. Braun and U. Sundararaj, Nanoscale, 2021, 13, 3572–3580 RSC.
- C. Lu, L. Yang, B. Yan, L. Sun, P. Zhang, W. Zhang and Z. Sun, Adv. Funct. Mater., 2020, 30, 2000852 CrossRef CAS.
- Y. Wei, P. Zhang, R. A. Soomro, Q. Zhu and B. Xu, Adv. Mater., 2021, 33, 2103148 CrossRef CAS PubMed.
- C. Liu, Y. Bai, W. Li, F. Yang, G. Zhang and H. Pang, Angew Chem. Int. Ed. Engl., 2022, 61, e202116282 CrossRef CAS PubMed.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Chem. Mater., 2017, 29, 7633–7644 CrossRef CAS.
- M. Benchakar, L. Loupias, C. Garnero, T. Bilyk, C. Morais, C. Canaff, N. Guignard, S. Morisset, H. Pazniak, S. Hurand, P. Chartier, J. Pacaud, V. Mauchamp, M. W. Barsoum, A. Habrioux and S. Célérier, Appl. Surf. Sci., 2020, 530 Search PubMed.
- I. Hussain, C. Lamiel, M. S. Javed, M. Ahmad, S. Sahoo, X. Chen, N. Qin, S. Iqbal, S. Gu, Y. Li, C. Chatzichristodoulou and K. Zhang, Prog. Energy Combust. Sci., 2023, 97, 101097 CrossRef.
- C. Lamiel, I. Hussain, J. H. Warner and K. Zhang, Mater. Today, 2023, 63, 313–338 CrossRef CAS.
- J. Li, X. Yuan, C. Lin, Y. Yang, L. Xu, X. Du, J. Xie, J. Lin and J. Sun, Adv. Energy Mater., 2017, 7, 1602725 CrossRef.
- H. Li, H. Zhou, L. Zhuang, T. Liu, W. Han and H. Huang, Adv. Energy Sustainability Res., 2022, 3, 2100216 CrossRef CAS.
- W. Cheng, J. Fu, H. Hu and D. Ho, Adv. Sci., 2021, 8, e2100775 CrossRef PubMed.
- Z. Cao, G. Liang, D. Ho, C. Zhi and H. Hu, Adv. Funct. Mater., 2023, 33 Search PubMed.
- A. Dey, S. Varagnolo, N. P. Power, N. Vangapally, Y. Elias, L. Damptey, B. N. Jaato, S. Gopalan, Z. Golrokhi, P. Sonar, V. Selvaraj, D. Aurbach and S. Krishnamurthy, Prog. Mater. Sci., 2023, 139 Search PubMed.
- M. R. Lukatskaya, S.-M. Bak, X. Yu, X.-Q. Yang, M. W. Barsoum and Y. Gogotsi, Adv. Energy Mater., 2015, 5, 1500589 CrossRef.
- H. Shao, K. Xu, Y.-C. Wu, A. Iadecola, L. Liu, H. Ma, L. Qu, E. Raymundo-Piñero, J. Zhu, Z. Lin, P.-L. Taberna and P. Simon, ACS Energy Lett., 2020, 5, 2873–2880 CrossRef CAS.
- Y. Wen, R. Li, J. Liu, Z. Wei, S. Li, L. Du, K. Zu, Z. Li, Y. Pan and H. Hu, J. Colloid Interface Sci., 2021, 604, 239–247 CrossRef CAS PubMed.
- X. Wei, M. Cai, F. Yuan, D. Lu, C. Li, H. Huang, S. Xu, X. Liang, W. Zhou and J. Guo, Appl. Surf. Sci., 2022, 606 Search PubMed.
- N. Gupta, R. K. Sahu, T. Mishra and P. Bhattacharya, J. Mater. Chem. A, 2022, 10, 15794–15810 RSC.
- L. Chen, Y. Bi, Y. Jing, J. Dai, Z. Li, C. Sun, A. Meng, H. Xie and M. Hu, Molecules, 2023, 28 CrossRef CAS.
- W. Bao, X. Tang, X. Guo, S. Choi, C. Wang, Y. Gogotsi and G. Wang, Joule, 2018, 2, 778–787 CrossRef CAS.
- M. Hu, R. Cheng, Z. Li, T. Hu, H. Zhang, C. Shi, J. Yang, C. Cui, C. Zhang, H. Wang, B. Fan, X. Wang and Q. H. Yang, Nanoscale, 2020, 12, 763–771 RSC.
- W. Liu, Y. Zheng, Z. Zhang, Y. Zhang, Y. Wu, H. Gao, J. Su and Y. Gao, J. Power Sources, 2022, 521 Search PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- H. Pazniak, M. Benchakar, T. Bilyk, A. Liedl, Y. Busby, C. Noel, P. Chartier, S. Hurand, M. Marteau, L. Houssiau, R. Larciprete, P. Lacovig, D. Lizzit, E. Tosi, S. Lizzit, J. Pacaud, S. Celerier, V. Mauchamp and M. L. David, ACS Nano, 2021, 15, 4245–4255 CrossRef CAS PubMed.
- J. L. Hart, K. Hantanasirisakul, A. C. Lang, B. Anasori, D. Pinto, Y. Pivak, J. T. van Omme, S. J. May, Y. Gogotsi and M. L. Taheri, Nat. Commun., 2019, 10, 522 CrossRef CAS PubMed.
- V. Natu, M. Benchakar, C. Canaff, A. Habrioux, S. Célérier and M. W. Barsoum, Matter, 2021, 4, 1224–1251 CrossRef CAS.
- F. Zhou, X.-S. Yang, J. Liu, J. Liu, R. Hu, L. Ouyang and M. Zhu, J. Power Sources, 2021, 485 Search PubMed.
- F. Zaman, G. Rooh, N. Chanthima, S. U. Khan, H. J. Kim, S. Kothan, N. Chanlek, M. Arshad and J. Kaewkhao, J. Alloys Compd., 2022, 893 Search PubMed.
- J. Halim, K. M. Cook, M. Naguib, P. Eklund, Y. Gogotsi, J. Rosen and M. W. Barsoum, Appl. Surf. Sci., 2016, 362, 406–417 CrossRef CAS.
- R. Meng, J. Huang, Y. Feng, L. Zu, C. Peng, L. Zheng, L. Zheng, Z. Chen, G. Liu, B. Chen, Y. Mi and J. Yang, Adv. Energy Mater., 2018, 8, 1801514 CrossRef.
- X. Chen, Y. Zhu, M. Zhang, J. Sui, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, ACS Nano, 2019, 13, 9449–9456 CrossRef CAS PubMed.
- A. Sarycheva and Y. Gogotsi, Chem. Mater., 2020, 32, 3480–3488 CrossRef CAS.
- N. Zhao, Y. Yang, D. Yi, Y. Xiao, K. Wang, W. Cui and X. Wang, Chem. Eng. J., 2021, 422 Search PubMed.
- Y. Wen, T. E. Rufford, X. Chen, N. Li, M. Lyu, L. Dai and L. Wang, Nano Energy, 2017, 38, 368–376 CrossRef CAS.
- K. Tao, Y. Gong and J. Lin, Nano Energy, 2019, 55, 65–81 CrossRef CAS.
- M. R. Lukatskaya, S. Kota, Z. Lin, M.-Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P.-L. Taberna, M. W. Barsoum, P. Simon and Y. Gogotsi, Nat. Energy, 2017, 2, 17105 CrossRef CAS.
- S. Fleischmann, Y. Zhang, X. Wang, P. T. Cummings, J. Wu, P. Simon, Y. Gogotsi, V. Presser and V. Augustyn, Nat. Energy, 2022, 7, 222–228 CrossRef CAS.
- X. Mu, D. Wang, F. Du, G. Chen, C. Wang, Y. Wei, Y. Gogotsi, Y. Gao and Y. Dall'Agnese, Adv. Funct. Mater., 2019, 29, 1902953 CrossRef.
- P. Salles, D. Pinto, K. Hantanasirisakul, K. Maleski, C. E. Shuck and Y. Gogotsi, Adv. Funct. Mater., 2019, 29, 1809223 CrossRef.
- Y. Tang, Y. Zhang, O. I. Malyi, N. Bucher, H. Xia, S. Xi, Z. Zhu, Z. Lv, W. Li, J. Wei, M. Srinivasan, A. Borgna, M. Antonietti, Y. Du and X. Chen, Adv. Mater., 2018, 30, 1802200 CrossRef PubMed.
- Y. Sun, C. Zhan, P. R. C. Kent, M. Naguib, Y. Gogotsi and D. E. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 763–770 CrossRef CAS PubMed.
- P. Zhang, Q. Zhu, R. A. Soomro, S. He, N. Sun, N. Qiao and B. Xu, Adv. Funct. Mater., 2020, 30 CAS.
- L. Liao, D. Jiang, K. Zheng, M. Zhang and J. Liu, Adv. Funct. Mater., 2021, 31, 2103960 CrossRef CAS.
- J. Zhang, N. Kong, S. Uzun, A. Levitt, S. Seyedin, P. A. Lynch, S. Qin, M. Han, W. Yang, J. Liu, X. Wang, Y. Gogotsi and J. M. Razal, Adv. Mater., 2020, 32, 2001093 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06032b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |