
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nitridochromate(IV) fluoride – LiCa8[CrN3]2N2F†
Natalia
Gloriozova
a,
Yurii
Prots
 a,
Mitja
Krnel
a,
Ulrich
Burkhardt
a,
Marcus
Schmidt
a,
Alim
Ormeci
a,
Franziska
Jach‡
ab,
Peter
Höhn
*a and
Yuri
Grin
a
a,
Mitja
Krnel
a,
Ulrich
Burkhardt
a,
Marcus
Schmidt
a,
Alim
Ormeci
a,
Franziska
Jach‡
ab,
Peter
Höhn
*a and
Yuri
Grin
a
aMax-Planck-Institut für Chemische Physik fester Stoffe, Nöthnitzer Straße 40, 01187, Dresden, Germany. E-mail: Peter.Hoehn@cpfs.mpg.de
bFaculty of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany
First published on 28th February 2024
Abstract
LiCa8[CrIVN3]2N2F (Pnnm (#58), a = 17.5230(13) Å, b = 7.3379(5) Å, c = 4.9433(4) Å) is an example of a multinary nitridochromate fluoride, that provides additional information on almost elusive tetravalent nitridochromates. Shorter Cr–N bond lengths compared to those in the previously reported nitridochromates(III), as well as diamagnetic behavior and vibrational spectroscopy data suggest Cr(IV), which is in good agreement with the charge balance and crystal structure refinement. According to band structure calculations, LiCa8[CrIVN3]2N2F is a semiconductor with a band gap of 1.1 eV. The compound features trigonal planar [CrN3]5− units of Cs symmetry, and lithium, calcium, nitrogen and fluorine atoms arranged in a fragment of the rock salt type structure.
Introduction
Multianionic compounds are materials containing several anionic species, usually with different properties such as charge, ionic radius, electronegativity, etc. Mixed anions in a single material lead to additional structural complexity and unexpected physical properties.1 A special class of such compounds are quaternary and multinary nitridometalate halides, containing alkali and/or alkaline earth metals or lanthanides as cations as well as isolated or condensed units [MxNy]z− (M is a transition metal) and halide as anions. These materials are scarcely investigated due to challenging synthesis procedures. To date, only a few quaternary and multinary nitridometalate halides, all fluorides, have been reported: LiSr2[TaVN3]F,2 LiBa5[GaIIIN3]F5,3 and Ce2[MnII,IIIN3]F2−δ,4 are known.Chromium exhibits a wide variety of oxidation states in ternary and higher alkaline-earth nitridochromates,5 ranging from Cr(III) to Cr(VI). The trivalent chromium is generally associated with its trigonal planar coordination and is represented among nitridochromates by Ca3[CrIIIN3],6 Sr3[CrIIIN3], and Ba3[CrIIIN3].7 Recently, Sr3[CrIIIN3] and Ba3[CrIIIN3]7 have been described as electrides Sr3[CrIVN3]·e−and Ba3[CrIVN3]·e−,8 able to react with hydrogen to form nitridochromate hydrides Sr3[CrIVN3]H8 and Ba3[CrIVN3]H.9 Trigonal coordination of chromium is not exclusive for Cr(III), it has also been observed in LiSr2[CrIVN3].10 Partial oxidation of Ca3[CrIIIN3]6 with hydrogen yields Ca6[CrIII,IV2N6]H11 with an ethane-like anion, containing interconnected mixed-valent/intermediate valent Cr(III) and Cr(IV) species. In addition, Cr–Cr bonding is observed in Ca4[CrV2N6], Sr4[CrV2N6]12 and Li4Sr2[CrV2N6],13 with CrVN4 tetrahedra condensed into edge-sharing pairs. In addition, Cr(V) and Cr(VI) in tetrahedral coordination are observed in Ba5[CrVN4]N14 and Sr3[CrVIN4],15 respectively. Apart from a few examples, such as LiSr2[CrIVN3]10 and nitridometalate hydrides, no other nitridochromates(IV) have been reported.
Here we present the preparation, crystal structure determination and characterization of a new phase LiCa8[CrIVN3]2N2F, which extends both previously discussed areas. It represents the first example of a nitridochromate halide and provides additional information on Cr(IV)-containing compounds.
Results and discussion
Crystal structure determination and refinement
The crystal structure solution was accomplished by applying direct methods.16 First, the positions of Cr and Ca were determined. In subsequent steps, the N positions were located from the difference Fourier maps and refined. Four crystallographic Ca positions, the CrN3 anion and one N atom located in the center of the Ca octahedron were identified. The refinement resulted in high residuals R1 = 0.10, wR2 = 0.30 and high residual electron density (−1.5/20.4 e Å−3). Evaluation of the difference Fourier map at this stage revealed two distinct maxima at (½, 0, ½) and (½, 0, 0) (Fig. 1a). In a further refinement cycle, these two positions were assigned as F1 and Li1, respectively, due to their environments. The refinement resulted in significantly lower residuals R1 = 0.04, wR2 = 0.09, and lower residual electron density (−1.8/2.2 e Å−3), however, the F1 position showed a high atomic displacement parameter (0.03 Å2), indicating either disorder or partial occupation, whereas the Li1 position showed a negative atomic displacement parameter (−0.001 Å2), indicating higher electron density at this position. Evaluation of the difference Fourier map revealed elongation of the electron density along [001] around both positions, indicating positional disorder in the Li/F substructure (Fig. 1b). In a subsequent refinement cycle, the occupancies of both F1 and Li1 positions were refined as free variables, resulting in values of 0.94(1) and 1.71(4), respectively. | ||
| Fig. 1 Difference Fourier maps at y = 0 during different stages of the refinement, (a) indicating two maxima between c = 0 and 1, (b) indicating elongation of maxima, (c) indicating elongation of one maximum and splitting of another maximum, (d) of the final crystal structure solution. Isolines in each panel are drawn in steps of 0.5 e Å−1. Detailed description of each step is given in the text. | ||
The corresponding difference Fourier map (Fig. 1c) clearly indicated the splitting of both positions. In a subsequent run, the F1 position was split into F1 (2c) and F1A (4f), with the total occupancy was set to two atoms. The splitting of the Li1 position could not be resolved, so the Li1 site was refined as mixed occupancy by Li or Ca5A species and voids (□). To maintain the charge neutrality, the occupancies of Ca5A and the voids were kept equal. The isotropic displacement parameters of F1/F1A and Li1/Ca5A/□ were constrained to be equal. The occupancies of F1 and F1A were found to be 0.75(1) (2c) and 0.123(5) (4f), and those of Li1 and Ca5A to be 0.75(1) (2d) and 0.123(5) (2d), respectively. The residuals decreased slightly (R1 = 0.031, wR2 = 0.073), leading to the composition LiCa8[CrIVN3]2N2F. The resulting difference Fourier map is shown in Fig. 1d. Due to the disorder and the constrains applied, anisotropic refinement of the displacement parameters of F1, F1A, Li1 and Ca5A was not performed.
Based on the results of EDX analysis on a freshly cleaved crystal surface showing no oxygen impurities, oxygen was not considered in the refinement (see Experimental for details). DTA-TG-MS showed the absence of hydrogen. Above 973 K, only the N species evolving from the heat treatment are observed in the gas phase. The masses of the N species were determined (see Experimental for details).
Crystal structure
LiCa8[CrIVN3]2N2F crystallizes in space group Pnnm (#58), with lattice parameters a = 17.5230(13) Å, b = 7.3379(5) Å, c = 4.9433(4) Å and represents a new structure type (Tables 1, 2, S1, and Fig. 2a, b, S1†). | ||
| Fig. 2 (a) The crystal structure of LiCa8[CrIVN3]2N2F. (b) SEM image of the cleaved crystal of LiCa8[CrIVN3]2N2F (SE contrast). (c) [CrN3]5− complex anion showing bond lengths (in Å, indicated in red) and N–Cr–N bond angles (in °, indicated in black). | ||
| Crystal system | Orthorhombic |
| space group | Pnnm (#58) |
| α (Å) | 17.5230(13) |
| b (Å) | 7.3379(5) |
| c (Å) | 4.9433(4) |
| V (Å3) | 635.62(7) |
| Z | 2 |
| ρ calc (g cm−3) | 2.87 |
| μ (mm−1) | 4.93 |
| 2θ range (°) | 4.65–63 |
| Diffractometer | RIGAKU AFC7 |
| Wavelength (Å) | 0.71073 (Mo Kα) |
| Monochromator | Graphite |
| Temperature (K) | 293(2) |
| Measured reflections | 4955 |
| Unique reflections | 1140 |
| R int | 0.030 |
| Observed reflections [Fo > 4σ(Fo)] | 1036 |
| number of refined parameters | 60 |
| R 1 [Fo > 4σ(Fo)] | 0.031 |
| R 1 (all data) | 0.036 |
| wR2 | 0.073 |
| Goodness-of-fit | 1.109 |
| residual peaks (e Å−3) | –1.17/+0.92 |
| Atom | Site | x/a | y/b | z/c | U eq/iso (Å2) | Occupancy |
|---|---|---|---|---|---|---|
| a U isotropic and constrained. b Constrained, for details see text. | ||||||
| Cr1 | 4g | 0.36332(3) | 0.18515(7) | 0 | 0.0096(1) | 1 |
| Ca1 | 4g | 0.40471(3) | 0.60304(9) | 0 | 0.0109(1) | 1 |
| Ca2 | 4g | 0.03216(3) | 0.80264(8) | 0 | 0.0093(1) | 1 |
| Ca3 | 4g | 0.14642(4) | 0.39502(9) | 0 | 0.0107(1) | 1 |
| Ca4 | 4g | 0.21390(3) | 0.9395(1) | 0 | 0.0136(1) | 1 |
| N1 | 4g | 0.4543(2) | 0.2765(4) | 0 | 0.0108(5) | 1 |
| N2 | 4g | 0.3548(2) | 0.9496(4) | 0 | 0.0124(5) | 1 |
| N3 | 4g | 0.2864(2) | 0.3325(4) | 0 | 0.0178(6) | 1 |
| N4 | 4g | 0.0977(2) | 0.1003(4) | 0 | 0.0106(5) | 1 |
| F1 | 2c | ½ | 0 | ½ | 0.0221(8)a | 0.75(1)b |
| F1A | 4f | ½ | 0 | 0.433(5) | 0.0221a | 0.123(5)b |
| Li1 | 2d | ½ | 0 | 0 | 0.0221a | 0.75(1)b |
| Ca5A | 2d | ½ | 0 | 0 | 0.0221a | 0.123(5)b |
The main structural feature of the title compound is a distorted planar [CrN3]5− anion with Cs symmetry (Fig. 2c). The symmetry reduction from ideal D3h to Cs can be explained by a highly inhomogeneous environment of nitrogen species in [CrN3]5− anions due to Li+ and Ca2+ cations with very different radii. These significantly different cationic environments around nitrogen atoms lead to a distortion of the 120° bond angle within the anion. Similar nitridochromate units, but with higher symmetry of D3h are observed in Sr3[CrIIIN3], Sr3[CrIVN3]H,8 Ba3[CrIIIN3],7 and Ba3[CrIVN3]H.9 Nitridochromate groups in Ca3[CrIIIN3]6 are distorted and exhibit C2v symmetry, while in LiSr2[CrIVN3]10 – pseudo C3v symmetry.
The average Cr–N distance in LiCa8[CrIVN3]2N2F is 1.731(2) Å, which is shorter than that in the previously reported nitridochromates(III), supports the presence of Cr(IV) in the structure (Table 3).
| Atoms | Compound | Distances/Å | Average distance/Å |
|---|---|---|---|
| Cr–N | LiCa8[CrIVN3]2N2F | 1.728(3)–1.735(3) | 1.731(2) |
| Ca3[CrIIIN3]6 | 1.766 (7)–1.864(8) | 1.799(7) | |
| Sr3[CrIIIN3]7 | 1.728(3) | ||
| Ba3[CrIIIN3]7 | 1.732(8) | ||
| Sr3[CrIVN3]H8 | 1.709(7) | ||
| Ba3[CrIVN3]H9 | 1.739(3) | ||
| LiSr2[CrIVN3]10 | 1.716(6)–1.724(6) | 1.719(4) | |
| Li–N | LiCa8[CrIVN3]2N2F | 2.181(3)–2.571(3) | 2.376(2) |
| LiMoN217 | 2.098(17)–2.179(18) | 2.138(7) | |
| LiBa2N18 | 2.28(2) | ||
| Li–F | LiCa8[CrIVN3]2N2F | 2.14(2)–2.4716(2) | 2.361(7) |
| LiF19 | 2.013 | ||
| Li2Ca6(GeO4)3F220 | 1.83(1) | ||
| LiBa5GaN3F53 | 1.976(9)–2.39(2) | 2.167(7) | |
| K2LiAlF621 | 1.960(4)–2.162(6) | 2.061(2) | |
| Ca–F | LiCa8[CrIVN3]2N2F | 2.14(2)–2.700(3) | 2.451(3) |
| Li2Ca6(GeO4)3F220 | 2.358(8)–2.624(1) | 2.445(2) | |
| Ca–N | LiCa8[CrIVN3]2N2F | 2.181(3)–3.152(3) | 2.5340(6) |
| Ca3[CrIIIN3]6 | 2.388(8)–2.728(6) | 2.535(5) | |
| Ca6[Cr2III,IVN6]H11 | 2.471(1)–2.535(1) | 2.5015(7) | |
| Ca6[Cr2III,IVN6]F22 | 2.473(3)–2.555(3) | 2.502(2) | |
| Ca4[Cr2VN6]12 | 2.4233(1)–3.0913(1) | 2.6205(7) | |
| Ca6[FeIIIN3]N223 | 2.413(2)–2.947(3) | 2.572(4) | |
| Ca4[FeIVN3]N24 | 2.4312(15)–2.773(6) | 2.596(1) | |
The coordination of the nitrogen species in the [CrN3]5− anion is present in two forms: NCa2/2Ca3Cr1/3 and NLi1/2Ca1/2Ca3Cr1/3 octahedra in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio (Fig. S2†). Similar coordination is found in Sr3[CrIIIN3]: three edge-sharing NSr2/2Sr3Cr1/3 octahedra.7 In Ca3[CrIIIN3], the nitrogen species are coordinated differently: one NCa2/2Ca3Cr1/3 and two NCa1/2Ca4Cr1/3 polyhedra sharing edges and forming an “open space” (Fig. S2†).6
:2 ratio (Fig. S2†). Similar coordination is found in Sr3[CrIIIN3]: three edge-sharing NSr2/2Sr3Cr1/3 octahedra.7 In Ca3[CrIIIN3], the nitrogen species are coordinated differently: one NCa2/2Ca3Cr1/3 and two NCa1/2Ca4Cr1/3 polyhedra sharing edges and forming an “open space” (Fig. S2†).6
In the title compound, NCa2/2Ca3Cr1/3 and NLi1/2Ca1/2Ca3Cr1/3 octahedra are arranged in a way, that large voids are formed above and below trigonal [CrN3]5− units. Nitridochromate anions and Ca, N, and F atoms form planar layers with a stacking sequence of …ABAB… along [001] (Fig. 3a). Similar coordination of [CrN3]6− groups, as well as voids, and stacking layers are observed in Sr3[CrIIIN3]7 (Fig. 3d, and S2†).
 | ||
| Fig. 3 (a) Two substructures in the crystal structure of LiCa8[CrIVN3]2N2F, viewed along [001]: the rock salt pattern (in yellow) and the “Sr3CrN3” blocks (transparent). (b) The rock salt fragments, highlighting NCa6 octahedra (in green). The nitridochromate [CrN3]x− anions in the structures of (c) LiCa8[CrIVN3]2N2F and (d) Sr3[CrIIIN3]. (e) The ordered channel in the ideal structure of LiCa8[CrIVN3]2N2F. (f) Examples of possible disorder in the channels in the structure of LiCa8[CrIVN3]2N2F. | ||
The structure of LiCa8[CrIVN3]2N2F can be described as a 3D network, consisting of two building blocks: the fragments of the rock salt structure and the “Sr3[CrN3]” blocks described above (Fig. 3a–c). The rock salt blocks are composed of nitrogen, fluorine, calcium and lithium atoms, and are parallel to [001] (Fig. 3b). Corner-sharing “NaCl” blocks are connected by lithium and fluorine atoms, arranged in chains, running along [001]. The chains are disordered from the ideal (–Li–F–) case: channels can be described as a sequence of (Li, Ca, voids) and F species (Fig. 3e and 3f). When the (½, 0, 0) site is occupied by Ca5A, the neighboring F atoms, occupying the (½, 0, 0.433(5)) site are shifted toward Ca5A; whereas when the cationic site is occupied by Li, the neighboring F occupies the (½, 0, ½) site. In the case of the void at the cationic site, the F site is shifted to towards the (½, 0, 0.567(5)) site (Fig. 3f). Such a distortion of the chains is in good agreement with the data and the structure remains charge neutral.
In addition to nitridochromate [CrN3]5− and F− anions, the title compound consists of isolated N4Ca6 octahedra running along [001] (Fig. 3b, and S3), with the remaining nitrogen atoms also 6-fold (Fig. S3†). The coordination of the lithium, calcium, nitrogen and fluorine atoms is shown in Fig. S3,† the corresponding distances are listed in Table 3.
Electronic structure
First-principles electronic structure calculations were performed on the model of the crystal structure of LiCa8[CrIVN3]2N2F with fully occupied F1 and Li1 positions. Spin-polarized calculations converged to the solution with zero magnetic moment on all atoms. Hence, the title compound is not expected to have a magnetic long-range order, which is consistent with the magnetization measurements. The electronic total and projected densities of states (DOS) for LiCa8[CrIVN3]2N2F show a band gap of 1.1 eV (Fig. 4a). The lowest lying three groups of states are all formed by N 2s electrons (middle panel). Here, N4 stands out in comparison to the other N types, because its 2s states between −11.0 and −10.5 eV are well separated from the others located in the ranges [−13.5, −13.0] and [−12.75, −12.25] eV. The reason for this is that N4 is only coordinated with six Ca atoms, while N1, N2 and N3 are bonded to the Cr atom (compare the top and middle panels in Fig. 4). The 2p states of the F atoms form very narrow bands, [−6.80, −6.71] and [−6.68, −6.64] eV due to (2px, 2py) and 2pz, respectively. Since F atoms are coordinated with Li, Ca2 and Ca3 atoms (2 times each), the only other significant contributions to DOS in these energy ranges come from these atoms (bottom panel for Ca2 and Ca3, Li not shown). The states between −4.2 and −2.0 eV consist mainly of contributions from N1, N2 and N3 2p and Cr 3d orbitals. The uppermost part of the occupied states, between −1.67 eV and the valence band maximum (VBM) set to 0 eV, has two interesting features. First, N4 2p states dominate this region. Second, the Cr contributions between −0.5 and 0 eV are significant because they form Cr-centered 4-atom bonds symmetrically located above and below the [CrN3] layer (cf. the following discussion on chemical bonding). The Cr states responsible for these bonds are due to the 3z2–r2 and 4s orbitals (Fig. 4b). This feature has been observed in other compounds with similar [CrN3] units.9,10 Since in the title compound these units lie in the (001) plane, the symmetry properties of the Cr and N orbital hybridizations are very clear.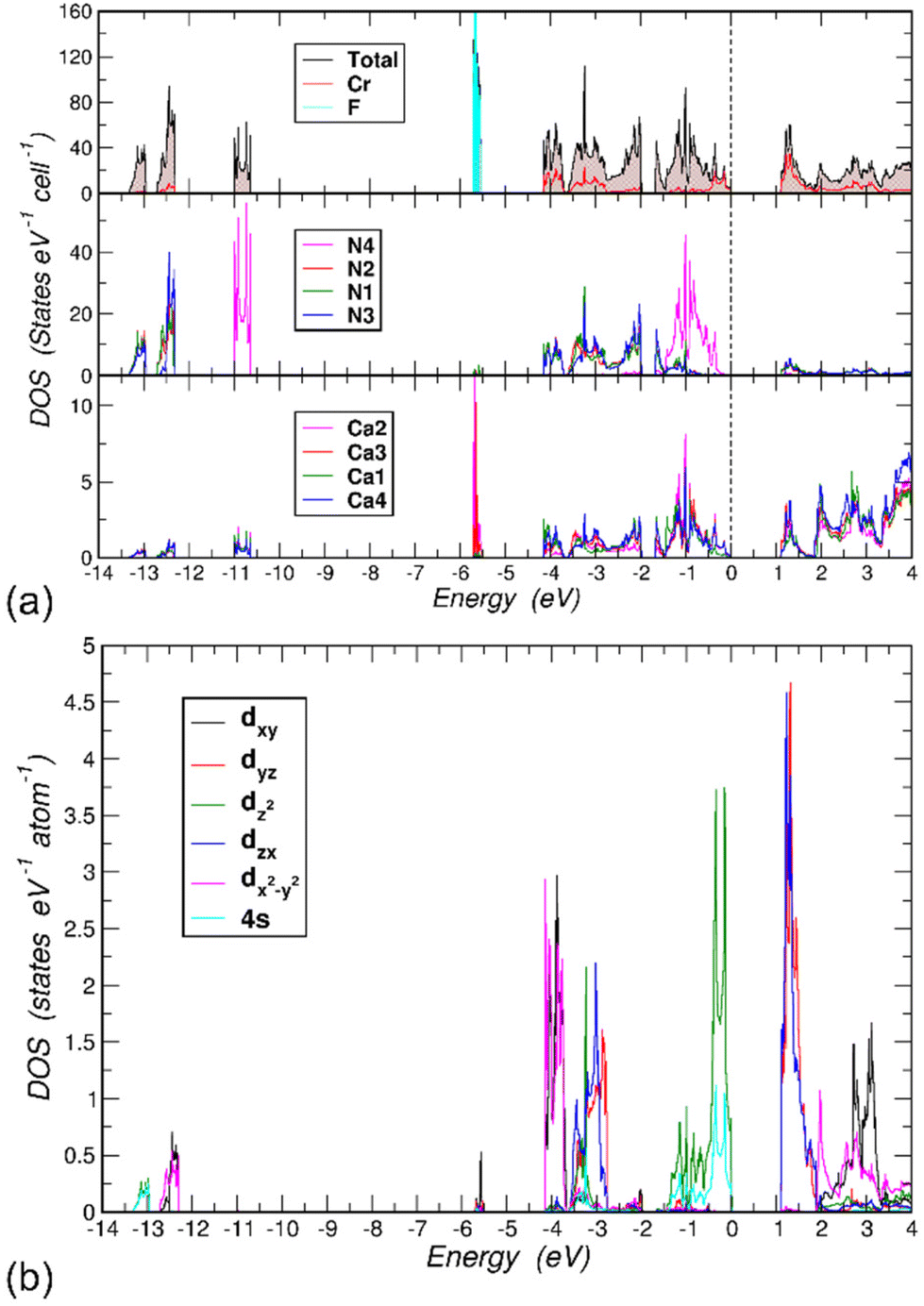 | ||
| Fig. 4 (a) Electronic density of states (DOS) for LiCa8[CrIVN3]2N2F. Top panel: total, Cr- and F-projected DOS, middle panel: projected DOS for N species, bottom panel: projected DOS for Ca species. (b) Cr 4s and (l,m)-decomposed 3d partial DOS with quantization axis being the crystallographic c-axis. Li contributions are not shown. | ||
The N 2s orbitals hybridize with the in-plane Cr orbitals: 3z2–r2, 4s (lower part, [−13.5, −13.0] eV) and xy, x2–y2 (upper part, [−12.75, −12.25] eV) orbitals. Note that the former orbitals have magnetic quantum number m = 0 and the latter orbitals have m = ±2. This different symmetry explains the observed split into two parts. Within the N 2p region, [−4.2, −2.75] eV, likewise the Cr orbitals with m = ±2 are largely decoupled from the other 3d orbitals with m = ±1 and m = 0 giving rise to two groups of states separated by a small gap of 0.03 eV at ∼ −3.65 eV.
Chemical bonding
The analysis of chemical bonding was performed on the model of the crystal structure of LiCa8[CrIVN3]2N2F with fully occupied F1 and Li1 positions. The topological analysis of the electron density yields the QTAIM basins for all atoms (Fig. 5a), and by integrating the ED within these regions their populations were obtained. Subtracting the atomic numbers from the latter results in the effective charges. According to the differences in electronegativity, Li, Cr and Ca atoms are positively charged, Li: +0.88, Cr: +0.96, Ca: between +1.37 and +1.40. F is negatively charged with −0.88, N atoms bonded to Cr have charges between −1.52 (×2) and −1.56, while the nitrogen N4 inside the Ca6 octahedron has a larger negative charge of −1.90. The round shape of the QTAIM basin of Li includes mainly the first shell, while that of F includes basically the entire second shell, signaling the ionic behavior of these species in LiCa8[CrIVN3]2N2F. This is in accordance with the very narrow F states in the electronic DOS (Fig. 4a, top panel), noted above. The QTAIM basin of Cr shows convex faces towards its bonding N partners, similar to those found in LiSr2[CrN3],10 implying polar covalent N–Cr bonds. The N4 atom at the center of the Ca6 octahedron has a cube-like QTAIM basin with concave faces toward the Ca ligands.In order to gain a deeper understanding of the bonding situation, the electron localizability approach was applied, employing combined analysis of electron density and electron localizability indicator in its ELI-D representation.25
A characteristic feature of ELI-D in the title compound is a low number of local maxima between atoms, indicating a high level of ionicity in the chemical bonding (Fig. 5b). When a dedicated maximum can be observed, the population of its basin is clearly formed by the most electronegative atom (Fig. 5c), e.g., even if the population of the Li–F basin is very low it is practically completely contributed by fluorine (pink in Fig. 5c), revealing for this bond a high polar character of 0.98 on the scale between 0 and 1 (calculated according to ref. 26 and 27). The bond basins around the [CrN3] anion form superbasins composed of the ‘lone-pairs’ and basins reflecting the Cr–N bonding. As in several other cases,10,28 the separation of these contributions is difficult. For N1 and N3 this was possible, for N2 only the superbasin was evaluated (green, Fig. 5c, right). The bond basin between N1 and Cr is formed with the minor support of the neighboring Ca3 species (reddish, Fig. 5c). As expected from the lower difference in electronegativity between chromium and nitrogen, the polarity of this bond is lower, amounting to 0.76. The bond basin for N3–Cr interaction is formed mainly by nitrogen and chromium, with minor contributions from Ca4 (orange, Fig. 5c). The population of the bond basins above and below the chromium nuclei (light pink, Fig. 5c) is formed mainly by chromium with contributions from three Ca neighbors. It can be understood as a ‘lone pair’ on chromium (cf. considerations above), which, due to the cationic environment, transforms to the four-atomic polar bond (polar character of 0.57). On the other hand, the ELI-D distribution around the nitrogen nuclei is non-spherical and markedly structured (Fig. 5b). This behavior is due to the interaction with the neighboring cations. So, four attractors are observed around the N4 nucleus. All of them represent five-atomic interactions of the central N4 with four of the six attached calcium ligands forming the octahedron. In the selected case (marked with an asterisk in Fig. 5c), the polar character of this multicenter bond is high (0.89, light green shape in the center of Fig. 5c). In total, the N4 atom provides 6.76 electrons out of the total bond population of 7.28 (∼93%), the rest being shared by the six Ca atoms forming the octahedron [N4Ca6]. The topological analysis of the ELI-D around the nitrogen species forming the trigonal planar [CrN3] anion reveals a similar behavior, e.g. of the total 5.88 electrons in the basin of the ‘lone pairs’ of N1, 5.51 are contributed by nitrogen, the remaining are coming from Ca2, Ca1 and Li; of the total 4.14 electrons in the ‘lone pairs’ of N3, 3.97 electrons are contributed by the nitrogen itself, the rest from the neighboring calcium (and chromium) atoms; from the total of 6.91 electrons in the ‘lone pairs’ of N2, 6.40 electrons originate from the N2, the rest from the neighboring calcium, lithium (and chromium) ligands. The ELI isosurface around the F atom is spherical (the orange ball in Fig. 5b), the basin of the corresponding ELI attractor contains 7.76 electrons, of which 7.70 are contributed by F. Note that this is very close to the full second shell occupancy value of 8. Among the 6 neighbors of the F atom, the two Ca1 atoms (at a distance of 2.679 Å) do not contribute to this F valence region basin, only the Ca3 (×2, at a distance of 2.292 Å) and Li (×2, at a distance of 2.471 Å) atoms do.
 | ||
| Fig. 5 Chemical bonding analysis in LiCa8[CrN3]2N2F. (a) The shapes and effective charges of the QTAIM atoms. (b) ELI-D distribution in the planes (100), approximately (10/4 00) and (002). (c) ELI-D bond basins, their atomicity (Na), participating atoms, total populations and partial contributions of participating atoms. The basin of the 5-atomic bond marked with an asterisk is an exemplary part of the superbasin around N4. | ||
Vibrational spectroscopy
Infrared (IR) and Raman spectra of LiCa8[CrIVN3]2N2F were used to investigate the nature of the [CrN3]5− anion (Fig. 6a). Considering an ideal trigonal-planar anion with point symmetry D3h, site symmetry Cs, and factor group D2h, six ν(CrN3) modes in IR and six in Raman spectra; four δ(NCrN) modes in IR and four in Raman spectra; and one γ(NCrN) mode in IR and two in Raman spectra are expected (Table S3†). Not all of these are observed in the experimental spectra.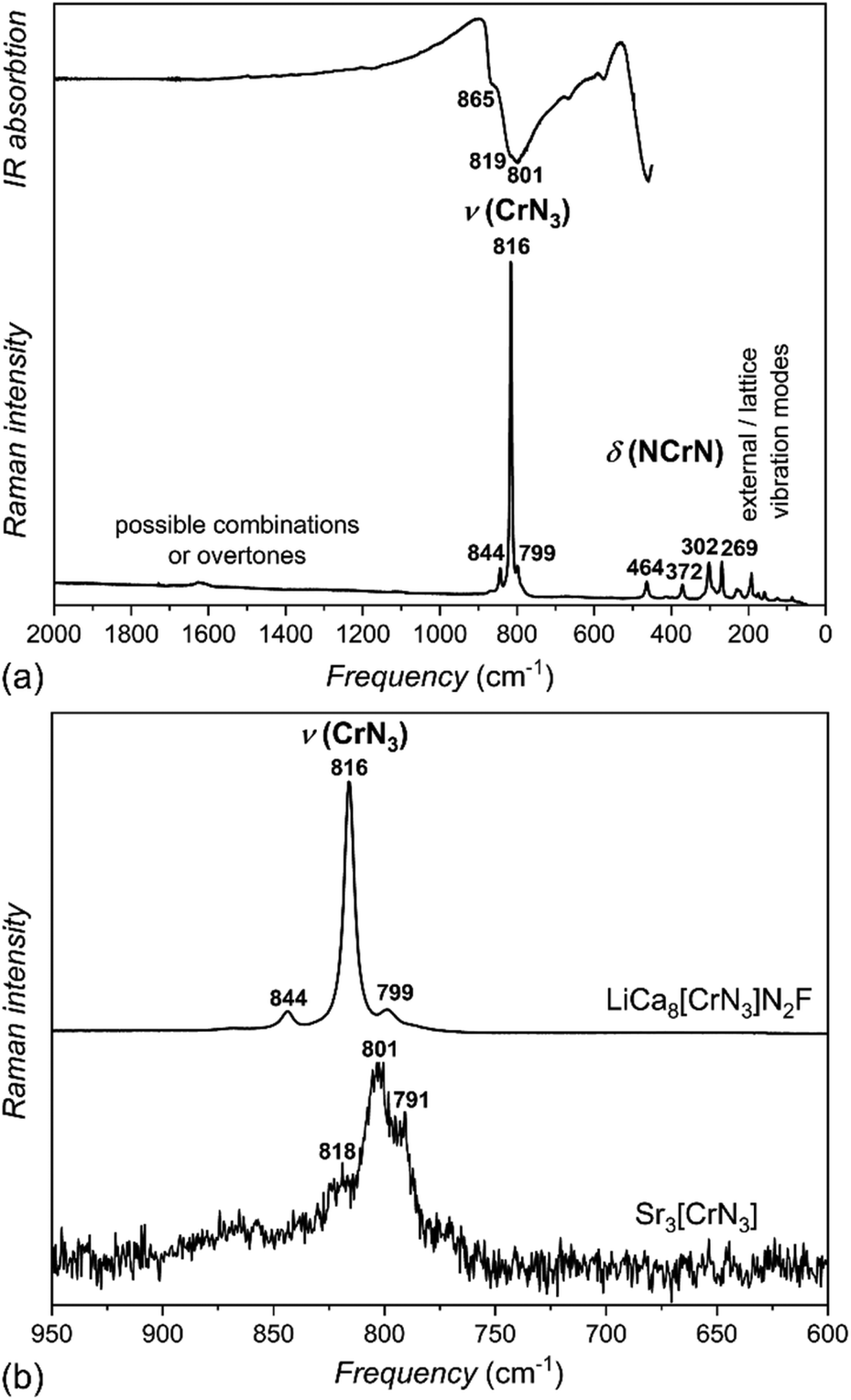 | ||
| Fig. 6 (a) Infrared (top) and Raman (bottom) spectra of LiCa8[CrIVN3]2N2F. (b) Comparison of the section between 950 cm−1 and 600 cm−1 of the Raman spectra of LiCa8[CrIVN3]2N2F (top) and Sr3[CrIIIN3] (bottom). | ||
Stretching modes are observed in both IR and Raman spectra between 800–860 cm−1, deformation modes below 500 cm−1; both typical values for nitridometalates.10,22,29
Additionally, vibrational spectra of Sr3[CrIIIN3] with ideal trigonal planar symmetry D3h, and a somewhat simpler cation environment were measured for comparison (Fig. 6b). With increasing oxidation state from Cr(III) in Sr3[CrIIIN3] to Cr(IV) in LiCa8[CrIVN3]2N2F, the stretching modes are shifted to higher frequencies, which correlates well with stronger Cr–N interactions in the Cr(IV) phase. This shift in the Raman spectra is an additional sign of the tetravalent metal center and agrees well with the results of crystal structure refinement.
Physical properties
The magnetic moment of LiCa8[CrIVN3]2N2F was measured in magnetic fields μ0H = 0.1, 1, and 3.5 T, at temperatures between 1.8 and 300 K. From the magnetic moment, the magnetic susceptibility χ = M/H was calculated. The temperature dependence of susceptibility χ(T) indicates a predominantly paramagnetic behavior (Fig. 7). The variation of χ(T) with field can be attributed to ferromagnetic impurities. The difference in susceptibility at high fields corresponds to an amount of 0.0006 at% Fe. After subtracting the contributions of the ferromagnetic impurities and the sample holder from the overall signal, the inverse magnetic susceptibility χ−1(T) was calculated. A fit of χ−1(T) with the Curie law, χ = C/T, where C is the Curie constant, yields C = 0.0138 emu K mol−1 and an effective magnetic moment (μeff) of 0.33μB per chromium atom (Fig. S4†). This value is too low to describe a paramagnetic ion and can only be attributed to a small amount of unidentified paramagnetic impurities close to the detection limit of PXRD. Therefore, we suggest that LiCa8[CrIVN3]2N2F is diamagnetic, which correlates well with the expected nonmagnetic ground state of chromium(IV), the results of crystal structure refinement, and vibrational spectroscopy data.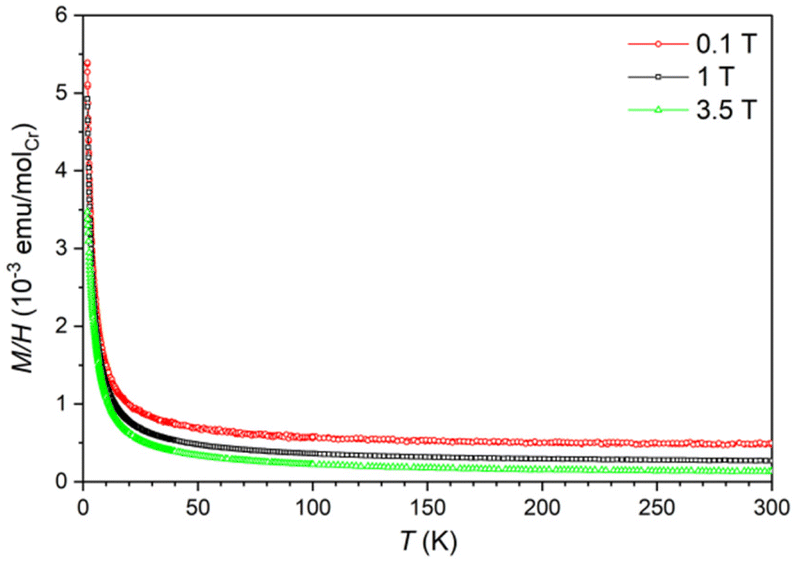 | ||
| Fig. 7 Temperature dependence of magnetic susceptibility of LiCa8[CrIVN3]2N2F under different fields. | ||
Electrical resistivity measurements have been performed on a crystal of LiCa8[CrIVN3]2N2F in the temperature range T = 120 K to 300 K in zero magnetic field. The compound exhibits semiconducting-like behavior (Fig. S5†), which is consistent with the electronic structure calculations. The energy gap Eg can be estimated by fitting the temperature dependence of the resistivity ρ(T) with ρ(T) = ρ0exp(Eg/2kBT) function, where the fit yields the value of Eg = 0.185 eV. The calculated band gap (1.1 eV) is larger than the value (0.185 eV) obtained from the resistivity measurements due to the simplified model employed in the calculations.
Conclusions
LiCa8[CrIVN3]2N2F is the first example of a multinary nitridochromate(IV) fluoride, which was synthesized using a modified high-temperature centrifugation-aided filtration technique using excess of Li. The compound features tetravalent chromium in trigonal planar coordination. The oxidation state of the transition metal was established by magnetization measurements, vibrational spectroscopy data, and analysis of Cr–N bond lengths in comparison with reported nitridochromates(III). According to band structure calculations and physical properties measurements, LiCa8[CrIVN3]2N2F is a diamagnet and a semiconductor. Consistent with the description as a nitridochromate, the compound exhibits highly polar N–Ca, F–Li and polar nitrogen-dominated triatomic N–Cr–Ca, as well as Cr-dominated multi-atomic Cr–Ca bonds.Experimental
Synthesis
Due to the extreme air and moisture sensitivity of both reagents and products, all manipulations were performed in an inert atmosphere using an argon-filled glove box (ρ(O2, H2O)/ρ0 < 0.1 ppm).The following elements and compounds were used as starting materials: calcium fluoride (Alfa, 99.99%), lithium rod (Alfa, 99.9%), chromium powder (Alfa, 99.97%), calcium nitride (Ca3N2) powder, dicalcium nitride (Ca2N) powder, lithium nitride (Li3N) powder.
Calcium nitride (Ca3N2) was prepared from calcium metal (Alfa, dendritic pieces, 99.8%) under nitrogen flow (Praxair, 99.9999%, additionally purified by molecular sieves and BTS catalyst) at 973 K for 12 h followed by cooling down to 373 K. Dicalcium nitride (Ca2N) was synthesized from the same starting materials at 1323 K for 12 h and cooled down to room temperature.
Lithium nitride (Li3N) was obtained from lithium rod (Alfa, 99.9%) under nitrogen flow (Praxair, 99.9999%, additionally purified by molecular sieves and BTS catalyst) at 573 K, followed by cooling down to room temperature.
Green crystals with metallic luster of LiCa8[CrN3]2N2F were grown using a modified high-temperature centrifugation-aided filtration technique (HTCAF)30,31 from Ca3N2, Ca2N, Cr, CaF2, Li3N and Li in a molar ratio of 0.6:1:0.9:0.5:1.5:22. The mixture of approximately 0.8 g was placed in a sealed tantalum ampoule with a sieve inside, heated at 1023 K for 2 hours and then centrifuged at 3000 min−1 to separate crystals from the flux. Besides a small amount of Li3N, crystals of LiCa8[CrIVN3]2N2F were obtained. The centrifuged material was not examined in detail.
To check for other phases in the Li–Ca–Cr–N–F system and for the phase width of the title compound, another experiment was performed using the same technique but with slightly different ratios of the starting components (Table S4†). Besides crystals of LiCa8[CrIVN3]2N2F, small amounts of Ca6[CrIII,IV2N6]F22 and single-crystalline Li3N were obtained. Experiments to produce single-phase powders of LiCa8[CrIVN3]2N2F from stoichiometric amounts of Ca3N2, Cr, CaF2 and Li3N were unsuccessful.
Single crystal X-ray diffraction
Single-crystal X-ray diffraction data were used to refine the crystal structure and composition of LiCa8[CrN3]2N2F. Suitable single crystals were mechanically selected under polybutene oil and sealed into glass capillaries. Diffraction data were collected at room temperature on a Rigaku AFC7 diffractometer equipped with a Saturn 724+ CCD area detector (Mo-Kα radiation). The crystal structure solution was performed using the SHELXS/SHELXL-2018 software package.16 Graphical representation of the structure was done using the software Diamond.32 Details of data collection and crystallographic information are listed in Tables 1, 2 and S1, S2.† Further details of the crystal structure investigations can be obtained from FIZ Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: (+49)7247-808-666; e-mail: crysdata@fiz-karlsruhe.de, on quoting the deposition number CSD 2326218†).X-ray powder diffraction
Powder X-ray diffraction (PXRD) data were recorded on a Huber G670 imaging plate Guinier camera using a curved germanium (111) monochromator and Cu-Kα1 radiation in the range 4° ≤ 2θ ≤ 100° with an increment of 0.005° at room temperature. Samples of finely ground crystals were placed between two Kapton foils sealed with vacuum grease for protection against oxidation. Preliminary data processing was performed using the WinXPow software package.33 The PXRD of the crystals is shown in Fig. S6.†EDX spectroscopy
Well-facetted crystals were examined by scanning electron microscopy (Jeol JSM 7800 F). A shuttle system was used to transfer selected crystals of the title compound from the argon glove box to the SEM specimen chamber without contact with air. The EDX spectra were recorded with an SDD (silicon drift detector, Bruker Quantax EDX system). An acceleration voltage of 10 kV and a beam current of less than 1 nA were used for the SEM and EDX investigations to avoid beam damage. The EDX spectra show the characteristic X-ray lines of Ca, Cr, N, and F. The Li K line with energy E = 54 eV is not present in the spectra due to the low energy cutoff at 100 eV. Several crystals were examined for the presence of oxygen. The oxygen concentration on the freshly cleaved surfaces of several crystals was below the detection limit of the EDX system. The low acceleration voltage and the underlined surface tilt do not lead to reliable element concentrations by evaluation of X-ray peak intensities.DTA-TG-MS measurements
The DTA-TG measurements were performed in STA 449 C (NETZSCH) in flowing argon atmosphere inside a glove box (Ar 99.999% 100 ml min−1 with subsequent drying and oxygen post-purification via a Big Oxygen Trap from Trigon Technologies). The sample of approximately 20 mg was measured in the temperature range 298–1173 K with a heating/cooling rate of 5 K min−1 in a tantalum DTA/TG crucible with perforated lid using a type S (PtRh/Pt) thermocouple. The determined temperature-dependent mass changes were subjected to a buoyancy correction according to the measurement conditions.The TG-MS measurements were accomplished utilizing a combination of a thermo balance STA 409 CD (NETZSCH) and a quadrupole mass spectrometer QMS 403 (Pfeiffer Vacuum) in an argon atmosphere inside a glove box (MBraun). The measurements were carried out in MID mode for the ions with m/z 1, 2, 14, 16, 17, 18, 28 and 32 and with 70 eV electron impact ionization. Approximately 20 mg of a sample was placed in a corundum Knudsen cell with a tantalum inlay and a lid with a small bore (0.3 mm) and heated up to 1173 K with a heating rate of 5 K min−1. The gases evolving from the heat treatment were mixed with a continuous stream of argon purge gas and directed to the ionization chamber of the mass spectrometer with the aid of a skimmer.
Vibrational spectroscopy
Raman spectra were recorded at room temperature in the 4000–50 cm−1 range using a HR Evolution spectrometer equipped with a Laser Quantum Ventus diode laser (532 mm) and a Synapse CCD detector. The spectral resolution was approximately 0.42 cm−1. Suitable crystals were sealed in silica capillaries to prevent oxidation.Infrared spectra were collected on a PerkinElmer UATR-Two FTIR spectrometer inside a glove box in the range 4000–450 cm−1 in Attenuated Total Reflectance (ATR) mode. Background was corrected by measuring with the empty setup. No signals indicating moisture were detected by IR or Raman spectroscopy in any of the samples.
Physical properties measurements
Magnetic measurements were conducted by a Quantum Design MPMS XL-7 SQUID magnetometer equipped with a 7 T magnet. To avoid contact with air and moisture during the measurements, crystals of the title compound with a mass equal to 39.46 mg were placed and sealed into a pre-calibrated quartz tube under 400 mbar of He. All measured data were corrected for the diamagnetic contribution of the quartz tube.Electrical resistivity was measured on a crystal with the following dimensions: 0.2 × 0.2 × 0.1 mm3 using the standard four-terminal technique. Four 25 μm thick platinum wires were bonded to the sample surface using DuPont 4922N silver conductive paint. The measurements were performed in an inert atmosphere using a home-built resistivity equipment. Although all preparative manipulations and measurements were performed inside a glove box, the surface of the crystal might be locally oxidized due to possible minor oxygen impurities in the silver paint.
Computational methods
First-principles electronic structure calculations were performed using the all-electron full-potential local orbital (FPLO) method34 within the local density approximation (LDA) of density functional theory. Perdew–Wang parametrization was employed.35 The experimentally determined crystal structure was adopted with mixed occupancy features ignored. A well-converged k-mesh of 12 × 5 × 18 was used to evaluate the Brillouin zone integrations. Analysis of chemical bonding in position space was based on the electron localizability approach.36–38 The topological analysis aspect of Bader's quantum theory of atoms in molecules (QTAIM)39 was applied to both electron density (ED) and electron localizability indicator (ELI); both quantities were calculated on a uniformly spaced three-dimensional grid in position space by a module implemented in the FPLO package.40 The program Dgrid41 was used to perform the topological analysis. The basin intersection method was utilized to determine the atoms contributing to each bond.42Author contributions
N. Gloriozova grew the single crystals, Yu. Prots collected and analyzed single crystal X-Ray diffraction data, N. Gloriozova, P. Höhn and Yu. Prots performed the refinement, M. Krnel performed the magnetization and electrical conductivity measurements and interpreted the data, M. Schmidt analyzed the DTA-TG-MS data, U. Burkhardt evaluated the EDX data, F. Jach and P. Höhn collected and interpreted IR and Raman data and A. Ormeci and Yu. Grin performed the electronic structure calculations and chemical bonding analysis. N. Gloriozova, Yu. Grin and P. Höhn wrote the paper with the help of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Steffen Hückmann for collecting the powder X-ray diffraction data, Petra Scheppan for collecting EDX data, and Susann Scharsach for thermoanalytical measurements. Open Access funding provided by the Max Planck Society.References
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 1–15 CrossRef CAS PubMed.
- P. Pust and W. Schnick, Z. Anorg. Allg. Chem., 2011, 637, 1486–1489 CrossRef CAS.
- F. Hintze and W. Schnick, Solid State Sci., 2010, 12, 1368–1373 CrossRef CAS.
- D. A. Headspith, E. Sullivan, C. Greaves and M. G. Francesconi, Dalton Trans., 2009, 42, 9273–9279 RSC.
- P. Höhn and R. Niewa, in Handbook of Solid State Chemistry, Wiley-VCH, 2017, pp. 251–359 Search PubMed.
- D. A. Vennos, M. E. Badding and F. J. DiSalvo, Inorg. Chem., 1990, 29, 4059–4062 CrossRef CAS.
- M. G. Barker, M. J. Begley, P. P. Edwards, D. H. Gregory and S. E. Smith, Dalton Trans., 1996, 1, 1–5 RSC.
- P. Chanhom, K. E. Fritz, L. A. Burton, J. Kloppenburg, Y. Filinchuk, A. Senyshyn, M. Wang, Z. Feng, N. Insin and J. Suntivich, J. Am. Chem. Soc., 2019, 141, 10595–10598 CrossRef CAS PubMed.
- N. W. Falb, J. N. Neu, T. Besara, J. B. Whalen, D. J. Singh and T. Siegrist, Inorg. Chem., 2019, 58, 3302–3307 CrossRef CAS PubMed.
- N. Gloriozova, Yu. Prots, F. Jach, M. Krnel, M. Bobnar, A. Ormeci, Yu. Grin and P. Höhn, Inorg. Chem., 2023, 62(32), 12940–12946 CrossRef CAS PubMed.
- M. S. Bailey, M. N. Obrovac, E. Baillet, T. K. Reynolds, D. B. Zax and F. J. DiSalvo, Inorg. Chem., 2003, 42, 5572–5578 CrossRef CAS PubMed.
- M. S. Bailey and F. J. DiSalvo, Dalton Trans., 2003, 12, 2621–2625 RSC.
- O. Hochrein, M. Kohout, W. Schnelle and R. Kniep, Z. Anorg. Allg. Chem., 2002, 628, 2738–2743 CrossRef CAS.
- A. Tennstedt, R. Kniep, M. Hüber and W. Haase, Z. Anorg. Allg. Chem., 1995, 621, 511–515 CrossRef CAS.
- R. Niewa, D. Zherebtsov and P. Höhn, Z. Kristallogr. - New Cryst. Struct., 2003, 218, 163–163 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- S. M. Hunter, PhD thesis, University of Glasgow, 2008.
- V. Smetana, V. Babizhetskyy, G. V. Vajenine and A. Simon, J. Solid State Chem., 2007, 180, 1889–1893 CrossRef CAS.
- P. F. Lang and B. C. Smith, Dalton Trans., 2010, 39, 7786–7791 RSC.
- S. Mitra, PhD thesis, University of South Carolina, 2013.
- J. Graulich, S. Drüeke and D. Babel, Z. Anorg. Allg. Chem., 1998, 624, 1460–1464 CrossRef CAS.
- N. Gloriozova and P. Höhn, unpublished work.
- G. Cordier, R. Kniep, P. Höhn and A. Rabenau, Z. Anorg. Allg. Chem., 1990, 591, 58–66 CrossRef CAS.
- S. D. Kloß, A. Haffner, P. Manuel, M. Goto, Y. Shimakawa and J. P. Attfield, Nat. Commun., 2021, 12, 1–6 CrossRef PubMed.
- F. R. Wagner and Yu. Grin, in Comprehensive Inorganic Chemistry III: Theory and Bonding of Inorganic Nonmolecular Systems, Elsevier, 2023, 3, 222–237 Search PubMed.
- F. Wagner, D. Bende and Yu. Grin, Dalton Trans., 2016, 45, 3236–3243 RSC.
- D. Bende, F. R. Wagner and Yu. Grin, Inorg. Chem., 2015, 54, 3970–3978 CrossRef CAS PubMed.
- L. Link, M. Pathak, F. Jach, P. Koželj, A. Ormeci, P. Höhn and R. Niewa, Angew. Chem., Int. Ed., 2021, 60, 7691–7696 CrossRef CAS PubMed.
- F. Jach, S. I. Brückner, A. Ovchinnikov, A. Isaeva, M. Bobnar, M. F. Groh, E. Brunner, P. Höhn and M. Ruck, Angew. Chem., 2017, 129, 2965–2968 CrossRef.
- M. Boström and S. Hovmöller, J. Alloys Compd., 2001, 314, 154–159 CrossRef.
- P. Höhn, T. J. Ballé, M. Fix, Yu. Prots and A. Jesche, Inorganics, 2016, 4, 42 CrossRef.
- Impact, C., Diamond-crystal and molecular structure visualization, Kreuzherrenstr. 102, 53227 Bonn, Germany, 2017 Search PubMed.
- WinXPOW, D. S. version 2.21, Stoe & Cie GmbH, Darmstadt, Germany, 2007 Search PubMed.
- K. Koepernik and H. Eschrig, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1743 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef PubMed.
- M. Kohout, Int. J. Quantum Chem., 2004, 97, 651–658 CrossRef CAS.
- M. Kohout, Faraday Discuss., 2007, 135, 43–54 RSC.
- F. R. Wagner, V. Bezugly, M. Kohout and Yu. Grin, Chem. – Eur. J., 2007, 13, 5724–5741 CrossRef CAS PubMed.
- R. F. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- A. Ormeci, H. Rosner, F. Wagner, M. Kohout and Yu. Grin, J. Phys. Chem. A, 2006, 110, 1100–1105 CrossRef CAS PubMed.
- M. Kohout, DGrid 4.6, Dresden, Germany, 2011 Search PubMed.
- S. Raub and G. Jansen, Theor. Chem. Acc., 2001, 106, 223–232 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CSD 2326218. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00283k |
| ‡ Current address: Fraunhofer Institute for Integrated Systems and Device Technology IISB, Schottkystraße 10, 91058 Erlangen, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |