
Triple para-substitution reactions of B(C6F5)3 and [(C6F5)3PF]+ with P(SiMe3)3†
Jordan
Thomson
,
Sofia
Michailovich
,
Linkun
Miao
 ,
Jeffrey M.
Farrell
,
Jillian A.
Murray
,
Alan
Lough
and
Douglas W.
Stephan
*
,
Jeffrey M.
Farrell
,
Jillian A.
Murray
,
Alan
Lough
and
Douglas W.
Stephan
*
Department of Chemistry, University of Toronto, 80 St. George St, Toronto, ON M5S3H6, Canada. E-mail: dstephan@chem.utoronto.ca
First published on 1st March 2024
Abstract
para-Substitution reactions on C6F5 rings of Lewis acids have been exploited to achieve triply substituted derivatives. The reaction of B(C6F5)3 with P(SiMe3)3 ultimately affords the Lewis acid B(C6F4P(SiMe3)2)31. This species binds Lewis bases affording the adducts LB(C6F4P(SiMe3)2)3 (L = MeCN 2, OPEt33, PMe34, PBu35) and reacts with LiMe to give the salt [Li][MeB(C6F4P(SiMe3)2)3]·3THF 6. It also reacts with H2O to give (L)B(C6F4PH2)3 (L = H2O 7, MeCN 8). In an analogous fashion, [(C6F5)3PF][B(C6F5)4] was converted to [FP(C6F4P(SiMe3)2)3] [B(C6F5)4] 9 and subsequently to [(MeO)P(C6F4PH2)3][B(C6F5)4] 10.
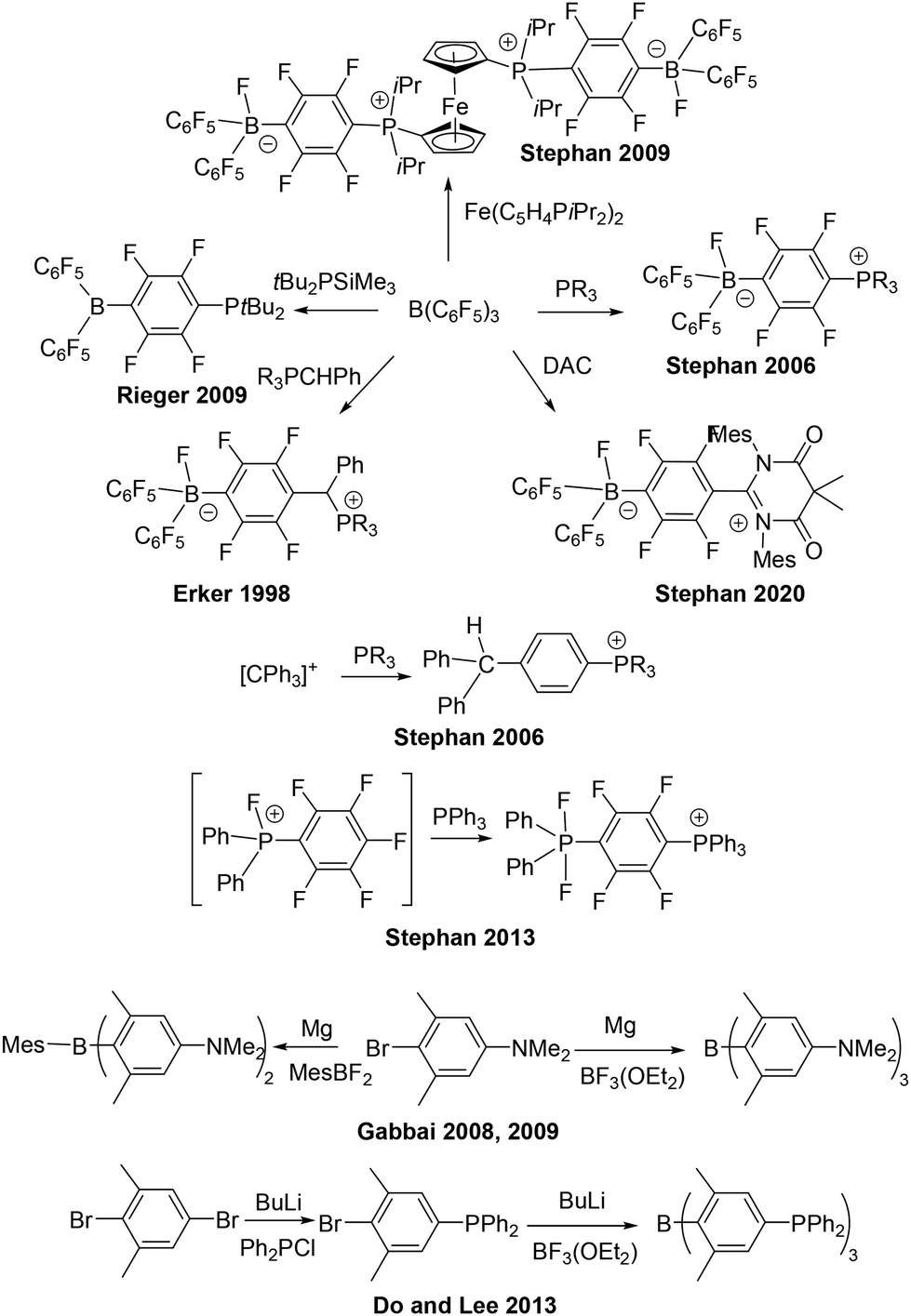
The discovery of frustrated Lewis pairs (FLPs) emerged from the initial exploration of the chemistry derived from the products of the reactions of B(C6F5)3 with bulky phosphines.1 In the first such systems, these reactions lead to para-substitution of the phosphine on a C6F5 ring with concurrent fluoride transfer to boron affording the zwitterions R3PC6F4B(C6F5)2F (Scheme 1). Use of secondary phosphines and replacement of the fluoride on boron by hydride, afforded R2PHC6F4B(C6F5)2H, which proved to be the first metal-free system capable of reversible activation of H2.1 This finding led to further studies revealing that bulky phosphines did not react with B(C6F5)3 at all, but the combination effected the homolytic cleavage of H2.2 These early findings set the stage for the plethora of FLP chemistry that has emerged in the last two decades.3–9
 | ||
| Scheme 1 Selected examples of routes to para-donor substitution on aryl-rings of Lewis acids. | ||
Focusing attention on the para-substitution reactions that provided the original FLP systems, it should be noted that Erker and coworkers10 had previously reported analogous para-substitution products as early as 1998. In those cases, while classical Lewis acid–base adducts were observed at room temperature, thermal reactions of ylides with B(C6F5)3 afforded the para-substitution products R3PCHRC6F4B(C6F5)2F (Scheme 1). Subsequent to our preparation of R2PHC6F4B(C6F5)2H and quite apart from FLP chemistry, we exploited such species as anionic ligands for transition metal chemistry.11,12 We also studied the analogous reaction of phosphines with trityl cations affording similar para-substitution products of the form [Ph2CHC6H4PR3]X.13 In 2010, Rieger and coworkers14 reported an alternative synthetic route, deriving tBu2PC6F4B(C6F5)2 from the reaction of tBu2PSiMe3 with B(C6F5)3 (Scheme 1). In 2013, we showed that analogous para-substitution of a C6F5 ring in fluorophosphonium cations provided an avenues to the species of the form [Ph3PC6F4PF2Ph2][X] (Scheme 1).15 More recently, we have described the reactions of B(C6F5)3 or the trityl cation with N,N′-dimesityldiamidocarbene (DAC) (Scheme 1).16 In the former case, Me2C(CONMes)2CC6F4BF(C6F5)2 was obtained. This species could be reduced to the radical borane derivative [Me2C(CONMes)2CC6F4B(C6F5)2]˙ while for the trityl analogue, the radical cation salt [Me2C(CONMes)2CC6H4CPh2˙][B(C6F5)4] was obtained directly.
The notion of multiple para-substitution reactions has been briefly probed. Using judiciously selected ferrocenyl-bisphosphine we showed that reaction with B(C6F5)3 affording the di-zwitterion Fe(C5H4P(iPr2)2C6F4B(C6F5)2F)2 (Scheme 1).17 Multiple substitutions on aryl boranes have drawn lesser attention. In 2008 and 2009, Gabbaï and coworkers18,19 reported the preparation of the bis- and tris-ammonium substituted boranes [MesB(C6H2Me2NMe3)2]2+ and [B(C6H2Me2NMe3)3]3+ derived from the amine-substituted precursors and their use in cyanide binding (Scheme 1). Subsequently, Do and Lee and coworkers,20 reported the synthesis of the tris-phosphine species affording the phosphonium borane [B(C6H2Me2PPh2Me)3]3+ (Scheme 1). In both studies, these species were prepared via the reaction of a donor substituted aryl lithium reagent with a boron halide. In this communication, we develop a synthetic route targeting species derived from para-substitution on fluoro-arene rings. Initially this is applied to B(C6F5)3 and subsequently to the fluorophosphonium cation, [FP(C6F4PR2)3]+. The Lewis acidity and chemistry of these derivatives is also explored.
Our initial efforts involved a modification of the Reiger protocol.14 Treatment of B(C6F5)3 with excess tBu2PSiMe3 was performed under various conditions, however this gave only the known species (C6F5)2B(C6F4PtBu2). This finding suggests that while this silyl phosphine is sufficiently bulky to effect para-substitution, the Lewis acidity of the product is sufficiently diminished precluding further reactivity.
Undaunted we next probed the reaction of B(C6F5)3 with one equivalent of P(SiMe3)3. This gave a 19F NMR spectrum consistent with a mixture of two products arising from para-substitution and release of Me3SiF. Addition of a further equivalent gave rise to yet another species with the consumption of one of the initial species. Finally, addition of three equivalents afforded signals consistent with one major product with a minor amount of a second species. These data were interpreted as the consecutive substitution at the para positions of the C6F5 rings of B(C6F5)3 (Scheme 2). Indeed, the reaction of B(C6F5)3 with a slight excess of 3 equivalents of P(SiMe3)3 afforded the new yellow product 1 after 4 days. This species was isolated in 82% yield. The 31P{1H} NMR spectrum displayed a resonance at δ −162.7 that is a singlet. The 19F NMR spectrum shows two multiplets at δ −125.0 and δ −130.0 ppm consistent with the presence of C6F4 fragments (Fig. 1). The 11B NMR spectrum showed only a broad resonance at δ 60.0 ppm consistent with a three coordinate B atom. These data together with elemental analysis affirm the formulation of 1 as B(C6F4P(SiMe3)2)3. Compound 1 proved to be soluble in all common organic solvents.
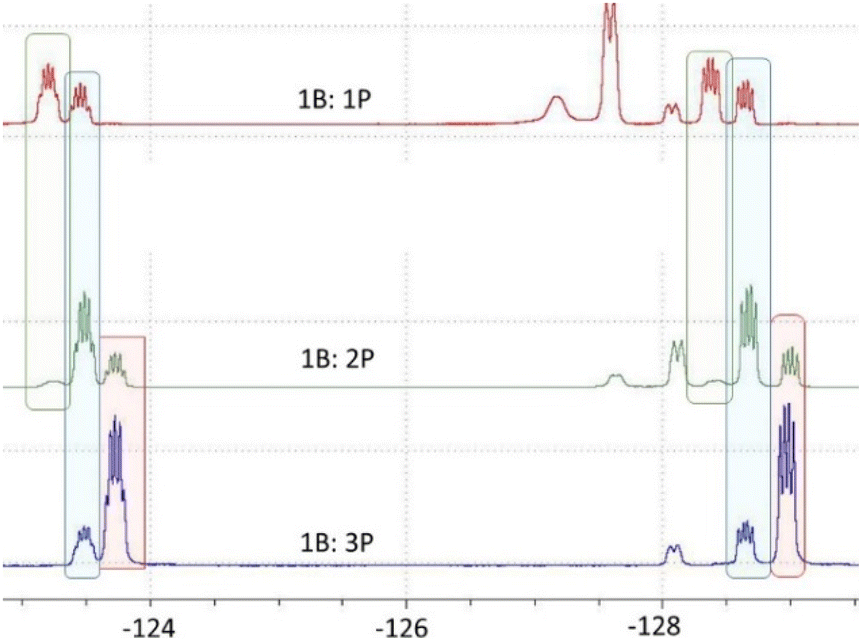 | ||
| Fig. 1 Portion of 19F NMR spectra of reaction mixtures of B(C6F5)3 and P(SiMe3)3. Signals arising from mono, di and tri substituted products shown in green, blue, and red shading respectively. | ||
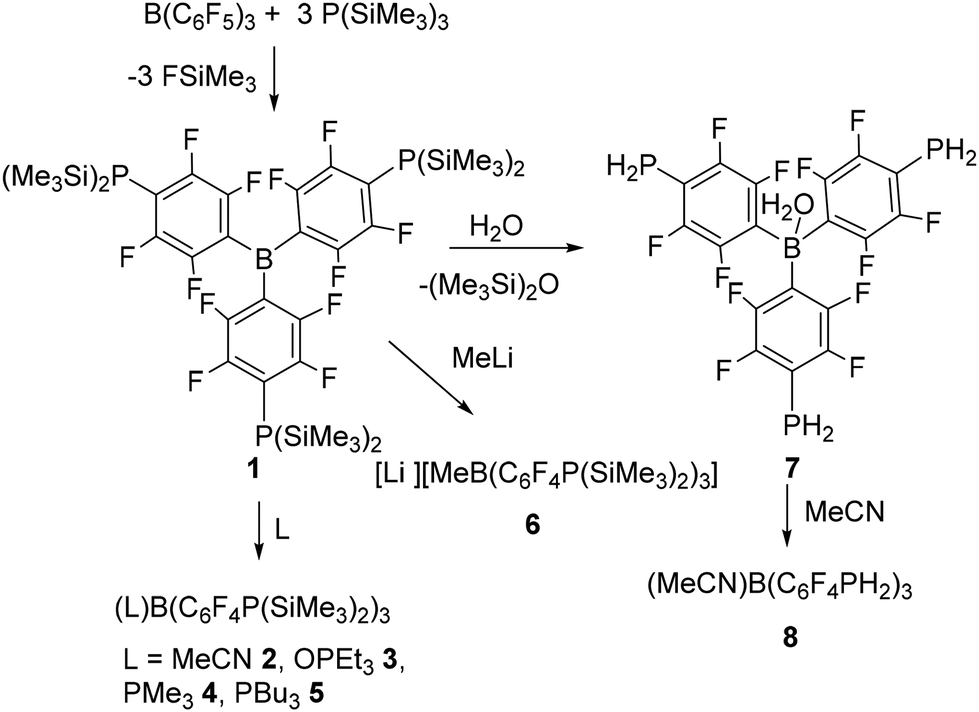 | ||
| Scheme 2 Synthesis of 1–8. | ||
The para-substitution reactions affording 1 hinge on the steric frustration that precludes B–P bond formation prompting instead attack at the para-carbon of a C6F5 ring. Loss of Me3SiF affords concurrent ring substitution. Interestingly the initially formed product (C6F5)2B(C6F4P(SiMe3)2) retains sufficient Lewis acidity to allow this process to be repeated on the remaining rings, ultimately affording 1. This stands in contrast to the known reaction where substitution with one PtBu2 group. In the present case, the installation of a P(SiMe3)2 fragment enhances the electrophilicity of the remaining rings, presumably via the beta-silicon effect, allowing subsequent substitutions to proceed.
Performing the Gutman–Beckett Lewis acidity test on 1 reveals an acceptor number of 72 based on the change in the 31P NMR chemical shift. As the corresponding acceptor number for B(C6F5)3 is 88, the replacement of the para-fluorine atoms with P(SiMe3)2 fragment reduces the Lewis acidity.
Nonetheless, compound 1 reacts with small, sterically unencumbered donors such as acetonitrile and PMe3 to form the classical Lewis acid–base adducts LB(C6F4P(SiMe3)2)3 (L = MeCN 2, OPEt33, PMe34, PBu35) (Scheme 2). The resulting colourless solutions showed 11B NMR resonances at δ −10.8, −2.4, −14.4, and −13.2 ppm respectively, suggest the presence of four coordinate boron. The 19F chemical shifts for these products differ only slightly. The 31P resonance for the P(SiMe3)2 groups of 2 was seen at −171.4 ppm. In the case of 4 and 5 the 31P resonances were observed at −6.4 and −171.5 ppm and −0.4, −171.9 ppm, respectively with the former signals being attributed to the phosphines coordinated to boron. In the case of 3 the 31P resonances were observed at 73.5 and −173.4 ppm, with the former signal arising from the coordinated OPEt3.
After protracted and exhaustive trails under various conditions, crystals of 4 and 5 were finally obtained via recrystallization from pentane using an excess of PMe3 and PBu3, respectively. Single crystal X-ray diffraction studies confirmed the formulation of 4 (Fig. 2) and 5 (Fig. 3). In the case of 4 the P–B bond was found to be 2.051(5) Å, while the C–B–P angles were 114.7(2)°, 112.7(2)°, and 116.8(2)°, affirming the pseudo-pyramidal geometry about boron. Three fluoroarene rings each substituted in the para-position with a P(SiMe3)2 fragment complete the coordination sphere of boron. The P–C for the P(SiMe3)2 substituents were found to be 1.841(4) Å, 1.844(5) Å, and 1.850(5) Å, while the P–Si bond lengths range from 2.255(2) Å to 2.266(2) Å. The Si–P–Si angles were found to be 105.54(7)°, 105.18(7)°, and 104.89(7)° while the Si–P–C angle range from to ranges from 98.6(1)° to 110.61(15)° consistent with a pseudo pyramidal geometry at the pendant phosphorus atoms. The corresponding data for 5 was very similar, although it is interesting to note that a threefold axis of symmetry was crystallographically imposed on the molecule in the solid state.
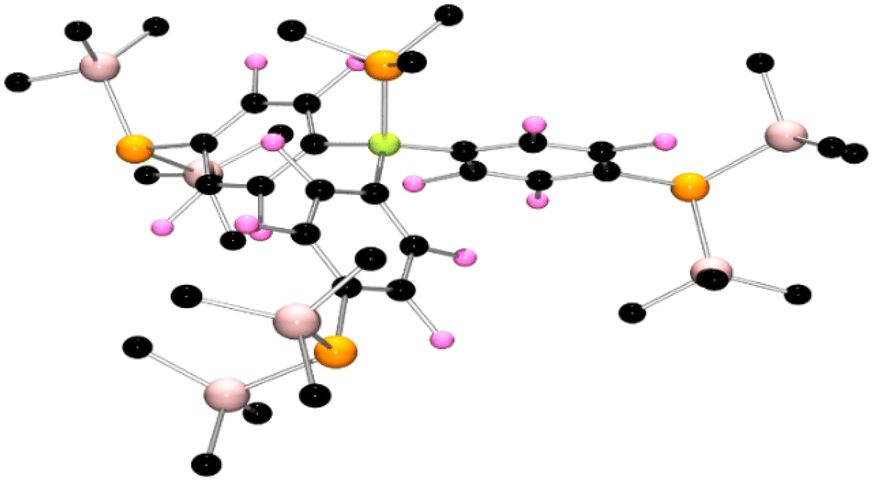 | ||
| Fig. 2 POV-ray depictions of the crystallographic structure of 4. Hydrogen atoms are omitted for clarity. C: black, B: yellow green, F; pink, P: orange, Si: salmon. | ||
 | ||
| Fig. 3 POV-ray depictions of the crystallographic structure of 5. Hydrogen atoms are omitted for clarity. C: black, B: yellow green, F; pink, P: orange, Si: salmon. | ||
In a similar fashion, reaction of 1 with methyllithium (1.6 M solution in diethyl ether) afforded the lithium methyl-borate salt [Li][MeB(C6F4P(SiMe3)2)3]·3THF 6 (Scheme 2). The 11B NMR spectrum was again consistent with the presence of a 4-coordinate boron species with a resonance at δ −14.0 ppm while 31P and 19F{1H} NMR resonances were seen at –177.2, and −130.7 and −131.5 ppm, respectively.
Compound 1 also showed a facile reaction with water or alcohol. In the case of water, the 31P NMR spectrum of the reaction mixture showed a triplet at −174.3 ppm with a 1JP–H of 209 Hz in C6H6. The 19F NMR spectrum shift slightly from that of 1 consistent with the retention of the C6F4 linking units. The corresponding 11B resonance was observed at −1.2 ppm consistent with coordination of water to the boron atom. These data are consistent with the new species 7 as (H2O)B(C6F4PH2)3 (Scheme 2). Interestingly, the corresponding reaction with D2O affording the analogous species (D2O)B(C6F4PD2)37d. This species exhibited a pentet in the 31P{1H} NMR spectrum at −179.7 ppm with P–D coupling of 32 Hz consistent with the presence of the PD2 fragments. Notably, trace amounts of the species (D2O)B(C6F4PHD)3 were also evident from the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet signal at −178.9 ppm in the 31P{1H} NMR spectrum.
:1:1 triplet signal at −178.9 ppm in the 31P{1H} NMR spectrum.
Dissolution of 7 in MeCN resulted in replacement of the water affording a new species 8. The observation of a 31P NMR triplet at −175.1 ppm with a 1JPH of 209 Hz and a 11B NMR signal at −10.8 ppm were consistent with the formulation of 8 as the adduct (MeCN)B(C6F4PH2)3 (Scheme 2). It is interesting to note that despite the presence of three primary phosphine sites in 7 and 8, no evidence of B–P bond formation is seen. This is presumed to be a result of the diminished Lewis basicity at phosphorus because of the BC6F4 substituent, and the steric congestion at boron.
To extend this reactivity to other systems, we recognized the fluorophosphonium Lewis acid cation [(C6F5)3PF][B(C6F5)4] has proved to be more Lewis acidic than B(C6F5)3.21 This prompted us to examine the corresponding reaction with an excess of three equivalents of P(SiMe3)3 in CH2Cl2 for 1 h at −35 °C. Pleasingly, this afforded the subsequent isolation of a new species 9 in 88% yield. This product gave rise to 19F NMR signals at 19F resonances at −116.5, −129.1, −133.1, −163.9 and −167.7 ppm consistent with the presence of C6F4 in the cation and C6F5 in the anion. In addition, a P–F doublet was observed at −115.9 ppm with a FP coupling constant of 1050 Hz. The corresponding 31P{1H} NMR showed a resonance at 62.6 ppm with a PF coupling of 1040 Hz as well as signal at −139.4 ppm. These data were consistent with the formulation of 9 as [PF(C6F4P(SiMe3)2)3][B(C6F5)4] (Scheme 3). Similar to the parent cation, efforts to use the Gutmann–Beckett method to assess the Lewis acidity of 9 were not possible due to an F/O exchange reaction.21
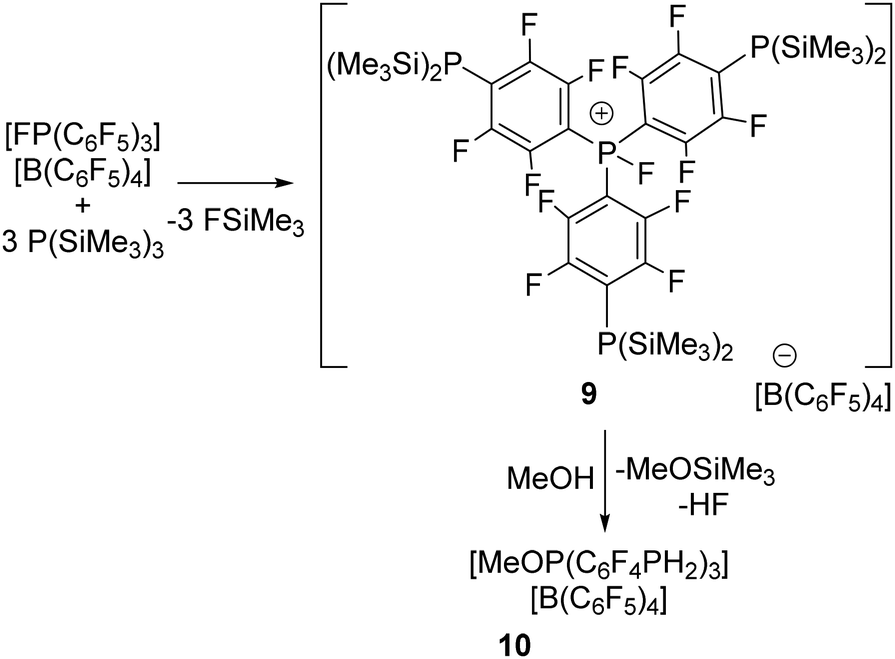 | ||
| Scheme 3 Synthesis of 9–10. | ||
Like the reaction generating 7, compound 9 was treated with excess methanol giving rise to the new product 10. This resulted in downfield shift of the 31P signal to 36.7 ppm and the appears of a triplet resonance at −164.4 ppm with P–H coupling of 215 Hz. The 19F resonances attributable to the C6F4 fragments were observed at −124.9 and −134.5 ppm. Collectively these data were consistent with the formulation of 10 as [(MeO)P(C6F4PH2)3][B(C6F5)4] (Scheme 3).
Conclusions
Exploiting the steric bulk and the basicity of P(SiMe3)3, it has been shown to effect triple para-substitution on the electron-deficient arene rings of B(C6F5)3 and [FP(C6F5)3]+, affording B(C6F4P(SiMe3)2)31 and [FP(C6F4P(SiMe3)2)3][B(C6F5)4] 9, respectively. Compound 1 remains Lewis acidic at boron forming adducts with sterically unencumbered donors or anions. Both compounds 1 and 9 react further with H2O or methanol to give the primary phosphine analogs (L)B(C6F4PH2)3 (L = H2O 7, MeCN 8) and [(MeO)P(C6F4PH2)3] [B(C6F5)4] 10, respectively. This synthetic methodology offers a new protocol to the incorporation of phosphine derived aryl substituents on Lewis acid centres. The potential of approach as an avenue tuning Lewis acidity as well as in formation of extended donor–acceptor arrays is the subject of ongoing study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NSERC of Canada is thanked for financial support.References
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef CAS.
- D. W. Stephan, Chem, 2020, 6, 1520–1526 CAS.
- S. Doering, G. Erker, R. Fröhlich, O. Meyer and K. Bergander, Organometallics, 1998, 17, 2183–2187 CrossRef CAS.
- S. L. Granville, G. C. Welch and D. W. Stephan, Inorg. Chem., 2012, 51, 4711–4721 CrossRef CAS PubMed.
- K. F. Firth, J. Möbus and D. W. Stephan, Chem. Commun., 2016, 52, 13967–13970 RSC.
- L. Cabrera, G. C. Welch, J. D. Masuda, P. R. Wei and D. W. Stephan, Inorg. Chim. Acta, 2006, 359, 3066–3071 CrossRef CAS.
- F. Schulz, V. Sumerin, M. Leskelä, T. Repo and B. Rieger, Dalton Trans., 2010, 39, 1920–1922 RSC.
- L. J. Hounjet, C. B. Caputo and D. W. Stephan, Dalton Trans., 2013, 42, 2629–2635 RSC.
- R. J. Andrews and D. W. Stephan, Chem. – Eur. J., 2020, 26, 7194–7198 CrossRef CAS PubMed.
- A. Ramos, A. J. Lough and D. W. Stephan, Chem. Commun., 2009, 1118–1120 RSC.
- C.-W. Chiu, Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 60–61 CrossRef CAS PubMed.
- C. W. Chiu and F. P. Gabbaï, Organometallics, 2008, 27, 1657–1659 CrossRef CAS.
- K. C. Song, K. M. Lee, N. V. Nghia, W. Y. Sung, Y. Do and M. H. Lee, Organometallics, 2013, 32, 817–823 CrossRef CAS.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and spectral details. CCDC 2328349–2328350. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00251b |
| This journal is © The Royal Society of Chemistry 2024 |