
Direct alkene functionalization via photocatalytic hydrogen atom transfer from C(sp3)–H compounds: a route to pharmaceutically important molecules
Hangqian
Fan
,
Yuxin
Fang
and
Jingbo
Yu
 *
*
Laboratory of Pharmaceutical Engineering of Zhejiang Province, Key Laboratory for Green Pharmaceutical Technologies and Related Equipment of Ministry of Education, Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou 310014, P. R. China. E-mail: yjb@zjut.edu.cn
First published on 5th November 2024
Abstract
Direct functionalization of alkenes with C(sp3)–H substrates offers unique opportunities for the rapid construction of pharmaceuticals and natural products. Although significant progress has been made over the past decades, the development of green, high step-economy methods to achieve these transformations under mild conditions without the need for pre-functionalization of C(sp3)–H bonds remains a substantial challenge. Therefore, the pursuit of such methodologies is highly desirable. Recently, the direct activation of C(sp3)–H bonds via photocatalytic hydrogen atom transfer (HAT), especially from unactivated alkanes, has shown great promise. Given the potential of this approach to generate a wide range of pharmaceutically relevant compounds, this review highlights the recent advancements in the direct functionalization of alkenes through photocatalytic HAT from C(sp3)–H compounds, as well as their applications in the synthesis and diversification of drugs, natural products, and bioactive molecules, aiming to provide medicinal chemists with a practical set of tools.
 Hangqian Fan | Hangqian Fan was born in Zhejiang, China in 2000. She achieved her BS from Zhejiang Chinese Medical University, major in Pharmacy in 2022. She is now working as a MS student under the supervision of Prof. Jingbo Yu. Her area of research is mainly focused on the C(sp3)–H functionalization reaction of alkanes based on hydrogen atom transfer. |
 Yuxin Fang | Yuxin Fang graduated from TaiShan University in 2022. She is now working as a MS student under the supervision of Prof. Jingbo Yu. Her area of research is mainly focused on the green pharmaceutical synthesis based on asymmetric founctionalization of C(sp3)–H bonds. |
 Jingbo Yu | Jingbo Yu is an Associate Professor at Zhejiang University of Technology (ZJUT). She obtained a BEng at Zhejiang University of Technology in Pharmaceutical Engineering (2008) and a PhD from ZJUT with Prof. Dr Weike Su on pharmaceutical synthesis (2013), followed by postdoctoral work with Hui Wu at HuaDong Medicine CO., LTD. for Pharmaceutical Chemistry. She began her independent career in 2013 as an Assistant Professor at ZJUT. Her research interests are in Mechanochemistry and green and sustainable methodologies in pharmaceutical synthesis. |
1. Introduction
Alkenes have garnered significant attention as a prominent class of abundant feedstock chemicals containing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, commonly utilized as fundamental elements in the synthesis of functional molecules and pharmaceutically relevant compounds.1–4 Hydrofunctionalization5–10 and dicarbofunctionalization11–16 reactions are recognized as reliable and potent methods for transforming alkenes by coupling them with one or two carbon electrophiles/nucleophiles to produce valuable compounds. In this scenario, the incorporation of ubiquitous alkyl groups into alkene stands out as a crucial and challenging task due to their high bond dissociation energies (BDEs, typically 90–105 kcal mol−1) (Scheme 1(A)), low acidity, and unreactive molecular orbital profile.17
C double bonds, commonly utilized as fundamental elements in the synthesis of functional molecules and pharmaceutically relevant compounds.1–4 Hydrofunctionalization5–10 and dicarbofunctionalization11–16 reactions are recognized as reliable and potent methods for transforming alkenes by coupling them with one or two carbon electrophiles/nucleophiles to produce valuable compounds. In this scenario, the incorporation of ubiquitous alkyl groups into alkene stands out as a crucial and challenging task due to their high bond dissociation energies (BDEs, typically 90–105 kcal mol−1) (Scheme 1(A)), low acidity, and unreactive molecular orbital profile.17
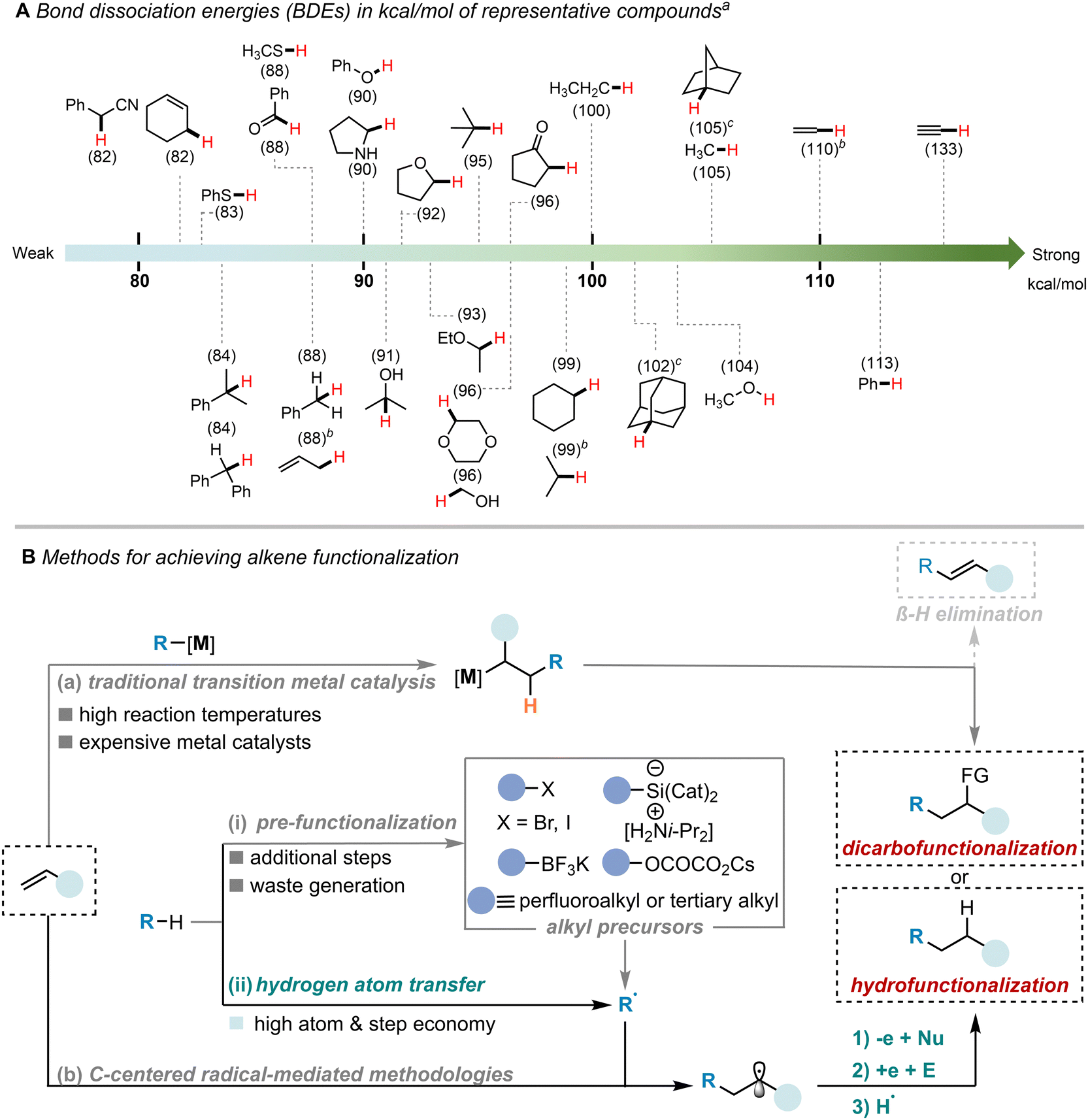 | ||
| Scheme 1 (A) Bond dissociation energies (BDEs) in kcal mol−1 of the X–H bonds (in red) of representative compounds. (B) Methods for achieving the functionalization of alkene. aBDEs values taken from ref. 18. bBDEs values taken from https://ibond.nankai.edu.cn. cBDEs values taken from ref. 19. | ||
Great progress has been made in this field through the utilization of transition metal catalysis strategies over the past decades.9,11,12,20–23 Traditional catalytic methods relying on double electron transfer23–25 often involve the use of organometallics, elevated reaction temperatures, or expensive metal catalysts, posing numerous challenges for the advancement of this area. Particularly in the case of non-activated alkene, alkylmetal C(sp3)–M species are prone to undergo β-hydride elimination, resulting in the generation of Heck products (Scheme 1(B), path a).26,27 The emergence of C-centered radical-mediated reactions has provided novel approaches to address these challenges.5,15,28–30 These methodologies avoid the formation of alkylmetal C(sp3)–M intermediates and exhibit enhanced tolerance towards functional groups, thereby complementing conventional catalytic modes effectively. Despite the advances, many of these approaches heavily rely on pre-activated alkyl substrates,31–44 such as alkyl halides, alkyl silicates, alkyl trifluoroborates, etc., leading to issues with additional steps and waste generation (Scheme 1(B), path b(i)). Furthermore, successful applications in the field are predominantly limited to perfluoroalkyl or tertiary alkyl precursors.45–49
Fortunately, the hydrogen atom transfer strategy (HAT, X˙ + H–Y → X–H + Y˙) (Scheme 1(B), path b(ii)) developed in recent decades provides an effective tool for directly activating native C(sp3)–H bonds.50–53 Through the careful choice of a suitable HAT agent, this approach allows for the direct conversion of C(sp3)–H compounds into reactive C-centered radicals, eliminating the prerequisite for pre-functionalizing substrates. Such streamlined process enhances the incorporation of alkyl groups into acceptor alkenes while maintaining a high level of atom economy. Given the high bond dissociation energies of unactivated C(sp3)–H bonds, oxygen-centered radicals (with a BDE around 106 kcal mol−1 for the –OH group in tert-butanol) are recognized as effective HAT agent.54 Notably, peroxides like di-tert-butyl peroxide (DTBP) are prominent among the array of oxygen radical precursors. However, the generation of oxygen-centered radicals at elevated reaction temperatures poses challenges in setting up reaction conditions. Recently, as a mild alternative, photocatalysis has made significant contributions to the development of this field.55–58 Based on the hydrogen abstractor involved, HAT processes can be divided into two classes: direct (d-), indirect (i-) HAT (Scheme 2).59–62 In the direct case, the excited state of the photocatalyst abstract a hydrogen atom from a substrate R–H, leading to the formation of C-centered radicals (R˙) and the deactivated form of the photocatalyst. The latter is subsequently recovered through reverse hydrogen atom transfer (RHAT) or continuous single electron transfer (SET) and proton transfer (PT) processes (Scheme 2(A)). Compared to direct HAT approach, indirect HAT methods display different selectivity and reactivity, offering an alternative tactic for the generation of C-centered radicals. In this process, additional HAT reagents are required to facilitate a single electron transfer with *PC, ultimately delivering a hydrogen atom abstractor to abstract the hydrogen atom and generate C-centered radicals (R˙) while regenerating X–H (Scheme 2(B)). In recent years, ligand-to-metal charge transfer (LMCT), an inner-sphere single-electron transfer process, that is independent of the oxidation–reduction potential, has emerged as a key strategy for generating radical intermediates (HAT reagents) in photocatalytic redox reaction (Scheme 3(a), left).63,64 Specifically, the ligand-centered radicals and single electron reduced metal centers are generated through homolysis of metal–ligand bonds upon light irradiation. This process is mainly used for iron, copper, bismuth, cerium, and titanium halide complexes. Additionally, radical intermediates can also be produced through photoredox catalyzed outer-sphere single-electron transfer, which relies on the use of photocatalysts with high reduction potential in excited states (Scheme 3(b)).65
 | ||
| Scheme 2 Two different photocatalytic HAT strategies to trigger the cleavage of a C–H bond in the substrate (R–H). | ||
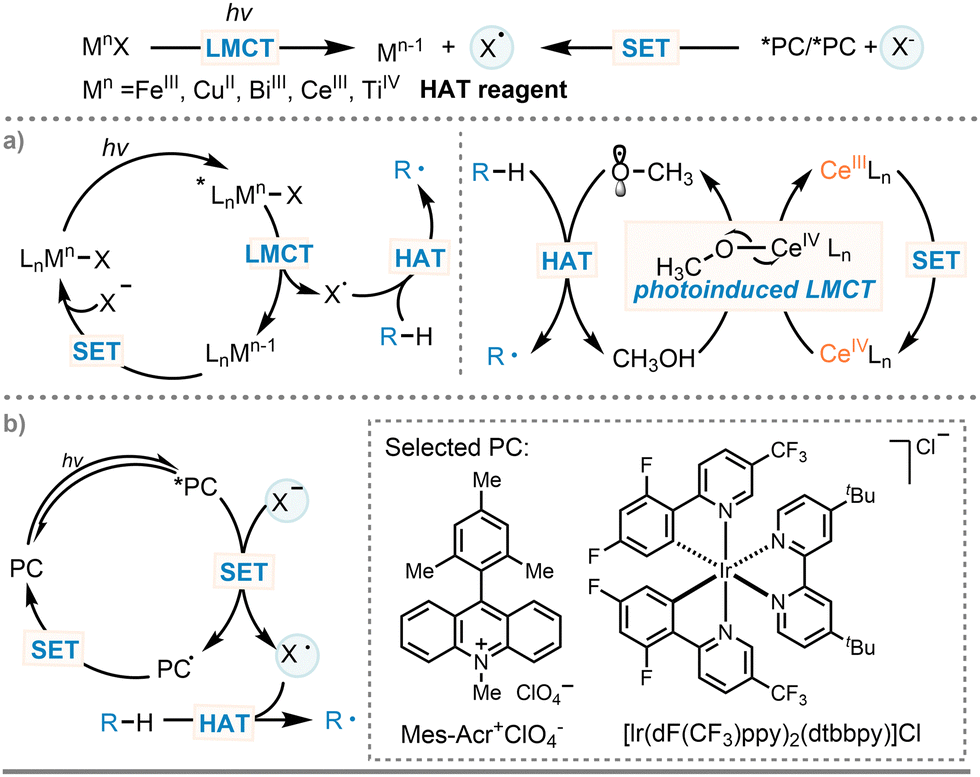 | ||
| Scheme 3 Indirect HAT methods for the generation of C-centered radicals. | ||
This highlight focuses on the recent advancements in alkene functionalization reactions, particularly through the activation of C(sp3)–H bonds using a photocatalytic hydrogen atom transfer (HAT) process. It provides an overview of the reaction conditions and mechanistic scenarios, as well as its application in synthesizing pharmaceutically important molecules. Relevant examples are categorized based on the types of alkene functionalizations: intermolecular dicarbofunctionalization and intermolecular hydroalkylation. Within each category, the reactions are further classified according to their HAT mechanisms, which include both direct and indirect pathways.
2. Intermolecular dicarbofunctionalization of alkenes
The intermolecular dicarbofunctionalization of alkenes, which precisely introduces two distinct carbon-based moieties across the CC bond, has established a highly versatile strategy for the rapid construction of bioactive compounds and natural products.16,27,47,58,66 Meanwhile, the adoption of a streamlined and eco-friendly process that directly transforms C(sp3)–H compounds into reactive C-centered radicals has spearheaded a revolutionary process in functionalizing this bond via diverse HAT pathways.
2.1. Intermolecular dicarbofunctionalization of alkenes via direct HAT (d-HAT)
The dicarbofunctionalization of alkenes with aryl halides and unactivated hydrocarbons offers a powerful platform for the synthesis of pharmaceutically relevant compounds. Notably, this approach has facilitated the production of important drugs like Piragliaptin.In 2021, the Molander and Gutierrez group unveiled a novel photochemical dual catalytic approach that employs diaryl ketone HAT catalysis to trigger the dicarbofunctionalization of alkenes by activating native C–H bonds (Scheme 4).67 This methodology proved compatible with a broad range of C–H precursors, including ethers, alcohols and amides, thereby presenting a more atom-economical and sustainable pathway in the realm of organic synthesis. It should be noted that the work includes the first photochemically mediated HAT example involving α-boronate C–H bond abstraction adjacent to apinacol boronate. Moreover, the rapid assembly of various complex molecular architectures, incorporating derivatives of glucose, homophenylalanine, as well as those derived from Isopinocampheol and Cholesterol, demonstrates the robust practicality and versatility of the approach. This highlights its potential in the streamlined synthesis of biologically pertinent compounds. Mechanistic study shows that the carbon-centered radical generated from HAT by the excited state of the photocatalyst first undergoes a regioselective Giese-type addition to an activated alkene, leading to the corresponding radical adduct. At the same time, the oxidative addition of an active catalyst Ni0 with an aryl halide gives an aryl–NiII species, which captures the radical adduct to generate an alkyl–NiIII species. Reductive elimination from alkyl–NiIII species produces the desired product and NiI complex. Density functional theory (DFT) calculations suggest that the hydrogen bonding between α-alkoxy radical and alkene components dramatically improve the rate of the Giese reaction process.
Piragliaptin, a potent glucokinase activator (GKA) for type 2 diabetes (T2D) treatment, progressed to phase 2 clinical trials.68 However, its racemic synthesis faced inefficiencies due to an extensive 12-step procedure with a low overall yield, highlighting the urgent need for more concise and productive synthesis pathways employing easily accessible starting materials.
 | ||
| Scheme 4 Three-component carboarylation of alkenes via synergistic diaryl ketone/Ni dual catalysis. Reprinted (“Adapted”) with permission from ref. 67. Copyright 2021 American Chemical Society. | ||
Kong disclosed an elegant method in 2021 for three-component alkylarylation of alkenes, facilitated by decatungstate photo-HAT coupled with nickel catalysis (Scheme 5), which was impressively employed in a streamlined three-step synthesis of Piragliaptin as well as a GKA lead compound.69 This protocol stands out for its unique capability to accommodate highly functionalized primary, secondary and tertiary alkyl radicals, which have proven uncooperative inprior studies. Of particular note, the protocol has successfully extended to the functionalization of several naturally occurring complex stereo-defined scaffolds, including Sclareolide (a sesquiterpene lactone), Tropinone, Camphene (a terminal-alkene-containing nature product) and an Estrone-derived acrylate, at carbon positions where traditional functionalization strategies have proven challenging. Starting with commercially available cyclopentane or cyclopentenone, benzyl acrylate and 4-bromo-2-chloro-1-fluorobenzene, the synthesis successfully yields a GKA lead compound and Piragliaptin with total yield of 27% and 26.88%, respectively. Further investigations into the mechanism uncovered that photo-excited tetrabutylammonium decatungstate (TBADT) abstract a hydrogen atom from the hydrocarbon substrates, giving rise to C-centered radicals alongside a reduced form of decatungstate.
 | ||
| Scheme 5 Three-component carboarylation of alkenes enabled by decatungstate photo-HAT/nickel dual catalysis. | ||
In addition to these two elegant three-component intermolecular cascades, an asymmetric three-component alkene dicarbofunctionalization was reported by Nevado and coworkers very recently via the combination of photocatalysed HAT and nickel catalysis (Scheme 6).70 Irradiating an acetone/PhCF3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution of substrates with a 390 nm Kessil lamp in the presence of 2 mol% TBADT, 10 mol% NiBr2·DME, 15 mol% chiral ligand Bilm L1, and 2 equivalents of K3PO4, yielded a series of products in moderate to good yields and excellent enantioselectivity. The protocol proved compatible with a wide array of functionalities—cycloalkanes of variable ring sizes, ethers, and alcohols—boosting its potential for post-functionalization and synthetic versatility. Modified conditions, using benzophenone-2 as the photocatalyst, Bilm L2 as the chiral ligand, and Na2CO3 as the base, enabled the participation of isopropanol and pentan-3-ol. The method also facilitated the straightforward synthesis of Flurbiprofen and Naproxen derivatives, both recognized non-steroidal anti-inflammatory drugs (NSAIDs). Aryl bromides derived from D-Glucose and (−)-Menthol successfully yielded the targeted three-component coupling products with satisfactory yields. Conversely, methyl tert-butyl ether, toluene and Boc-protected pyrrolidine led to two-component aryl–alkyl couplings. This strategy further encompassed the synthesis of the pharmaceutically important molecules, such as a Piragliatin lead compound, and the enantioenriched glucokinase activator RO28-1675.
:1) solution of substrates with a 390 nm Kessil lamp in the presence of 2 mol% TBADT, 10 mol% NiBr2·DME, 15 mol% chiral ligand Bilm L1, and 2 equivalents of K3PO4, yielded a series of products in moderate to good yields and excellent enantioselectivity. The protocol proved compatible with a wide array of functionalities—cycloalkanes of variable ring sizes, ethers, and alcohols—boosting its potential for post-functionalization and synthetic versatility. Modified conditions, using benzophenone-2 as the photocatalyst, Bilm L2 as the chiral ligand, and Na2CO3 as the base, enabled the participation of isopropanol and pentan-3-ol. The method also facilitated the straightforward synthesis of Flurbiprofen and Naproxen derivatives, both recognized non-steroidal anti-inflammatory drugs (NSAIDs). Aryl bromides derived from D-Glucose and (−)-Menthol successfully yielded the targeted three-component coupling products with satisfactory yields. Conversely, methyl tert-butyl ether, toluene and Boc-protected pyrrolidine led to two-component aryl–alkyl couplings. This strategy further encompassed the synthesis of the pharmaceutically important molecules, such as a Piragliatin lead compound, and the enantioenriched glucokinase activator RO28-1675.
 | ||
| Scheme 6 Enantioselective carboarylation of alkenes by photocatalysed HAT/nickel dual catalysis. | ||
Mesembrine is an archetypal psycho-active Sceletium alkaloid that possesses a 3a-(aryl)octahydroindole core structure with a synthetically challenging quaternary benzylic carbon.71 Its intricate structure, coupled with diverse biological activities (including anti-anxiety and anti-addiction), has consistently piqued the interest of synthetic chemists as both a target and source of inspiration. Following the pioneering racemic total synthesis of (±)-Mesembrine accomplished by Shamma and Rodriguez in 1965 through a 21-step process, numerous alternative synthesis routes for Mesembrine have since emerged in the literature.72–76
Recently, Wang and coworker innovated a rapid assembly strategy of an alkene, an aliphatic C(sp3)–H feedstock, and allyl carbonate by merging HAT photocatalyst-mediated nucleophile generation and palladium catalyzed allylic alkylation, which instrumental in the synthesis of (±)-Mesembrine (Scheme 7).77 The synthesis commenced with 2-aryl acrylate, tert-butyl dimethylcarbamate, and allyl methyl carbonate as starting materials, converging to form the target allylation product in a single, streamlined step. Subsequently, merely six straightforward functional group transformations culminated in a concise formal synthesis of (±)-Mesembrine, emphasizing the atom- and step-economy of this method. Mechanistic insights reveal that the protocol operates through a radical/ionic relay process, eco-friendly producing the non-stabilized carbanionic intermediate via C(sp3)–H bond addition to terminal alkene, proceeding to nucleophilic attack on π-allylpalladium via a classic two electron allylation pathway. The protocol's versatility was evidenced by its compatibility with a wide range of terminal alkenes, diverse hydrocarbons and α-heteroatom C(sp3)–H bonds, as well as substituted allyl carbonates.
 | ||
| Scheme 7 Pd-catalyzed allylic alkylation reaction via HAT photocatalyst-mediated nucleophile generation strategy. | ||
Allyl and hydroxyl functionalities are common structures in functional organic molecules, hold significant value for their versatility.78,79 Consequently, the concomitant incorporation of these groups as fundamental structural units in the assembly of complex structures is of great significance across numerous disciplines. In 2024, the independent research by the Wang group80 and the Zhu group81 introduced a dual catalytic system that combines photo-induced HAT with chromium catalysis, enabling the efficient accomplishment of three-component Nozaki–Hiyama–Kishi (NHK) type reactions of 1,3-butadienes (11 or 14) with C(sp3)–H partners (5 or 1) and aldehydes (12 or 15) (Scheme 8). Unlike CrCl2/photocatalyzed two-component reactions involving alkanes and aldehydes that activate C(sp3)–H compounds via ligand-to-metal charge transfer (LMCT)82,83 or by excited aldehyde,82 this method markedly differs, initiating transformation through direct H-abstraction from radical precursors by an excited TBADT or a triplet state diaryl ketone, producing both a C-centered radical and the reduced decatungstate [W10O32]5− or a ketyl radical. The C-centered radical further engaged in an addition reaction with 1,3-butadiene, thereby yielding an allyl radical intermediate, which is then trapped by a CrII catalyst to give rise to π-allyl chromium intermediate. Subsequently, the oxidative addition of aldehydes into this intermediate take place, followed by a protonation step to give the final product and release of CrIII catalyst. The latter is reduced by [W10O32]5− or ketyl radical to CrII. This SET process releases a proton and recovers [W10O32]4− or diaryl ketone photocatalyst.
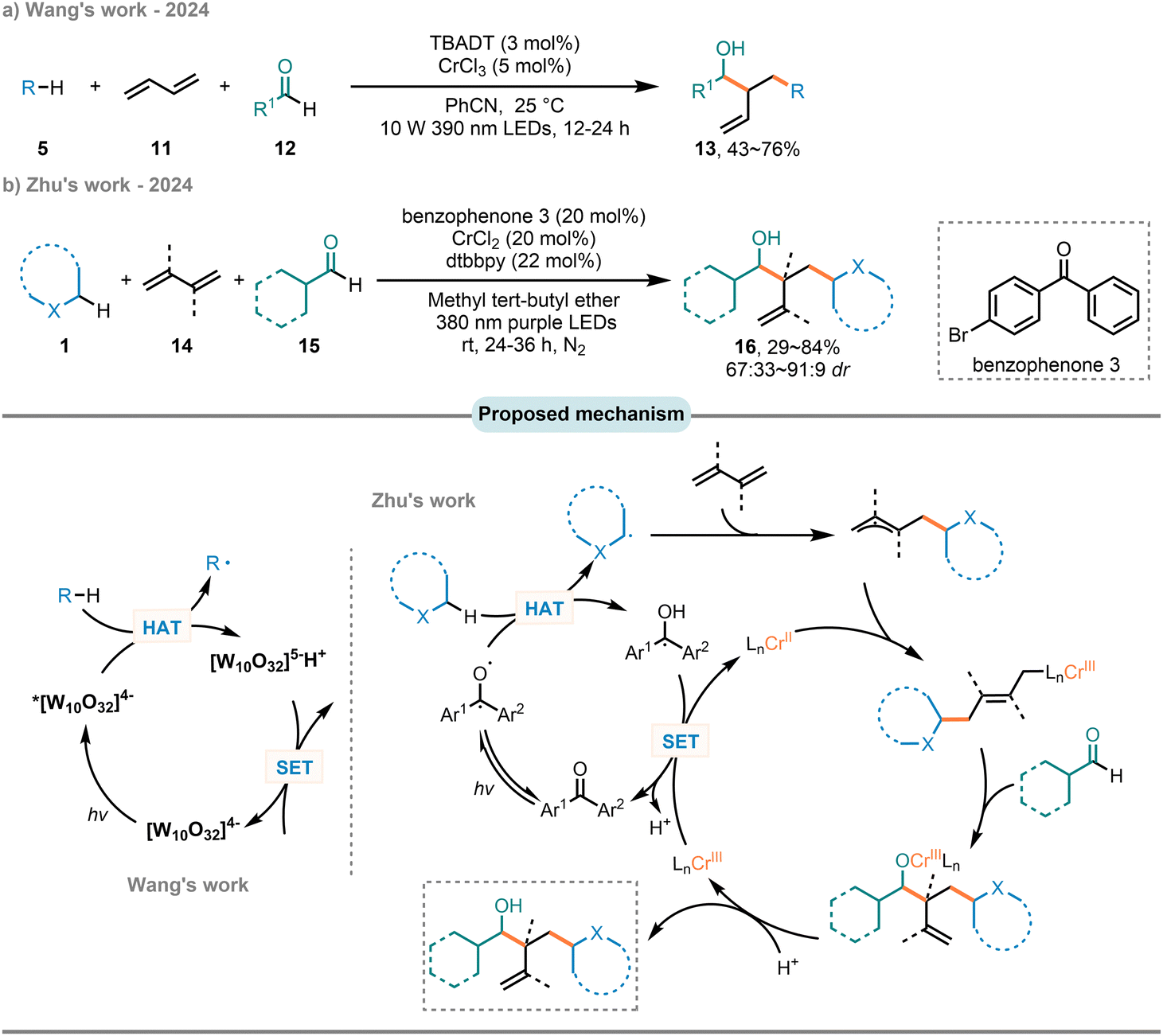 | ||
| Scheme 8 Three-component carboallylation of alkenes via photo-HAT/chromium dual catalysis. | ||
The direct 1,2-carboacylation of alkenes enables the synthesis of carbonyl-containing compounds, which are pivotal structural features prevalent in numerous pharmaceuticals and natural products, exemplified by Ketoprofen, Lovastatin and Ouabagenin.84,85 Conventional approaches, however, depend on preformed organometallic reagents, leading to diminished synthesis efficiency and compromised resource utilization. Given these drawbacks, it is of great importance to establish a highly efficient and eco-friendly methodology to accomplish the direct 1,2-carboacylation of alkenes, notably with the simultaneous installation of alkyl and acyl groups.
In 2022, Ackermann's group pioneered a three-component carboacylation of alkenes, facilitated by cooperative nickel–photoredox catalysis (Scheme 9).86 This innovative method employed a catalytic system comprised of 2 mol% sodium decatungstate (NaDT) and 10 mol% of Ni(dtbbpy)Br2, and utilized 2.0 equivalents of K2HPO4 as the base, effectively producing the desired carboacylation products. Using this method, a wide range of C(sp3)–H compounds were successfully employed as radical precursors, providing products in moderate to good yields. Notably, in order to showcase the applicability of this approach for diverse late-stage functionalizations, natural terpenoid (−)-Ambroxide, Sclareolide, and medically relevant coupling partner Camphene were selectively installed into the carbon framework containing carbonyl groups to afford the target products in good yields.
 | ||
| Scheme 9 Three-component carboacylation of alkenes via cooperative nickelaphotoredox catalysis. | ||
In addition to electron-deficient alkenes, vinylarenes have also provided access to unique motifs through difunctionalization, thereby directly contributing to the synthesis of pharmaceutical molecules. Recently, recognizing the significant potential of vinylarenes as a key component in the assembly of saturated heterocycles and aromatic heterocycles such as pyridines in drug scaffolds, Liu and colleagues reported a metal-free benzophenone-catalyzed functionalization of vinylarenes with these heterocycles (Scheme 10).87 In this process, the saturated heterocycle first undergoes a HAT event with an excited benzophenone to give a C-centered radical and a ketyl radical. The ketyl radical then undergoes single electron transfer (SET) with cyanopyridine, regenerating benzophenone and forming a persistent radical anion (19-int). Subsequently, the C-centered radical adds selectively to vinylarene, generating a new benzyl radical that can participate in further radical–radical coupling with 19-int. Notably, this method was successfully applied to the late-stage functionalization of a range of pharmaceutical reagents and natural products, including the non-steroidal NSAIDs Ibuprofen and Oxaprozin, the lipid-lowering drug Gemfibrozil, several steroids (Cholesterol, Estrone, Tigogenin, Epiandrosterone), Diacetone-D-glucose, and amino acid derivatives (Boc-D-Phe-OH).
 | ||
| Scheme 10 Three-component functionalization of vinylarenes with saturated heterocycles and pyridines via metal-free benzophenone catalysis. | ||
2.2. Intermolecular dicarbofunctionalization of alkenes via indirect HAT (i-HAT)
In the past decade, with the rapid development of electrochemistry and photochemistry, photoelectrocatalysis has attracted significant interest because of its unique reaction conditions, high resource economy, and high practicality.88Recently, Lu and coworkers developed a method for the dicarbo-functionalization of alkenes through an intermolecular three-component coupling involving alkanes, electron-deficient alkenes, and aryl bromides, which combines electrocatalysis with photoredox catalysis (Scheme 11(a)).89 This approach allows for the catalytic cycles of iron and nickel catalysts to proceed without the requirement for additional oxidants. Notably, the process operates at an ultra-low anodic potential (∼0.23V vs. Ag/AgCl), enhancing its compatibility with a variety of complex substrates, including natural products (D)-Allofuranose and (L)-Menthol, pharmaceutical derivatives Naproxen and Ibuprofen. Mechanistically, the chlorine radicals generated from the excited state of the photocatalyst [FeIIICl4]−via a LMCT process, abstracting a hydrogen atom from the C–H compound to form a carbon-centered radical intermediate, and this species subsequently undergoes a Giese-type addition to an alkene and a nickel-catalyzed coupling with an aryl halide. The [FeIICl3]− then undergoes anodic oxidation with chloride ions to regenerate the [FeIIICl4]−. One year later, the same group reported the first example of enantioselective alkylarylation of alkenes with aliphatic C–H bonds, achieved through asymmetric paired oxidative and reductive catalysis under similar reaction conditions (Scheme 11(b)).90 This system demonstrates a broad substrate scope for aryl halides, encompassing aryl bromides, aryl iodides, and even aryl chlorides.
 | ||
| Scheme 11 Enantioselective carboarylation of alkenes by photocatalysed HAT/nickel dual catalysis. | ||
In 2024, the Wang group reported the first example of the photoelectrochemical nickel-catalyzed three-component carboacylation of alkenes with different C–H-bearing substrates via the LMCT process by using the cheap and readily available FeCl3 as photocatalyst (Scheme 12).91 A series of acyl chlorides, featuring different substituents at the para-positions of the aromatic ring as well as various heteroaromatic acyl chlorides, were applicable in this reaction with good to high efficiency. Furthermore, a variety of unactivated C–H bond precursorswith different skeletal structures exhibited favorable reactivity. Notably, a complex acrylate derivative sourced from the natural product D-Menthol was also successfully incorporated, giving moderate yield.
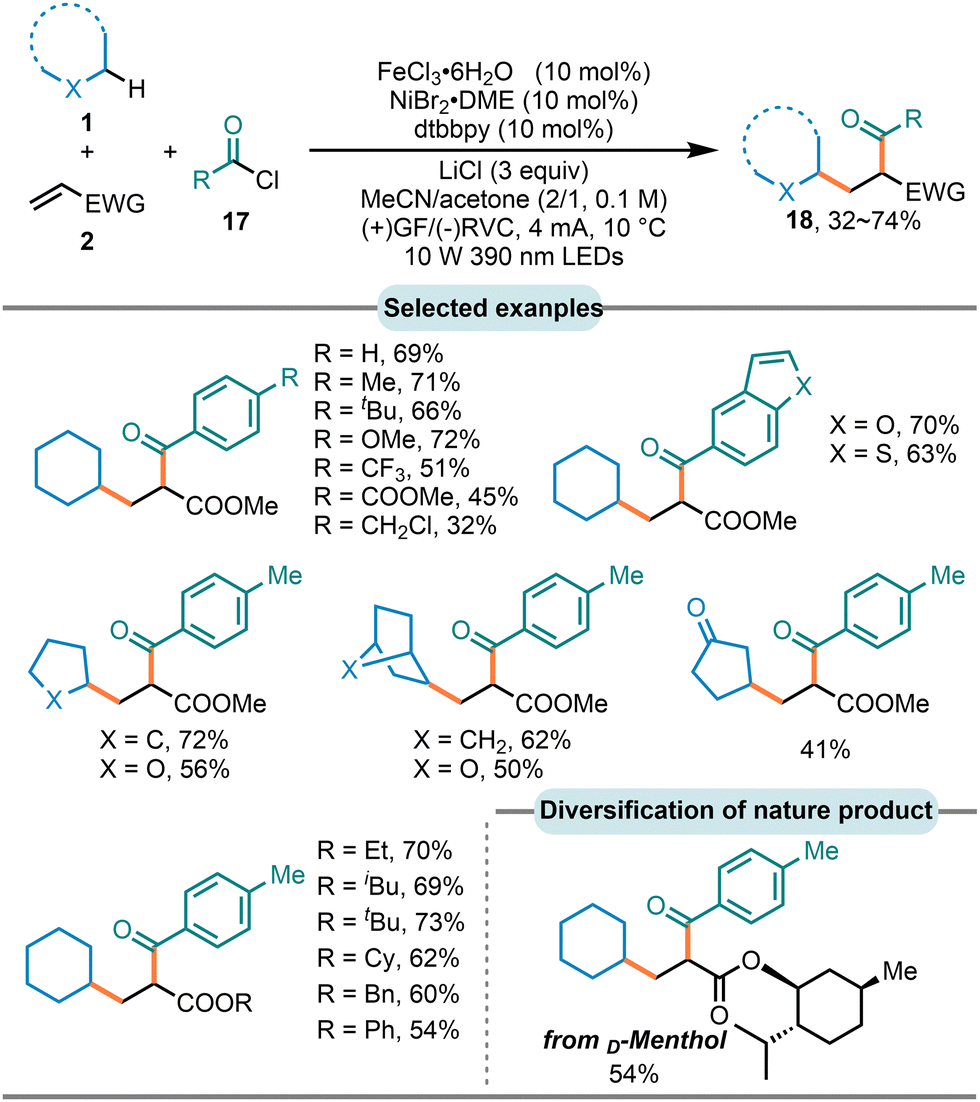 | ||
| Scheme 12 Three-component carboacylation of alkenes via photoelectrochemical LMCT process. | ||
3. Intermolecular hydroalkylation of alkenes
3.1. Intermolecular hydroalkylation of alkenes via direct HAT (d-HAT)
Researchers have explored using various classes of electronically biased alkenes as radical acceptors in the hydroalkylation of alkenes via direct HAT. Over the past decade, the groups of Albini,92–94 Fagnoni,95 and Ravelli96 have employed decatungstate as a HAT photocatalyst to achieve the hydroalkylation of electron-poor alkenes with a range of C(sp3)–H substrates, including 1,3-benzodioxoles, cycloalkanes, and oxetanes, thereby demonstrating the robust HAT capability of this heterogeneous photocatalyst. In this context, a carbon-centered radical is generated via a HAT event with excited decatungstate anion. This radical then readily undergoes a Giese-type addition to electron-poor alkenes to form an adduct radical. A back-hydrogen atom transfer from the reduced decatungstate anion gives the alkylated product and regenerates the starting decatungstate anion.In 2015, Ryu and coworkers used aliphatic nitriles (22), which are potentially useful compounds for late-stage functionalization due to their ease of conversion into functionalized compounds such as carboxylic acids, aldehydes, esters, amines, and amides,97–101 as radical precursors to achieve β- or γ-site-selective C–H alkylation of aliphatic nitriles with electron-poor alkenes (Scheme 13(a)).102 Previously, the C–H conversion in aliphatic nitriles had occurred predominantly at the activated α-position of nitriles. The site-selectivity in Ryu's work was attributed to a radical polar effect103 in the transition states for hydrogen abstraction. Due to the intrinsic inertness of gaseous hydrocarbons, the cleavage of their C–H bonds pose a significant challenge in synthetic chemistry. In 2020, the Noël group introduced a general and mild approach to activate C(sp3)–H bonds in light hydrocarbons, using TBADT as a HAT photocatalyst in a microflow reactor at room temperature (Scheme 13(b)).104 By increasing the reaction pressure up to 10 bar to liquefy the gas, they were able to synthesize a series of hydroalkylated compounds (25). Later the same year, the same research group illustrated the utility of a continuous oscillatory millistructured photoreactor (HANU) for optimizing the TBADT-catalyzed C(sp3)–H alkylation of 2-benzylidenemalononitrile (2-A), showcasing its potential for scaling up photocatalytic transformations (Scheme 13(c)).105
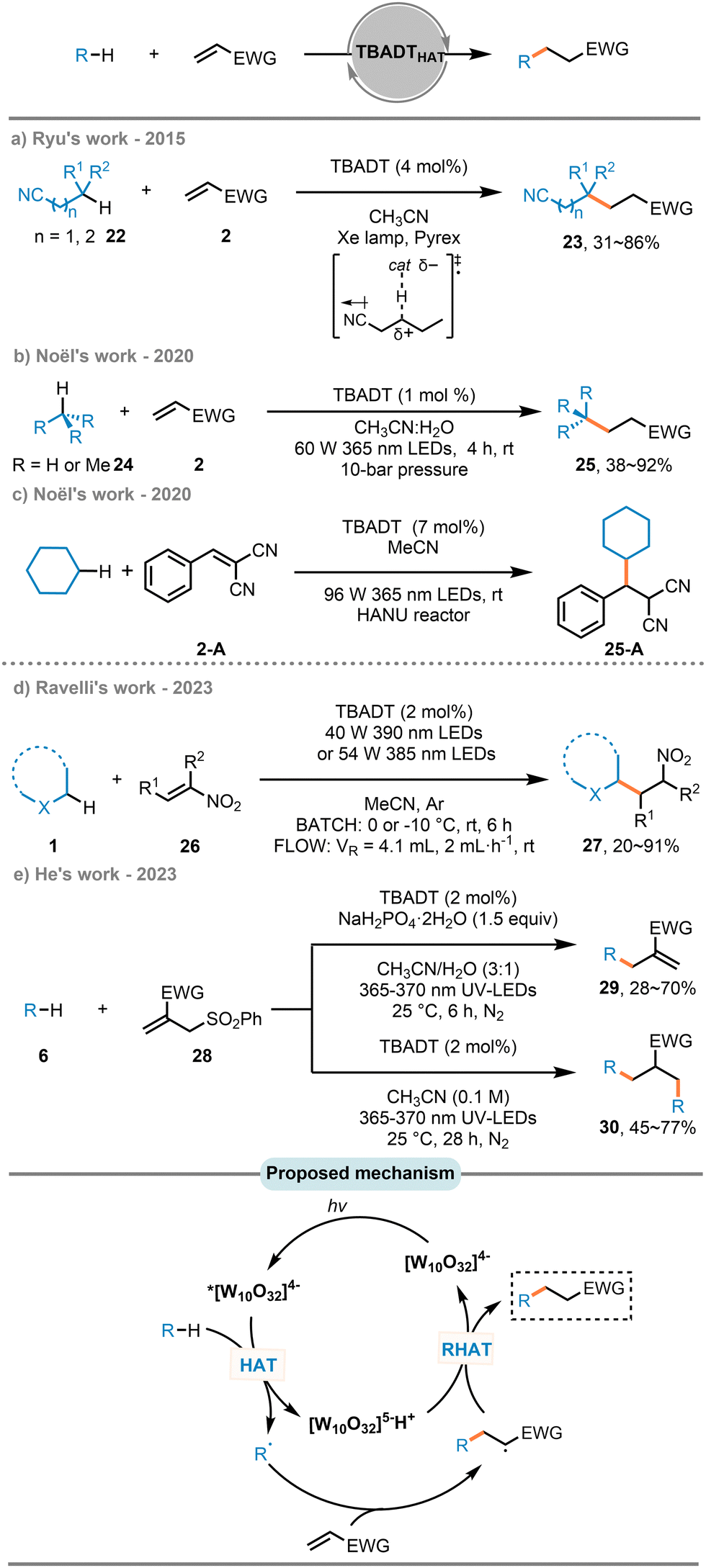 | ||
| Scheme 13 Hydroalkylation of electron-poor alkenes with TBADT as a d-HAT photocatalyst. | ||
Among different categories of electron-poor alkenes, nitroalkenes have attracted the attention of researchers as key intermediates leading to highly functionalized molecules and biologically active targets.106 In 2023, Ravelli and co-workers reported a photocatalytic strategy for the hydroalkylation of nitroalkenes and β-nitroacrylates (26) under both batch and continuous flow conditions (Scheme 13(d)).107
As reliable allylating sources, allyl sulfones have emerged as effective and promising precursors for providing high-value allylic groups.108,109 Recently, a general protocol for one-step desulfonylative allylation of C–H-bearing substrates was reported by He and co-workers, using TBADT as a HAT reagent to generate C-centered radicals from various C–H coupling partners (Scheme 13(e)).110 This methodology requires only a single equivalent of C–H substrates and allows for the difunctionalization of allyl sulfones (28) by increasing the amount of the C–H substrates.
Besides TBADT, inexpensive xanthene dyes were first employed by Wu and coworkers as organic direct HAT photocatalysts in 2018 to achieve the alkylation of C–H bonds with electron-deficient alkenes (Scheme 14(a)).111 Amongst the tested xanthene dyes, Eosin Y, which possesses heavy atoms that facilitates intersystem crossing for a long-lived triplet excited state, gave the best performance. Using this method, a common spice compound like Ambroxide was successfully modified with the alkyl substituent installed at the equatorial position in the major isomers. Later in 2022, the same group used a Brønsted acid to enhance the HAT reactivity of Eosin Y for the functionalization of unactivated C(sp3)–H bonds (BDE > 96 kcal mol−1). This strategy enables the site-selective alkylation of complex pharmaceutically important molecules, including Pinane (spice compound), Leucine, Memantine (N-methyl-D-aspartate (NMDA) blocker), the complex derivatives of the immunomodulator Thalidomide, Pristane (natural saturated terpenoid), and Androsterone (hormone-derived steroid metabolite), with electron-poor alkenes (Scheme 14(b)).112 In 2019, Singh and co-workers showed that styrene, in addition to electron-deficient alkenes, can act as a radical acceptor when Eosin Y is used as a direct HAT photocatalyst for the alkylation of amine substrates (Scheme 14(c)).113
 | ||
| Scheme 14 Alkylation of C(sp3)–H bonds with Eosin Y as a d-HAT photocatalyst. | ||
Detailed spectroscopic and computational studies show that the selective protonation of sp3 oxygen atoms on Eosin Y results in significantly enhanced HAT efficiency. The mechanism was proposed starts with a HAT of the radical precursor by the excited photocatalyst *Eosin Y to generate a carbon-centered radical. This carbon-centered radical then was trapped by an electron-deficient alkene or styrene to selectively generate radical adduct, which undergo a reverse hydrogen atom transfer (RHAT) with Eosin Y-H (path a) or another radical precursor (path b) to deliver the desired alkylation product.
In the same year, Vincent and coworkers developed a dual photoredox/copper catalytic strategy, enabling the hydroalkylation of highly polymerizable alkenes with C(sp3)–H donors such as amines, alcohols, ethers, and cycloalkanes (Scheme 15).114 They discovered that the polymerization of alkene acceptors can be effectively limited or suppressed when the photocatalytic activity of benzophenone (BP) is combined with a catalytic amount of Cu(OAc)2. In this process, the radical precursor undergoes a direct HAT event with excited BP photocatalyst to give a C-centered radical. This radical then adds to the alkene to form a new C(sp3) radical (31-int), which can react with Cu(OAc)2 to generate an alkylcopper species as a masked radical in the early stage of the reaction. This step is crucial for limiting the polymerization of alkenes, especially when the CuII concentration is high. The CuII can be reduced by the BP ketyl radical to generate low-valent copper species (CuI and/or Cu0), which subsequently reduce 36-int, followed by protonation to yield the desired Giese reaction product. However, significant side reactions such as rapid accumulation of copper nanoparticles (NPs) and polymerization of BP, may occur at low alkene concentrations.
 | ||
| Scheme 15 Alkylation of C(sp3)–H bonds via dual BP/copper catalysis. | ||
Unlike existing catalysts that primarily rely on molecular entities containing oxo groups, especially aromatic ketones and inorganic polyoxometalates, Ooi and co-workers developed a zwitterionic acridinium amidate as a nitrogen-centered catalyst for photoinduced direct HAT, which is suitable for the cleavage of various C–H bonds (Scheme 16).115 Experimental and theoretical studies have shown that the hydrogen-bonding interaction between the anionic amidate nitrogen and a pertinent hydrogen-bond donor, such as hexafluoroisopropanol, is essential for generating catalytically active triplet state species efficiently. These species can undergo HAT with C(sp3)–H compounds to produce the amide acridinyl radical and a C-centered radical. This design concept provides a novel approach for the development of direct HAT catalysts and opens up new possibilities for their application in pharmaceutically important molecules in the future.
 | ||
| Scheme 16 Alkylation of C(sp3)–H bonds using zwitterionic acridinium amidate as a d-HAT photocatalyst. | ||
The synthesis of chiral molecules holds significant potential for the discovery and development of pharmaceuticals.116 Recently, elegant methods that combine photocatalysis and asymmetric catalysis, directly utilizing abundant alkanes as carbon radical precursors for catalytic enantioselective functionalization of alkenes, have been developed. In 2016, Melchiorre's group successfully designed a chiral amine catalyst bearing a redox-active carbazole moiety, which enabled the asymmetric Giese radical addition of benzodioxole to β-substituted cyclic enones. (Scheme 17(a)).117 The stable tetrafluoroborate salts of the chiral iminium ion, generated upon condensation of encumbered primary amine (Cat 1) and β-methyl cyclohexenone, govern the stereocontrol of the radical conjugate addition (RCA). The bulky carbazole unit is positioned to effectively shield the diastereotopic Si face of the iminium ion, thereby exposing the Re face exposed for enantioselective bond formation.
 | ||
| Scheme 17 Asymmetric Giese reactions with various C(sp3)–H bonds via the synergistic catalysis of HAT photocatalysts and chiral catalysts. | ||
By using chiral spiro phosphoric acid as a chiral proton-transfer shuttle (CPTS) and TBADT as a HAT catalyst, Wang and colleagues delivered a light-mediated asymmetric C–H functionalization of unactivated hydrocarbons with exocyclic enones and α-substituted acrylates (Scheme 17(b)).118,119 As a cocatalyst, CPTS assists in the process of subsequent radical addition/hydrogen abstraction/enantioselective protonation process to furnish the desired products. Later, the same dual catalytic system was utilized by Jang's group to asymmetrically synthesize a series of azaarene-based compounds (Scheme 17(c)).120 In addition, some metal complex catalysts have demonstrated significant efficacy in asymmetric Giese radical reactions. In 2019, the Wu group realized the addition of β-substituted acrylamides by combining neutral Eosin Y with a chiral-at-metal bis-cyclometalated rhodium(III) complex Λ-RhS (Scheme 17(d)).121 Recently, in 2024, Feng and co-workers disclosed a quinone-initiated photocatalytic asymmetric Giese radical addition of the C(sp3) radical to α-substituted acrylamides. This process utilized simple quinones as HAT photocatalysts in combination with a chiral N,N′-dioxide/Pr(OTf)3 catalyst, enabling the synthesis of diverse chiral α-aryl amide derivatives, and the late-stage C–H functionalization of small-molecule drugs, including a lipid lowering agent Gemfibrozil and an anti-inflammatory Celecoxib (Scheme 17(e)).122 DFT-based conformational analysis indicated that the interaction between quinone and chiral Lewis acid was essential for enantio-induction in the asymmetric back hydrogen atom transfer process. Very recently, the Li group disclosed a new chiral HAT reagent 8H-BINOL/DTs that was generated easily from 8H-BINOL, potassium carbonate, and TBADT under irradiation (Scheme 17(f)).123 Mechanistic investigations confirmed that the chiral H donor functionalities of this new complex, which could deliver a chiral hydrogen atom to a prochiral carbon radical.
3.2. Intermolecular hydroalkylation of alkenes via indirect HAT (i-HAT)
The hydroalkylation of alkenes through i-HAT pathway commonly used additives include tertiary amines (like heterocyclic quinine rings), alcohols, peroxides (like diacylperoxides),124 N-oxides, azides and halide salts to facilitate a single electron transfer with *PC, ultimately delivering a hydrogen atom abstractor (Scheme 18). | ||
| Scheme 18 Different HAT reagents used for hydroalkylation of alkenes via indirect HAT. | ||
Due to the relatively high BDE of HCl (103 kcal mol−1125), the use of the chlorine radical as a HAT reagent for alkanes in the hydroalkylation of alkenes has been explored. In 2018, Wu and coworkers described the use of HCl as an effective HAT catalyst precursor, activated by 9-mesityl-10-methylacridinium ion (Mes-Acr+) under visible-light irradiation. This process was conducted in a “stop-flow” microtubing (SFMT) reactor, which proved more efficient than batch reactors, achieving complete conversion with fewer equivalents of alkanes (2 vs. 10 equiv.) and a lower molar percentage of HCl (5 vs. 20 mol%) (Scheme 19(a)).126 Later, the same group reported the first example of selective activation of unactivated alkanes using bromine radicals in a catalytic and metal-free process. This method has been successfully applied to the synthesis of natural products, including an amino acid derivative and a 3β-androsterone derivative (Scheme 19(a), right).127 Notably, the bromine-radical-based photo-HAT protocol showed no reactivity toward primary C–H bonds but displayed excellent reactivity and selectivity for tertiary C–H bonds. In 2018, Barriault and co-workers realized the generation of highly reactive chlorine atoms for the redox-neutral Giese-type addition of C(sp3) radicals to activated alkenes by photoredox-mediated activation of the [Ir(dF(CF3)ppy)2(dtbbpy)]Cl complex (Scheme 19(b)).128 Contrary to conventional wisdom regarding chlorine atom reactivity, this methodology demonstrated that a chlorine atom can be rendered less reactive yet more selective toward tertiary C–H bonds compared to a bromine atom, thanks to the controlling influence of a coordinating solvent such as pyridine. By making simple modifications to the reaction conditions such as temperature and concentration, or by selecting electron-deficient acceptors with a directing effect, Rovis and coworkers developed an iron-catalyzed, photocatalytic LMCT method for the divergent alkylation of electron-deficient C(sp3)–H bonds via a 1,2-skeletal rearrangement in 2021 (Scheme 19(d)).129 The authors observed that increasing the temperature and decreasing the effective concentration of the acceptor led to a higher proportion of the rearranged product. They proposed a mechanism in which the addition of the initially formed primary radical to the acceptor is in direct competition with the 1,2-migration, which forms the more stable tertiary radical. Subsequently, the same group reported further studies on Giese radical reactions involving strong electron-rich C(sp3)–H bonds with electron-deficient alkenes, utilizing CuCl2-catalyzed130 or FeCl3-catalyzed131 photoinduced LMCT methods (Scheme 19(d)). Although the BDE of H–Cl bonds is slightly lower than that of CH3–H bonds (which can be up to 105.3 kcal mol−1), methane, ethane, and heavier gaseous alkanes have been successfully employed in the hydroalkylation of alkenes via a chlorine radical generated through the LMCT process of FeCl3·6H2O (Schemes 19(e) and (g)).132 The catalytic efficiency of the iron catalyst can be increased to achieve up to 8000 turnover numbers (TON) by extending the reaction time. This methodology can also be applied to the functionalization of diverse naturally sourced compounds, such as the dipeptide Glycylglycine, the monoterpenoid Geraniol and the diterpenoid (±)-dehydroabietylamine, using heavier gaseous alkanes, with moderate reaction yields. Building on these works, in 2022, Gong and co-workers introduced hydrochloric acid as a promotor to significantly enhance the catalytic activity of the FeCl3 catalyst. This enhancement was achieved through the formation of a H+[FeIIICl4]− complex, which efficiently generated C(sp3) radicals from unactivated alkanes, achieving high levels of efficiency with a TON of up to 9900 (Scheme 19(f)).133 Using FeCl3 as the photocatalyst, alcohols can serve as C-centered radical precursors through a chlorine radical mediated O–H HAT and subsequent C–C bond cleavage, enabling their participation in Giese-type addition reactions (Scheme 19(h)).134 Specifically, chlorine radicals are generated from an excited iron chloride complex and facilitate the HAT of the OH group, assisted by a base. This process leads to the formation of various alkoxy intermediates, which then undergo β-scission to produce C(sp3) radicals from both cyclic and linear α-OH C–H substrates. In addition, various metal salts, such as cerium(IV) chloride,135 titanium(IV) chloride136,137 and bismuth(II) chloride,138 are likewise exploited to facilitate LMCT in conjunction with alcohols or on their own. This enables the generation of a O-centered radical (Scheme 19(c)) or chlorine radicals (Scheme 19(i)–(k)).
 | ||
| Scheme 19 Hydroalkylation of alkenes with various C(sp3)–H bonds via chlorine/bromine/alkoxy radicals mediated HAT process. | ||
In 2023, Wu and co-workers developed a novel synergistic catalytic approach that differs from conventional LMCT processes. This method combines organophotoredox catalysis, thianthrene, and methanol to generate C-centered radicals via a HAT process (Scheme 20).139 In this system, the thianthrene radical cation is generated by SET oxidation from the photoexcited 4CzIPN. Simultaneously, with the assistance of PhCO2Na− for deprotonation, the reaction of methanol with two molecules of the thianthrene radical cations results in the formation of 5-methoxythianthrenium and the regeneration of one molecule of thianthrene. A SET from the reduced 4CzIPN˙− to the 5-alkoxythianthrenium triggers S–O bond homolysis, producing an oxygen-centered methoxy radical which then undergoes HAT with an alkane substrate to generate a C-centered radical. This radical participated in a radical conjugate addition to the alkene, then undergoes SET from the reduced photocatalyst 4CzIPN˙− and anion-shuttled protonation from benzoate leads to the product.
 | ||
| Scheme 20 Hydroalkylation of alkenes with various C(sp3)–H bonds via alkoxy radicals mediated HAT process. | ||
Quinuclidine has also proven to be an effective HAT mediator in the hydroalkylation of electron-deficient alkenes. This process typically involves the one-electron oxidation of quinuclidine by an excited-state photocatalyst, leading to the formation of a reactive intermediate that abstracts a hydrogen atom from the substrate. Following the reaction of the substrate-derived radical, the catalytic cycle is completed through reduction and proton transfer steps.
The approach of site-selective catalysis is crucial for late-stage modification of complex natural products.140 In 2015, MacMillan and co-workers pioneered quinuclidine-mediated, highly site-selective Giese-type reactions with alcohols, using Ir[dF(CF3)ppy]2(dtbbpy)PF6 as the photocatalyst and tetrabutylammonium dihydrogen phosphate (TBAP) as the hydrogen-bonding catalyst (Scheme 21(a)).141 Mechanistically, hydrogen bonding from the OH group to the H2PO4− anions enhances the hydridicity of the α-C–H bond, thereby accelerating HAT to the electrophilic quinuclidine radical cation through polarity matching. Later, Minnaard and co-workers demonstrated that the “hydrogen bonding-induced” selective HAT reaction, as proposed by the MacMillan group, could be effectively applied to the formation of site-selective carbon–carbon bonds in unprotected monosaccharides with electron-deficient alkenes (Scheme 21(b)).142
 | ||
| Scheme 21 Hydroalkylation of alkenes with various C(sp3)–H bonds via quinuclidines mediated HAT process. | ||
Similarly, in 2019, the Taylor research group reported the use of diphenylborinic acid as a cocatalyst to enhance the hydridic character of the α-C–H bonds. This approach enabled stereo- and site-selective C–H alkylation of carbohydrate derivatives with methyl acrylate, thereby expanding the utility of these readily available starting materials as pharmacophores and related molecules (Scheme 21(c)).143 Computational modeling showed that the HAT process between a tetracoordinate diarylborinic ester and the quinuclidine radical cation is accelerated due to polarity matching and/or ion pairing effects. Additionally, Rovis and co-workers utilized quinuclidine as a HAT mediator and introduced a trifluoromethanesulfonyl group on nitrogen to allow for the formation of a C–C bond at the position of primary amine derivatives by coupling α-amino radicals with electron-deficient alkenes. This method facilitated the functionalization of sulfonamides, including a lysine-derived triflimide, which bear additional potential sites of activation (Scheme 21(d)).144 Building on this study,144 Su and collaborators subsequently proposed a multistep mechanism involving proton transfer (PT), SET, HAT, and radical addition, based on theoretical calculations.145
In 2019, Martin and co-workers designed a new HAT catalyst based on the quinuclidine scaffold (HAT 1), which enabled the highly selective activation of strong tertiary C–H bonds in adamantanes (Scheme 21(e)).146 The enhanced polarity effect of sulfonylated quinuclidinol was shown to increase the driving force for HAT by strengthening the N–H bond of the ammonium group, thus improving both the reaction rate and product yield. Subsequently, Kanai and co-workers reported the use of Martin's spirosilane as an innovative bond-weakening catalyst. This catalyst reduced the BDE of the α-C–H bond in alcohols, thereby accelerating the HAT process and facilitating the synthesis of a variety of lactones and alcohols (Scheme 21(f)).147
Furthermore, in 2021, Ye and colleagues disclosed a dual HAT protocol for the hydroalkylation of unactivated alkenes with substrates containing electron-deficient C–H bonds. This method uses catalytic amounts of an amine–borane and an in situ generated thiol as the hydrogen atom abstractor and donor, respectively, complementing the established HAT-initiated Giese-type hydroalkylation (Scheme 22).148 The protocol is completely atom-economical and exhibited a broad scope, including structurally complex alkenes derived from drug molecules (blood lipid regulating drug Gemfibrozil, NSAIDs Oxaprozin and Ibuprofen), natural products (L-Menthone, L-Mentho), and materials precursors (preservative Ethylparaben, antioxidant (+)-Nootkatone).
Beyond the use of quinuclidine, HAT reagents have also been extended to azide ions, zwitterionic 1,2,3-triazolium amidate, and N-oxides. These reagents can undergo a SET event with an excited photocatalyst, generating reactive radicals that can abstract a hydrogen atom from a C–H substrate. In 2020, Cresswell and coworkers elegantly combined 4CzIPN and azide ion as a HAT precursor, realizing visible-light photocatalyzed α-C–H alkylation of primary aliphatic amines with electrophilic alkenes for the first time, without any in situ protection of the amino group (Scheme 23(a)).149 This protocol is applicable to the decagram-scale preparation of previously difficult-to-access α-tertiary amines and aza(spiro)cyclic building blocks in continuous flow, providing a scalable approach for drug discovery. One year later, the research group extended this chemistry to include styrenes (Scheme 23(b))150 and vinyl phosphonates (Scheme 23(c))151 as radical acceptors. Notably, when optimized conditions were applied to serinol and 4-octylstyrene, Fingolimod—a sphingosine 1-phosphate (S1P1) receptor modulator for treating relapsing-remitting multiple sclerosis (MS), which achieved worldwide sales of $3 billion in 2020152—was synthesized with a 43% isolated yield. This protocol showcased complete atom economy in the key step and eliminated the necessity for protecting groups. The versatility and reliability of these methodologies, were further evidenced by the successful synthesis of the analogues of Dopamine, Tryptamine, Baclofen, and the antiviral drug Oseltamivir, even in the presence of amino functionalities. In 2023, the same research group employed transient electronic absorption spectroscopy (TEAS) and transient vibrational absorption spectroscopy (TVAS) to provide kinetic data on the intermediates involved in three critical steps (excitation of the photocatalyst 4CzIPN; oxidation of azide ion by the photo-excited 4CzIPN; HAT from the relatively weak α-C–H bond of the primary amine by azidyl radical) of the previously proposed reaction mechanism,150 which led to some important refinements in the understanding of the mechanism.153
 | ||
| Scheme 22 Hydroalkylation of unactivated alkenes with various C(sp3)–H bonds via quinuclidines mediated HAT process. | ||
 | ||
| Scheme 23 Hydroalkylation of alkenes with various C(sp3)–H bonds via azide ion, pyridine N-oxide or 1,2,3-triazolium amidate mediated HAT process. | ||
Recently, a zwitterionic 1,2,3-triazolium amidate has been developed by Ooi and coworkers as a HAT catalyst for the hydroalkylation of electron-deficient alkenes with a wide range of nitrogen- or oxygen-containing substrates (Scheme 23(d)).154 Following this, the Deng155 and Nicewicz156 groups independently introduced another strategy using pyridine N-oxides as HAT catalyst under photoredox conditions (Schemes 23(e) and (f)). The key intermediates in this process are oxygen-centered radicals, generated by single-electron oxidation of the N-oxides, whose reactivity can be easily modified through structural adjustments.
Building on these advancements, Duan and coworkers achieved the late-stage functionalization of natural products and drug molecules with electron-deficient alkenes in 2021, utilizing sulfonamides as efficient HAT catalysts (Scheme 24).157 This method was successfully applied to the antidepressant Prozac and (−)-Ambroxide, which reacted smoothly to give moderate yields. The mechanism starts with a SET oxidation of thesulfonamide by the excited *4CzIPN in the presence of a base, facilitated by a concerted proton-coupled electron transfer (PCET) event. This generates an electrophilic sulfamidyl radical, which then undergoes HAT with a C–H substrate to form a C-centered radical.
 | ||
| Scheme 24 Hydroalkylation of alkenes with various C(sp3)–H bonds via sulfonamide mediated HAT process. | ||
4. Conclusions
In this review, we have highlighted the recent progress in the direct functionalization of alkenes via photocatalytic hydrogen atom transfer from C(sp3)–H compounds and their application in synthesizing pharmaceutically important molecules. We categorize these alkene functionalizations into three- or two-component systems based on distinct HAT mechanisms. Despite substantial progress in advancing the direct functionalization of alkenes for pharmaceuticals, several challenges remain, emphasizing the need for further investigation and innovation to unlock the full potential of this approach in drug discovery. Firstly, side reactions, including radical dimerization and decomposition, are common with HAT reagents and certain photocatalysts, such as aromatic ketones, often necessitating high catalyst loads, sometimes even (super) stoichiometric amounts.55,114,158 A second challenge is achieving good regioselectivity when dealing with alkanes that possess multiple C–H bonds, especially in the context of complex natural frameworks. Lastly, although methods for synthesizing chiral molecules have been established, their practical application to the synthesis of complex drug compounds remains limited. We look forward to further advances in the direct functionalization of alkenes through photocatalytic HAT, particularly focusing on sustainable and environmentally friendly catalysis, which will provide additional pathways for the synthesis of key pharmaceuticals.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (22478354, 21978270), the Zhejiang Provincial Natural Science Foundation of China (LY23B060005), and the National Key R&D Program of China (2021YFC2101000) for financial support.Notes and references
- J. Smidt, W. Hafner, R. Jira, J. Sedlmeier, R. Sieber, R. Rüttinger and H. Kojer, Angew. Chem., Int. Ed. Engl., 1959, 71, 176–182 CrossRef.
- J. Smidt, W. Hafner, R. Jira, R. Sieber, J. Sedlmeier and A. Sabel, Angew. Chem., Int. Ed. Engl., 1962, 1, 80–88 Search PubMed.
- J. A. Keith and P. M. Henry, Angew. Chem., Int. Ed., 2009, 48, 9038–9049 CrossRef PubMed.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degreve, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953 Search PubMed.
- S. W. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 Search PubMed.
- J. R. Carney, B. R. Dillon, L. Campbell and S. P. Thomas, Angew. Chem., Int. Ed., 2018, 57, 10620–10624 CrossRef PubMed.
- C. Lin and L. Shen, ChemCatChem, 2019, 11, 961–968 Search PubMed.
- Y. Ping, Y. Li, J. Zhu and W. Kong, Angew. Chem., Int. Ed., 2019, 58, 1562–1573 Search PubMed.
- X.-X. Wang, X. Lu, Y. Li, J.-W. Wang and Y. Fu, Sci. China: Chem., 2020, 63, 1586–1600 CrossRef.
- H. Yang and Y. Ye, Top. Curr. Chem., 2023, 381, 23 CrossRef PubMed.
- G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413–2423 CrossRef.
- J. Derosa, V. A. van der Puyl, V. T. Tran, M. Liu and K. M. Engle, Chem. Sci., 2018, 9, 5278–5283 RSC.
- R. K. Dhungana, S. Kc, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314–1340 CrossRef.
- G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef.
- J. Lin, R. J. Song, M. Hu and J. H. Li, Chem. Rec., 2019, 19, 440–451 CrossRef.
- S. O. Badir and G. A. Molander, Chem, 2020, 6, 1327–1339 Search PubMed.
- X. S. Xue, P. Ji, B. Zhou and J. P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef PubMed.
- A. Fattahi, L. Lis, Z. A. Tehrani, S. S. Marimanikkuppam and S. R. Kass, J. Org. Chem., 2012, 77, 1909–1914 CrossRef.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 Search PubMed.
- J. S. Zhang, L. Liu, T. Chen and L. B. Han, Chem. – Asian J., 2018, 13, 2277–2291 CrossRef.
- H. Wang, C. F. Liu, R. T. Martin, O. Gutierrez and M. J. Koh, Nat. Chem., 2022, 14, 188–195 CrossRef.
- Y. C. Luo, C. Xu and X. Zhang, Chin. J. Chem., 2020, 38, 1371–1394 CrossRef.
- R. Giri and S. Kc, J. Org. Chem., 2018, 83, 3013–3022 CrossRef PubMed.
- X. Qi and T. Diao, ACS Catal., 2020, 10, 8542–8556 CrossRef PubMed.
- J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed.
- L. M. Wickham and R. Giri, Acc. Chem. Res., 2021, 54, 3415–3437 Search PubMed.
- E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC.
- J. Diccianni, Q. Lin and T. Diao, Acc. Chem. Res., 2020, 53, 906–919 CrossRef.
- Z. L. Li, G. C. Fang, Q. S. Gu and X. Y. Liu, Chem. Soc. Rev., 2020, 49, 32–48 RSC.
- A. Garcia-Dominguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835–6838 CrossRef PubMed.
- S. Kc, R. K. Dhungana, B. Shrestha, S. Thapa, N. Khanal, P. Basnet, R. W. Lebrun and R. Giri, J. Am. Chem. Soc., 2018, 140, 9801–9805 Search PubMed.
- M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef.
- A. Garcia-Dominguez, R. Mondal and C. Nevado, Angew. Chem., Int. Ed., 2019, 58, 12286–12290 CrossRef.
- L. Guo, H. Y. Tu, S. Zhu and L. Chu, Org. Lett., 2019, 21, 4771–4776 CrossRef PubMed.
- W. Shu, A. Garcia-Dominguez, M. T. Quiros, R. Mondal, D. J. Cardenas and C. Nevado, J. Am. Chem. Soc., 2019, 141, 13812–13821 CrossRef PubMed.
- L. Guo, M. Yuan, Y. Zhang, F. Wang, S. Zhu, O. Gutierrez and L. Chu, J. Am. Chem. Soc., 2020, 142, 20390–20399 CrossRef.
- H. M. Huang, P. Bellotti, P. M. Pfluger, J. L. Schwarz, B. Heidrich and F. Glorius, J. Am. Chem. Soc., 2020, 142, 10173–10183 CrossRef PubMed.
- S. Z. Sun, Y. Duan, R. S. Mega, R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 4370–4374 CrossRef CAS.
- X. Wei, W. Shu, A. Garcia-Dominguez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2020, 142, 13515–13522 CrossRef CAS PubMed.
- J. Yang, S. Zhu, F. Wang, F. L. Qing and L. Chu, Angew. Chem., Int. Ed., 2021, 60, 4300–4306 Search PubMed.
- A. Luridiana, D. Mazzarella, L. Capaldo, J. A. Rincon, P. Garcia-Losada, C. Mateos, M. O. Frederick, M. Nuno, W. Jan Buma and T. Noel, ACS Catal., 2022, 12, 11216–11225 CrossRef CAS.
- W. H. Sun, J. Y. Zou, X. J. Xu, J. L. Wang, M. L. Liu and X. Y. Liu, Adv. Synth. Catal., 2022, 364, 2260–2265 CrossRef CAS.
- Z. K. Wang, Y. P. Wang, Z. W. Rao, C. Y. Liu, X. H. Pan and L. Guo, Org. Lett., 2023, 25, 1673–1677 CrossRef CAS PubMed.
- X. Zhao, H. Y. Tu, L. Guo, S. Zhu, F. L. Qing and L. Chu, Nat. Commun., 2018, 9, 3488 Search PubMed.
- K. F. Zhang, K. J. Bian, C. Li, J. Sheng, Y. Li and X. S. Wang, Angew. Chem., Int. Ed., 2019, 58, 5069–5074 CrossRef CAS PubMed.
- J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287–4296 RSC.
- H. Y. Tu, F. Wang, L. Huo, Y. Li, S. Zhu, X. Zhao, H. Li, F. L. Qing and L. Chu, J. Am. Chem. Soc., 2020, 142, 9604–9611 CrossRef CAS PubMed.
- F. Wang, S. Pan, S. Zhu and L. Chu, ACS Catal., 2022, 12, 9779–9789 CrossRef CAS.
- J. T. Hynes, J. P. Klinman, H. H. Limbach and R. L. Schowen, Hydrogen-Transfer Reactions, Wiley-VCH, Weinheim, 2007 Search PubMed.
- W. Lai, C. Li, H. Chen and S. Shaik, Angew. Chem., Int. Ed., 2012, 51, 5556–5578 CrossRef CAS.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed.
- C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef CAS PubMed.
- L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS.
- I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC.
- C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051–4064 RSC.
- S. Protti, M. Fagnoni and D. Ravelli, ChemCatChem, 2015, 7, 1516–1523 CrossRef CAS.
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed.
- L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056–2071 CrossRef CAS.
- L. Capaldo, L. L. Quadri and D. Ravelli, Green Chem., 2020, 22, 3376–3396 RSC.
- F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef.
- A. M. May and J. L. Dempsey, Chem. Sci., 2024, 15, 6661–6678 RSC.
- S. Bonciolini, T. Noël and L. Capaldo, Eur. J. Org. Chem., 2022, e202200417 CrossRef CAS.
- F.-D. Lu, G.-F. He, L.-Q. Lu and W.-J. Xiao, Green Chem., 2021, 23, 5379–5393 RSC.
- M. W. Campbell, M. Yuan, V. C. Polites, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 3901–3910 CrossRef CAS PubMed.
- R. Sarabu, F. T. Bizzarro, W. L. Corbett, M. T. Dvorozniak, W. Geng, J. F. Grippo, N. E. Haynes, S. Hutchings, L. Garofalo, K. R. Guertin, D. W. Hilliard, M. Kabat, R. F. Kester, W. Ka, Z. Liang, P. E. Mahaney, L. Marcus, F. M. Matschinsky, D. Moore, J. Racha, R. Radinov, Y. Ren, L. Qi, M. Pignatello, C. L. Spence, T. Steele, J. Tengi and J. Grimsby, J. Med. Chem., 2012, 55, 7021–7036 CrossRef CAS.
- S. Xu, H. Chen, Z. Zhou and W. Kong, Angew. Chem., Int. Ed., 2021, 60, 7405–7411 CrossRef CAS PubMed.
- X. Hu, I. Cheng-Sanchez, W. Kong, G. A. Molander and C. Nevado, Nat. Catal., 2024, 7, 655–665 CrossRef CAS PubMed.
- F. Makolo, A. Viljoen and C. G. L. Veale, Phytochemistry, 2019, 166, 112061 CrossRef CAS.
- Y. Zhao, Y. Zhou, L. Liang, X. Yang, F. Du, L. Li and H. Zhang, Org. Lett., 2009, 11, 555–558 CrossRef CAS.
- K. Geoghegan and P. Evans, J. Org. Chem., 2013, 78, 3410–3415 CrossRef CAS.
- H. Kotsuki, S. Nunokawa, M. Minamisawa, K. Nakano and Y. Ichikawa, Synlett, 2015, 26, 2301–2305 CrossRef.
- P. Gan, M. W. Smith, N. R. Braffman and S. A. Snyder, Angew. Chem., Int. Ed., 2016, 55, 3625–3630 CrossRef CAS PubMed.
- H. Kim, H. Choi and K. Lee, Synlett, 2018, 29, 1203–1206 CrossRef CAS.
- Y. Shen, Z.-Y. Dai, C. Zhang and P.-S. Wang, ACS Catal., 2021, 11, 6757–6762 CrossRef CAS.
- G. C. Hargaden and P. J. Guiry, Adv. Synth. Catal., 2007, 349, 2407–2424 CrossRef CAS.
- G. Zhang and Q. Tian, Synthesis, 2016, 48, 4038–4049 CrossRef.
- P.-S. Wang and D. Ding, Synthesis, 2024, 56, 2077–2083 CrossRef.
- Q. Hu, S. Song, T. Zeng, L. Wang, Z. Li, J. Wu and J. Zhu, Org. Lett., 2024, 26, 1550–1555 CrossRef CAS PubMed.
- P. Peng, Y. Zhong, C. Zhou, Y. Tao, D. Li and Q. Lu, ACS Cent. Sci., 2023, 9, 756–762 CrossRef CAS.
- X. Zeng, F.-H. Zhang, R. Lai, X. Lin and Z. Wang, Sci. China: Chem., 2024, 67, 1589–1595 CrossRef CAS.
- H. Renata, Q. Zhou, G. Dunstl, J. Felding, R. R. Merchant, C. H. Yeh and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 1330–1340 CrossRef CAS.
- J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D. H. Bao, F. L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213–218 CrossRef CAS PubMed.
- D. Wang and L. Ackermann, Chem. Sci., 2022, 13, 7256–7263 RSC.
- J. Xu and B. Liu, Chem. – Eur. J., 2024, 30, e202400612 CrossRef CAS.
- Y. Qiu, Y. Wang and G. Yang, Chin. J. Org. Chem., 2021, 41, 3935–3947 CrossRef.
- L. Zou, S. Xiang, R. Sun and Q. Lu, Nat. Commun., 2023, 14, 7992 CrossRef CAS.
- L. Zou, X. Zheng, X. Yi and Q. Lu, Nat. Commun., 2024, 15, 7826 CrossRef CAS.
- L. Wang, X. Huo, X. He, L. Ackermann and D. Wang, Green Chem., 2024, 26, 8315–8322 RSC.
- D. Dondi, M. Fagnoni, A. Molinari, A. Maldotti and A. Albini, Chem. – Eur. J., 2004, 10, 142–148 CrossRef CAS PubMed.
- D. Dondi, M. Fagnoni and A. Albini, Chem. – Eur. J., 2006, 12, 4153–4163 CrossRef CAS PubMed.
- S. Protti, D. Ravelli, M. Fagnoni and A. Albini, Chem. Commun., 2009, 7351–7353 RSC.
- D. Ravelli, A. Albini and M. Fagnoni, Chem. – Eur. J., 2011, 17, 572–579 CrossRef CAS.
- D. Ravelli, M. Zoccolillo, M. Mella and M. Fagnoni, Adv. Synth. Catal., 2014, 356, 2781–2786 CrossRef CAS.
- T. Naota, Y. Shichijo and S.-L. Murahashi, J. Chem. Soc., Chem. Commun., 1994, 1359–1360 RSC.
- A. Roy, F. G. Roberts, P. R. Wilderman, K. Zhou, R. J. Peters and R. M. Coates, J. Am. Chem. Soc., 2007, 129, 12453–12460 CrossRef CAS PubMed.
- A. Goto, K. Endo and S. Saito, Angew. Chem., Int. Ed., 2008, 47, 3607–3609 CrossRef CAS PubMed.
- S. A. Snyder, D. S. Treitler, A. P. Brucks and W. Sattler, J. Am. Chem. Soc., 2011, 133, 15898–15901 CrossRef CAS PubMed.
- S. Liu, Y. Yang, X. Zhen, J. Li, H. He, J. Feng and A. Whiting, Org. Biomol. Chem., 2012, 10, 663–670 RSC.
- K. Yamada, M. Okada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, Org. Lett., 2015, 17, 1292–1295 CrossRef CAS.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed.
- Z. Wen, A. Maheshwari, C. Sambiagio, Y. Deng, G. Laudadio, K. Van Aken, Y. Sun, H. P. L. Gemoets and T. Noel, Org. Process Res. Dev., 2020, 24, 2356–2361 CrossRef CAS.
- S. Gabrielli, E. Chiurchiù and A. Palmieri, Adv. Synth. Catal., 2018, 361, 630–653 CrossRef.
- A. Jorea, B. Bassetti, K. Gervasoni, S. Protti, A. Palmieri and D. Ravelli, Adv. Synth. Catal., 2023, 365, 722–727 CrossRef CAS.
- P. Hünemörder and E. Mejía, Catal. Sci. Technol., 2020, 10, 6754–6768 RSC.
- J. Corpas, S.-H. Kim-Lee, P. Mauleón, R. G. Arrayás and J. C. Carretero, Chem. Soc. Rev., 2022, 51, 6774–6823 RSC.
- J.-F. Zhao, H. Wang, H.-B. Wang, Q.-Q. Tian, Y.-Q. Zhang, H.-T. Feng and W. He, Org. Chem. Front., 2023, 10, 348–354 RSC.
- X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. D. Tan, C. W. Xue, L. Z. Wu, H. R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS.
- H. Cao, D. Kong, L.-C. Yang, S. Chanmungkalakul, T. Liu, J. L. Piper, Z. Peng, L. Gao, X. Liu, X. Hong and J. Wu, Nat. Synth., 2022, 1, 794–803 CrossRef.
- V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron Lett., 2019, 60, 1333–1336 CrossRef CAS.
- B. Abadie, D. Jardel, G. Pozzi, P. Toullec and J. M. Vincent, Chem. – Eur. J., 2019, 25, 16120–16127 CrossRef CAS.
- L.-M. Entgelmeier, S. Mori, S. Sendo, R. Yamaguchi, R. Suzuki, T. Yanai, O. García Mancheño, K. Ohmatsu and T. Ooi, Angew. Chem., Int. Ed., 2024, 63, e202404890 CAS.
- X. Chu, Y. Bu and X. Yang, Front. Oncol., 2021, 11, 785855 CrossRef CAS PubMed.
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS.
- Z.-Y. Dai, Z.-S. Nong and P.-S. Wang, ACS Catal., 2020, 10, 4786–4790 CrossRef CAS.
- Z. Y. Dai, Z. S. Nong, S. Song and P. S. Wang, Org. Lett., 2021, 23, 3157–3161 CrossRef CAS PubMed.
- Y. Tan, Y. Yin, S. Cao, X. Zhao, G. Qu and Z. Jiang, Chin. J. Catal., 2022, 43, 558–563 CrossRef CAS.
- Y. Kuang, K. Wang, X. Shi, X. Huang, E. Meggers and J. Wu, Angew. Chem., Int. Ed., 2019, 58, 16859–16863 CrossRef CAS PubMed.
- Y. Luo, Y. Zhou, F. Xiao, X. He, Z. Zhong, Q.-L. Zhou, W. Cao, X. Liu and X. Feng, ACS Catal., 2024, 14, 12031–12041 CrossRef CAS.
- L. Li, S.-Q. Zhang, X. Cui, G. Zhao, Z. Tang and G.-X. Li, Org. Lett., 2024, 26, 8371–8376 CrossRef CAS PubMed.
- J. Fang, W.-L. Dong, G.-Q. Xu and P.-F. Xu, Org. Lett., 2019, 21, 4480–4485 CrossRef CAS PubMed.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Ratón, FL, 2007 Search PubMed.
- H.-P. Deng, Q. Zhou and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 12661–12665 CrossRef CAS PubMed.
- P. Jia, Q. Li, W. C. Poh, H. Jiang, H. Liu, H. Deng and J. Wu, Chem, 2020, 6, 1766–1776 CAS.
- S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed., 2018, 57, 15664–15669 CrossRef CAS PubMed.
- Y. C. Kang, S. M. Treacy and T. Rovis, ACS Catal., 2021, 11, 7442–7449 CrossRef CAS.
- S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729–2735 CrossRef CAS PubMed.
- T. Rovis, Y. C. Kang and S. M. Treacy, Synlett, 2021, 32, 1767–1771 CrossRef.
- Y. Jin, Q. Zhang, L. Wang, X. Wang, C. Meng and C. Duan, Green Chem., 2021, 23, 6984–6989 RSC.
- Z.-Y. Dai, S.-Q. Zhang, X. Hong, P.-S. Wang and L.-Z. Gong, Chem. Catal., 2022, 2, 1211–1222 CrossRef CAS.
- Q. Wu, W. Liu, M. Wang, Y. Huang and P. Hu, Chem. Commun., 2022, 58, 9886–9889 RSC.
- Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu and Z. Zuo, J. Am. Chem. Soc., 2020, 142, 6216–6226 CrossRef CAS PubMed.
- G. B. Panetti, Q. Yang, M. R. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem. Catal., 2022, 2, 853–866 CrossRef CAS.
- Q. Zhang, S. Liu, J. Lei, Y. Zhang, C. Meng, C. Duan and Y. Jin, Org. Lett., 2022, 24, 1901–1906 CrossRef CAS.
- D. Birnthaler, R. Narobe, E. Lopez-Berguno, C. Haag and B. König, ACS Catal., 2023, 13, 1125–1132 CrossRef CAS.
- X. Zhang, S. Ning, Y. Li, Y. Xiong and X. Wu, ChemCatChem, 2023, 15, e202300311 CrossRef CAS.
- A. Fürstner, Acc. Chem. Res., 2014, 47, 925–938 CrossRef.
- J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed.
- I. C. S. Wan, M. D. Witte and A. J. Minnaard, Chem. Commun., 2017, 53, 4926–4929 RSC.
- V. Dimakos, H. Y. Su, G. E. Garrett and M. S. Taylor, J. Am. Chem. Soc., 2019, 141, 5149–5153 CrossRef CAS PubMed.
- M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed., 2019, 58, 4002–4006 CrossRef CAS PubMed.
- Z. W. Zhao, Y. J. Dong, Y. Geng, R. H. Li, W. Guan and Z. M. Su, J. Org. Chem., 2021, 86, 484–492 CrossRef CAS.
- H.-B. Yang, A. Feceu and D. B. C. Martin, ACS Catal., 2019, 9, 5708–5715 CrossRef CAS.
- K. Sakai, K. Oisaki and M. Kanai, Adv. Synth. Catal., 2019, 362, 337–343 CrossRef.
- G. Lei, M. Xu, R. Chang, I. Funes-Ardoiz and J. Ye, J. Am. Chem. Soc., 2021, 143, 11251–11261 CrossRef CAS.
- A. S. H. Ryder, W. B. Cunningham, G. Ballantyne, T. Mules, A. G. Kinsella, J. Turner-Dore, C. M. Alder, L. J. Edwards, B. S. J. McKay, M. N. Grayson and A. J. Cresswell, Angew. Chem., Int. Ed., 2020, 59, 14986–14991 CrossRef CAS PubMed.
- H. E. Askey, J. D. Grayson, J. D. Tibbetts, J. C. Turner-Dore, J. M. Holmes, G. Kociok-Kohn, G. L. Wrigley and A. J. Cresswell, J. Am. Chem. Soc., 2021, 143, 15936–15945 CrossRef CAS PubMed.
- J. D. Grayson and A. J. Cresswell, Tetrahedron, 2021, 81, 131896 CrossRef CAS.
- M. Sanford, Drugs, 2014, 74, 1411–1433 CrossRef CAS.
- M. Sneha, G. L. Thornton, L. Lewis-Borrell, A. S. H. Ryder, S. G. Espley, I. P. Clark, A. J. Cresswell, M. N. Grayson and A. J. Orr-Ewing, ACS Catal., 2023, 13, 8004–8013 CrossRef CAS.
- K. Ohmatsu, R. Suzuki, Y. Furukawa, M. Sato and T. Ooi, ACS Catal., 2019, 10, 2627–2632 CrossRef.
- B. Wang, C. Ascenzi Pettenuzzo, J. Singh, G. E. McCabe, L. Clark, R. Young, J. Pu and Y. Deng, ACS Catal., 2022, 12, 10441–10448 CrossRef CAS.
- M. Schlegel, S. Qian and D. A. Nicewicz, ACS Catal., 2022, 12, 10499–10505 CrossRef CAS.
- Z. Y. Ma, M. Li, L. N. Guo, L. Liu, D. Wang and X. H. Duan, Org. Lett., 2021, 23, 474–479 CrossRef CAS.
- D. Dondi, A. M. Cardarelli, M. Fagnoni and A. Albini, Tetrahedron, 2006, 62, 5527–5535 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |