
Operando Auger/XPS using an electron beam to reveal the dynamics/morphology of Li plating and interphase formation in solid-state batteries†
Julien
Morey
 *a,
Jean-Bernard
Ledeuil
a,
Hervé
Martinez
abc and
Lénaïc
Madec
*ab
*a,
Jean-Bernard
Ledeuil
a,
Hervé
Martinez
abc and
Lénaïc
Madec
*ab
aUniversite de Pau et des Pays de l'Adour, E2S UPPA, CNRS, IPREM, Pau, France. E-mail: julien.morey@univ-pau.fr; lenaic.madec@univ-pau.fr
bRéseau sur le Stockage Electrochimique de l'Energie, CNRS FR3459, Amiens, France
cCentrale Casablanca, Centre de Recherche Systèmes Complexes et Interactions, Ville Verte, Bouskoura, Morocco
First published on 7th April 2023
Abstract
Interfaces and their understanding/control are the key to pave the way for the development of solid-state batteries. This work focuses on the development of operando Auger cycling using an electron beam to investigate the Li/solid electrolyte (SE) interphases. To do so, the fully tunable electron gun of the Auger was applied on top of a model Li/Li6PS5Cl(Arg) stack, allowing charge build up at the Arg surface and Li+ migration from the lithium electrode followed by SE interphase formation and Li plating. Overall, it is found that (i) Li6PS5Cl is first reduced to Li2S, LiCl and Li3P while (ii) Li plating occurs almost concomitantly and (iii) proceeds until the end of the operando cycling. These results were then confirmed by operando XPS using an electron beam. Importantly, this study highlights that operando Auger is more powerful than operando XPS as it provides visual observation of the dynamics/morphology of both Li/solid electrolyte interphase formation and Li plating together with reliable chemical information. This study thus opens the door for future development of operando Auger cycling using an electron beam as a powerful approach to better understand the interfaces in solid-state batteries.
Introduction
Lithium-ion batteries play a key role in today's society. Indeed, they are used to power portable electronic devices, tools and electric/hybrid-electric vehicles as well as for grid storage of renewable wind/solar energies. However, further improvements in terms of energy and/or power densities together with a longer lifetime and improved safety at an affordable cost are mandatory to meet future requirements,1 especially for electric vehicles. In that direction, all-solid-state batteries (ASSBs) offer a promising solution2,3 as they can increase the volumetric energy density up to 50% and the gravimetric energy density up to 150% compared with conventional liquid electrolyte-based Li-ion batteries. ASSBs are based on solid electrolytes (SEs), which are composed of quasi non-flammable materials such as ceramics, polymers or a combination of them and act as both the ion conduction medium and the separator. The expected energy density increase originates from (i) the use of metallic lithium at the anode, (ii) the increase of the electrochemical voltage stability window and (iii) the more flexible/compact cell packaging. Nevertheless, numerous challenges need to be solved in order to develop a new market based on this technology.First, the ionic conductivity of solid electrolytes (SEs) still remains relatively low, especially for polymer-based SEs. More importantly, the electro-chemo-mechanical stability of the numerous solid electrode materials/SE interfaces is still unsatisfactory.4,5 SE materials are also difficult to process,6 especially for ceramic solid-state batteries, for which it is difficult to achieve good Li ion transport between the ceramics and electrode materials.7 Second, at the anode, the best possible material is Li metal, but its use remains limited by possible dendrite formation and SE degradation at its surface, which can lead to rapid rollover of the cell.8–10 Beyond the choice of SEs and electrode materials, true battery performance in terms of cycle and calendar life, such as voltage window/profile and impedance, is often lower/worse than expected due to parasitic reactions at the electrode materials/SE interfaces. The origin of these decomposition reactions is thermodynamic. More specifically, if the chemical potentials μA (anode) and μC (cathode) are within the SE electrochemical stability window, the active materials/SE interfaces are stable.11 However, this ideal case almost never happens therefore solid electrolyte interphases (SEIs) are formed, similar to liquid Li-ion batteries. In ASSBs, SEIs can originate from cell storage/cycling as well as from the sequential manufacturing processes, especially for ceramic based SEs.6 The ideal SEI should, however, meet several requirements such as high ionic conductivity, compact structure and sufficient mechanical properties (both high elastic and shear strength) to suppress lithium dendrite formation. In practice, SEIs are often unstable and/or inhomogeneous and become thicker over time, which increase the electrode material/SE resistance and thus decrease the electrochemical performance. Therefore, in ASSBs, it is better to limit the mixing of components and stack preparation steps to hinder the formation of electrode materials/SE interphases and thus preserve the intrinsic properties of each material. Despite the numerous combinations of materials proposed for ASSBs in the literature, the electrode material/SE interface studies remain very sparse compared to the hundreds of papers reported on liquid Li-ion batteries. This is explained by the challenge to reveal and analyse the interfacial regions with high reliability as they are buried in the entire ASSB stacks.
In that direction, state-of-the-art cross-section preparation techniques such as focused ion beam (FIB) or broad ion beam (BIB) are often necessary before analytical techniques can be used to characterize the buried interfaces. In that case, ex situ cycling is used as the most common method and consists in electrochemically cycling the ASSB cell, disassembling it, then revealing the buried interfaces prior to the analysis using so far: optical microscopy (OM),12–17 scanning electron microscopy (SEM),18–20 high-resolution transmission electron microscopy (HR-TEM)18,21–23 combined with chemical/structural analyses by electron energy loss spectroscopy (EELS)18 or energy dispersive spectroscopy (EDS),24–26 scanning electron microscopy (SEM),26–30 Raman spectroscopy,31 time of flight secondary ion mass spectrometry (ToF-SIMS),32–35 Auger electron spectroscopy25 and X-ray photoelectron spectroscopy (XPS).24,36–42 Electrochemical impedance spectroscopy (EIS) has also been extensively used to investigate electrochemical interfacial changes43–45 but careful precaution should be taken as this technique examines all interfaces of the ASSB stacks and does not necessarily make distinction among the different interfaces. Furthermore, in situ and operando approaches are very attractive as they allow analysing “in real time” and limit possible sample pollution/interface degradation induced by the cell disassembling and sample preparation. During in situ analysis, the ASSB cell is sequentially cycled and analysed inside an analysis chamber. During operando analysis, the cell is cycled at the same time the analysis is performed. For in situ cycling, dedicated cells need to be designed to fit in the analytical machine used. For operando cycling, the dynamic evolution of the electrode materials and electrode materials/SE interfaces can be directly followed with minimal sample preparation and concomitant possible alteration. Note that considering the numerous in situ/operando cell setup reported so far, it is difficult to discuss about their respective advantage and limitation. Therefore, the standardization of the cell setups and also of the experimental setups will need to be addressed in the future in order to get more reliable and reproducible experiments between laboratories. While different operando cycling cells are described in the literature,46 the major interest of operando is that it can be performed without any specific dedicated cell. Indeed, the electron and/or UV sources can be used instead to discharge/charge the ASSB stack.47–49 Typically, the charge compensation gun present in XPS and ToF-SIMS apparatus is used in the electron flooding mode while the primary electron beam is used for SEM/Auger analysis. Moreover, the UV source, often present in XPS equipment can be used to reversibly cycle the ASSB stack by removing valence band electrons. This operando approach thus suppresses the need for cross-section preparation as well as the need to adapt the ASSB stack to the format/dimension of the electrochemical cell and so on. In that case, the main drawback is still the lack of pressure control on the ASSB stack, which can be critical only if high current is used, especially for ceramic SE. Indeed, the most common use and interest of this approach so far is to study the Li/SE interface formation that cannot be accessed by ex situ or in situ cycling/analysis.50–52 Note that to our knowledge, this operando approach using an electron gun has been proposed two times for XPS,47,49 only one time for Auger49 and remains to be developed for ToF-SIMS. Moreover, the previous operando Auger study only reported elemental mapping evolution without chemical/spectral information while such data are of high interest and are even essential to get a reliable understanding.
To fill this gap, the present work focuses on the use of operando Auger cycling using an electron beam to illustrate the high interest of this approach with a comparison with operando XPS also using an electron beam. To do so, the electrochemical stability of a model Li(M)/Li6PS5Cl system is investigated during Li plating. Note that Li6PS5Cl is well known to be reduced as follows:24,56,58 Li6PS5Cl + 8Li+ + 8e− → LiCl + Li3P + 5Li2S. Li6PS5Cl is thus used here to validate the operando Auger approach and to make a reliable comparison with operando XPS. It is shown that operando Auger allows mapping and analysing the dynamic Li/SE interface formation with the chemical environment as well as visualizing the Li plating at both micro- and nano-scales. Operando XPS shows similar chemical environment evolution but at the micro-scale and with no optical information. Overall, this work opens the door for future developments of operando Auger cycling using an electron beam and paves the way for a better understanding of interphase formation in ASSBs.
Materials and methods
Li6PS5Cl (99.9%, Ampcera, denoted as Arg hereafter) was used as received. Inside an Ar filled-glovebox (H2O < 0.1 ppm and O2 < 0.1 ppm), pellets of 10 mm diameter were made by pressing 120 mg of Arg under 375 MPa (3T) for 5 min, which led to about 600 μm thick pellets. The Arg pellets were placed on top of a lithium foil (GoodFellow, 99.9%, thickness of 0.2 mm) then deposited on an Auger/XPS sample holder. For comparison, Arg pellets were also used without Li (as the reference) to evaluate any possible damage induced by the electron beam in Auger and XPS. The Li/Arg stacks or Arg pellets were transferred into the machines using a transfer vessel (Auger) or an Ar filled-glovebox directly connected (XPS) to prevent any air contamination. Once inside the Auger/XPS analysis chambers, sample holders were grounded so that the electron flow applied on top of the Li/Arg stacks will create a potential difference by charge build up, which will then induce Li+ migration from the lithium electrode, SEI formation if any then Li plating (Fig. 1).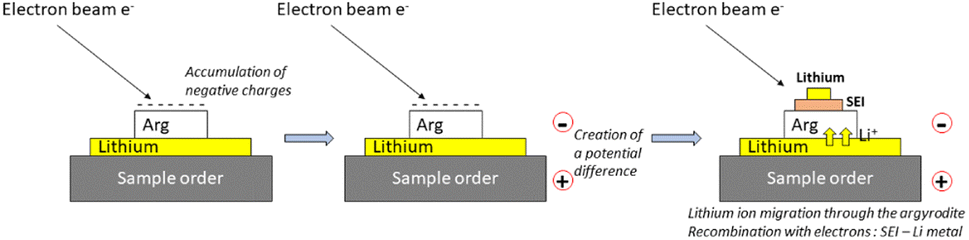 | ||
| Fig. 1 Schematic view of the operando Auger cycling method used in this work via an electron beam. | ||
Operando SEM/Auger cycling using an electron beam was performed using a JEOL JAMP95000F working under ultra-high vacuum (1.5 × 10−9 mbar) during analysis. Typically, the sample was tilted to 45° (an optimal angle under such operating conditions) to prevent any possible charging effect. The electron beam was fixed at 5 × 10−8 A and 10 keV, corresponding to a cycling rate of about 0.8 mA cm−2. Operando SEM/Auger cycling using an electron beam was performed by alternatively recording (60 times) a SEM image (magnification of about ×800, duration < 10 s) and an Auger survey spectrum (large area scan of 30 × 20 μm, i.e., average spectrum obtained from 256 × 256 points, with dE/E = constant = 0.5%, kinetic energy from 20 to 600 eV, duration 30 s). Thus, the sample charging is induced by both the Auger/SEM analysis while short time acquisition SEM images (<10 s) are used to follow the morphological evolution.
The total duration of the experiment was about 40 min. A “probe tracking” correction was used after each SEM/survey spectra step to control and compensate any drift. Additionally, for one experiment, scanning Auger microscopy (SAM, elemental 2D distribution) images of Li KVV (from Li2S and Li metal) and S LVV transitions were also recorded. This acquisition was performed after half of the SEM/Auger spectra steps using the same large area with a 10 eV pass energy and 2 ms dwell time in a fixed energy resolution (dE = constant or constant analyzer energy mode). Duration of the SAM was about 10 min. The contrast intensity of the images is represented using the “peak minus background”. Peak fitting processing was performed using JEOL's peak deconvolution software based on real reference spectra.
Operando XPS cycling using an electron beam was performed using a THERMO FISHER ESCALAB 250 Xi with a monochromatized Al Kα radiation (hν = 1486.6 eV) working under ultra-high vacuum (9.0 × 10−9 mbar). Operando XPS was performed by recording (12 times) core level spectra every 55 min (elliptic 325 × 650 μm X-ray beam spot, 20 eV pass energy, 0.1 eV step size and short time iterative acquisition scans to follow any possible degradation, duration 5 min) while using the charge neutralizer (flood gun) in electron mode (in lens only: beam = 0.1 V, emission = 100 μA, focus = 20 V and extractor = 15 V corresponding to a current applied to the sample of about 3 μA for a surface area of about 0.03 cm2). The total duration of the experiment was 12 h. Peak fitting was performed using CasaXPS software. The binding energy scale was calibrated from the 285 eV peak of adventitious carbon. A non-linear Shirley-type background was used for core peak analysis while 70% Gaussian–30% Lorentzian–Voigt peak shapes and full width at half-maximum constraint ranges were selected to optimize areas and peak positions. XPS quantification was performed using the relative sensitivity factor provided by the Thermo database (based on modified Scofield cross-sections).
Results and discussion
Operando Auger cycling using an electron beam
Note first that when using only an Arg pellet (i.e., without Li foil), no change was observed in SEM images even in Auger analysis (Fig. S1†), which highlights that no electron beam damage will occur during the operando Auger cycling on the Li/Arg stack. Fig. 2 shows typical SEM image evolution from the beginning to the end of the operando cycling procedure. The area where the electron beam is scanned is highlighted by the dotted rectangle. Fig. S2† also provides a short video of the full evolution of the SEM images during the operando. The reproducibility was checked by repeating the experiment on another pellet (Fig. S3†). At the beginning of the experiment, a slight volume expansion of the Arg surface was first observed with a secondary electron white contrast (Fig. 2b) due to the SEI formation. It is explained by the creation of reduction compounds from the Arg more likely with different density than the Arg. Note that this phenomenon would be an issue in a real cell as it could lead to contact loss at the Li/SE interfaces and thus to possible dendrite formation. Then, the apparition of a thicker layer (∼2 to 5 μm) with a secondary electron black contrast was observed (Fig. 2c and d) and is attributed to the Li plating. These results thus suggest that the SEI is first formed followed by Li plating. Regarding the Li plating morphology, it appears relatively rough, i.e., it did not take place evenly over the entire surface, leading to an inhomogeneous thickness (see Fig. 6 for a closer view), which can be attributed to the relatively high current density used for the experiment (about 0.8 mA cm−2). Porous Li may also form under such high current. Also note that measuring precisely the Li thickness over the entire area remains a challenge. In any case, the thickness of the Li expected to be plated at the end of the operando Auger is about 2 micron while it appears thicker (up to ∼5 micron) in some areas even after only 20 min of experiment (Fig. 2c and d). In addition, the Li plating (black contrast) occurred on a larger area than the scanned one for the experiment (dotted rectangle) more likely due to an electron flow at the Arg surface supported by the well-known electronic conductivity53 of Arg (∼10−6 S cm−1 at 25 °C from the datasheet supplier54). | ||
| Fig. 2 SEM images of an Arg pellet surface (a) before the operando Auger cycling using an electron beam, (b) after 10 min, (c) 20 min and (d) at the end of operando cycling. The dotted rectangle indicates where the operando Auger cycling was performed. Note that the Li expected to be plated is about 0.7 μm, 1.3 μm and 2 μm after 10 min, 20 min and at the end of the operando Auger, respectively. | ||
To confirm these hypotheses, Auger spectra and SAM image analysis were then used. Fig. 3 shows the evolution of the Auger spectra recorded as a function of the time during the operando cycling on a duplicate sample. Auger survey spectra showed the decrease of the P LVV, S LVV and Cl LVV transition peaks from the Arg and the increase of the Li transition peaks (Fig. 3a), indicating the covering of the Arg by the SEI and/or Li platting. This was confirmed by the evolution of the shape and position of the Li KVV transition that showed the appearance almost simultaneously of Li2S and Li metal KVV transition peaks after only 2 min (Fig. 3b), according to the JEOL reference database. This was further confirmed by the fit of the 6 min Li KVV spectrum in derivative representation (Fig. 3c). These results thus confirm the SEI formation from the Arg reduction during the Li plating, in agreement with the well-known Arg reduction pathway.55–59 Note that here, the appearance of LiCl should also be observed but it was impossible to confirm it by Auger spectra peak fitting as it would be within the uncertainty of the fitting. It is also possible that the starting Li6PS5Cl was already in partial equilibrium with Li3PS4 and LiCl so that further reduction would lead only to Li2S formation.
 | ||
| Fig. 3 (a) Evolution of the Auger survey spectra in derivative representation as acquired at the Arg pellet surface during the operando Auger cycling using an electron beam, (b) zoom on the Li KLL transition of the Auger survey spectra during the beginning of the experiment, (c) comparison of the Auger spectrum acquired after 6 min of operando cycling with Auger spectra of Li metal, Li2S and LiCl from JEOL references as well as the corresponding fitting. | ||
To further visualize the SEI and Li plating formation, additional energy resolved SAM (elemental 2D distribution) images of Limetal KVV, LiLi2S KVV and S LVV transitions were taken in the middle of the SEM/Auger spectra acquisition (see the experimental part) as reported in (Fig. 4). The energy resolved SAM image overlay of all transitions clearly shows the Li2S formation and Li plating. It is also observed that the Li metal covers mostly the Li2S surface as seen by comparison between the two SAM images. These results further suggest that Li2S is formed first from the Arg reduction then Li plating occurs.
 | ||
| Fig. 4 (a) SEM image of an Arg pellet surface in the middle of the operando Auger cycling using an electron beam with (b) energy resolved SAM (elemental 2D distribution) images of Limetal KVV, LiLi2S KVV and S LVV transitions and their overlays. | ||
Operando XPS cycling using an electron beam
In this part, operando XPS was performed on a duplicate Li/Arg stack to confirm the results of the operando Auger cycling. Note first that when using only an Arg pellet (i.e., without Li foil), no change was observed in the XPS core level spectra (Fig. S1†), which highlights that no electron beam damage will occur during the operando XPS cycling on the Li/Arg stack. Fig. 5 shows the evolution of the Li 1s, S 2p and O 1s XPS core level spectra recorded as a function of the time during the operando cycling. Fig. S4† shows the corresponding evolution of the Cl 2p, C 1s and P 2p XPS core level spectra. Table S1† shows the full XPS quantification (in at%) while Table 1 shows the corresponding XPS quantification (in at%) for the main compounds. Note first that the starting Arg contains about 2 at% of P2S5 and 6.5 at% of LixSO3 as well as 25.2 at% of Li2CO3, more likely originating from the synthesis (residual P2S5) and the storage conditions of the commercial Arg. Overall, these impurities showed a continuous decrease during the operando cycling, indicating their covering by new compounds, including the reduction products of the Arg as well as the Li plating. Also, note that such a relatively high amount of Li2CO3 is often observed at the Arg surface in the literature while authors often neglect it. However, this could increase the contact resistance between Arg and active materials including Li metal and it could thus lead to poorer performance as well as possible Li dendrite formation. | ||
| Fig. 5 XPS core level spectra of Li 1s, S 2p and O 1s as recorded at the Arg pellet surface during operando XPS cycling using an electron beam. | ||
| Compounds | 0 h | 1 h | 3 h | 7 h | 12 h |
|---|---|---|---|---|---|
| Li6PS5Cl | 37.2 | 21.6 | 14.4 | 10.8 | 7.2 |
| Li2S | 5.1 | 10.8 | 10.8 | 9 | 7.8 |
| LiCl | 2.2 | 2.6 | 2.6 | 2.2 | 1.8 |
| Li2O | — | 9.9 | 21.6 | 35.7 | 45.3 |
| LixP | — | 3.0 | 3.0 | 2.8 | 2.1 |
| Li2CO3 | 24 | 16.8 | 13.8 | 10.8 | 7.8 |
| P2S5 | 2.1 | 1.1 | 1.1 | 0.7 | 0.4 |
| LixSO3 | 7.5 | 3.5 | 3 | 1.5 | 1.5 |
Regarding the evolution of the S 2p doublet of the Arg at about 161.8 eV,39,56 XPS data showed a continuous decrease during the operando cycling. Simultaneously, an increase first of the O 1s peak of LiOH (∼531.3 eV) then of the O 1s peak of Li2O (∼528.8 eV) were observed. The continuous shift of the Li 1s spectra towards lower binding energy further confirms the LiOH then Li2O increase. These phenomena are attributed to Li plating with, however, a quick conversion into LiOH then Li2O, more likely due to the reaction of Li with the residual oxygen present inside the XPS analysis chamber. This point will be discussed in more detail in the following part. In addition, a large increase of Li2S (about 5 at%) and a slight increase of LiCl/Li3P (about 1 at% both) were observed after 1 h, followed by their continuous decrease until the end of the cycling. Overall, these results thus confirm the operando Auger cycling: (i) Arg is first reduced following the well-known Li6PS5Cl + 8 Li+ + 8e− → LiCl + Li3P + 5Li2S reaction pathway; (ii) almost concomitantly, Li plating occurs and (iii) then Li plating continues until the end of the operando cycling.
Comparison between operando Auger and XPS
The combination of Auger and XPS allows validating the operando cycling method based on the use of an electron gun applied on a Li/Argpellet stack to evaluate the reduction pathway of Arg during Li plating. Interestingly, these spectroscopies provide complementary information. While operando XPS gave the evolution of the degradation species of the Arg solid electrolyte with reliable quantification, Auger allowed visualizing the dynamics of all the processes (volume expansion during Arg reduction, Li plating and its morphology) together with chemical information.Beyond that, operando XPS showed the formation of LiOH then Li2O instead of Li metal while operando Auger showed Li metal plating. Note that previous operando XPS studies already reported the formation of Li2O instead of Li metal but did not discuss it.47,60,61 At this point, both the analysis time and the analysis chamber pressure (i.e., residual O2 pressure) can govern the Li2O formation. In the present case, the duration of the operando Auger was 40 min while for operando XPS, it was 12 hours. Regarding the analysis chamber pressure, it was 1.5 × 10−9 mbar for Auger and almost one decade higher for XPS (9 × 10−9 mbar). To evaluate these parameters, the evolution of the Li metal plating (observed in Auger) was followed by recording additional SEM images and Auger spectra 20 min and 12 h after the end of the operando Auger cycling procedure. At the end of the operando Auger, the SEM image showed the black contrast attributed to the Li metal plating with an additional white contrast in some areas (Fig. 6a), attributed to Li2O by comparison with the JEOL reference database (Fig. 6b). After 20 more min or 12 h in the analysis chamber, the white areas significantly increased, indicating that Li metal continues to be converted into Li2O (Fig. 6c and d) but at a slow rate as Li metal is still observed (black areas). For comparison, Li metal was never observed by XPS. These results highlight that both the analysis time and chamber pressure govern the Li2O formation, as expected. Therefore, operando Auger cycling and its fully tunable electron gun (both current and probe size) are of high interest to investigate both Li/solid electrolyte interphase formation and Li plating with its morphology as it could be done as a function of the cycling rate. In operando XPS cycling, however, careful precaution should be taken if one wants to strip the Li metal/Li2O using the often-available UV source as it could lead to misinterpretation of the results when only Li2O is observed. In that case, in order to limit Li2O formation, either the analysis time should be decreased by increasing the current and/or the chamber pressure should be lowered.
 | ||
| Fig. 6 SEM images as observed (a) at the end of the operando Auger using an electron beam at the Arg pellet surface, (c) 20 min later and (d) 12 h later after the end of the operando Auger cycling procedure. (b) Auger core level spectra of the Li KVV transition as recorded on two different points (black and white areas) as obtained at the end of the operando Auger, as indicated. | ||
Conclusion
This work focuses on the use of operando Auger cycling to illustrate the high interest of this approach in the case of the Li/solid electrolyte (SE) interphase study. This approach is based on the charge build up on top of an Li/SE stack using the fully tunable electron gun of Auger (current and probe size), which creates a potential difference then leads to the Li+ migration from the lithium electrode and through the solid electrolyte, to the SEI formation, if any, then to Li plating. Here, a model Li/Li6PS5Cl stack was used. Overall, it was shown that: (i) Arg is first reduced following the well-known Li6PS5Cl + 8Li+ + 8e− → LiCl + Li3P + 5Li2S pathway; (ii) and Li plating occurs almost concomitantly; (iii) then Li plating continues until the end of the operando cycling. These results were confirmed by operando XPS cycling on a duplicate Li/Li6PS5Cl stack. Interestingly, while operando XPS allows following the Li/SE interface formation with reliable quantification, Auger allows visualizing the dynamics of both interphase formation and Li plating with their morphologies together with chemical information. In the future, the fully tunable electron gun of Auger will be used to investigate both Li/SE interface formation and Li plating as a function of the cycling rate as it is of high concern in the literature. Overall, this work paves the way for future development of operando Auger cycling and for a better understanding of solid electrolyte reactivity (including halides, oxides etc. as well as polymer-based electrolytes) in lithium solid-state batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This manuscript was produced as part of the RAISE2024 project. This project has received funding from the Excellence Initiative of Université de Pau et des Pays de l'Adour (I-Site E2S UPPA), a French “Investissement d'Avenir” program.Notes and references
- D. Andre, S. J. Kim, P. Lamp, S. F. Lux, F. Maglia, O. Paschos and B. Stiaszny, J. Mater. Chem. A, 2015, 3, 6709–6732 RSC.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- Q. Zhang, K. Liu, F. Ding and X. Liu, Nano Res., 2017, 10, 4139–4174 CrossRef.
- T. T. Zuo, R. Rueß, R. Pan, F. Walther, M. Rohnke, S. Hori, R. Kanno, D. Schröder and J. Janek, Nat. Commun., 2021, 12, 6669 CrossRef CAS PubMed.
- I. López, J. Morey, J. B. Ledeuil, L. Madec and H. Martinez, J. Mater. Chem. A, 2021, 9, 25341–25368 RSC.
- W. Zaman and K. B. Hatzell, Curr. Opin. Solid State Mater. Sci., 2022, 26, 101003 CrossRef CAS.
- T. Abe, F. Sagane, M. Ohtsuka, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2005, 152, A2151 CrossRef.
- S. S. Zhang, ACS Appl. Energy Mater., 2018, 1, 910–920 CrossRef CAS.
- T. Krauskopf, F. H. Richter, W. G. Zeier and J. Janek, Chem. Rev., 2020, 120, 7745–7794 CrossRef CAS PubMed.
- T. Krauskopf, H. Hartmann, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2019, 11, 14463–14477 CrossRef CAS PubMed.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- M. Otoyama, H. Kowada, A. Sakuda, M. Tatsumisago and A. Hayashi, J. Phys. Chem. Lett., 2020, 11, 900–904 CrossRef CAS PubMed.
- S. Kim, C. Jung, H. Kim, K. E. Thomas-Alyea, G. Yoon, B. Kim, M. E. Badding, Z. Song, J. M. Chang, J. Kim, D. Im and K. Kang, Adv. Energy Mater., 2020, 10, 1–11 Search PubMed.
- M. Sun, T. Liu, Y. Yuan, M. Ling, N. Xu, Y. Liu, L. Yan, H. Li, C. Liu, Y. Lu, Y. Shi, Y. He, Y. Guo, X. Tao, C. Liang and J. Lu, ACS Energy Lett., 2021, 6, 451–458 CrossRef CAS.
- Y. X. Song, Y. Shi, J. Wan, S. Y. Lang, X. C. Hu, H. J. Yan, B. Liu, Y. G. Guo, R. Wen and L. J. Wan, Energy Environ. Sci., 2019, 12, 2496–2506 RSC.
- W. Manalastas, J. Rikarte, R. J. Chater, R. Brugge, A. Aguadero, L. Buannic, A. Llordés, F. Aguesse and J. Kilner, J. Power Sources, 2019, 412, 287–293 CrossRef CAS.
- E. Kazyak, R. Garcia-Mendez, W. S. LePage, A. Sharafi, A. L. Davis, A. J. Sanchez, K. H. Chen, C. Haslam, J. Sakamoto and N. P. Dasgupta, Matter, 2020, 2, 1025–1048 CrossRef.
- D. Santhanagopalan, D. Qian, T. McGilvray, Z. Wang, F. Wang, F. Camino, J. Graetz, N. Dudney and Y. S. Meng, J. Phys. Chem. Lett., 2014, 5, 298–303 CrossRef CAS PubMed.
- A. Aboulaich, R. Bouchet, G. Delaizir, V. Seznec, L. Tortet, M. Morcrette, P. Rozier, J. M. Tarascon, V. Viallet and M. Dollé, Adv. Energy Mater., 2011, 1, 179–183 CrossRef CAS.
- A. Brazier, L. Dupont, L. Dantras-Laffont, N. Kuwata, J. Kawamura and J. M. Tarascon, Chem. Mater., 2008, 20, 2352–2359 CrossRef CAS.
- J. Z. Lee, T. A. Wynn, Y. S. Meng and D. Santhanagopalan, J. Visualized Exp., 2018, 2018, 1–9 Search PubMed.
- A. Brazier, L. Dupont, L. Dantras-Laffont, N. Kuwata, J. Kawamura and J. M. Tarascon, Chem. Mater., 2008, 20, 2352–2359 CrossRef CAS.
- K. Yamamoto, R. Yoshida, T. Sato, H. Matsumoto, H. Kurobe, T. Hamanaka, T. Kato, Y. Iriyama and T. Hirayama, J. Power Sources, 2014, 266, 414–421 CrossRef CAS.
- J. Auvergniot, A. Cassel, D. Foix, V. Viallet, V. Seznec and R. Dedryvère, Solid State Ionics, 2017, 300, 78–85 CrossRef CAS.
- A. Uhart, J. B. Ledeuil, B. Pecquenard, F. Le Cras, M. Proust and H. Martinez, ACS Appl. Mater. Interfaces, 2017, 9, 33238–33249 CrossRef CAS PubMed.
- M. Yamamoto, Y. Terauchi, A. Sakuda, A. Kato and M. Takahashi, J. Power Sources, 2020, 473, 228595 CrossRef CAS.
- J. Zagórski, B. Silván, D. Saurel, F. Aguesse and A. Llordés, ACS Appl. Energy Mater., 2020, 3, 8344–8355 CrossRef.
- D. Cheng, T. A. Wynn, X. Wang, S. Wang, M. Zhang, R. Shimizu, S. Bai, H. Nguyen, C. Fang, M. cheol Kim, W. Li, B. Lu, S. J. Kim and Y. S. Meng, Joule, 2020, 4, 2484–2500 CrossRef CAS.
- X. Li, Z. Ren, M. Norouzi Banis, S. Deng, Y. Zhao, Q. Sun, C. Wang, X. Yang, W. Li, J. Liang, X. Li, Y. Sun, K. Adair, R. Li, Y. Hu, T. K. Sham, H. Huang, L. Zhang, S. Lu, J. Luo and X. Sun, ACS Energy Lett., 2019, 4, 2480–2488 CrossRef CAS.
- J. B. Ledeuil, A. Uhart, S. Soulé, J. Allouche, J. C. Dupin and H. Martinez, Nanoscale, 2014, 6, 11130–11140 RSC.
- M. Otoyama, Y. Ito, A. Hayashi and M. Tatsumisago, J. Power Sources, 2016, 302, 419–425 CrossRef CAS.
- F. Walther, R. Koerver, T. Fuchs, S. Ohno, J. Sann, M. Rohnke, W. G. Zeier and J. Janek, Chem. Mater., 2019, 31, 3745–3755 CrossRef CAS.
- F. Walther, S. Randau, Y. Schneider, J. Sann, M. Rohnke, F. H. Richter, W. G. Zeier and J. Janek, Chem. Mater., 2020, 32, 6123–6136 CrossRef CAS.
- F. Walther, F. Strauss, X. Wu, B. Mogwitz, J. Hertle, J. Sann, M. Rohnke, T. Brezesinski and J. Janek, Chem. Mater., 2021, 33, 2110–2125 CrossRef CAS.
- N. Wu, P. H. Chien, Y. Li, A. Dolocan, H. Xu, B. Xu, N. S. Grundish, H. Jin, Y. Y. Hu and J. B. Goodenough, J. Am. Chem. Soc., 2020, 142, 2497–2505 CrossRef CAS PubMed.
- J. Liang, S. Hwang, S. Li, J. Luo, Y. Sun, Y. Zhao, Q. Sun, W. Li, M. Li, M. N. Banis, X. Li, R. Li, L. Zhang, S. Zhao, S. Lu, H. Huang, D. Su and X. Sun, Nano Energy, 2020, 78, 105107 CrossRef CAS.
- R. Schlenker, D. Stȩpień, P. Koch, T. Hupfer, S. Indris, B. Roling, V. Miß, A. Fuchs, M. Wilhelmi and H. Ehrenberg, ACS Appl. Mater. Interfaces, 2020, 12, 20012–20025 CrossRef CAS PubMed.
- C. Xu, B. Sun, T. Gustafsson, K. Edström, D. Brandell and M. Hahlin, J. Mater. Chem. A, 2014, 2, 7256–7264 RSC.
- J. Auvergniot, A. Cassel, J. B. Ledeuil, V. Viallet, V. Seznec and R. Dedryvère, Chem. Mater., 2017, 29, 3883–3890 CrossRef CAS.
- B. Sun, C. Xu, J. Mindemark, T. Gustafsson, K. Edström and D. Brandell, J. Mater. Chem. A, 2015, 3, 13994–14000 RSC.
- S. A. Pervez, B. P. Vinayan, M. A. Cambaz, G. Melinte, T. Diemant, T. Braun, G. Karkera, R. J. Behm and M. Fichtner, J. Mater. Chem. A, 2020, 8, 16451–16462 RSC.
- J. Qiu, X. Liu, R. Chen, Q. Li, Y. Wang, P. Chen, L. Gan, S. J. Lee, D. Nordlund, Y. Liu, X. Yu, X. Bai, H. Li and L. Chen, Adv. Funct. Mater., 2020, 30, 1–8 Search PubMed.
- S. Larfaillou, D. Guy-Bouyssou, F. Le Cras and S. Franger, ECS Trans., 2014, 61, 165–171 CrossRef CAS.
- N. Grillon, E. Bouyssou, S. Jacques and G. Gautier, J. Electrochem. Soc., 2015, 162, A2847–A2853 CrossRef CAS.
- S. Larfaillou, D. Guy-Bouyssou, F. Le Cras and S. Franger, J. Power Sources, 2016, 319, 139–146 CrossRef CAS.
- F. Strauss, D. Kitsche, Y. Ma, J. H. Teo, D. Goonetilleke, J. Janek, M. Bianchini and T. Brezesinski, Adv. Energy Sustainability Res., 2021, 2, 2100004 CrossRef CAS.
- A. L. Davis, E. Kazyak, J. Sakamoto, N. P. Dasgupta, R. Garcia-Mendez, K. H. Chen, J. Sakamoto, K. N. Wood, G. Teeter and K. N. Wood, J. Mater. Chem. A, 2020, 8, 6291–6302 RSC.
- M. Golozar, A. Paolella, H. Demers, S. Savoie, G. Girard, N. Delaporte, R. Gauvin, A. Guerfi, H. Lorrmann and K. Zaghib, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
- K. N. Wood, K. X. Steirer, S. E. Hafner, C. Ban, S. Santhanagopalan, S. H. Lee and G. Teeter, Nat. Commun., 2018, 9, 1–10 CrossRef CAS PubMed.
- Y. K. Sun and P. V. Kamat, ACS Energy Lett., 2021, 6, 2356–2358 CrossRef CAS.
- L. Yang, W. You, X. Zhao, H. Guo, X. Li, J. Zhang, Y. Wang and R. Che, Nanoscale, 2019, 11, 17557–17562 RSC.
- X. Liu, D. Wang, G. Liu, V. Srinivasan, Z. Liu, Z. Hussain and W. Yang, Nat. Commun., 2013, 4, 1–8 Search PubMed.
- J. M. Lee, Y. S. Park, J. W. Moon and H. Hwang, Front. Chem., 2021, 9, 1–8 CAS.
- AMPCERA Argyrdite Li6PS5CL Sulfide Solid Electrolyte, Pass 150 Mesh (Below 100 μm) Coarse Powder, https://www.msesupplies.com/collections/electrolyte-materials/products/ampcera-sulfide-solid-electrolyte-argyrodite-li6ps5cl-powder?variant=22924472614970 Search PubMed.
- Y. Zhou, C. Doerrer, J. Kasemchainan, P. G. Bruce, M. Pasta and L. J. Hardwick, Batteries Supercaps, 2020, 3, 647–652 CrossRef CAS.
- D. H. S. Tan, E. A. Wu, H. Nguyen, Z. Chen, M. A. T. Marple, J. M. Doux, X. Wang, H. Yang, A. Banerjee and Y. S. Meng, ACS Energy Lett., 2019, 2418–2427 CrossRef CAS.
- S. Wenzel, T. Leichtweiss, D. Krüger, J. Sann and J. Janek, Solid State Ionics, 2015, 278, 98–105 CrossRef CAS.
- S. Wenzel, S. J. Sedlmaier, C. Dietrich, W. G. Zeier and J. Janek, Solid State Ionics, 2018, 318, 102–112 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- M. I. Nandasiri, L. E. Camacho-Forero, A. M. Schwarz, V. Shutthanandan, S. Thevuthasan, P. B. Balbuena, K. T. Mueller and V. Murugesan, Chem. Mater., 2017, 29, 4728–4737 CrossRef CAS.
- M. Mirolo, X. Wu, C. A. F. Vaz, P. Novák and M. El Kazzi, ACS Appl. Mater. Interfaces, 2021, 13, 2547–2557 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta00386h |
| This journal is © The Royal Society of Chemistry 2023 |