
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully-fused boron-doped olympicenes: modular synthesis, tunable optoelectronic properties, and one-electron reduction†
Jing
Guo‡
ab,
Kaihua
Zhang‡
ab,
Yanpei
Wang
ab,
Haipeng
Wei
 ab,
Wang
Xiao
ab,
Kun
Yang
*ab and
Zebing
Zeng
*ab
ab,
Wang
Xiao
ab,
Kun
Yang
*ab and
Zebing
Zeng
*ab
aState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, P. R. China
bShenzhen Research Institute of Hunan University, Shenzhen 518000, P. R. China. E-mail: zbzeng@hnu.edu.cn; yangk@hnu.edu.cn
First published on 7th March 2023
Abstract
We report here a novel family of boraolympicenes, structurally featuring boron-doping at the concave 11a-position of their π-skeletons and synthetically prepared via a facile one-pot triply borylation-based double-fold borocyclization reaction. Despite having no bulky protecting groups, these boraolympicenes exhibit excellent chemical stability against air and moisture, ascribed to the significant π-electron delocalization over the vacant pz orbitals of boron atoms as evidenced by both single-crystallographic and theoretical analyses. More importantly, the modular synthesis of these boraolympicenes allows the fine-tuning of their physicochemical properties, endowing them with intriguing electronic features, such as intense visible-to-NIR absorption and low-lying LUMO energy levels (∼−3.8 eV) as well as tunable molecular stacking characteristics in the crystalline state. As a model compound, a radical-anion salt of 6-phenyl-11a-boraolympicene was further generated through chemical reduction and well characterized by UV-vis-NIR absorption, ESR, and IR spectroscopy. This radical anion salt is sensitive to air and moisture but shows persistent stability under inert conditions benefiting from its stable borataalkene-containing resonant form.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) have captivated chemists for decades with their intriguing physicochemical properties and promising applications in organic (opto)electronics and spintronics.1 In designing new PAH systems, heteroatom doping has proved to be a versatile strategy to enrich the structure diversity and tune the key properties, e.g., redox behaviors, charge transport characteristics, and self-assembly profiles.2 Specifically, the incorporation of three-coordinate boron atoms is particularly robust for devising novel PAHs with unique features such as low-lying lowest unoccupied molecular orbital (LUMO) energy levels, intensified luminescence, strong Lewis acidity, etc, by virtue of its vacant pz orbital.3 Therefore, a large number of B-doped PAHs with varied B-doping patterns and satisfactory ambient stability have been developed to date, yielding promising performances in metal-free catalysis,4 electron transporting materials,5 and light-emitting materials.6 Among the various B-doped PAHs, those with boron atoms embedded in their inner positions, namely fully-fused B-PAHs (Fig. 1a), are attracting increasing attention in light of their high excellent chemical stability as well as strong π-stacking capability derived from their less crowded and planar geometries. For instance, by introducing well-tailored structural constraints, Yamaguchi and co-workers have reported a series of B-doped PAHs with boron atoms embedded at the non-edge position of their fully-conjugated skeletons by dehydrogenative aromatic C–C coupling reactions.7 Although lacking bulky protecting groups, these PAHs not only exhibited much-improved chemical stability against oxygen and moisture but also showed substantial π-stacking capability and robust Lewis acidity to form intriguing Lewis acid–base adducts. Wagner,8 Hatakeyama,9 and Ingleson10 also developed diverse fully-fused B-doped PAHs based on different sophisticated synthetic strategies. However, despite these encouraging advances, currently the material library of ambient-stable and protection-free fully-fused B-PAHs is still limited, mainly owing to the severe lack of facile and modular synthetic methodologies.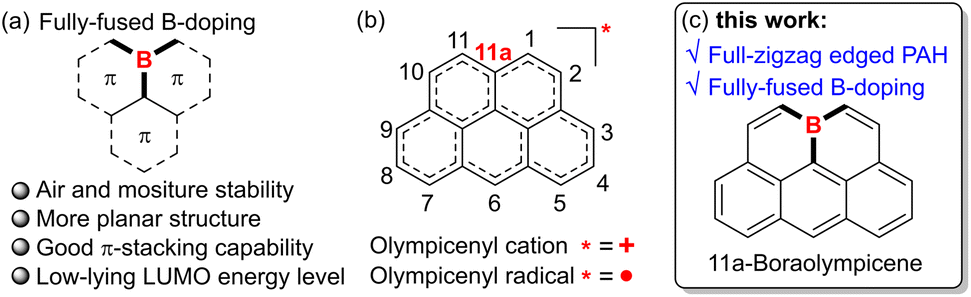 | ||
| Fig. 1 (a) Illustration of fully-fused B-doping and potential unique properties. (b) Skeletal structures of carbon-based olympicenes. (c) Full-zigzag edged and fully-fused B-doped 11a-boraolympicene reported in this work. | ||
With their skeletons closely resembling the famous Olympic rings, olympicene and its full-conjugated counterparts, such as the olympicenyl radical and cation (Fig. 1b), have attracted much attention from researchers since the 1960s as a class of odd-alternant hydrocarbons possessing unique non-bonding molecular orbitals and potential applications in the expanding field of organic semiconductors.11 Meanwhile, many efforts have been devoted to exploring their preparation and physicochemical properties by means of either experimental investigation or theoretical calculations. It is until recently that the pure olympicenyl radicals were first isolated in the crystalline state, showing π-dimeric structures assembled by multicenter pancake bonds.12 Nevertheless, owing to their open-shell electronic structure-associated high reactivity, those olymeicenyl radicals need to attach bulky protecting groups but are still slowly decomposed when dissolved in solution, thus severely hampering their further applications. In this regard, we envisaged that the skeletal doping with electron-withdrawing boron atoms into the fully-fused positions of the electron-rich olympicenyl frameworks would allow access to benchtop-stable isosteres of the highly reactive carbonaceous olympicenyl derivatives which are extremely appealing as potential optoelectronic, conducting, and magnetic materials. In addition, the inherent electron-deficient nature of incorporated boron atoms might further endow these boraolympicenyl derivatives with extra fascinating electronic properties/functions like electron-accepting and/or transporting characteristics, high Lewis acidity to form labile adducts with charge-neutral Lewis bases, and so on.7–10 However, as mentioned above, due to the lack of efficient synthetic methodologies, to the best of our knowledge, to date the preparation of such boraolympicenyl derivatives with fully-fused boron atoms is still remaining unexplored, not to speak of the illustration and modulation of boron-substitution effect on the physicochemical and molecular packing properties of these novel graphenoid PAHs.
Herein, by devising an efficient one-pot triply borylation-enabled double-fold borocyclization strategy, we report a class of novel boron-doped olympicenyl derivatives, namely 11a-boraolympicenes (Fig. 1c), as a type of new fully-fused B-PAH. These new boraolympicenes possess several beneficial features: (1) their synthesis is facile (starting from the commercial available anthracene-1,8-diyl bis(OTf), more details in the ESI†) and highly modular as it allows broad structural modulations at multiple positions (such as 2, 6, and 11-positions) of the olympicenyl π-frameworks; (2) despite having no protecting groups on their highly Lewis acidic boron atoms, excellent chemical stability against oxygen, moisture, and heat was still observed for these compounds, thus allowing the further post-functionalization on the basis of their bromo-functionalities to yield homologues with more complex structures and extended π-systems; (3) benefitting from such great structural diversity, these boraolympicenes also exhibited various intriguing electronic features, such as intense visible-to-NIR absorption, low-lying LUMO energy levels (∼−3.8 eV), as well as significantly varied molecular packing behaviors in the crystalline state that completely differ from their carbonaceous olympicenyl radicals;12 (4) persistently stable radical anions can be prepared via controlled chemical reduction, thus providing an ideal platform for investigating the boron-doping and electron-withdrawing effect on the electronic structures of these fully zigzag-edged PAHs.
Results and discussion
Electrophilic borylation of well-designed alkyne-incorporated arenes with BX3 (X = Cl or Br) as boron sources, followed by an intramolecular Friedel–Crafts cyclization, has been widely adopted for constructing boracycles and boron-doped PAHs.3,13 While two new C–B bonds are formed upon the ring-closure process in this approach, the remaining B–X bond on the newly formed boracycle typically requires extra post-protection modifications with bulky substituents due to its instability toward moisture. To this end, we rationalized that the further utilization of the remaining dangling B–X bond for a successive electrophilic borylation/intramolecular Friedel–Crafts cyclization would not only avoid the formation of liable dangling B–X bonds which need further conversion to bulky protecting groups but also result in a double-fold boracycle formation to yield a fully-fused B-doped PAH by generating three C–B bonds in a one-pot reaction, as long as an extra alkynyl moiety is appropriately placed in close proximity. To our delight, by adopting an anthracene substrate bearing two alkynyl moieties closely positioned on its 1- and 8-sites, we found that boraolympicene BO-1, namely 2,11-dibromo-1,10-diphenyl-11a-boraolympicene, which features a boron-embedded olympicene-like π-backbone, was successfully generated upon a triply borylation process with a moderate yield of 45% (Scheme 1).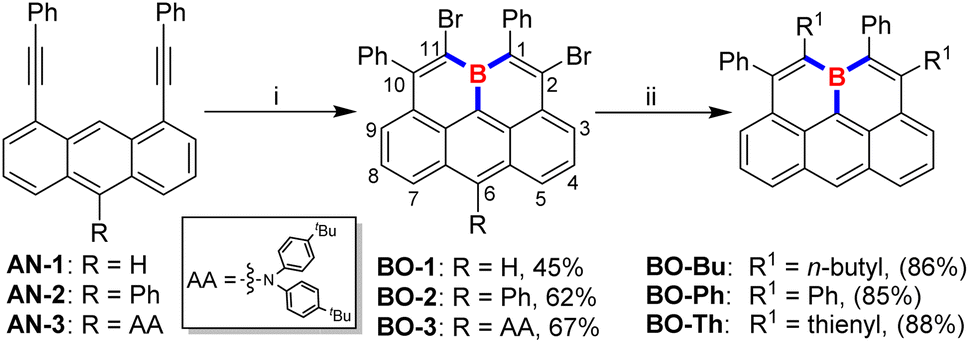 | ||
| Scheme 1 Synthetic routes toward 11a-boraolympicenes. Conditions: (i) BBr3, toluene, reflux; (ii) tetrabutyltin or tributylphenylstannane or 2-(tributylstannyl)thiophene, Pd(PPh3)4, CuI, toluene, reflux. | ||
With the aim to broaden the scope and further prove the inherent modularity of this triply borylated double-fold cyclization strategy, we next targeted structural derivatization at the anthracene moiety. We prepared new substrates AN-2/3, decorated with functional groups of phenyl ring or aromatic amine (AA: N,N-bis(4-(tert-butyl)phenyl)amine) at the 10-position of the anthryl backbone, respectively. Then, double-fold borolative ring-cyclization of AN-2/3 was efficiently accomplished to provide 11a-boraolympicene BO-2 and BO-3 in a yield of 62% and 67%, respectively. Notably, despite the absence of steric groups directly attached at the boron atom, the resulting 11a-boraolympicenes BO-1–3 exhibited excellent stability and without any decomposition against air, water, and silica gel, thus allowing the further post-synthetic introduction of additional functionalities onto the boraolympicene framework to access more complex B-doped structures based on its aryl bromide functionalities. For instance, by the palladium-catalyzed Stille coupling reactions, the dibrominated BO-1 can be efficiently converted into various alkyl or aryl-attached boraolympicenes BO-Bu/Ph/Th in high yields (85%∼88%) (Scheme 1). 1H NMR spectroscopy reveals that all the BO-1/Bu/Ph/Th exhibited low-field proton resonances located at δ = ∼9.2 ppm which can be assigned to the protons at 6-positions of π-skeletons (more details in the ESI†), ascribed to the high electron deficiency of these resulting boraolympicenes upon B-doping. Besides, the 11B NMR resonances of these three 11a-boraolympicenes are centred at δ = 42.3–45.4 ppm, which was typical for a π-conjugated boracyclic system.7–10
Single crystals of BO-1/2 suitable for X-ray diffraction analysis were grown by slowly diffusing hexane into their chloroform solutions, revealing a nearly planar structure for the pentacyclic π-skeletons of the 11a-boraolympicenes with ∑(∠(C–B–C)) ≈ 360° (Fig. 2a, b and S1†).14 Such planar boron-doping should favor the conjugation of the vacant pz orbital of the B atom with its adjacent aromatic segment and contribute to the high stability of these boraolympicenes without steric B-protecting groups. For the pendant moieties, while all the benzene rings of BO-1/2 were perpendicularly oriented against the boraolympicenyl cores regardless of their substituting positions, it was found that the presence of phenyl substitution at the 6-position of boraolympicenes played a significant role in determining the packing manners of these compounds. For instance, for the unsubstituted BO-1, while an infinite cofacial columnar packing mode was formed along the a-axis direction, the boraolympicene molecules were found to be cofacially π-stacked in a repeated ABC stacking sequence (Fig. 2a). Meanwhile, the π-stacking patterns between adjacent BO-1 molecules in each repeated ABC assembly varied significantly from a head-to-tail cofacial packing for AB and BC bilayers to a 55°-rotated head-to-head cofacial stacking for CA bilayers; the packing distances between all the AB/BC/CA bilayers are similar and averaged to be ∼3.401 Å with the shortest distance to be 3.270 Å. These values are in close proximity with the π-stacking distances as found for the π-dimers of carbonaceous olympicenyl radicals (3.25–3.29 Å).12 Clearly, such cofacial molecular alignment resulted from the maximization of intermolecular π–π overlaps between these unsymmetric boraolympicenyl molecules, which was also rather different from the head-to-tail type dimeric stacking mode as observed for the case of all carbon-based olympicenyl radicals.
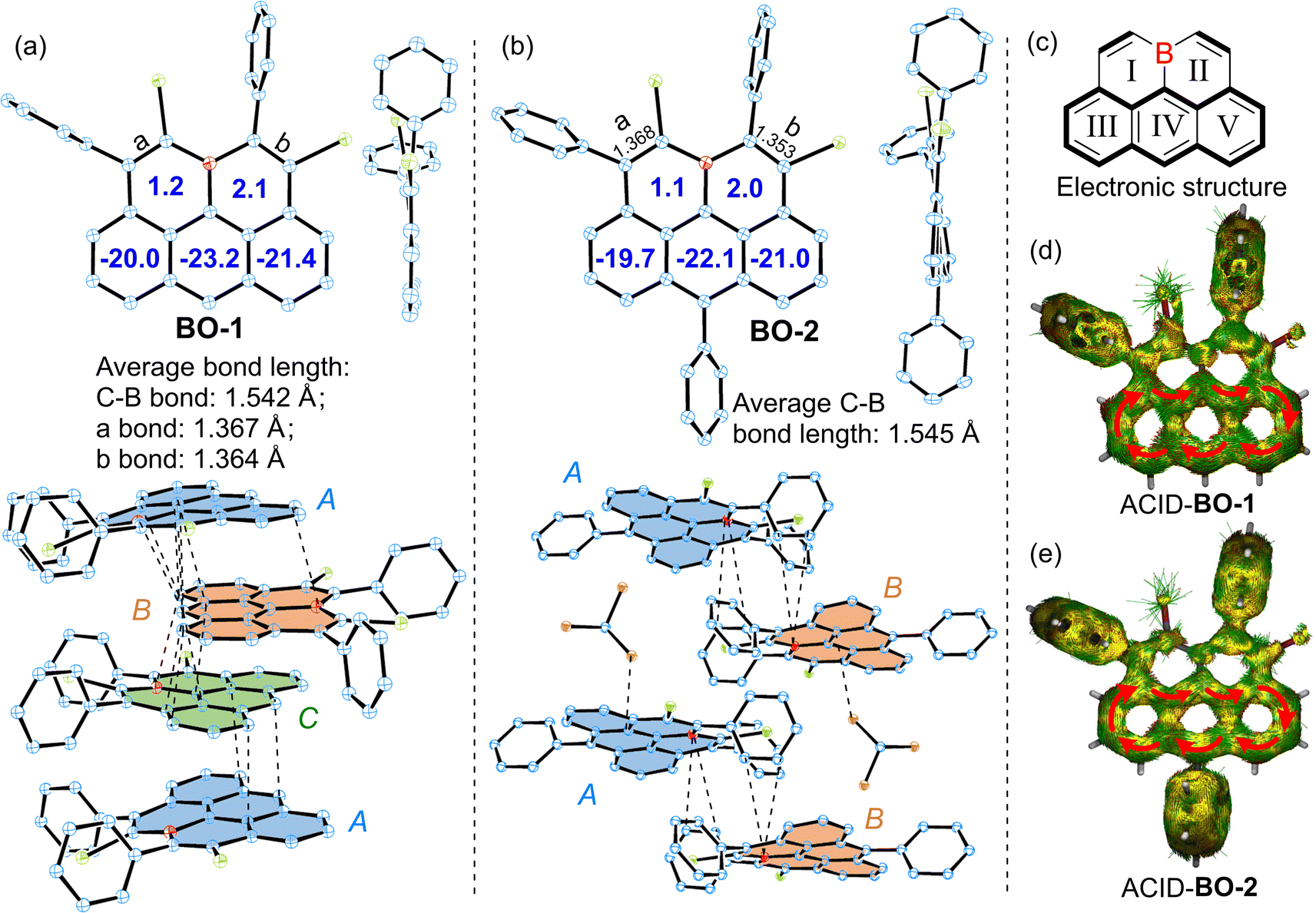 | ||
| Fig. 2 ORTEP drawing of (a) BO-1 and (b) BO-2 with selected bond lengths, and (c) graphic illustration of the electronic structure of BO. ACID plots of BO-1 and BO-2 (isovalue = 0.02). Thermal ellipsoids are shown at 50% probability; hydrogen atoms are omitted for clarity. Colors: blue, carbon; red, boron; green, bromine; orange, chlorine (thermal ellipsoids are shown at 50%; hydrogen atoms are omitted for clarity). | ||
In sharp contrast to the closely π-stacked BO-1 molecules, strikingly, BO-2 adopted a parallel-displaced tail-to-tail stacking pattern in a repeated AB sequence that was interrupted by two equivalents of solvent molecules (CHCl3) and exhibited no obvious π–π interactions between neighboring molecules. Only weak B…C bondings as well as the π (boraolypienic)…Cl (solvent CHCl3 molecule) interactions were found to be the major inter-layer interactions responsible for the parallel-displaced orientations of BO-2 molecules (Fig. 2b). Therefore, it seems that the installation of a bulky substituent at the 6-position of boraolympicene can result in large steric resistance that hampers the efficient π-staking of those boraolympicenyl molecules, which might also explain well that our attempts for growing single crystals of BO-3 ultimately failed as a more bulky aromatic amine substituent at the 6-position might significantly interrupt the ordered packing of BO-3 molecules during the crystallization growth process.
Bond length analysis of BO-1/2 crystals further disclosed much shorter averaged C–B bond lengths of BO-1 (∼1.542 Å) and BO-2 (∼1.545 Å) compared to that of triarylboranes (such as 1.574–1.589 Å for Ph3B),15 thus suggesting that vacant pz-orbitals of B atoms in BO-1/2 have strong p–π conjugation to their adjacent π-bonds. Notably, bonds a/b in both BO-1 and BO-2 exhibited typical olefic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond character (1.353–1.368 Å), thus furnishing a typical alkene-B-alkene motif in these molecules (Fig. 2a–c and S2†). In addition, two B-embedded six-membered rings (rings I and II) were revealed to be slightly antiaromatic in view of their positive nucleus-independent chemical shift (NICS)16 values (ca. NICS(1)zz: 1.2/2.1 ppm for BO-1 and 1.1/2.0 ppm for BO-1). In contrast, rings III/IV/V exhibit strong aromaticity for both BO-1 and BO-2 (NICS(1)zz: −20.0/−23.2/−21.4 ppm for BO-1 and −19.7/−22.1/−21.0 ppm for BO-1), resembling an anthracene unit (Fig. 2a and b). Anisotropy of the induced current density (ACID)17 plots further confirmed the high aromaticity of the anthracene segment as evidenced by the clockwise diatropic ring current along their periphery edges (Fig. 2d and e).
C bond character (1.353–1.368 Å), thus furnishing a typical alkene-B-alkene motif in these molecules (Fig. 2a–c and S2†). In addition, two B-embedded six-membered rings (rings I and II) were revealed to be slightly antiaromatic in view of their positive nucleus-independent chemical shift (NICS)16 values (ca. NICS(1)zz: 1.2/2.1 ppm for BO-1 and 1.1/2.0 ppm for BO-1). In contrast, rings III/IV/V exhibit strong aromaticity for both BO-1 and BO-2 (NICS(1)zz: −20.0/−23.2/−21.4 ppm for BO-1 and −19.7/−22.1/−21.0 ppm for BO-1), resembling an anthracene unit (Fig. 2a and b). Anisotropy of the induced current density (ACID)17 plots further confirmed the high aromaticity of the anthracene segment as evidenced by the clockwise diatropic ring current along their periphery edges (Fig. 2d and e).
The UV-vis-NIR spectra of the as-prepared boraolympicenes are presented in Fig. 3a. BO-1 and BO-2 showed similar absorption spectra with two major absorption bands in the region of 350–420 nm and 420–600 nm. The lower energy absorption bands with a respective maximum of 479 nm and 492 nm (ε = 0.78 × 104 M−1 cm−1 and 1.11 × 104 M−1 cm−1), together with a corresponding shoulder at 506/561 nm and 517/565 nm for BO-1 and BO-2, could be assigned to the HOMO-2 → LUMO and HOMO → LUMO transitions, respectively, according to the time-dependent density functional theory (TD-DFT) calculations at the B3LYP/6-31G(d,p) level (Fig. S3–S8†). BO-3 showed similar dual absorption bands in the range of 450–600 nm, but with an extra red-shifted electronic transition extending to the near-infrared region (NIR) (λmax = 663 nm, ε = 0.58 × 104 M−1 cm−1), ascribed to the strong intramolecular charge transfer (ICT) effect between its electron-donating diaryl-amine moiety and electron-accepting boraolympicenic skeleton. No such ICT bands were observed for the 2/11-dialkyl or -diaryl incorporated BO-Bu/Ph/Th (Fig. S9†), in accordance with the limited conjugation between these perpendicularly arranged pendant groups with the π-planes of boraolympicene skeletons. Accordingly, based on their onset absorption wavelengths, the optical bandgaps of these boraolypicenes can be calculated to be 2.03/2.02 eV and 1.45 eV for BO-1/2 and BO-3, respectively. Furthermore, DFT calculations also revealed that the LUMOs of this 11a-boraolympicene are well delocalized over the whole backbone, suggesting a good conjugation of the boron atoms in the π-system (more details in Fig. S4, S6, and S8†).
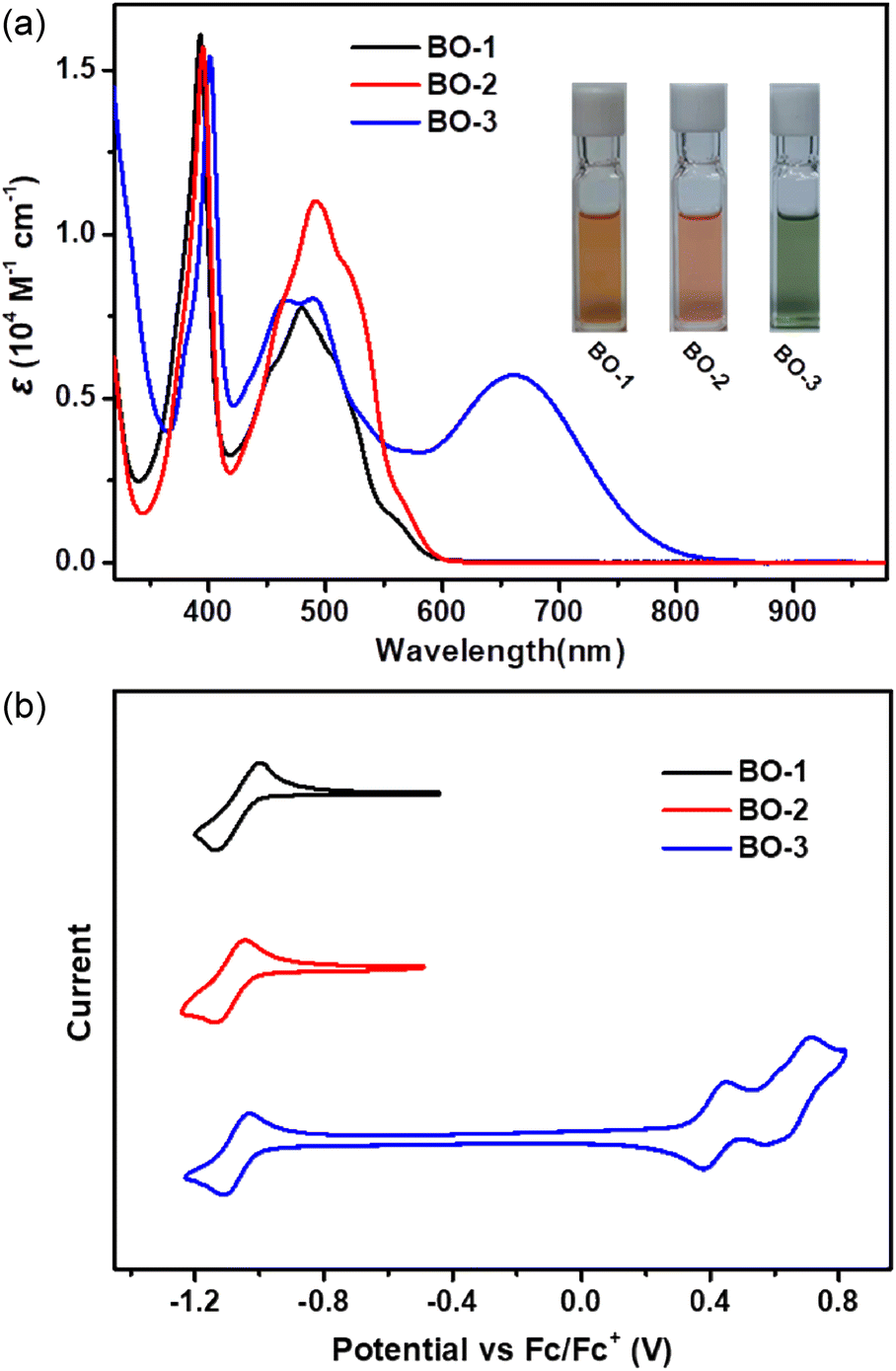 | ||
| Fig. 3 (a) UV-vis-NIR absorption spectra of BO-1–3 in DCM (10−5 M). Inset: photo image of BO-1–3 in DCM solution. (b) Cyclic voltammogram of BO-1–3 (1.0 mM in DCM) with 0.10 M TBA-PF6 as the supporting electrolyte. | ||
The redox properties of BO-1–3 were characterized by cyclic voltammetry (CV) measurements that were carried out in dichloromethane at room temperature. As depicted in Fig. 4a, all three compounds showed similar reversible reduction waves with almost identical onsets of the reduction potentials of −1.01, −1.03, and −1.00 V vs. Fc/Fc+ for BO-1–3, corresponding to low-lying LUMO energy levels of −3.79, −3.77, and −3.80 eV, respectively. BO-3 also displayed two extra reversible oxidation waves with the onset of the oxidation potentials of 0.39 and 0.59 V, pointing to the high HOMO energy levels of this molecule upon the installation of a strong electron-donating diaryl amine moiety. The absence of such oxidation behaviors of BO-1 and BO-2 revealed the overall high electron deficiency of these two compounds upon B-doping. To the best of our knowledge, the low-lying LUMO energy of these B-doped molecules is amongst the deepest ones reported for single B-doped PAHs. In addition, when compared to the other two B-fused boraolympicene analogues having a non-fully-fused boron atom in their molecular skeletons (5-boraolympicene: −3.14 eV, 6-boraolympicene: −2.74 eV),18 our 11a-boraolympicenes showed much lower LUMO energy levels, thus suggesting the great advantages of using the fully-fused B-doping strategy for the construction of low-lying LUMO materials.
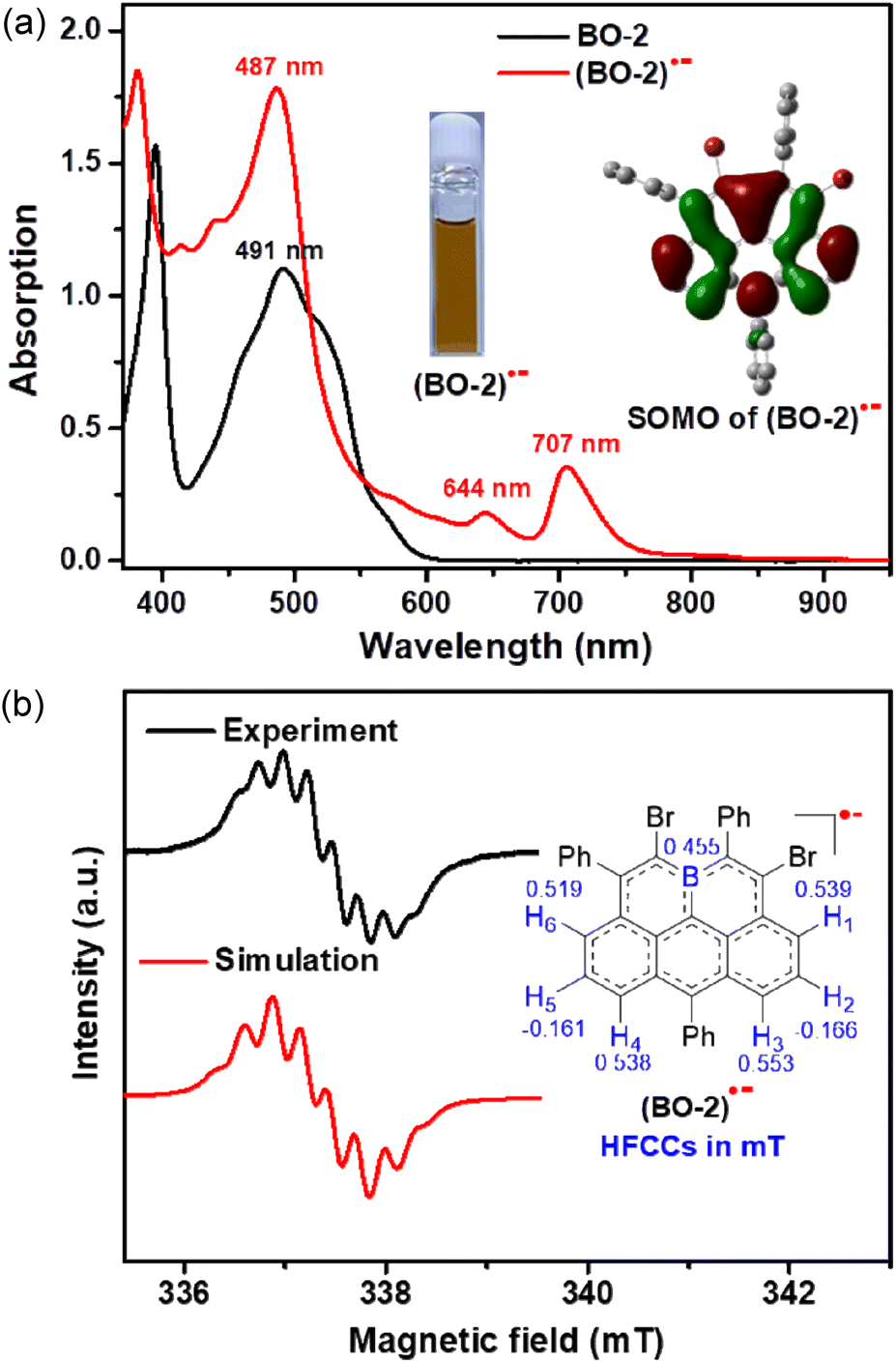 | ||
| Fig. 4 (a) UV-vis-NIR absorption spectra of BO-2 and (BO-2)˙− in DCM (10−5 M). Inset: photo image of (BO-2)˙− in DCM and DFT calculated SOMO distribution (UB3LYP/6-31G(d,p)). (b) Observed and simulated ESR spectra for (BO-2)˙− in DCM at 293 K with hyperfine coupling constants (HFCCs). | ||
In view of their strong electron-accepting character and reversible reduction transitions, the radical anions of these boraolympicenes, which are isoelectronic to the carbonaceous olympicenyl charge-neutral radicals, are expected to be accessible by means of controlled chemical reduction for providing valuable insights into the electron disturbing effect of the boron atom fully-fused in the zigzag-edged PAHs. Therefore, with the 11-position kinetically protected BO-2 as the model compound, we successfully obtained radical anion (BO-2)˙− by using cobaltocene (CoCp2) as the one-electron reducing agent. (BO-2)˙− was stable without detectable spectral variations for one week when kept in a glovebox, but extremely sensitive to air and moisture. Compared to its parent neutral molecule, (BO-2)˙− exhibited two extra low-energy absorptions at λmax = 644 nm and 707 nm, which can be ascribed to the SOMO-associated electron transitions according to the TD-DFT calculated at the UPBEPBE/6-31G(d,p) level19 (Fig. S10†). These two low-energy absorptions at λmax = 644 nm and 707 nm of (BO-2)˙− resemble closely to the absorption bands of the carbon-based isoelectronic olympicenyl radicals,12 thus indicating the similar electronic structures of both (bora)olympicenyl species.
The X-band electron spin resonance (ESR) spectrum of (BO-2)˙− in DCM solution was then recorded, which afforded a well-resolved multiline curve with hyperfine splitting and a g-tensor (ge) determined to be 2.0042 (Fig. 4b), pointing to the formation of open-shell species. The ge value is larger than those of the localized carbonic radical (∼2.002),20 but slightly smaller than those of the localized boron radical (∼2.005),21 possibly arising from the delocalization of the unpaired electron in the conjugated system. Due to the asymmetric molecular architecture originating from the irregular pattern of substituents, the spectral simulation afforded six sets of protons together with one boron atom hyperfine coupling constants (HFCCs, Fig. 4b), suggesting that both protons and boron atoms have contributed to the splitting of the ESR curve. Specifically, H1/H3/H4/H6 have relatively larger HFCC values (0.539 mT for H1, 0.553 mT for H3, 0.538 mT for H4, and 0.519 mT for H6) than H2/H5 (−0.166 mT for H2, −0.161 mT for H5). The observed HFCCs of (BO-2)˙− also correlated well with its calculated Mulliken spin density (MSD) distributions (Fig. 5a), which showed that B/C1/C3/C4/C6 have relatively large MSD values compared to that of C2/C5. MSD calculations also disclosed a well-delocalized spin over the whole B-doped π-framework, in which the 6-position has the largest MSD value of 0.38, similar to the carbonaceous olympicenyl radicals. In addition, by comparing the relative portions of the MSD value at the B-doped 11a-position to that at the 6-positions between the reduced boraolympicene (BO-2)˙− (0.15/0.38) and its carbon-based isoelectronic counterpart (0.13/0.44), it can be found that relatively higher spin densities were located at the 11a-position of (BO-2)˙− when compared to the carbonaceous olympicenyl radicals, which should be ascribed to the high electron-withdrawing nature of the B atom and the strong p-π conjugation in the B-doped compounds. The high spin density of the 11a-position in (BO-2)˙− can also be reflected by electrostatic surface potentials (ESP, Fig. 5b), which clearly showed a more negative charge distribution of the B atom when compared to other sites in the π-skeleton.
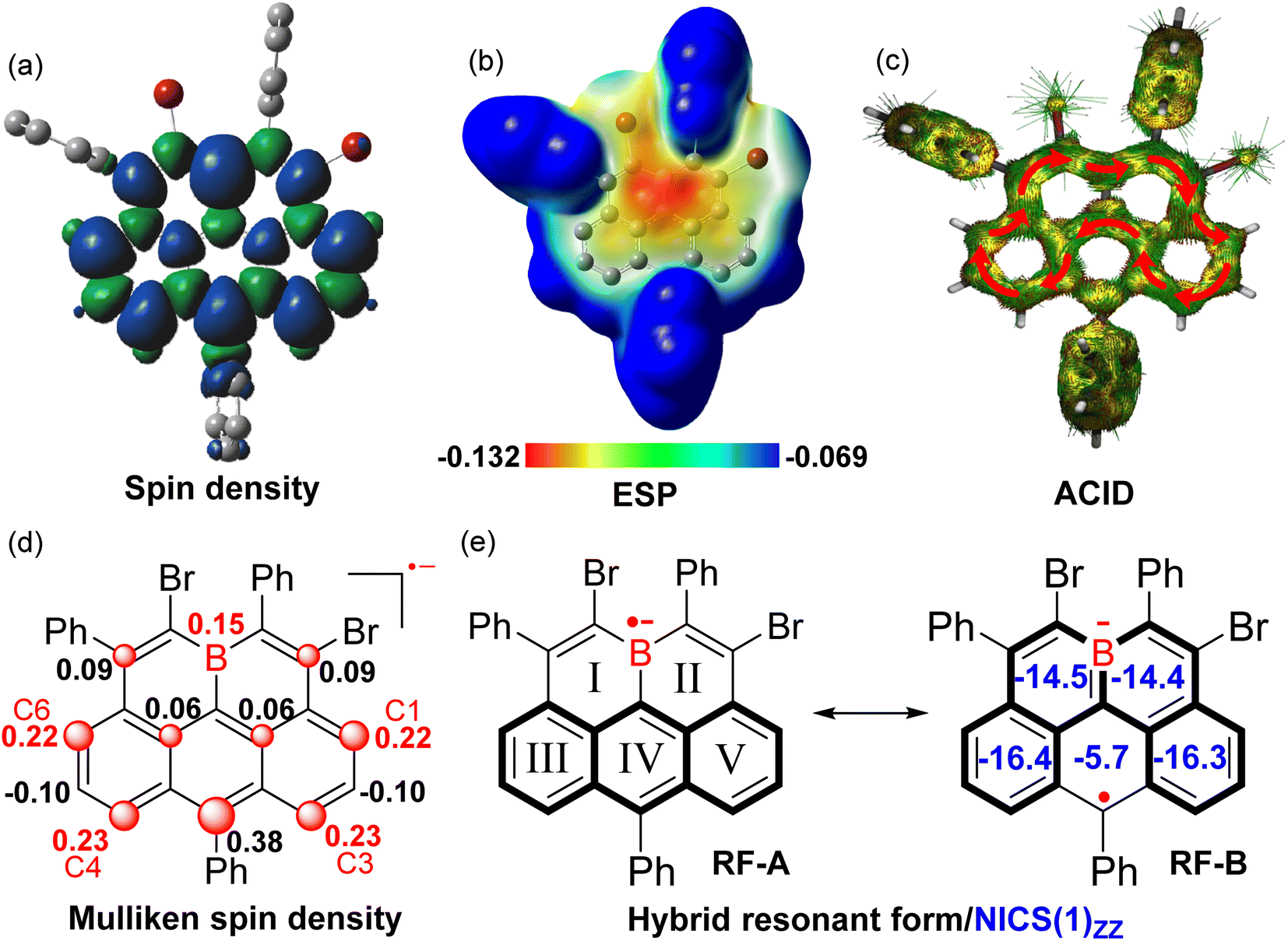 | ||
| Fig. 5 Computed spin density distribution (a) and ESP map (b) of (BO-2)˙−. (c) ACID plots of (BO-2)˙− (isovalue = 0.02). (d) Selected MSD distribution (>0.05) on the molecular backbone (BO-2)˙−. (e) The hybrid resonant forms of (BO-2)˙− and calculated NICS(1)ZZ values of the rings. | ||
To gain more insight into the effect of reduction on the electronic structure evolution upon reduction, NICS values of (BO-2)˙− were further calculated (Fig. 5e). The results revealed that two B-containing six-membered rings (I and II) in (BO-2)˙− turned out to be strongly aromatic compared to its parent neutral molecule (NICS(1)zz = −14.5/−14.4 ppm in (BO-2)˙−vs. NICS(1)zz = +1.1/2.0 ppm in BO-2), suggesting that π-electrons were rearranged from a C–B single bond character in the neutral state to the corresponding borataalkene-like CB anion subunit upon the reduction process. In contrast, ring IV becomes poorly aromatic (NICS(1)zz = −5.7 vs. −22.1 ppm) in (BO-2)˙− compared with the parent BO-2, due to the large spin density distribution at the 6-site. Anisotropy of the induced current density (ACID) of (BO-2)˙− shows a similar clockwise diatropic ring current compared to carbonous olympicenyl radicals, which flew along the periphery of the [4]helicene subunit containing ring I/II/III/V and bypassed the ring IV. Therefore, given the high spin density at the 6-position as well as the aromaticity variations within the rings in the B-doped pentacyclic system, it can be speculated that the major resonant form (RF) of (BO-2)˙− is best described as the form of RF-B, with a greater tendency to become aromatic Borabenzene moieties, rather than aromatic anthracene unit-based RF-A.
To further investigate the bonding character between C and B atoms in the boron-doped anion radical (BO-2)˙−, Fourier transform infrared (FT-IR) measurements of BO-2 and the in situ generated (BO-2)˙− are performed (Fig. S13 and S14†). While the neutral BO-2 (having three C–B bonds) exhibited an intense stretching peak at 1021 cm−1 which can be assigned to the C–B bond stretching,22 in sharp contrast, this stretching became very weak and slightly shifted to a higher frequency of 1023 cm−1 for (BO-2)˙−, thus revealing the poor contribution of C–B bond character in the reduced species. This in fact correlated well with NICS results which have shown that the two borabenzene rings in (BO-2)˙− are more likely aromatic with significant CB character. Indeed, (BO-2)˙− does exhibited a higher stretching frequency and a broad peak appears at 1073 cm−1, which can be reasonably ascribed to the CB bond stretching.23 In addition, the shift from C–B stretching (1023 cm−1) to CB stretching (1073 cm−1) was also closely resembling to the stretching changes as observed in similar cases as reported in the literature.23 To a certain degree, the stable borataalkene-like CB anion substructure,24 which is an isoelectronic structure to the CC motif, together with the recovered aromaticity of two borabenzene rings upon the reduction process, increases the molecular stability compared to the boron radicals. Besides, the IR spectrum of BO-2 showed an intense stretching frequency at 1719 cm−1, which can be assigned to the CC bond stretch. This stretching frequency is slightly higher than the reported CC bond stretch (1600 cm−1–1700 cm−1),25 probably due to the electron-withdrawing character of B atoms according to Hook's Law.26 These findings agree well with the observed alkene-B-alkene motifs in its single-crystal structures. However, this typical CC bond stretching is extremely weak in (BO-2)˙−, thus further confirming that the electronic structure evolution from BO-2 to (BO-2)˙− in which the character of the CC bond was significantly weakened along with the recovery of the aromaticity in rings I and II upon the radical anion formation.
Conclusions
In summary, by devising a novel mild one-pot triply borylation protocol, a class of planar boraolympicenes featuring boron-doping at their concave 11a-positions were successfully synthesized. Thanks to the strong p–π conjugation between the vacant boron pz orbitals and their π-systems, these boraolympicenes not only possessed various intriguing electronic features, such as intense visible-to-NIR absorption and low-lying LUMO energy levels (∼−3.8 eV), but also showed excellent chemical stability against air and moisture despite having no bulky protecting groups, thus allowing further post-modification on the peripheral substituents to give homologues in high yields. Notably, depending on the size of their 6-position substituents, those boraolympicenes also exhibited distinct molecular stacking motifs from a trimeric π-stacked structure for BO-1 to a dimeric non-π-stacking structure in their one-dimensional infinite π-stacking columns due to the variation of their intermolecular close contacts, which is completely different from the packing behaviors of the recently reported carbonaceous olympicenyl radical. Finally, apart from the neutral derivatives, our approach also provides access to the radical anion (BO-2)˙−, which can be prepared via one-electron reduction of BO-2 with CoCp2 and well-characterized by UV-vis-NIR absorption, ESR, and IR spectroscopy. The B-embedded olympicenyl radical showed reasonable stability under inert conditions due to the preferable borataalkene-containing resonant form. We believe that the high stability, low-lying LUMOs, wide-range absorbance, and regulable close intermolecular contacts/packing will lead to promising applications of these novel hetero PAHs in organic electronics. Further structural modulation of BOs and investigation of their electron-conducting behaviors are currently in progress.Data availability
Thanks, additional experimental or computational data have summarized in ESI.†Author contributions
J. G., K. Z. conducted all the experiments. Y. W. and W. X. helped to analyze the data. H. W. solved and refined the X-ray structures. Z. Z. conceived and supervised the project. The manuscript was written by J. G., K. Y. and Z. Z. All authors approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (51922039 and 22005133), the Shenzhen Science and Technology Program (Grant No. RCJC20200714114434015), Science and Technology Innovation Program of Hunan Province (No. 2020RC5033), National Key Research and Development Program of China (No. 2020YFC1807302) and the China Postdoctoral Science Foundation (2022M711107) for financial support.Notes and references
- (a) A. Narita, X. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC; (b) Y. Gu, Z. Qiu and K. Müllen, J. Am. Chem. Soc., 2022, 144, 11499–11524 CrossRef CAS PubMed; (c) Z. Liu, S. Fu, X. Liu, A. Narita, P. Samorì, M. Bonn and H. I. Wang, Adv. Sci., 2022, 9, 2106055 CrossRef PubMed.
- (a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed; (b) A. Borissov, Y. K. Maurya, L. Moshniaha, W. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS PubMed; (c) Y. Wang, S. Qiu, S. Xie, L. Zhou, Y. Hong, J. Chang, J. Wu and Z. Zeng, J. Am. Chem. Soc., 2019, 141, 2169–2176 CrossRef CAS PubMed; (d) T. Luo, Y. Wang, J. Hao, P. Chen, Y. Hu, B. Chen, J. Zhang, K. Yang and Z. Zeng, Angew. Chem., Int. Ed., 2023, 62, e202214653 CAS; (e) Z. Li, Y. Tang, J. Guo, J. Zhang, M. Deng, W. Xiao, F. Li, Y. Yao, S. Xie, K. Yang and Z. Zeng, Chem, 2023, 9 DOI:10.1016/j.chempr.2023.01.019.
- (a) C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed; (b) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC; (c) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed; (d) X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed.
- (a) B. R. Sathe, X. Zou and T. Asefa, Catal. Sci. Technol., 2014, 4, 2023–2030 RSC; (b) T. V. Tam, S. G. Kang, K. F. Babu, E.-S. Oh, S. G. Lee and W. M. Choi, J. Mater. Chem. A, 2017, 5, 10537–10543 RSC; (c) J. Chen, B. Gao, X. Feng, W. Meng and H. Du, Org. Lett., 2021, 23, 8565–8569 CrossRef CAS PubMed.
- (a) T. Kushida, S. Shirai, N. Ando, T. Okamoto, H. Ishii, H. Matsui, M. Yamagishi, T. Uemura, J. Tsurumi, S. Watanabe, J. Takeya and S. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 14336–14339 CrossRef CAS PubMed; (b) F. Zhuang, Z. Sun, Z. Yao, Q. Chen, Z. Huang, J. Yang, J. Wang and J. Pei, Angew. Chem., Int. Ed., 2019, 58, 10708–10712 CrossRef CAS PubMed; (c) W. Li, C. Du, X. Chen, L. Fu, R. Gao, Z. Yao, J. Wang, W. Hu, J. Pei and X. Wang, Angew. Chem., Int. Ed., 2022, e202201464 CAS; (d) W. Sun, J. Guo, Z. Fan, L. Yuan, K. Ye, C. Dou and Y. Wang, Angew. Chem., Int. Ed., 2022, e202209271 CAS.
- (a) K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 1195–1198 CrossRef CAS PubMed; (b) A. John, M. Bolte, H.-W. Lerner, G. Meng, S. Wang, T. Peng and M. Wagner, J. Mater. Chem. C, 2018, 6, 10881–10887 RSC; (c) X. Liang, Z. Yan, H. Han, Z. Wu, Y. Zheng, H. Meng, J. Zuo and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 11316–11320 CrossRef CAS PubMed; (d) P. Qiang, Z. Sun, M. Wan, X. Wang, P. Thiruvengadam, C. Bing, W. Wei, W. Zhu, D. Wu and F. Zhang, Org. Lett., 2019, 21, 4575–4579 CrossRef CAS PubMed; (e) S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bässler, D. Beljonne, M. Buck, Y. Olivier, A. Köhler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef CAS PubMed; (f) J. U. Kim, I. S. Park, C.-Y. Chan, M. Tanaka, Y. Tsuchiya, H. Nakanotani and C. Adachi, Nat. Commun., 2020, 11, 1765–1772 CrossRef CAS PubMed; (g) M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468–19472 CrossRef CAS PubMed; (h) Y. Xia, M. Zhang, S. Ren, J. Song, J. Ye, M. G. Humphrey, C. Zheng, K. Wang and X. Zhang, Org. Lett., 2020, 22, 7942–7946 CrossRef CAS PubMed; (i) J. Kashida, Y. Shoji, Y. Ikabata, H. Taka, H. Sakai, T. Hasobe, H. Nakai and T. Fukushima, Angew. Chem., Int. Ed., 2021, 60, 23812–23818 CrossRef CAS PubMed; (j) M. Liu, M. Cui, L. Zhang, Y. Guo, X. Xu, W. Li, Y. Li, B. Zhen, X. Wu and X. Liu, Org. Chem. Front., 2022, 9, 3328–3334 RSC; (k) S. Uemura, S. Oda, M. Hayakawa, R. Kawasumi, N. Ikeda, Y.-T. Lee, C.-Y. Chan, Y. Tsuchiya, C. Adachi and T. Hatakeyama, J. Am. Chem. Soc., 2023, 145, 1505–1511 CrossRef CAS PubMed; (l) G. Meng, H. Dai, J. Zhou, T. Huang, X. Zeng, Q. Wang, X. Wang, Y. Zhang, T. Fan, D. Yang, D. Ma, D. Zhang and L. Duan, Chem. Sci., 2023, 14, 979–986 RSC.
- (a) S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130–9133 CrossRef CAS PubMed; (b) K. Matsuo, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 12580–12583 CrossRef CAS PubMed; (c) K. Matsuo, S. Saito and S. Yamaguchi, Angew. Chem., Int. Ed., 2016, 55, 11984–11988 CrossRef CAS PubMed; (d) N. Ando, T. Yamada, H. Narita, N. N. Oehlmann, M. Wagner and S. Yamaguchi, J. Am. Chem. Soc., 2021, 143, 9944–9951 CrossRef CAS PubMed; (e) H. Narita, H. Choi, M. Ito, N. Ando, S. Ogi and S. Yamaguchi, Chem. Sci., 2022, 13, 1484–1491 RSC.
- K. Schickedanz, T. Trageser, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2015, 51, 15808–15810 RSC.
- F. Miyamoto, S. Nakatsuka, K. Yamada, K. Nakayama and T. Hatakeyama, Org. Lett., 2015, 17, 6158–6161 CrossRef CAS PubMed.
- A. Escande, D. L. Crossley, J. Cid, I. A. Cade, I. Vitorica-Yrezabal and M. J. Ingleson, Dalton Trans., 2016, 45, 17160–17167 RSC.
- (a) D. H. Reid and W. Bonthrone, J. Chem. Soc., 1965, 5920–5926 RSC; (b) A. H. Reddoch and D. H. Paskovich, Chem. Phys. Lett., 1969, 3, 351–352 CrossRef CAS; (c) I. C. Lewis and L. S. Singer, Magn. Reson. Chem., 1985, 23, 698–704 CrossRef CAS; (d) A. Mistry, B. Moreton, B. Schuler, F. Mohn, G. Meyer, L. Gross, A. Williams, P. Scott, G. Costantini and D. J. Fox, Chem. − Eur. J., 2015, 21, 2011–2018 CrossRef CAS PubMed.
- Q. Xiang, J. Guo, J. Xu, S. Ding, Z. Li, G. Li, H. Phan, Y. Gu, Y. Dang, Z. Xu, Z. Gong, W. Hu, Z. Zeng, J. Wu and Z. Sun, J. Am. Chem. Soc., 2020, 142, 11022–11031 CrossRef CAS PubMed.
- (a) S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Chem. Soc. Rev., 2020, 49, 4564–4591 RSC; (b) D. L. Crossley, R. J. Kahan, S. Endres, A. J. Warner, R. A. Smith, J. Cid, J. J. Dunsford, J. E. Jones, I. Vitorica-Yrezabal and M. J. Ingleson, Chem. Sci., 2017, 8, 7969–7977 RSC.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed.
- F. Zettler, H. D. Hausen and H. Hess, J. Organomet. Chem., 1974, 72, 157–162 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- (a) V. M. Hertz, H.-W. Lerner and M. Wagner, Org. Lett., 2015, 17, 5240–5243 CrossRef CAS PubMed; (b) K. Yuan, R. J. Kahan, C. Si, A. Williams, S. Kirschner, M. Uzelac, E. Zysman-Colman and M. J. Ingleson, Chem. Sci., 2020, 11, 3258–3267 RSC.
- G. Liao, X. Chen, Y. Qiao, K. Liu, N. Wang, P. Chen and X. Yin, Org. Lett., 2021, 23, 5836–5841 CrossRef CAS PubMed.
- (a) S. Qiu, C. Wang, S. Xie, X. Huang, L. Chen, Y. Zhao and Z. Zeng, Chem. Commun., 2018, 54, 11383–11386 RSC; (b) J. Guo, C. Zhou, S. Xie, S. Luo, T. Y. Gopalakrishna, Z. Sun, J. Jouha, J. Wu and Z. Zeng, Chem. Mater., 2020, 32, 5927–5936 CrossRef CAS; (c) B. Li, C. Yang, X. Wang, G. Li, W. Peng, H. Xiao, S. Luo, S. Xie, J. Wu and Z. Zeng, Angew. Chem., Int. Ed., 2021, 60, 19790–19796 CrossRef CAS PubMed; (d) J. Guo, Z. Li, J. Zhang, B. Li, Y. Liang, Y. Wang, S. Xie, H. Phan, T. S. Herng, J. Ding, J. Wu, B. Z. Tang and Z. Zeng, CCS Chem., 2021, 3, 399–407 Search PubMed; (e) Y. Wang, Y. Huang, T. Huang, J. Zhang, T. Luo, Y. Ni, B. Li, S. Xie and Z. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202200855 CAS.
- (a) P. P. Power, Chem. Rev., 2003, 103, 789–809 CrossRef CAS PubMed; (b) H. Wei, Y. Liu, T. Y. Gopalakrishna, H. Phan, X. Huang, L. Bao, J. Guo, J. Zhou, S. Luo, J. Wu and Z. Zeng, J. Am. Chem. Soc., 2017, 139, 15760–15767 CrossRef CAS PubMed.
- (a) E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef CAS PubMed; (b) T. A. Bartholome, J. J. Martinez, A. Kaur, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Organometallics, 2021, 40, 1966–1973 CrossRef CAS.
- T. A. Bartholome, A. Kaur, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Angew. Chem., Int. Ed., 2020, 59, 11470–11476 CrossRef CAS PubMed.
- (a) G. C. Fu, Adv. Organomet. Chem., 2001, 47, 101–119 CrossRef CAS; (b) P. A. Brown, C. D. Martin and K. L. Shuford, Phys. Chem. Chem. Phys., 2019, 21, 18458–18466 RSC.
- (a) C. F. Zinola, J. Rodríguez, M. C. Arévalo and E. Pastor, J. Electroanal. Chem., 2005, 585, 230–239 CrossRef CAS; (b) B. Jin, P. Liu, Y. Wang, Z. Zhang, Y. Tian, J. Yang, S. Zhang and F. Cheng, J. Phys. Chem. B, 2007, 111, 1517–1522 CrossRef CAS PubMed.
- A. Zhang, W. Jiang and Z. Wang, Angew. Chem., Int. Ed., 2020, 59, 752–757 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1905586 and 2235180. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00342f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |