
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn unusual autocatalysis with an air-stable Pd complex to promote enantioselective synthesis of Si-stereogenic enynes†
Fang-Ying
Ling
a,
Fei
Ye
a,
Xiao-Jun
Fang
a,
Xiao-Hua
Zhou
a,
Wei-Sheng
Huang
a,
Zheng
Xu
a and
Li-Wen
Xu‡
 *ab
*ab
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Key Laboratory of Organosilicon Material Technology of Zhejiang Province, Hangzhou Normal University, No. 2318, Yuhangtang Road, Hangzhou 311121, P. R. China. E-mail: liwenxu@hznu.edu.cn
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute and Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, P. R. China
First published on 21st December 2022
Abstract
Given the powerful potential of chiral-at-silicon chemistry, enantioselective synthesis of Si-stereogenic centers has attracted substantial research interest in recent years. However, the catalytic asymmetric synthesis of Si-stereogenic organosilicon compounds remains an appealing venture and is a challenging subject because of the difficulty in achieving high reactivity and stereoselectivity for “silicon-center” transformations. Herein, we disclose a highly enantioselective palladium-catalyzed hydrosilylation of 1,3-diynes with dihydrosilanes, which enables the facile preparation of Si-stereogenic enynes and an enyne-linked chiral polymer (polyenyne) in good yields and excellent ees (up to >99%) by desymmetrization. The unusual stereoselectivity in this reaction is achieved by precisely controlling the steric hindrance and electronic effect of the newly developed chiral ligands, resulting in a wide range of chiral silanes and a Si-containing polymer bearing a Si-stereogenic center which is otherwise difficult to access. The key to the high enantioselectivity relies on catalyst aggregation-induced non-covalent interaction, which exerts a remarkably positive influence on the Si–H bond activation and enhancement of enantioselectivity, in which the palladium/P-ligand complex was proved to be air-stable and moisture-insensitive in this reaction.
Introduction
Organosilicon compounds belong to one of the most important class of molecules and are indispensable building blocks in synthetic chemistry, which have been widely used in the synthesis of industrial and agricultural chemicals, biologically active molecules and silicon-containing polymers as well as functional materials.1 The related synthetic methods not only have the potential to promote the development of new chiral Si-containing drugs and advanced materials,2 but also to greatly enrich the strategy of silicon-mediated organic synthesis (SiMOS).3 Especially, the catalytic asymmetric synthesis of chiral organosilicon compounds bearing a silicon-stereogenic center have attracted more and more attention in recent years.4 However, since there is no chiral source related to silicon in nature, the synthesis of chiral organosilicon compounds containing Si-stereogenic centers is recognized as one of the most challenging and continuously researched problems in past decades.5 Traditionally, the construction of Si-stereogenic centers was mainly completed by kinetic resolution-based transformations of chlorosilanes.5,6 In the past ten years, the strategy of desymmetrization through Si–C or Si–H bond functionalization of silacycles and hydrosilanes has gradually become one of the mainstream methods,4,7 including the ring expansion or ring opening of silacyclobutanes,8 C–H silylation reaction,9 Si–H carbene insertion reaction,10 hydrosilylation reaction11 and other reactions.12 In the past years, we also reported catalytic asymmetric hydrosilylation and other strategies to access Si-stereogenic compounds containing different skeletons.13Notably, hydrosilylation is one of the most important reactions to convert hydrosilanes into chiral organosilicon compounds, including silicon-stereogenic silanes (Fig. 1a).11,14 However, there are still some shortcomings in the previously reported methods including: (1) most organosilanes bearing a silicon-stereogenic center are mainly silicon-containing heterocyclic compounds and the synthesis of non-cyclic silicon-stereogenic silanes remains a difficult task to date. For example, neither the synthesis of linear silicon-containing diborylalkenes bearing a silicone stereocenter or the desymmetric construction of silicon-stereogenic silylmethanols was successful in our group.15 (2) A high level of stereoselectivity and regio- or chemoselectivity cannot be obtained because of the poor compatibility between silicon-center chirality and functional groups.16 Considering the importance of the catalytic construction of functional silanes bearing a silicon-stereogenic center, we expect to further develop new reaction systems of catalytic asymmetric hydrosilylation, which can be applied to more types of functional alkynes. Notably, there is no report on the catalytic asymmetric synthesis of silicon-stereogenic enynes via hydrosilylation of 1,3-diynes.17 We now hypothesize that the development of a chiral ligand with a large cavity might discriminate two proximal Si–H bonds in prochiral hydrosilanes through oxidative addition of a metal species, allowing enantioselective synthesis of silicon-stereogenic silanes.
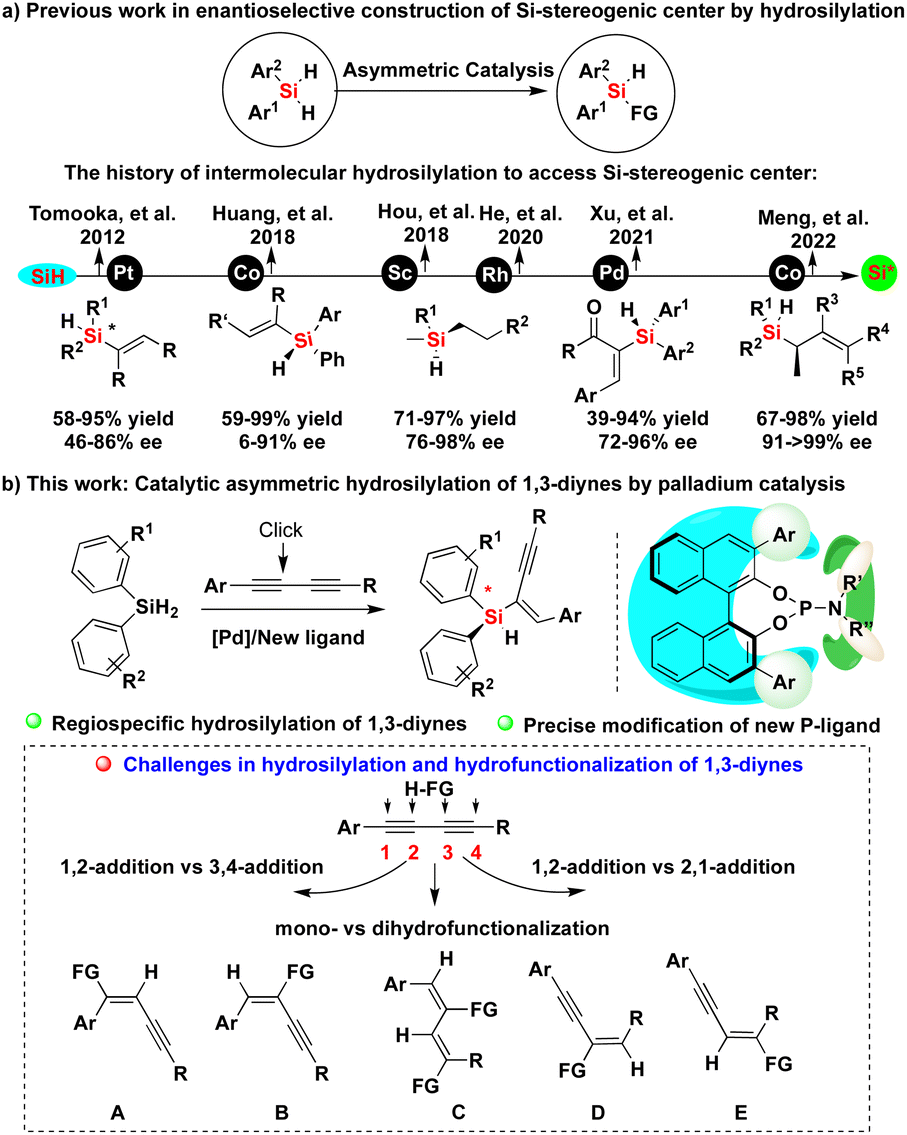 | ||
| Fig. 1 Introducing regioselectivity for (a) hydrosilylation of 1,3-diynes and the history as well as (b) this work of catalytic asymmetric hydrosilylation to access silicon-stereogenic center. | ||
Herein, we show our efforts in the development of enantioselective palladium-catalyzed hydrosilylation of diynes with the aid of a newly developed chiral P-ligand (Fig. 1b). On the basis of precise modification of a novel phosphoramidite ligand and its structure–activity relationship, this work has established a new method for the synthesis of silicon-stereogenic enynes with good regio- and enantioselectivity, solving the problem of controlling the regioselectivity due to the existence of four different reactive sites (the lower scheme of Fig. 1b).17a Importantly, the chiral BINOL-derived phosphoramidite ligand employed in this reaction acts cooperatively with the product, Si-stereogenic silane, to exhibit a remarkably positive influence on the Si–H bond activation and enhancement of enantioselectivity.18
Results and discussion
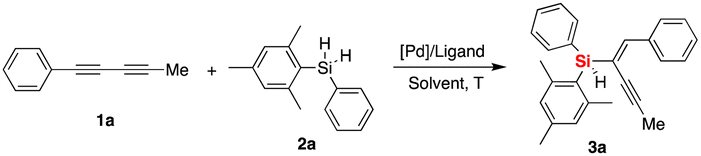
Initially, we investigated the asymmetric palladium-catalyzed hydrosilylation of unsymmetric 1,3-diyne 1a with hydrosilane 2a in the presence of a series of phosphoramidite ligands. The model reaction was performed first using Pd2(dba)3 as a precatalyst and L1 as the chiral ligand in dichloromethane (DCM) at room temperature (25 °C), and it was proved to be operationally simple and to possibly realize stereoselective control with promising enantioselectivity (74% ee) and moderate yield (50%) as well as excellent regioselectivity (97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 rr). Thus, the exciting finding in the chiral P-ligand controlled Pd-catalyzed desymmetric hydrosilylation of dihydrosilane shown in entry 1 (Table 1) prompted us to evaluate this reaction by applying structurally diverse chiral phosphine and phosphoramidite ligands (see Fig. 2, the chemical structures of L2–L37 were provided in the ESI†). By examining a set of chiral P-ligands and their analogues, we are pleased to find that most P-ligands enabled enantioselective synthesis of chiral enyne 3a bearing a silicon-stereogenic center, albeit with different regio- and enantioselectivity. For example, the TADDOL-derived phosphoramidite ligand L7 and its analogues exhibited inferior regioselectivity and decreased enantioselectivity in comparison to that of L1, which revealed that the BINOL-derived phosphoramidite ligand would be an ideal backbone for the improvement of enantioselective control of Pd-catalyzed asymmetric hydrosilylation. As shown in Fig. 2 and Table S1 of ESI,† we carried out a deep modification of the chemical structure of L1 and synthesized a series of chiral BINOL-derived phosphoramidite ligands by changing the phenyl substituents on the binaphthyl backbone or the two butyl groups on the nitrogen atom to other groups.
:3 rr). Thus, the exciting finding in the chiral P-ligand controlled Pd-catalyzed desymmetric hydrosilylation of dihydrosilane shown in entry 1 (Table 1) prompted us to evaluate this reaction by applying structurally diverse chiral phosphine and phosphoramidite ligands (see Fig. 2, the chemical structures of L2–L37 were provided in the ESI†). By examining a set of chiral P-ligands and their analogues, we are pleased to find that most P-ligands enabled enantioselective synthesis of chiral enyne 3a bearing a silicon-stereogenic center, albeit with different regio- and enantioselectivity. For example, the TADDOL-derived phosphoramidite ligand L7 and its analogues exhibited inferior regioselectivity and decreased enantioselectivity in comparison to that of L1, which revealed that the BINOL-derived phosphoramidite ligand would be an ideal backbone for the improvement of enantioselective control of Pd-catalyzed asymmetric hydrosilylation. As shown in Fig. 2 and Table S1 of ESI,† we carried out a deep modification of the chemical structure of L1 and synthesized a series of chiral BINOL-derived phosphoramidite ligands by changing the phenyl substituents on the binaphthyl backbone or the two butyl groups on the nitrogen atom to other groups.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | T (°C) | Solvent | Yield (%) | rrb | eec (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol), Pd2(dba)3 (2 mol%), chiral ligand (8 mol%), and solvent (2 mL) for 12 h. The data of yield is determined by NMR analysis. All the ligands (L1–L37) are provided in Table S1 of the ESI. b The ratio of regioselectivity (rr) is determined by GC-MS analysis. c The ee values were determined by HPLC using chiral columns. d With Pd(C3H5)2Cl2. e With Pd(OAc)2. f With 1 mol% of Pd2(dba)3 and 4 mol% of chiral ligand. | ||||||
| 1 | L1 | 25 | DCM | 50 | 97:3 |
74 |
| 2 | L7 | 25 | DCM | 24 | 82:18 |
12 |
| 3 | L10 | 25 | DCM | 16 | 88:12 |
32 |
| 4 | L12 | 25 | DCM | NR | — | — |
| 5 | L14 | 25 | DCM | 67 | 96:4 |
72 |
| 6 | L16 | 25 | DCM | 10 | 92:8 |
5 |
| 7 | L31 | 25 | DCM | 60 | 86:14 |
76 |
| 8 | L33 | 25 | DCM | 63 | 96:4 |
90 |
| 9d | L33 | 25 | DCM | 10 | 86:14 |
91 |
| 10e | L33 | 25 | DCM | 56 | 88:12 |
91 |
| 11 | L33 | 25 | THF | 68 | 97:3 |
89 |
| 12 | L33 | 25 | DCE | 60 | 95:5 |
90 |
| 13 | L33 | 25 | CH3CN | 19 | 97:3 |
86 |
| 14 | L33 | 25 | c-Hex | 65 | 98:2 |
91 |
| 15 | L33 | 0 | c-Hex | 74 | 99:1 |
93 |
| 16 | L33 | 40 | c-Hex | 63 | 97:3 |
89 |
| 17f | L33 | 0 | c-Hex | 60 | 99:1 |
93 |
|
||||||
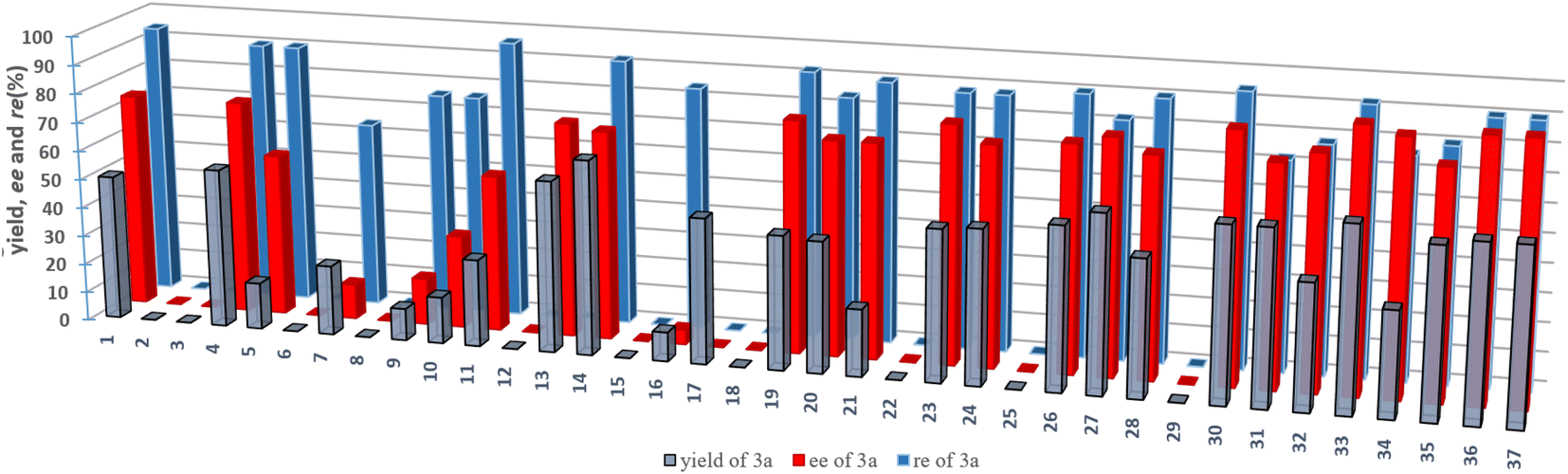 | ||
| Fig. 2 The activity and enantioselective induction of various chiral ligands in the Pd-catalyzed asymmetric hydrosilylation of an unsymmetric 1,3-diyne. The detailed structure of these chiral ligands is provided in the ESI.† | ||
Interestingly, both the aryl substituents and amine moiety played crucial roles in the precise control of the steric repulsion and electronic properties of the P-ligand. The L10 or L16 without phenyl substituents exhibited a poor ability in this reaction because of the low yield and enantioselectivity of 3a (entries 3 and 5). In addition, L12 bearing a silicon-based bulky group (-TMS) inhibited the palladium catalysis (entry 4), which further supported the importance of aromatic interaction in the stereoselective transformation. In addition, the implementation of these chiral P-ligands-controlled Pd-catalyzed asymmetric hydrosilylation presents onerous challenges on the exploration of an effective phosphoramidite ligands.
To circumvent the limitation of desymmetrization in enantioselective Pd-catalyzed hydrosilylation, we continued to explore the modular synthesis of aryl-substituted BINOL-derived phosphoramidite ligands, leading to the determination of the best ligand L33 that promoted the Pd-catalyzed hydrosilylation with excellent regio- and enantioselectivity (96:4 rr and 90% ee). Other P-ligands, such as L31, exhibited an inferior performance in this reaction (entry 7). Unexpectedly and magically, the ligand L25 bearing a silicon-based bulky group at the para-position of the phenyl substituent on the binaphthyl backbone resulted in no reaction at room temperature. It is extremely rare that a palladium complex with this chiral ligand L25 has no catalytic activity due to such a small difference in the Si–C bond of the TMS group and the C–C bond of the t-butyl group on the para-position of the phenyl substituent on the binaphthyl backbone. To look for the relationship between the molecular structure of chiral P-ligands and enantioselectivity, we calculated the ligand-based π-donor orbitals (HOMO) and the high energy anti-bonding orbitals (LUMO). However, the electronic effect of the chiral ligand on enantioselectivity is not very clear (Fig. S1 of ESI†). The t-butyl group at the para-position of the phenyl ring and Bn/Me-substituted amine on the P-ligand manipulates a suitable cavity arising from steric repulsion to affect the enantioselective Pd-catalyzed hydrosilylation.
Encouraged by these aforementioned results, we subsequently investigated the solvent effect and other reaction parameters (entries 9–17 of Table 1), such as palladium salts, temperature and the loading of catalyst or ligand (detailed information is provided in Tables S2–S6 of the ESI†). Finally, it was found that the reaction occurred smoothly in cyclohexane at 0 °C to give the best enantioselectivity and yield (entry 15). It is worth mentioning that the previously reported platinum12g or cobalt12f catalytic system resulted in no reaction, which showed the necessity of developing a new palladium catalyst system for this reaction.
After successfully determining the L33-controlled Pd-catalyzed asymmetric hydrosilylation with optimal reaction conditions, we then examined the substrate scopes for this desymmetric hydrosilylation of diynes 1 and dihydrosilanes 2. We prepared a series of unsymmetric aryl 1,3-diynes bearing electron-withdrawing and electron-donating groups on the phenyl ring. As shown in Scheme 1, the palladium-catalyzed asymmetric hydrosilylation of various 1,3-diynes 1a–1p took place with excellent regio- and enantioselectivity to give the desired silicon-stereogenic enynes 3a–3p (90–97% ee). The regioselectivity is generally high between 98:2 and >99:1 rr, which revealed that the reactivity of two different alkynes on the unsymmetric 1,3-diynes could be distinguished by this Pd/L33 catalyst system. Notably, this reaction could tolerate heterocycle, alkyl chloride, and bromide groups. In fact, it is impossible to isolate the trace side-isomer and thus the chemo- and regioselectivity are almost perfect in this reaction. In addition, we also evaluated the Pd-catalyzed asymmetric hydrosilylation of symmetric 1,3-diynes and found that there is only one regiospecific product for each case. All the diaryl 1,3-diynes proved to be suitable substrates to transform into the corresponding products 3q–3z with moderate to good yields (45–91%) and excellent enantioselectivities (91–95% ee). In the reaction, the stereoselectivity is not affected by the heterocycles or electron-withdrawing substituents. Notably, dialkyl 1,3-diyne could be used in this reaction, which represented as product 3p with quite a high enantioselectivity (96% ee). We also found that the steric repulsion on dihydrosilane is crucial to the enhancement of enantioselectivity. As shown in Scheme 1, the Pd-catalyzed hydrosilylation with ortho-ethyl substituted hydrosilane can achieve perfect desymmetrization because of its optically pure product (>99% ee for product 3aa). Finally, it should be noted that the absolute configuration and chemical structure of chiral 3ab (91% ee) was unambiguously analyzed and confirmed by single-crystal X-ray analysis.19
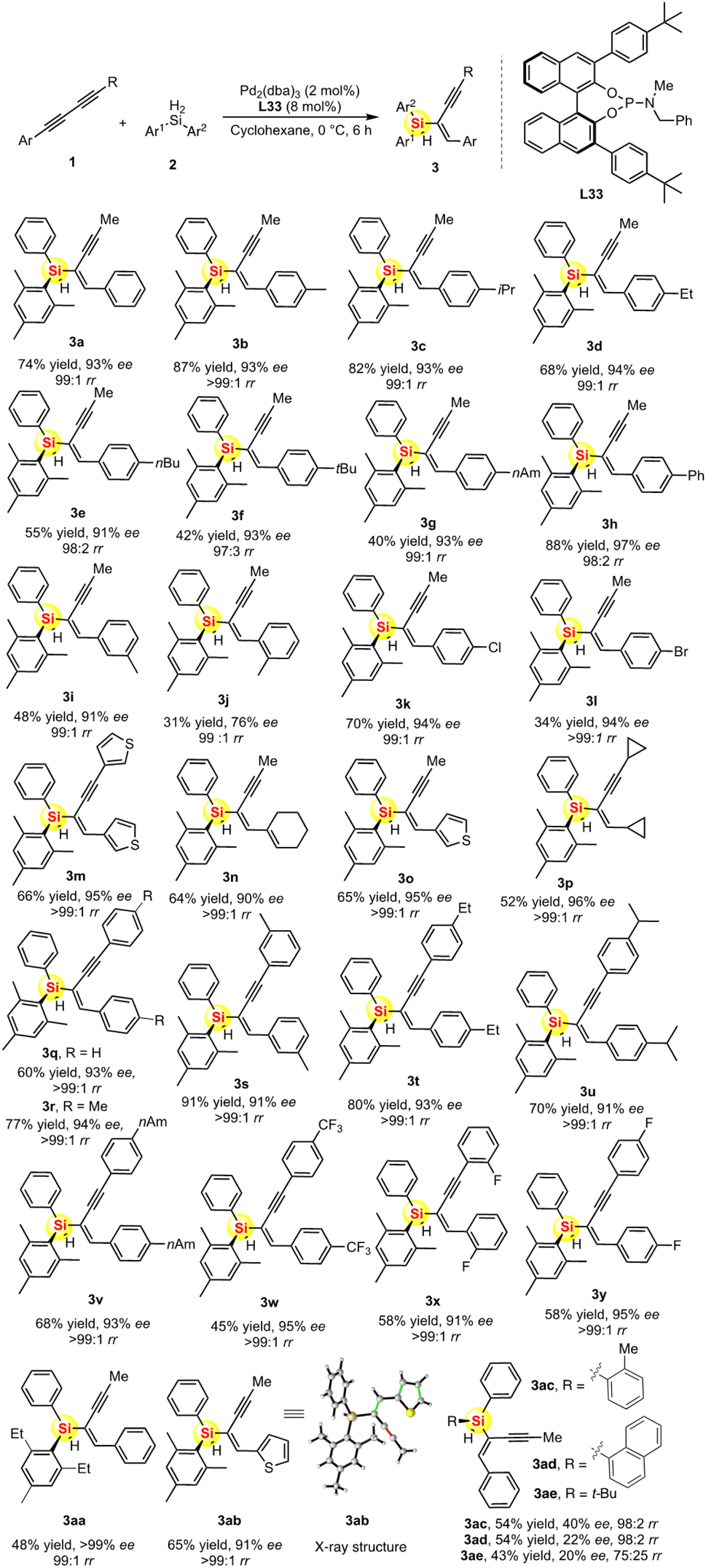 | ||
| Scheme 1 The substrate scope of Pd-catalyzed hydrosilylation of 1,3-diynes to access silicon-stereogenic enynes. | ||
The gram-scale synthesis of silicon-stereogenic enyne 3a was evaluated to support the synthetic potential of this enantioselective Pd-catalyzed hydrosilylation in this reaction, giving the corresponding product in 70% yield and the same excellent enantioselectivity (see eqn (1) of Scheme 2, 93% ee). Moreover, the hydrosilylation reaction was also suitable for the synthesis of drug-like complex molecule 5, affording the desired product bearing an additional silicon-stereogenic center with 83% ee (eqn (2)). In addition, we also checked the downstream transformations of silicon-stereogenic enyne 3a to the synthesis of 6 and 7 by methylation or alcoholysis of the Si–H bond (eqn (3) and (4) of Scheme 2), in which the corresponding products were achieved by maintaining a slightly decreasing point chirality of the silicon center. It should be noted that the highly enantioselective hydrosilylation of diynes inspired us to expand it as a new springboard reaction for the synthesis of a chiral polymer bearing a silicon-stereogenic pendant (Fig. 3). As expected, the spectra analysis confirmed that the newly synthesized chiral Si-containing polyenyne 9 could be easily prepared as an insoluble and solid material in cyclohexane with good yields. We also checked the palladium-catalyzed hydrosilylation of polyalkyne 8 by different chiral P-ligands (S)-L33, (R)-L33, and (S)-L14. The corresponding polyenyne with a Si-stereogenic pendant would be a useful chiral Si-functional material that has been characterized by IR, SEM, TGA, and circular dichroism spectra (see Fig. S13–S16 of the ESI†). For example, the chiral polyenyne bearing a pendant Si-stereogenic center with reverse chiral-at-silicon configuration exhibited an obvious Cotton effect during the circular dichroism (CD) analysis (Fig. 3). The CD spectra showed that the Cotton effect is mainly caused by the fragments of silicon-stereogenic silanes, which is basically consistent with the CD spectrum of Si-stereogenic enyne 3q (Fig. S13 of ESI†).
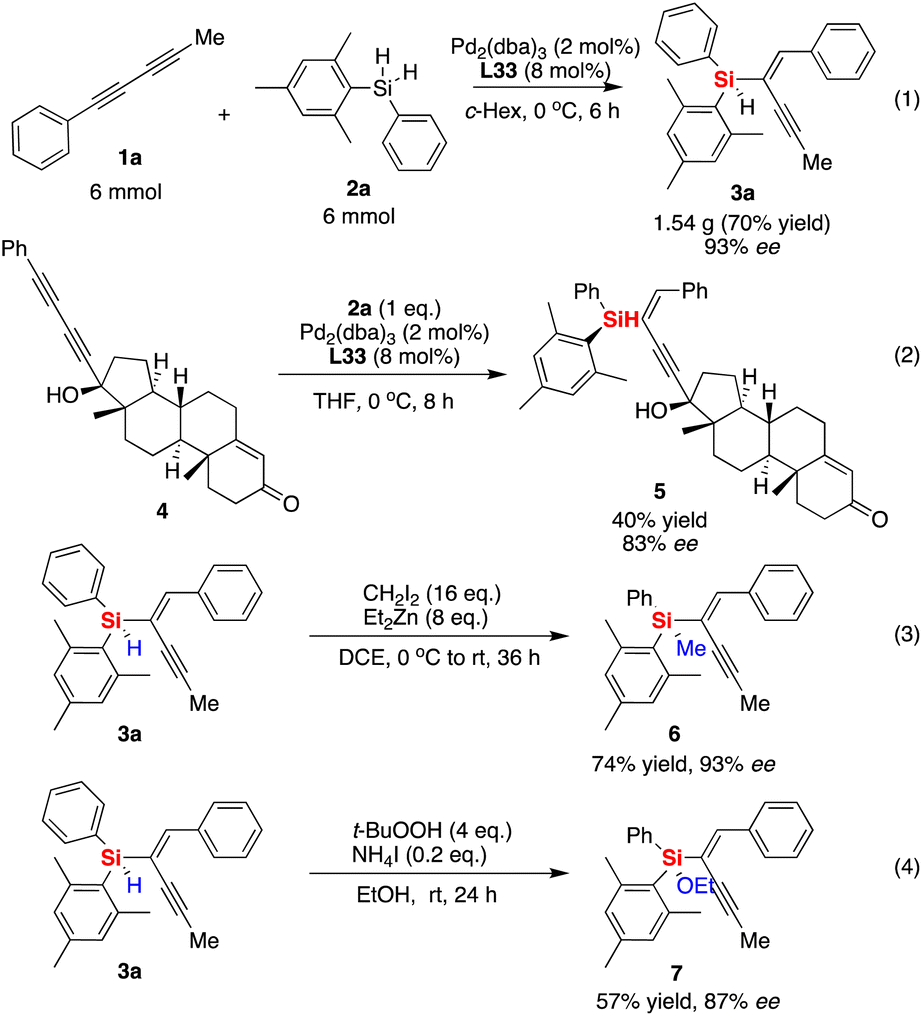 | ||
| Scheme 2 Gram-scale synthesis of silicon-stereogenic enyne 3a and its downstream transformations or synthesis of a drug-like complex molecule and a chiral Si-containing polyenyne. | ||
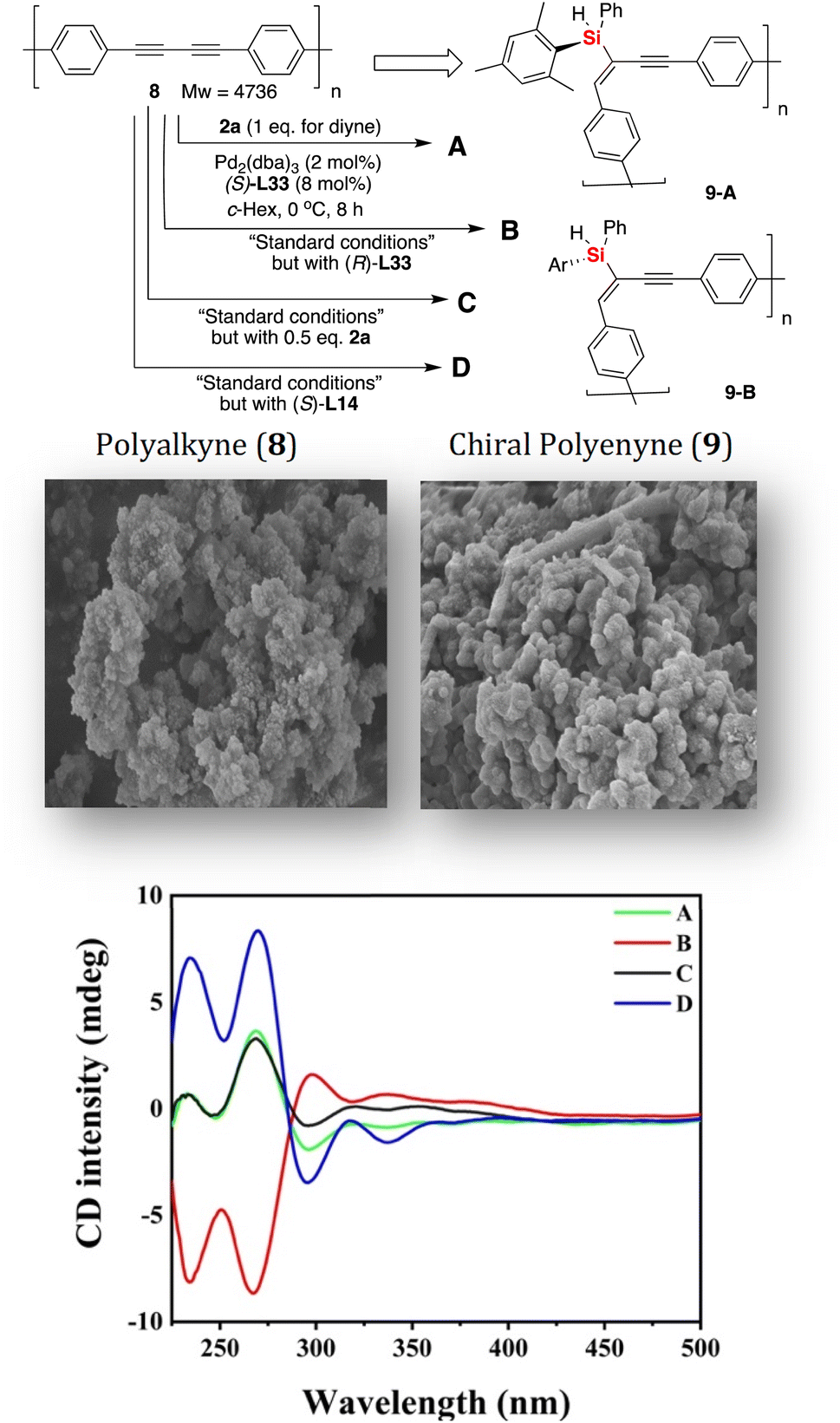 | ||
| Fig. 3 The synthesis of chiral Si-stereogenic polyenyne 9 from polyalkyne and its CD analysis in THF (c = 2 g L−1). | ||
To gain insight into the mechanism of Pd-catalyzed hydrosilylation, we performed detailed kinetic studies that revealed an unusual effect of ligand L33 on the enantioselectivity and conversion of this reaction of 1a with 2a. Indeed, the hydrosilylation reaction was found to be first order in the initial stage under the optimized reaction conditions (see line M1 of Fig. 4a). In sharp contrast, the reaction was accelerated suddenly between 1.5 and 2 hours (the second stage could be recognized as the sudden stage) after the yield exceeded 10% and then occurred slowly until the end of the reaction (the third stage would be the stable stage). In the sudden stage, it is very likely that this reaction features autocatalytic action or formation of a new active Pd species by aggregation of the catalyst with the substrate or product. In order to show the effect of the chiral product on the reaction progress, we use 10 mol% of chiral product 3a as an additive to verify whether it has autocatalytic performance.20 As expected, the Pd-catalyzed hydrosilylation could be accelerated by chiral product 3a due to rapid formation of the corresponding product with about 60% yield in one hour (see line M2 of Fig. 4a). In addition, using (S)-3a as an additive obtained from the reaction with (S)-L33 as the ligand, we investigated (S)-3a-promoted Pd-catalyzed hydrosilylation with (R)-L33 or (R)-L11 as the ligand (Scheme 3). The reaction results showed that the (S)-3a had an important effect on the chiral induction of the Pd catalyst system. For example, when (R)-L33 or (R)-L11 was used as the chiral ligand in this reaction, the ee value desired product should be 58% or 18% respectively, however, better enantioselectivity was observed as 88% ee or 51% ee, which indirectly revealed the possibility of product-involved catalyst aggregation and the existence of autocatalysis in this reaction.
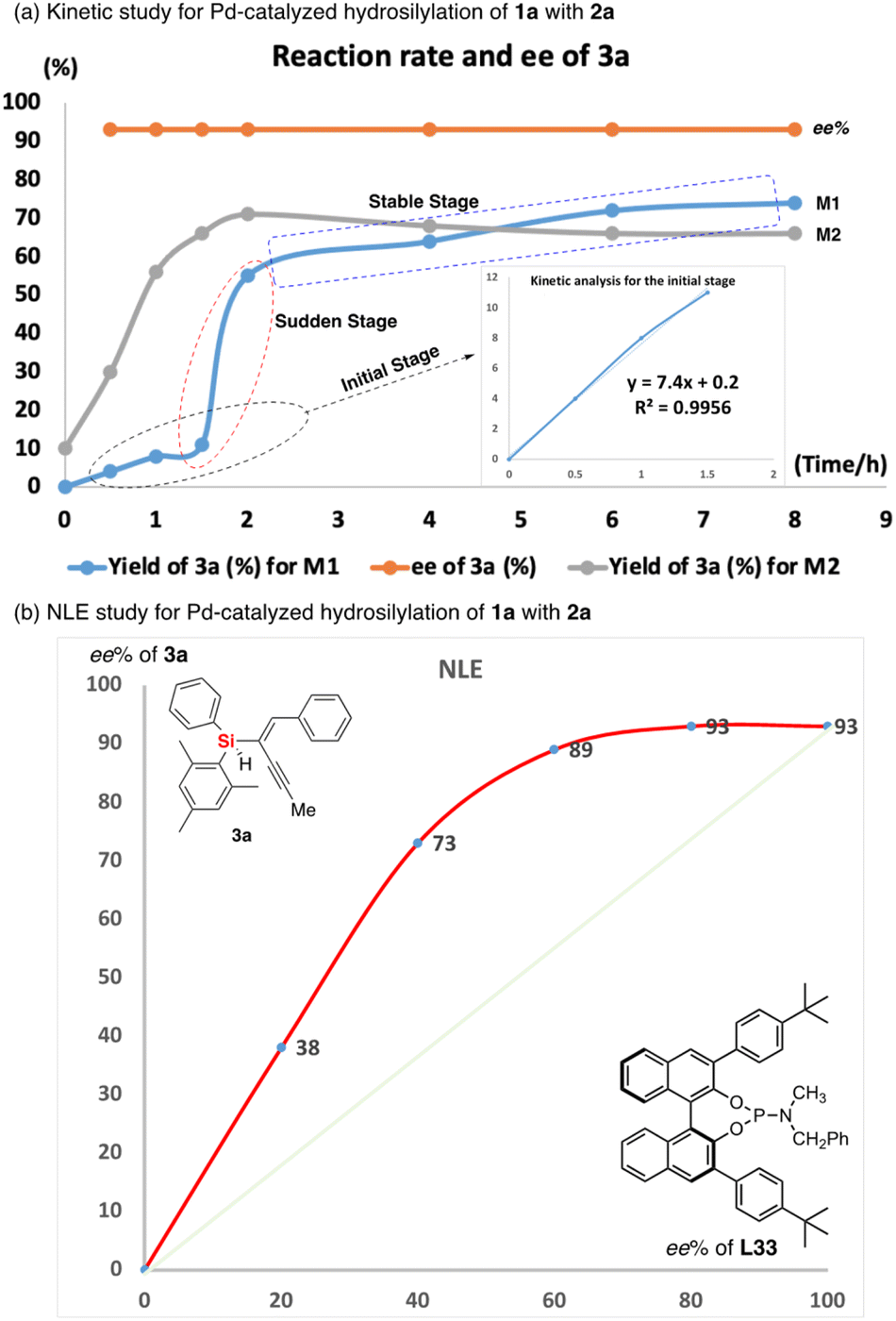 | ||
| Fig. 4 (a) Kinetic analysis for the enantioselective Pd-catalyzed hydrosilylation of 1,3-diyne 1a with 2a. Line M1 is the yield of 3a in this reaction (without additive) and Line M2 is the yield of 3a in the additive (with 10% of product 3a as additive)-assisted hydrosilylation reaction. And (b) non-linear effect (NLE) study for Pd-catalyzed hydrosilylation of 1a with 2a. | ||
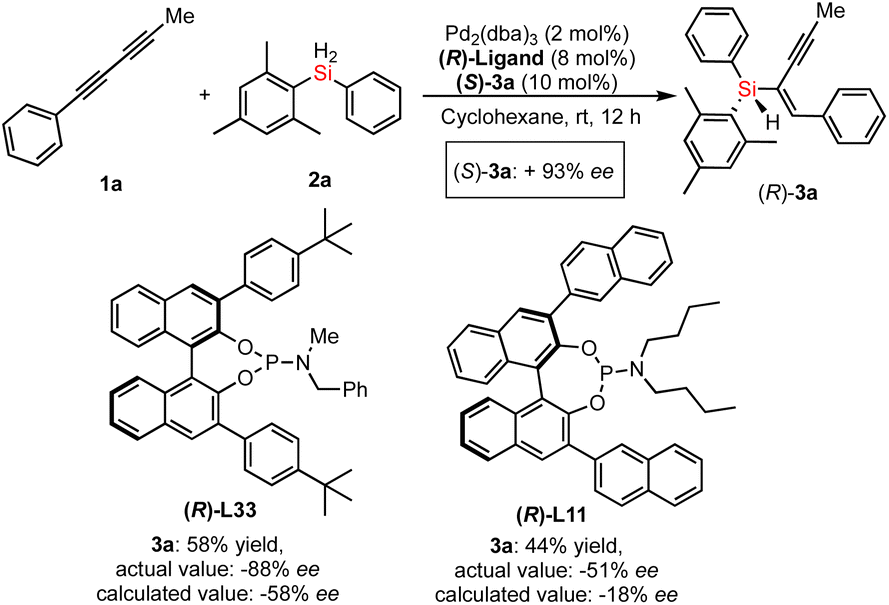 | ||
| Scheme 3 The determination of product-promoted palladium catalysis (autocatalysis) by chirality matching between the chiral additive (3a) and chiral ligand in the Pd-catalyzed hydrosilylation. | ||
Moreover, we unraveled a considerable positive non-linear effect (NLE)21 in the presence of ligand L33 (Fig. 4b and S5 and S6† with cyclohexane or DCM respectively) under the optimized reaction conditions. These findings can be rationalized by catalyst aggregation-induced asymmetric amplification,18 as was substantiated by detailed 31P-NMR, UV, and circular dichroism (CD) spectroscopy (see Fig. S7–S10 of the ESI†). For example, the 31P-NMR of chiral ligand L33 is largely different from that of the Pd/L33 complex (143.78 ppm versus 11.99 ppm). Interestingly, there is no change for the 31P-NMR of mixtures of Pd/L33 complex with 1,3-dialkyne or hydrosilane (11.99 ppm, see Fig. S7 of ESI†), even in the reaction system. Furthermore, different from the 1H-NMR spectrum of the pure ligand L33, the 1H-NMR analysis shows that there is only one peak for the tert-butyl group of the Pd/L33 complex (Fig. S8 of ESI†) and the 1H-NMR chemical shift for the aromatic ring of the complex is difficult to identify (Fig. S9 of ESI†), which revealed the possibility of the aggregation of the catalyst with the substrate. In addition, the UV-vis and CD analysis support that the addition of the substrate did not change the peak shape but resulted in a slight difference in intensity (Fig. 5). These experimental results on spectra analysis revealed that the catalyst aggregation would be possibly formed by chiral ligand-controlled molecular interaction between catalyst and substrate/product in this reaction. In order to verify the advantages of catalyst aggregation,22 we also try to conduct the reaction in air atmosphere or aqueous solvent. The experimental results showed that both reactions can be successfully carried out to achieve the desired products with the same high ee value (91% ee and 93% ee respectively, see Scheme 4). In addition, we also checked the reactivity of the less hindered diethylsilane which will not give a stereogenic center. As expected, we found that almost no reaction occurred to give the desired product and this negative result revealed the importance of aromatic interaction between the catalyst and silane in this reaction.
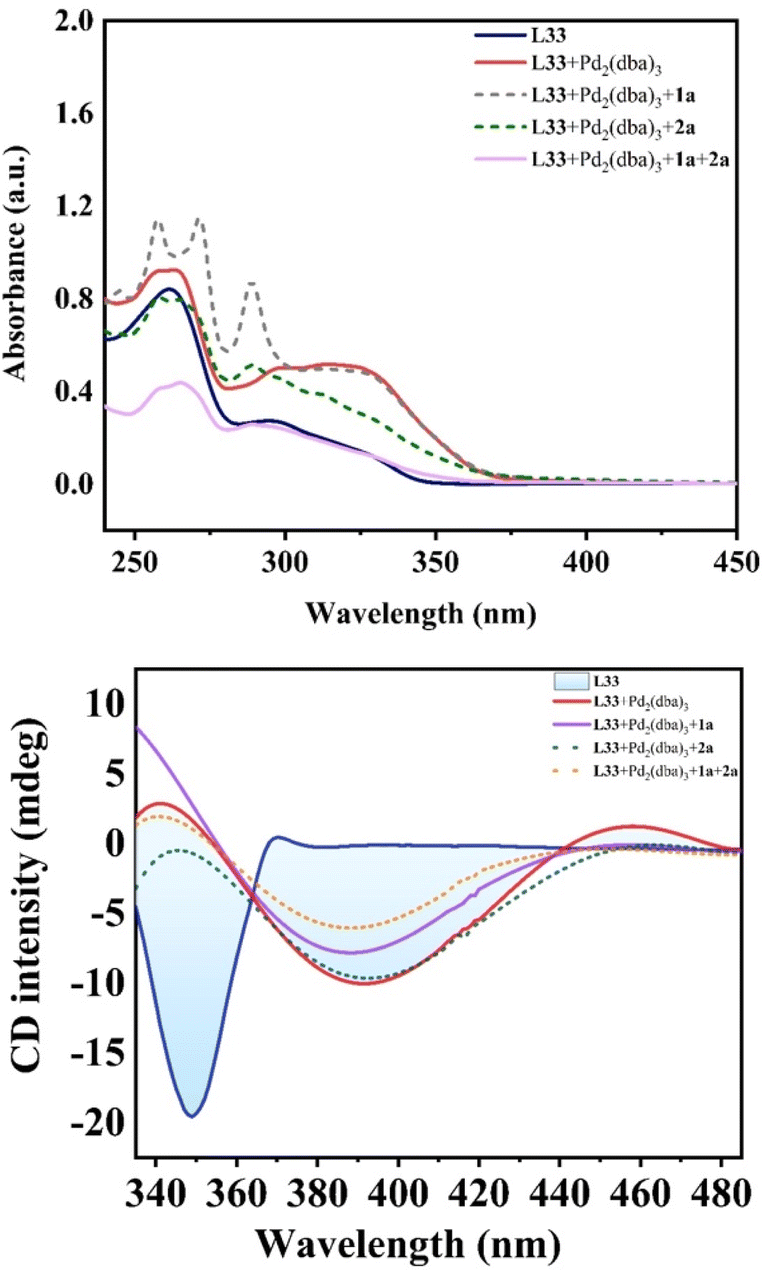 | ||
| Fig. 5 UV-vis absorption spectrum (upper figure) and CD analysis (lower figure) for the molecular interaction between Pd catalyst and substrate. | ||
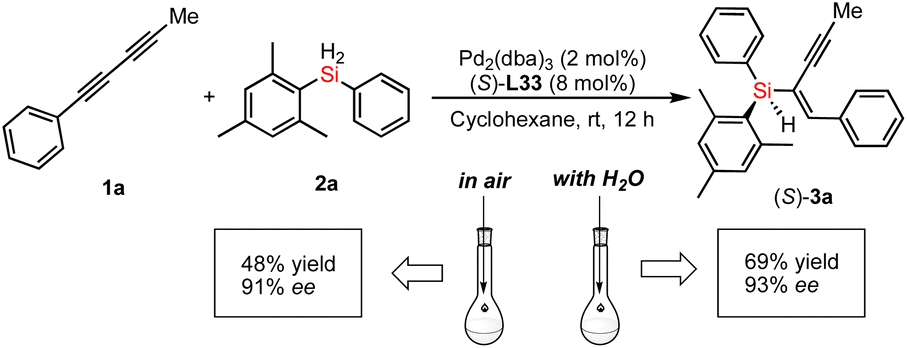 | ||
| Scheme 4 Palladium-catalyzed hydrosilylation of 1a and 2a under an air atmosphere or in water. | ||
Thereafter, except the effect of catalyst aggregation on the enantioselective Pd-catalyzed hydrosilylation, the key point of the reaction mechanism is the Si–H bond activation. On the basis of the experimental results and previously reported mechanism with the palladium-catalyzed hydrosilylation of alkynes,15 we proposed three different pathways for corresponding stages shown in Fig. 4a and a detailed reaction mechanism for the initial stage (Fig. 6). At first, the 1,3-diyne-coordinated Pd species was generated from a Pd-L33 complex and probably transformed into its dimer-like species (A) coordinated with a 1,3-diyne through a pre-activation process with ligand exchange and substrate coordination, which is consistent with the experimental results of the positive NLE and NMR analysis. Then the Si–H activation and hydrometallation which was accelerated by catalyst aggregation occurred to give the intermediate B. The reaction continues with isomerization of intermediate C after the α-selective alkyne insertion of the hydrogen atom and subsequent reductive elimination of the Pd species D towards the formation of the desired silicon-stereogenic enyne 3a and the palladium catalyst is re-generated for the next catalytic cycles. In this reaction mechanism, the function of product-involved aggregation or autocatalysis occurs in the (A→B) step to exchange another ligand on the palladium catalyst to coordinate with (S)-3a as a ligand-like molecule. This phenomenon of aggregation is consistent with the experimental results during the rapid increase of reaction rate (the second stage), as shown in Fig. 4a.
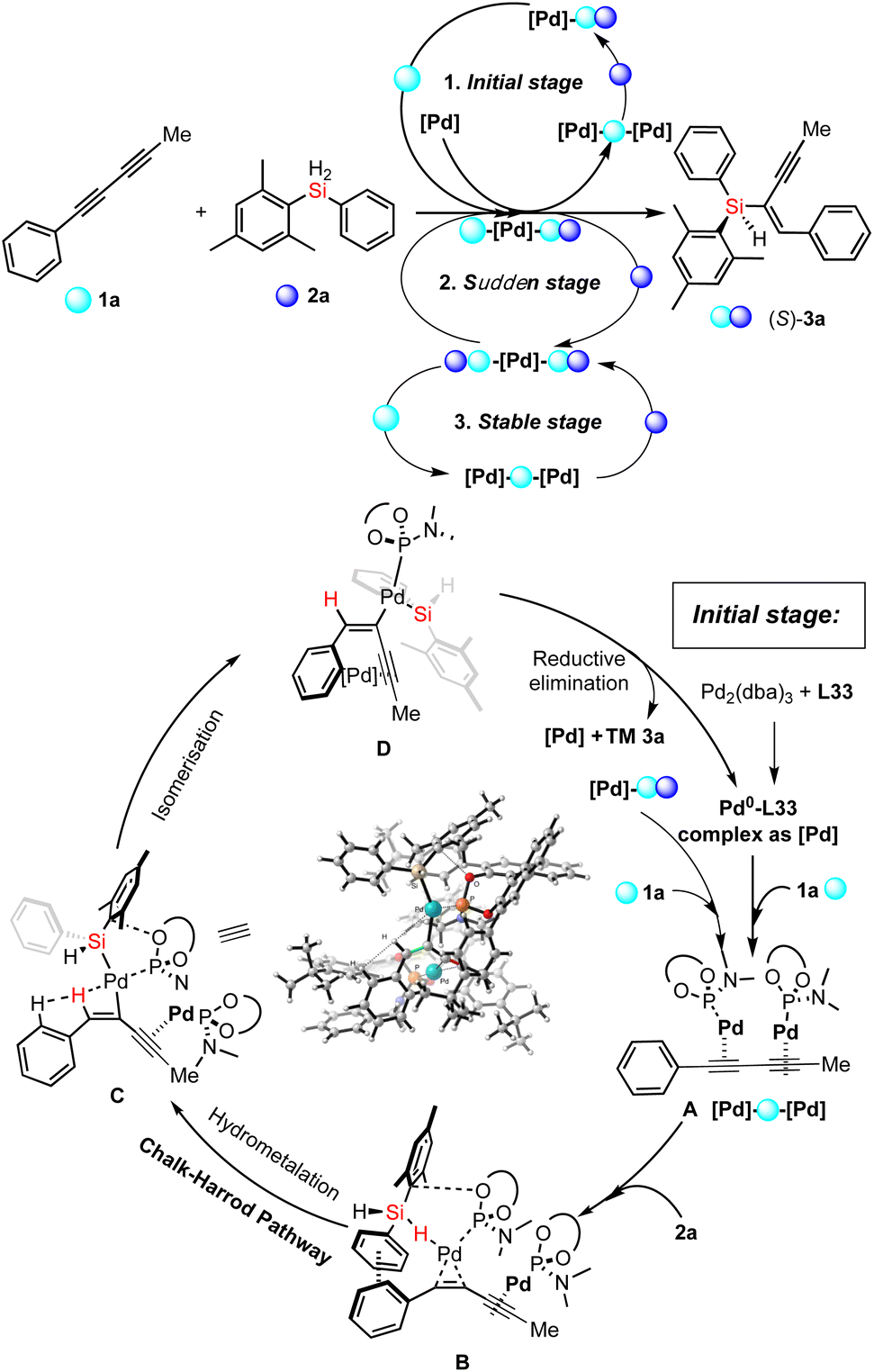 | ||
| Fig. 6 Proposed mechanism for the Pd-catalyzed hydrosilylation of 1,3-diyne. | ||
Conclusions
In summary, we have reported the first enantioselective Pd-catalyzed hydrosilylation of 1,3-diynes by using the strategy of desymmetrization. The catalytic asymmetric hydrosilylation was characterized by a wide scope under exceedingly mild conditions, in which the design of a novel chiral ligand enabled Pd-catalyzed synthesis of silicon-stereogenic enynes with high enantioselectivities (>99% ee). In addition, this reaction could be expanded to the synthesis of chiral Si-containing polyenyne bearing silicon-stereogenic pendants. Detailed kinetic studies on the palladium catalysis uncovered a considerable autocatalysis as to the kinetics and a positive non-linear effect due to the pronounced effect of product and ligand-induced catalyst aggregation. Further studies on the chiral polyenynes and the detailed reaction mechanism are ongoing in our laboratory and will be reported elsewhere in due course.Data availability
For all the data generated and analysed in this study, including the experimental details and spectra for all unknown compounds, see the ESI Files.† All data underlying the findings of this work are available from the corresponding author upon reasonable request.Author contributions
L. W. X. conceived the concept. F. Y. L. developed the asymmetric Pd-catalyzed hydrosilylation of 1,3-diynes. F. Y. and X. J. F. carried out the synthesis of the substrates and spectral analysis of the new products. X. H. Z., W. S. H. and Z. X. assisted with preparation of the substrates, structural analysis of the new product and theoretical studies of the reaction pathway. All authors discussed the results and participated in writing the manuscript. L. W. X. supervised the project.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the grants of National Natural Science Foundation of China (NSFC No. 22072035), Special Support Program for High-level Talents of Zhejiang Province (2021R51005), “Ten-Thousand Talents Plan” of Hangzhou city, Hangzhou Innovation Team (TD2020015), Zhejiang Provincial Natural Science Foundation of China (Nos. LZ23B020002, LY21B030007 and LY22B020006), which are gratefully acknowledged. The authors also thank Dr J. Cao, Dr Y. M. Cui, Dr K. Z. Jiang and Dr L. Li for their assistance on the spectral analysis.Notes and references
- (a) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC; (b) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS; (c) Q. C. Mu, J. Chen, C. G. Xia and L. W. Xu, Coord. Chem. Rev., 2018, 374, 93–113 CrossRef CAS; (d) N. S. Sarai, B. J. Levin, J. M. Roberts, D. E. Katsoulis and F. H. Arnold, ACS Cent. Sci., 2021, 7, 944–953 CrossRef CAS PubMed; (e) F. Ye and L. W. Xu, Synlett, 2021, 32, 1281–1288 CrossRef; (f) D. D. Roberts and M. G. McLaughlin, Adv. Synth. Catal., 2022, 364, 2307–2332 CrossRef; (g) K. Kucinski, H. Stachwoiak-Dluzynska and G. Hreczycho, Coord. Chem. Rev., 2022, 459, 214456 CrossRef; (h) W. S. Huang, Q. Wang, H. Yang and L. W. Xu, Synthesis, 2022, 54, 5400–5408 CrossRef.
- (a) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef PubMed; (b) R. L. N. Hailes, A. M. Oliver, J. Gwyther, G. R. Whittell and I. Manners, Chem. Soc. Rev., 2016, 45, 5358–5407 RSC; (c) Y. Zuo, X. Liang, J. Yin, Z. Gou and W. Lin, Coord. Chem. Rev., 2021, 447, 214166 CrossRef; (d) L. H. Wang, G. X. Zhang, Y. Li and L. W. Xu, Angew. Chem., Int. Ed., 2022, 61, e202210851 Search PubMed.
- (a) L. Li, Y. L. Wei and L. W. Xu, Synlett, 2020, 31, 21–34 CrossRef; (b) J. H. Ma, L. Li, Y. L. Sun, Z. Xu, X. F. Bai, K.-F. Yang, J. Cao, Y. M. Cui, G.-W. Yin and L.-W. Xu, Sci. China: Chem., 2020, 63, 1082–1090 CrossRef; (c) Q. Wang, K.-B. Zhong, H. Xu, S.-N. Li, W.-K. Zhu, F. Ye, Z. Xu, Y. Lan and L. W. Xu, ACS Catal., 2022, 12, 4571–4580 CrossRef.
- (a) Y. C. Wu and P. Wang, Angew. Chem., Int. Ed., 2022, 61, e202205382 Search PubMed; (b) W. Yuan and C. He, Synthesis, 2022, 54, 1939–1950 CrossRef; (c) R. Shintani, Synlett, 2018, 29, 388–396 CrossRef; (d) J. O. Bauer and C. Strohmann, Eur. J. Inorg. Chem., 2016, 2868–2881 CrossRef; (e) R. Shintani, Asian J. Org. Chem., 2015, 4, 510–514 CrossRef.
- (a) L. W. Xu, L. Li, G. Q. Lai and J. X. Jiang, Chem. Soc. Rev., 2011, 40, 1777–1790 RSC; (b) M. Oestreich, Synlett, 2007, 1629–1643 CrossRef.
- S. Rendler, G. Auer, M. Keller and M. Oestreich, Adv. Synth. Catal., 2006, 348, 1171–1182 CrossRef.
- L. W. Xu, Angew. Chem., Int. Ed., 2012, 51, 12932–12934 CrossRef PubMed.
- (a) X. C. Wang, B. Li, C. W. Ju and D. Zhao, Nat. Commun., 2022, 13, 3392 CrossRef PubMed; (b) X. X. Tang, Y. Zhang, Y. L. Tang, Y. Li, J. J. Zhou, D. Y. Wang, L. Gao, Z. S. Su and Z. L. Song, ACS Catal., 2022, 12, 5185–5196 CrossRef; (c) J. Zhang, N. Yan, C. W. Ju and D. Zhao, Angew. Chem., Int. Ed., 2021, 60, 25723–25728 CrossRef; (d) X. Wang, S. S. Huang, F. J. Zhang, J. L. Xie, Z. Li, Z. Xu, F. Ye and L. W. Xu, Org. Chem. Front., 2021, 8, 6577–6584 RSC; (e) H. Chen, Y. Chen, X. Tang, S. Liu, R. Wang, T. Hu, L. Gao and Z. Song, Angew. Chem., Int. Ed., 2019, 58, 4695–4699 CrossRef; (f) R. Shintani, K. Moriya and T. Hayashi, Org. Lett., 2012, 14, 2902–2905 CrossRef CAS PubMed; (g) R. Shintani, K. Moriya and T. Hayashi, J. Am. Chem. Soc., 2011, 133, 16440–16443 CrossRef.
- (a) S. Chen, J. Zhu, J. Ke, Y. Li and C. He, Angew. Chem., Int. Ed., 2022, 61, e202117820 Search PubMed; (b) H. Zhang and D. Zhao, ACS Catal., 2021, 11, 10748–10753 CrossRef; (c) Y. Guo, M. M. Liu, X. Zhu, L. Zhu and C. He, Angew. Chem., Int. Ed., 2021, 60, 13887–13891 CrossRef; (d) S. Chen, D. Mu, P. L. Mai, J. Ke, Y. Li and C. He, Nat. Commun., 2021, 12, 1249 CrossRef; (e) B. Yang, W. Yang, Y. Guo, L. You and C. He, Angew. Chem., Int. Ed., 2020, 59, 22217–22222 CrossRef PubMed; (f) Q. W. Zhang, K. An, L.-C. Liu, Q. Zhang, H. Guo and W. He, Angew. Chem., Int. Ed., 2017, 56, 1125–1129 CrossRef CAS PubMed.
- (a) J. R. Jagannathan, J. C. Fettinger, J. T. Shaw and A. K. Franz, J. Am. Chem. Soc., 2020, 142, 11674–11679 CrossRef CAS PubMed; (b) Y. Yasutomi, H. Suematsu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 4510–4511 CrossRef CAS PubMed.
- (a) L. Wang, W. Lu, J. Zhang, Q. Chong and F. Meng, Angew. Chem., Int. Ed., 2022, 61, e202205624 CAS; (b) W. X. Lu, Y. M. Zhao and F. K. Meng, J. Am. Chem. Soc., 2022, 14, 5233–5240 CrossRef; (c) Y. H. Huang, Y. Wu, S. Zheng, Z. Ye, Q. Peng and P. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113052 CAS; (d) Y. E. You and S. Z. Ge, Angew. Chem., Int. Ed., 2021, 60, 12046–12052 CrossRef CAS; (e) W. Ma, L. C. Liu, K. An, T. He and W. He, Angew. Chem., Int. Ed., 2021, 60, 4245–4251 CrossRef CAS; (f) J. L. Xie, Z. Xu, H. Q. Zhou, Y. X. Nie, J. Cao, G. W. Yin, J. P. Bouillon and L. W. Xu, Sci. China: Chem., 2021, 64, 761–769 CrossRef CAS; (g) G. Zhan, H. L. Teng, Y. Luo, S. J. Lou, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2018, 57, 12342–12346 CrossRef CAS; (h) H. Wen, X. Wan and Z. Huang, Angew. Chem., Int. Ed., 2018, 57, 6319–6323 CrossRef CAS PubMed; (i) K. Igawa, D. Yoshihiro, N. Ichikawa, N. Kokan and K. Tomooka, Angew. Chem., Int. Ed., 2012, 51, 12745–12748 CrossRef CAS.
- (a) H. Zhou, J. T. Han, N. Nöthling, M. M. Lindner, J. Jenniches, C. Kühn, N. Tsuji, L. Zhang and B. List, J. Am. Chem. Soc., 2022, 144, 10156–10161 CrossRef CAS; (b) H. Liu, P. He, X. Liao, Y. Zhou, X. Chen, W. Ou, Z. Wu, C. Luo, L. Yang and J. Xu, ACS Catal., 2022, 12, 9864–9871 CrossRef CAS; (c) W. Yang, L. Liu, J. Guo, S. G. Wang, J.-Y. Zhang, L. W. Fan, Y. Tian, L. L. Wang, C. Luan, Z. L. Li, C. He, X. Wang, Q. S. Gu and X. Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202205743 CAS; (d) J. Gao, P. L. Mai, Y. Ge, W. Yuan, Y. Li and C. He, ACS Catal., 2022, 12, 8476–8483 CrossRef CAS; (e) W. Yuan, X. Zhu, Y. Xu and C. He, Angew. Chem., Int. Ed., 2022, 61, e202204912 CAS; (f) M. Zhou, J. Liu, R. Deng, Q. Wang, S. Wu, P. Zheng and Y. R. Chi, ACS Catal., 2022, 12, 7781–7788 CrossRef CAS; (g) J. Zhu, S. Chen and C. He, J. Am. Chem. Soc., 2021, 143, 5301–5307 CrossRef CAS PubMed; (h) X. Bi, J. Feng, X. Xue and Z. Gu, Org. Lett., 2021, 23, 3201–3206 CrossRef CAS PubMed; (i) D. L. Mu, W. Yang, S. Y. Chen, N. Wang, B. Yang, L. J. You, B. Zu, P. Y. Yu and C. He, J. Am. Chem. Soc., 2020, 142, 13459–13468 CrossRef CAS PubMed; (j) G. Zhang, Y. Li, Y. Wang, Q. Zhang, T. Xiong and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 11927–11931 CrossRef CAS; (k) E. R. Barth, A. Krupp, F. Langenohl, L. Brieger and C. Strohmann, Chem. Commun., 2019, 55, 6882–6885 RSC; (l) Y. Sato, C. Takagi, R. Shintani and K. Nozaki, Angew. Chem., Int. Ed., 2017, 56, 9211–9216 CrossRef; (m) X. F. Bai, J. F. Zou, M. Y. Chen, Z. Xu, L. Li, Y.-M. Cui, Z. J. Zheng and L. W. Xu, Chem.–Asian J., 2017, 12, 1730–1735 CrossRef PubMed.
- (a) R. H. Tang, Z. Xu, Y. X. Nie, X. Q. Xiao, K. F. Yang, J. L. Xie, B. Guo, G. W. Yin, X. M. Yang and L. W. Xu, iScience, 2020, 23, 101268 CrossRef PubMed; (b) X. B. Wang, Z. J. Zheng, J. L. Xie, X. W. Gu, Q. C. Mu, G. W. Yin, F. Ye, Z. Xu and L. W. Xu, Angew. Chem., Int. Ed., 2020, 59, 790–797 CrossRef PubMed; (c) Y. Zeng, X. J. Fang, R. H. Tang, J. Y. Xie, F. J. Zhang, Z. Xu, Y. X. Nie and L. W. Xu, Angew. Chem., Int. Ed., 2022, 61, e202214147 Search PubMed.
- (a) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS; (b) L. D. De Almeida, H. Wang, K. Junge, X. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550–565 CrossRef CAS PubMed.
- (a) P. W. Long, J. L. Xie, J. J. Yang, S. Q. Lu, Z. Xu, F. Ye and L. W. Xu, Chem. Commun., 2020, 56, 4188–4191 RSC; (b) Q. Wang, F. Ye, J. Cao, Z. Xu, Z. J. Zheng and L. W. Xu, Catal. Commun., 2020, 138, 105950 CrossRef CAS.
- (a) F. Ye, Z. Xu and L. W. Xu, Acc. Chem. Res., 2021, 54, 452–470 CrossRef CAS; (b) H. Q. Zhou, F. Y. Ling, X. J. Fang, H. J. Zhu, L. Li, F. Ye, Z. Xu and L. W. Xu, Org. Chem. Front., 2022 10.1039/d2qo01688e , ASAP..
- (a) J. Walkowiak, J. Szyling, A. Franczyk and R. L. Melen, Chem. Soc. Rev., 2022, 51, 869–994 RSC; (b) J. Walkowiak, K. Salamon, A. Franszyk, K. Stefanowska, J. Szyling and I. Kownacki, J. Org. Chem., 2019, 84, 2358–2365 CrossRef CAS; (c) H. L. Sang, Y. Hu and S. Ge, Org. Lett., 2019, 21, 5234–5237 CrossRef CAS.
- (a) L. Chen, J. B. Huang, Z. Xu, Z. J. Zheng, K. F. Yang, Y. M. Cui, J. Cao and L.-W. Xu, RSC Adv., 2016, 6, 67113–67117 RSC; (b) J. J. Yang, Z. Xu, Y. X. Nie, S. Q. Lu, J. Zhang and L. W. Xu, J. Org. Chem., 2020, 85, 14360–14368 CrossRef CAS PubMed.
- Deposition number for product 3ab is 2209523. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- (a) K. Soai, T. Shibata and I. Sato, Acc. Chem. Res., 2000, 33, 382–390 CrossRef CAS PubMed; (b) A. J. Bissette and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 12800–12826 CrossRef CAS; (c) K. Soai, T. Kawasaki and A. Matsumoto, Chem. Rec., 2014, 14, 70–83 CrossRef CAS.
- (a) C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922–2959 CrossRef; (b) T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456–494 CrossRef CAS; (c) Y. Geiger and S. Bellemin-Laponnaz, ChemCatChem, 2022, 14, e202200165 CrossRef CAS.
- (a) R. Jiang, C. Liu, O. Yang and S. You, Prog. Chem., 2022, 34, 1537–1547 Search PubMed; (b) H. Du, X. Zhang, Z. Wang, H. Bao, T. You and K. Ding, Eur. J. Org. Chem., 2008, 2248–2254 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2209523. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06181c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |