
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiple C–C bond formation upon electrocatalytic reduction of CO2 by an iron-based molecular macrocycle†
Si-Thanh
Dong
,
Chen
Xu
and
Benedikt
Lassalle-Kaiser
 *
*
Synchrotron SOLEIL, Route Départementale 128, l’Orme des Merisiers, 91190 Saint-Aubin, France. E-mail: benedikt.lassalle@synchrotron-soleil.fr
First published on 15th December 2022
Abstract
Molecular macrocycles are very promising electrocatalysts for the reduction of carbon dioxide into value-added chemicals. Up to now, most of these catalysts produced only C1 products. We report here that iron phthalocyanine, a commercially available molecule based on earth-abundant elements, can produce light hydrocarbons upon electrocatalytic reduction of CO2 in aqueous conditions and neutral pH. Under applied electrochemical potential, C1 to C4 saturated and unsaturated products are evolved. Isotopic labelling experiments unambiguously show that these products stem from CO2. Control experiments and in situ X-ray spectroscopic analysis show that the molecular catalyst remains intact during catalysis and is responsible for the reaction. On the basis of experiments with alternate substrates, a mechanism is proposed for the C–C bond formation step.
Introduction
Atmospheric carbon dioxide (CO2) has been used as a feedstock to produce all biomass,1 including fossil fuels, since photosynthesis developed on Earth more than two billion years ago. Industrialized societies are eagerly searching for such a solar-to-chemical conversion process, which would help mitigate their carbon dioxide emissions while providing alternative ways to produce staple chemicals.2 The electrochemical CO2 reduction reaction (CO2RR) has been proposed as a viable technology to convert this gas into valuable chemicals. At present, the most common products of CO2 reduction are carbon monoxide (CO) and formic acid (HCOOH). Although these C1 products are near the market-ready level, they only reflect a few of the chemicals that can be generated from CO2.3 Great efforts have been directed to further reduce CO2 to higher value products, particularly those including C–C bonds. Starting from the work of Hori and co-workers4 in the 80s, copper-based catalysts have shown remarkable activity towards the formation of light hydrocarbons, such as methane and ethylene.5 Despite these performances, the precise nature of the active sites involved remains debatable,6 partly because of the restructuring of copper during catalysis.7,8 Recently, single-atom catalysts (SACs) have gained significant attention in the field of CO2 reduction due to their low metal content and high activity.9 While the reduction of CO2 on SACs yields mostly CO, other more reduced products have been obtained,10,11 including C2+ products such as ethanol12 and acetone.13 Although SACs properties are expected to be favorable to the reduction of CO2 beyond CO,14 the heterogeneity of their structures9 prevents establishing detailed reaction mechanisms.Molecular complexes catalyze the reduction of CO2 to CO with high efficiency and stability,15,16 and the homogeneity of their chemical structure allows establishing precise reaction mechanisms.17,18 Transition metal macrocycles such as porphyrins19 or phthalocyanines20–23 have shown very promising results in the electrochemical reduction of CO2, both as homogeneous23 and heterogeneous systems.24 Beside their efficiency and robustness, these molecular catalysts also offer chemical tunability, which allows orienting their reactivity towards higher turnover numbers (TONs),25 higher selectivity,26 or the synthesis of a specific compound.27 While most molecular species catalyzed the CO2 to CO reduction, a handful of examples have shown that C1 products such as methane28,29 or methanol21,30 can be produced by catalysts with M–N4 structure. Ethanol and acetic acid were found as a product of CO2 reduction on cobalt and manganese corrole species, respectively.31,32 There is, however, no example of C2+ compounds produced from CO2 reduction by a catalyst for which the molecular nature during catalysis is demonstrated.
We report here on the electrocatalytic formation of saturated and unsaturated light hydrocarbons (C1–C4) from CO2 using a heterogenized molecular iron phthalocyanine catalyst in a flowing system (see Fig. 1 and S1†). The analysis of the products formed under applied electrochemical potential shows that, besides CO and H2, small amounts of methane, ethylene, ethane, propene, propane, and n-butane are detected along with traces of C5 products. Electrocatalytic experiments using alternate substrates such as carbon monoxide (CO) or formaldehyde (HCHO) support a mechanism involving a terminal carbene as a critical species on the way to C2+ compounds.
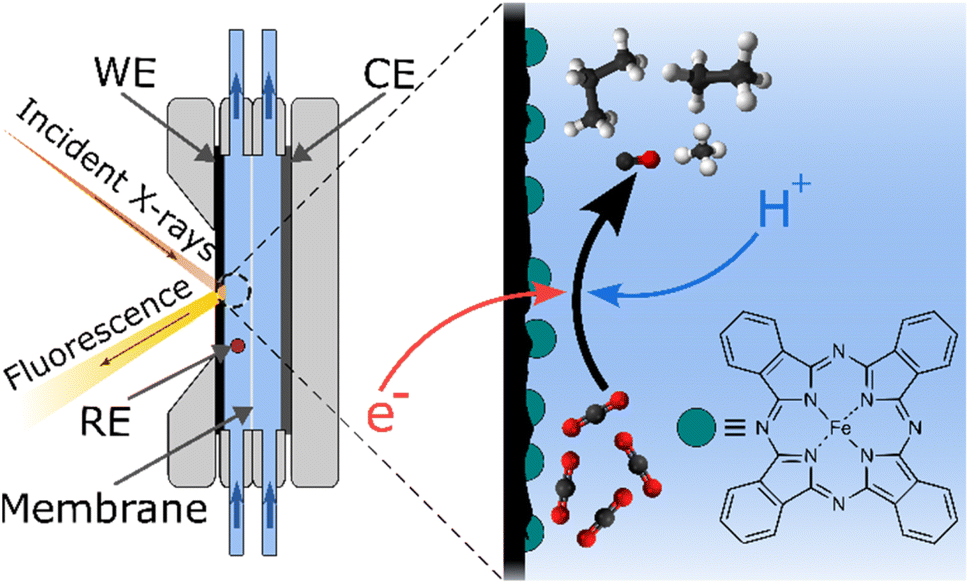 | ||
| Fig. 1 Schematic side view of the electrochemical cell and simplified CO2 reduction process on the surface of the working electrode. The dark green circles represent iron phthalocyanine (FePc), the molecular catalyst used in this study, whose structure is shown in the right corner. WE, CE, and RE stand for “working electrode”, “counter electrode”, and “reference electrode”, respectively. The scheme is not drawn to scale. | ||
Results and discussion
Electrocatalysis and distribution of products
Based on the design of Ager and co-workers,33 CO2 reduction experiments were performed in an electrocatalytic flow cell consisting of two chambers for the cathodic and anodic reactions, separated by a Nafion® membrane (see Fig. 1 and S1†). The cathode consists of a glassy carbon electrode, on which an ink containing iron phthalocyanine (FePc) and a carbon powder is drop-casted with a surface concentration of around 80 nmol cm−2 (see ESI† for electrode preparation procedure). A reference electrode is arranged in the cathode chamber, and the anode consists of a nickel plate. Two independent reservoirs contain the catholyte and anolyte (KHCO3 0.1 M), which are flown independently in the anodic and cathodic chambers. The headspace of the cathodic compartment is analyzed during the experiment by an online gas chromatograph that performs automatic sampling every thirty minutes (see Fig. S2A†).Fig. 2A shows the chronoamperometric curves of constant potential electrolysis under Ar or CO2 for two hours. After poising an FePc-modified electrode at −1.1 V vs. RHE under a CO2 atmosphere, peaks corresponding to ethylene (C2H4), ethane (C2H6), propene (C3H6), propane (C3H8) and n-butane (C4H10) clearly stand out in the chromatogram (see Fig. 2B). Other peaks corresponding to unsaturated C4 as well as n-pentane and unsaturated C5 products can also be observed, although with a lower intensity. Methane (CH4) is also clearly present on another module of the gas chromatograph (see Fig. S3†). The catholyte was analyzed by 1H NMR to check for liquid products, but none was detected (see Fig. S4†). Control experiments were performed to ensure that FePc catalyzed the reduction of CO2 into the products observed. When a FePc-modified electrode was set under catalytic conditions in an Ar-saturated KHCO3 electrolyte, hydrogen was the major product detected after two hours of electrolysis, with a trace amount of CO and hydrocarbons (see Fig. S5†). The observed carbon products can be attributed to the small amount of CO2 that exists in equilibrium with the KHCO3 electrolyte. In fact, a similar FePc-modified electrode, at the same potential, under a CO2-free environment (Ar-saturated 0.1 M KCl electrolyte), produces only H2 (see Fig. 2B and S3†). The current density observed is higher than under CO2 (see Fig. 2A), indicating a high activity of FePc towards hydrogen evolution in a CO2-free environment at high overpotential. When an H2Pc-modified electrode was poised at −1.1 V in a CO2-saturated KHCO3 electrolyte, a twenty-fold decrease in current density was observed as compared to an FePc-modified electrode, indicating a negligible catalytic activity without the Fe center. In agreement with the low catalytic activity, the main product detected was H2, with a miniscule amount of CO (Fig. S3†). These results strongly indicate CO2 as the source of the carbon products observed and underline the essential role of the metal center in the catalytic process.
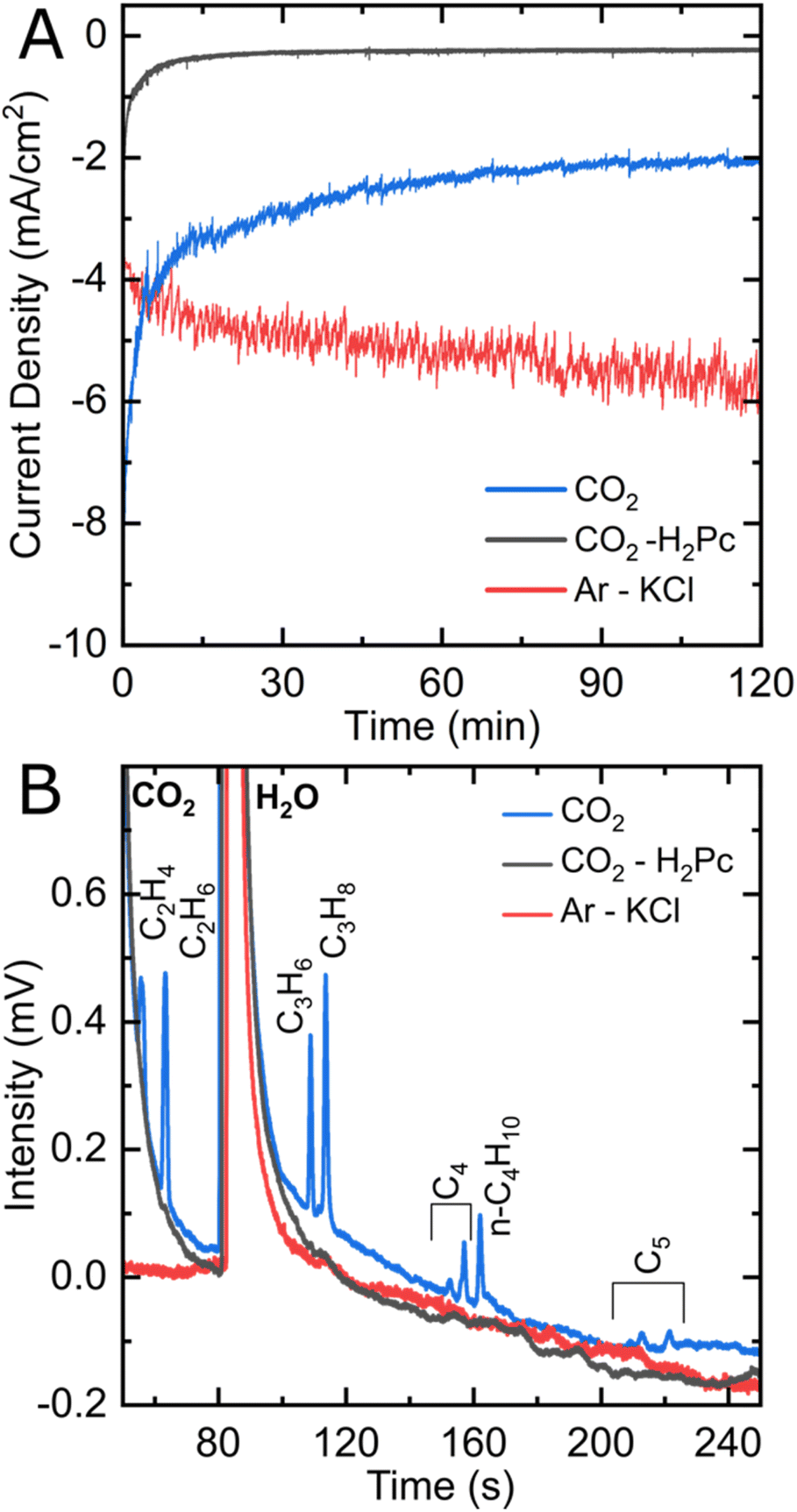 | ||
| Fig. 2 (A) Chronoamperometric curves and (B) module B gas chromatograms of FePc-modified electrodes under Ar in 0.1 M KCl (red) and CO2 in 0.1 M KHCO3 (blue) and an H2Pc-modified electrode under CO2 in KHCO3 (black). Chronoamperometric experiments are performed at −1.1 V vs. RHE for two hours, after which a gas chromatograph (GC) samples their resulting gaseous products. | ||
To confirm the origin of the carbon atoms in the C2+ products observed in the chromatograms, gas chromatography coupled mass spectrometry (GC-MS) experiments were performed. Fig. 3 shows the GC-MS chromatograms corresponding to the retention time of C4 products (190–240 s). We focus on the C4H8 signal since it has the highest intensity of all C4 products without overlapping the signal of CO2 or CO. The m/z = 56 mass, which corresponds to a C4H8 species containing four 12C atoms, shows a peak when 12CO2 is used as the substrate, while it does not appear in the presence of 13CO2. The opposite behavior is observed with the m/z = 60 mass, which corresponds to a C4H8 species with four 13C atoms. The GC-MS chromatogram of the m/z = 41 mass at the region of the retention time of C3 and C4 products is shown on Fig. S6.† The m/z = 41 mass, which corresponds to a C3H5 fragment (originating from both C3 and C4 products) shows two distinguishable peaks when 12CO2 is used, which disappear with 13CO2. These isotopic labelling experiments demonstrate that the origin of the carbon atoms in the hydrocarbons observed is indeed CO2.
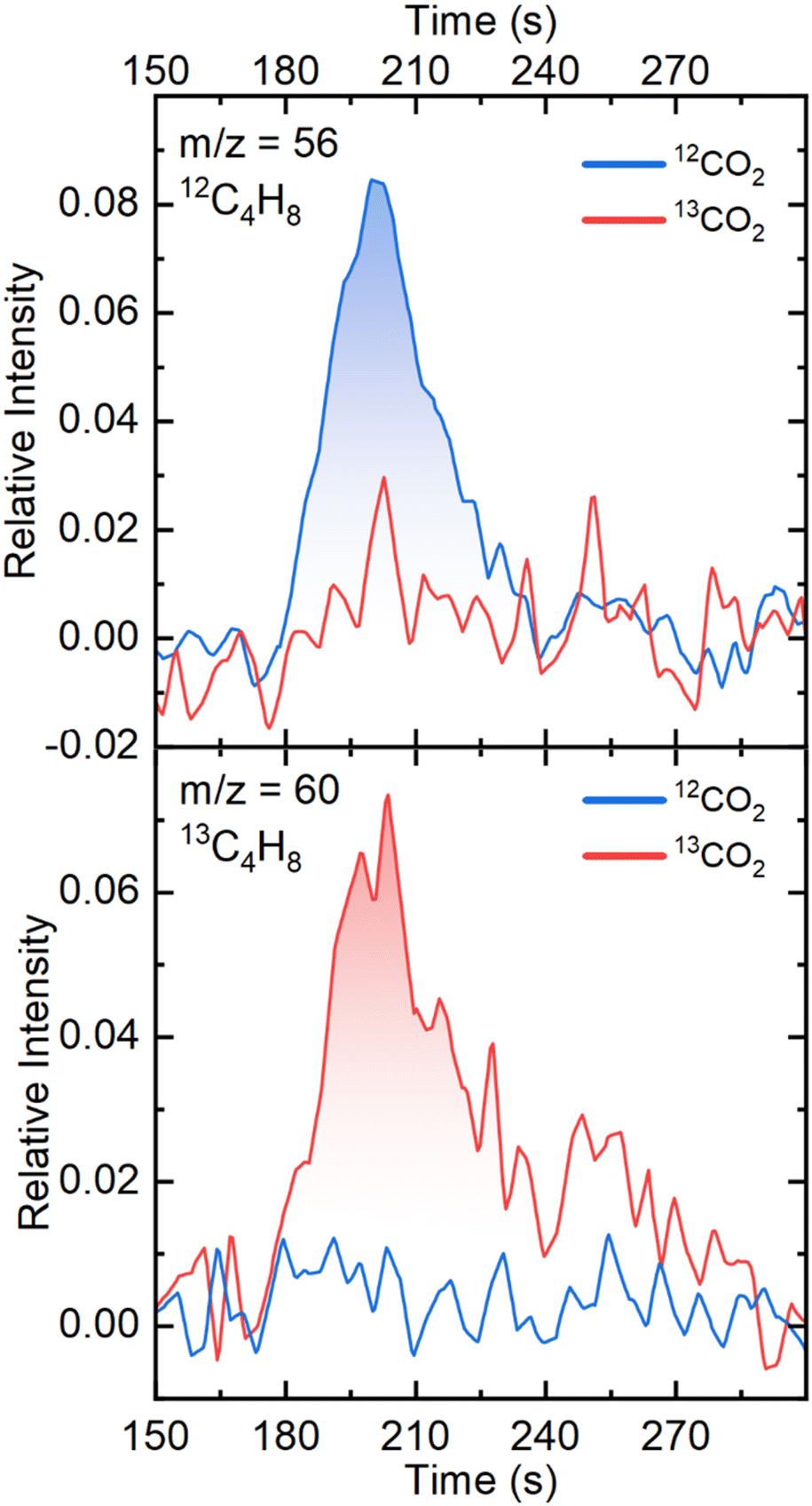 | ||
| Fig. 3 GC-MS chromatograms for mass m/z = 56 of 12C4H8 and mass m/z = 60 of 13C4H8 at the retention time of C4 products. Chronoamperometric experiments were performed on FePc-modified electrodes at −1.1 V vs. RHE for two hours, after which the GC-MS sampled the resulting gaseous products. The chromatograms were averaged from three independent experiments. | ||
Fig. 4 shows the product distribution in terms of faradaic efficiency (FE) for each of the products obtained after a 2 hours electrocatalytic experiment under CO2. The potential range is chosen where there is significant CO2 reduction activity without generating hydrogen bubbles that would make the system unstable. Between −0.7 V and −0.9 V, CO and H2 are the only products observed. Although the current density at high overpotential slightly decreases over time, the selectivity of all products remains constant (see Fig. S7†). The amount of hydrogen increases steadily from −0.7 V to −1.1 V up to 65% FE at the expense of CO. Noticeable amounts of hydrocarbons can be observed starting from −0.9 V, methane being the main of them with up to 4% at −1.0 and −1.1 V. Ethylene, ethane, propene, propane and n-butane are produced with faradaic efficiencies of 0.1–0.25% each, making the total of C2+ products to be around 1%. It should be noted that the amount of ethylene is underestimated because of the high background generated by CO2 in the chromatograph column. Despite the low amounts of products generated, their distribution was reproducible, as shown by the replicates performed on three different electrodes.
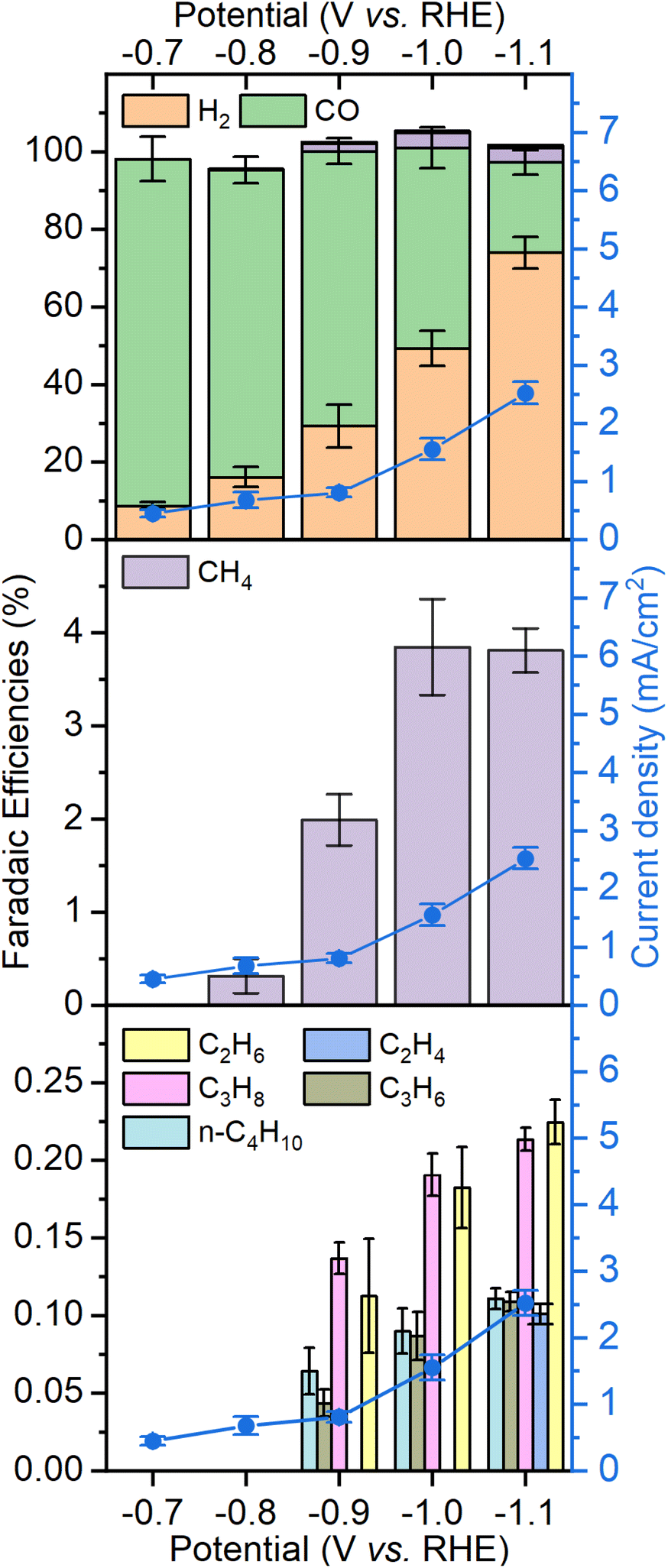 | ||
| Fig. 4 Product distribution observed as faradaic efficiencies with FePc-modified electrodes as a function of applied potential. The total current density is plotted for each potential as the blue lines. The amount of ethylene (C2H4) is underestimated due to the high background generated by CO2 in the chromatograph column. | ||
Catalyst characterization
In order to determine the nature of the catalyst under operating conditions and check its integrity, we performed in situ and operando X-ray absorption spectroscopy (XAS) experiments at the iron K-edge in the exact same electrochemical cell (see Fig. 1, S1 and S2B†). Fig. 5 shows the X-ray absorption near-edge structure (XANES) spectra of the FePc-modified electrode under open circuit potential (OCP) and under catalytic conditions, in a CO2 atmosphere, with the corresponding non-normalized spectra in Fig. S8.† The spectrum of the electrode under resting state presents the same features as the pure, starting compound, showing the chemical robustness of the FePc catalyst after dropcasting and in the presence of an electrolyte. When poising the electrode at a potential of −1.1 V (at which C2+ products are observed), the spectrum shows a shift to lower energies (−2.7 eV at half-edge jump) while keeping the features that are characteristic of phthalocyanines,34i.e. two pre-edge peaks at 7114 eV and 7118 eV. A XANES spectrum was also recorded in situ after performing electrocatalytic experiments under a CO2 atmosphere, and it was remarkably similar to the one collected on the starting electrode. The non-normalized spectra indicate a 10% material loss after 4.5 hours of continuous flowing, which is standard in this type of experiment.35 The comparison between the spectra collected under in situ and operando conditions and those of reference compounds (metallic iron, Fe3O4) clearly rule out the presence of metallic iron or iron oxide nanoparticles that would be produced by the decomposition of the molecular catalyst. Altogether, these spectroscopic data collected under in situ conditions clearly demonstrate the molecular nature and the stability of the catalyst throughout the experiment.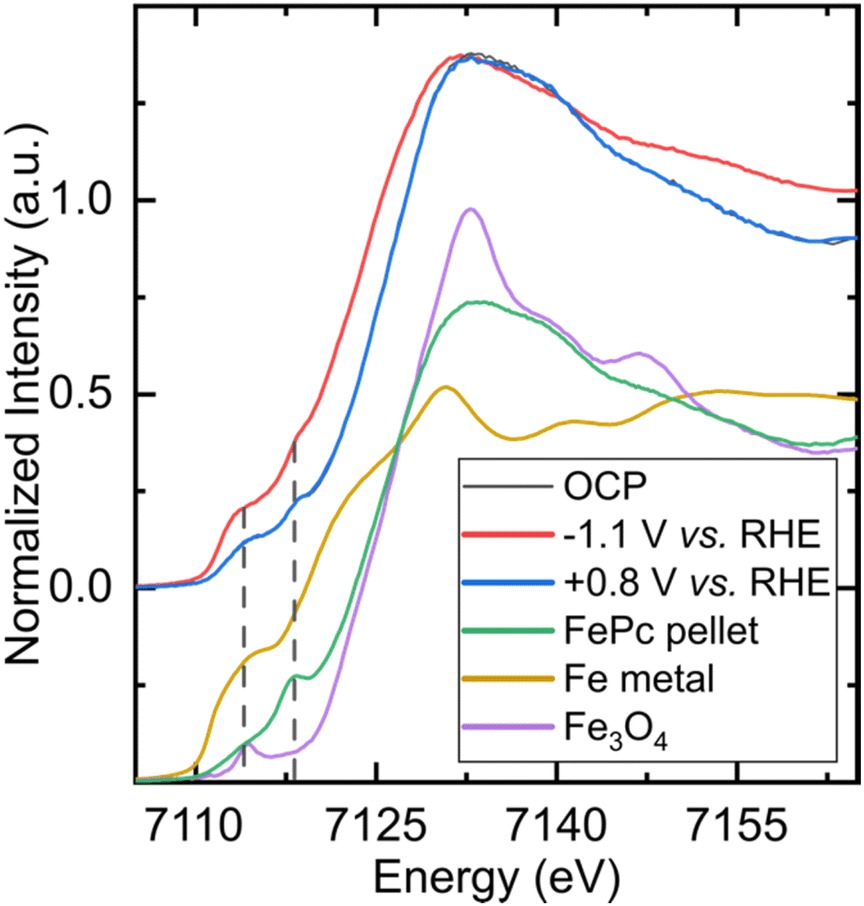 | ||
| Fig. 5 In situ and operando X-ray absorption spectra collected on an FePc-modified electrode under different conditions and on reference samples. The spectra at the top correspond to an FePc-modified electrode under a CO2 atmosphere poised at of FePc (green), on a metallic sheet of iron (gold), and on a pellet of Fe3O4 (purple). The two dashed lines indicate the pre-edge features of FePc at 7114 eV and 7118 eV. OCP (black), at −1.1 V vs. RHE (red) and going back at +0.8 V vs. RHE (blue). The spectra at the bottom correspond to ex situ reference spectra collected on a pellet. | ||
In addition, infrared (IR) spectra were recorded on the FePc catalyst powder and FePc-modified electrodes before and after electrolysis at −1.1 V vs. RHE (see Fig. S9†). The spectra recorded show close similarity, agreeing with the XAS results concerning the stability of the catalyst during electrolysis.
C–C bond formation mechanism
The demonstration that a molecular electrocatalyst can form several consecutive C–C bonds poses the question of their formation mechanism. Since the major product observed upon reduction of CO2 is CO, we postulated that the first intermediate for the formation of C–C bonds could be an FePc–CO species (see Fig. 6). Indeed, electrolysis with CO as substrate shows the formation of similar hydrocarbon products as observed for CO2, albeit with faradaic efficiencies approximately twice as low (see Fig. S10†). We attribute this lower faradaic efficiency to the low solubility of CO in water (1 mM) as compared to that of CO2 (33 mM). As observed in other studies,33,36 we surmised that the difference in initial concentration of substrate was responsible for this difference in efficiency and performed finite element calculations to simulate concentration profiles upon CO2 reduction. The results show that the concentration of CO generated by CO2 reduction within 5 μm from the working electrode ranges from 1.5 to 4.5 mM (see ESI†), supporting a relationship between the amount of C2+ products and the local concentration of CO. These experiments confirm that FePc–CO is indeed an intermediate on the way to C2+ products.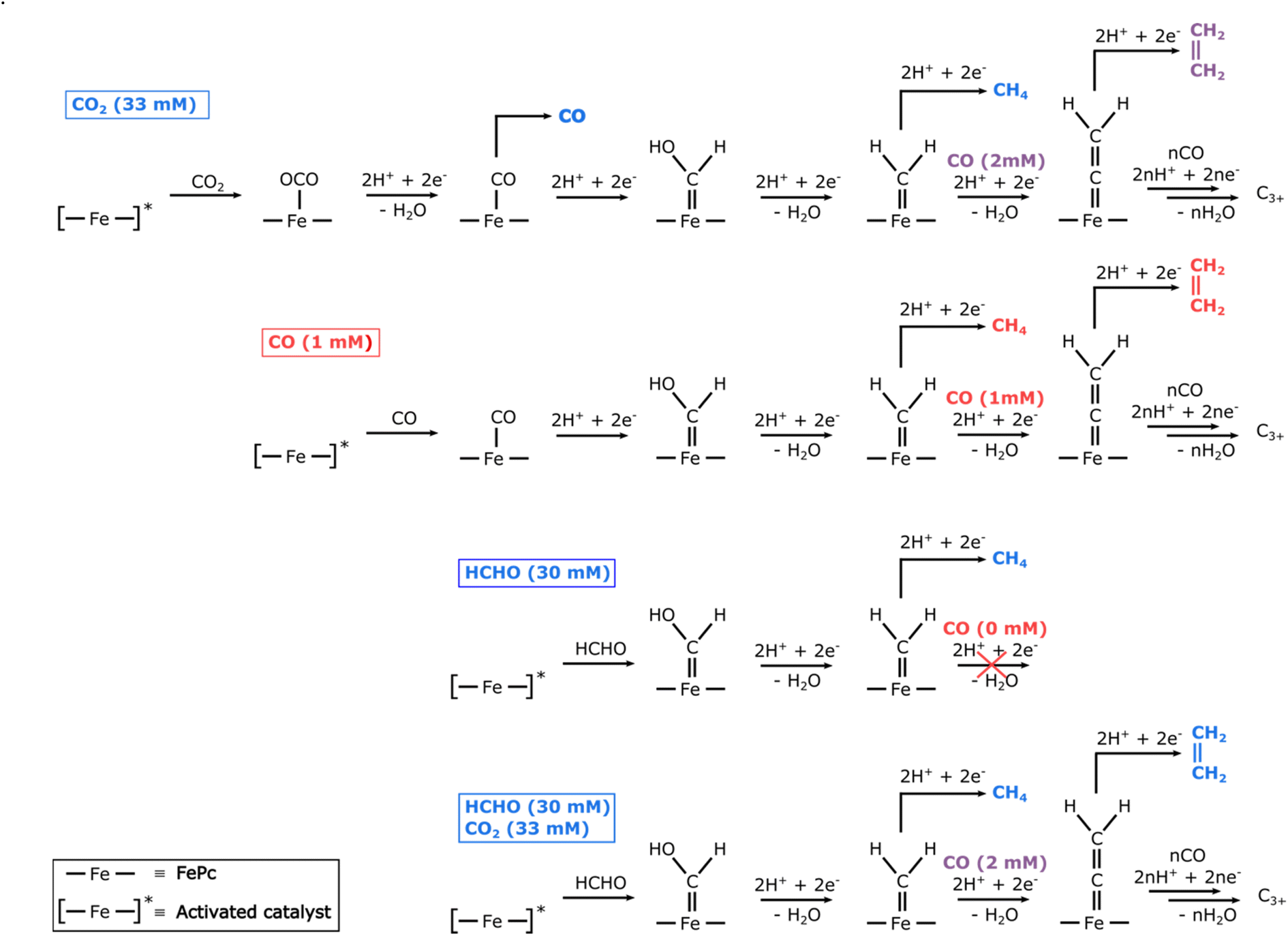 | ||
| Fig. 6 Simplified mechanisms proposed for the formation of C–C bonds by FePc with either CO2, CO, HCHO, or a mixture of HCHO and CO2 as substrates. The colour code for substrates and products is as follow: blue, purple, and red for high, intermediate, and low concentrations, respectively. | ||
By analogy with what happens during the Fischer–Tropsch synthesis of long-chain hydrocarbons, we hypothesized that a terminal FePc![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 carbene species could be the precursor to all C2+ products. This transient species would be formed, via a hydroxycarbene species, by reaction of an FePc–CO adduct with either H2 formed locally or with protons and electrons available in large amounts. This species would then further react either with more H2 (or protons and electrons) to form methane. To form a C–C bond, the iron carbene species undergo CO insertion similar to Fischer–Tropsch mechanism. This CO insertion pathway is different from the carbene coupling involved in the formation of ethylene on metallic copper since there are no adjacent active sites in iron phthalocyanine. The process could continue a few times following various pathways and lead to the formation of longer saturated or unsaturated hydrocarbon chains (see Fig. 6).
CH2 carbene species could be the precursor to all C2+ products. This transient species would be formed, via a hydroxycarbene species, by reaction of an FePc–CO adduct with either H2 formed locally or with protons and electrons available in large amounts. This species would then further react either with more H2 (or protons and electrons) to form methane. To form a C–C bond, the iron carbene species undergo CO insertion similar to Fischer–Tropsch mechanism. This CO insertion pathway is different from the carbene coupling involved in the formation of ethylene on metallic copper since there are no adjacent active sites in iron phthalocyanine. The process could continue a few times following various pathways and lead to the formation of longer saturated or unsaturated hydrocarbon chains (see Fig. 6).
To verify this hypothesis, we used formaldehyde as a substrate, expecting it to form an FePcCH(OH) hydroxycarbene species by direct reaction with the activated form of the catalyst37 (see Fig. 6). In these conditions, the product distribution shows similar multi-carbon products as with CO2 or CO as substrates, although in much lower amounts (Fig. S10†), as well as methane (2%) and traces of methanol (Fig. S10 and S13†). While the concentration of formaldehyde (30 mM) is similar to that of saturated CO2 in water at 25 °C (33 mM), the very low faradaic efficiencies observed for C2+ products can be explained by the absence of any CO to be inserted in the FePcC bond of the putative carbene species. Without any CO to react further, the carbene species is preferentially reduced into methane, leading to a 6-fold increase in the faradaic yield of methane as compared to the CO substrate. In order to confirm the reaction of CO with an FePc carbene species, we performed electrolysis with HCHO in a CO2 atmosphere, as CO2 reduction could provide the necessary CO to increase the C2+ formation with the carbene species generated from HCHO. Indeed, at −1.1 V vs. RHE, the total faradaic efficiency of C2+ products is one order of magnitude higher than observed in HCHO without CO2 (see Fig. S11 and S12†), thus confirming our hypothesis of a reaction between CO and a carbene species obtained from formaldehyde. At less negative potentials, where the formation of CO from CO2 is more favorable, the total C2+ faradaic efficiency increases further, reaching a maximum value of 1.5% at −0.9 V vs. RHE, ca. three times higher than when only CO2 is used as substrate (see Fig. S12†). The higher selectivity of C2+ products after electrolysis of HCHO under CO2 clearly supports the hypothesis that the formation of C2+ species involves CO insertion on an intermediate FePc carbene species, as described in Fig. 6.
The existence of an FePcCH2 intermediate on the way to C2+ products is supported by the fact that several carbene species of iron porphyrin (a macrocycle similar to FePc) have been reported, either as transient38,39 or stable species,40 and that FePc itself has been reported as an efficient catalyst for the cyclopropanation of alkenes,41 which involves FePc–carbene intermediates. Iron porphyrins are also known for catalyzing the insertion of carbenes into N–H, S–H, or C–H bonds.42,43 These precedents, as well as the experiments described here with alternate substrates, strongly support the involvement of an FePc carbene species on the way to C2+ hydrocarbons.
Conclusions
We reported in this paper on the electrocatalytic activity of iron phthalocyanine, a molecular macrocycle based on earth-abundant elements, towards the reduction of CO2 into light hydrocarbons with up to three C–C bonds. Using operando X-ray absorption spectroscopy, we showed that the catalyst keeps its molecular structure throughout the electrocatalytic process. Based on experiments involving carbon monoxide or formaldehyde, we showed that the local concentration of CO is key to the formation of C–C bonds and propose a terminal carbene as an intermediate on the way to C2+ products. These results indicate that the formation of several consecutive carbon–carbon bonds can be catalyzed by single-site transition metal macrocycles under mild electrochemical conditions. The mechanistic insights that we provided open a perspective for the design of both molecular and material-based catalysts with enhanced selectivity towards C2+ products.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI material.† Raw data that support the findings of this study are available from the corresponding author, upon reasonable request.Author contributions
Conceptualization: STD, BLK investigation: STD, CX funding acquisition: BLK supervision: BLK writing – original draft: STD, BLK writing – review & editing: STD, BLK.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We express our gratitude to Dr Jean-Blaise Brubach of the AILES beamline of Synchrotron SOLEIL for his help during infrared spectra collection. We are indebted to Ms. Chanjuan Zhang and Dr Zakaria Halime at the Institut de Chimie Moléculaire et des Matériaux d’Orsay, Université Paris-Saclay for their assistance in performing 1H-NMR measurements. The Agence Nationale de la recherche is acknowledged for a young researcher grant to B. L.-K. (grant no. 18-CE05-0007).References
- A. A. Benson, J. A. Bassham, M. Calvin, T. C. Goodale, V. A. Haas and W. Stepka, J. Am. Chem. Soc., 1950, 72, 1710–1718 CrossRef CAS.
- J. Cheon, J. Y. Yang, M. Koper and O. Ishitani, Acc. Chem. Res., 2022, 55, 931–932 CrossRef CAS PubMed.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 15, 897–898 CrossRef.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS.
- J. Vavra, T.-H. Shen, D. Stoian, V. Tileli and R. Buonsanti, Angew. Chem., 2021, 133, 1367–1374 CrossRef.
- Y.-G. Kim, J. H. Baricuatro and M. P. Soriaga, Electrocatalysis, 2018, 9, 526–530 CrossRef CAS.
- J. Zhang, W. Cai, F. Xin Hu, H. Yang and B. Liu, Chem. Sci., 2021, 12, 6800–6819 RSC.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- W. Ju, A. Bagger, X. Wang, Y. Tsai, F. Luo, T. Möller, H. Wang, J. Rossmeisl, A. S. Varela and P. Strasser, ACS Energy Lett., 2019, 4, 1663–1671 CrossRef CAS.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Catal. Today, 2017, 288, 74–78 CrossRef CAS.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- J. Bonin, A. Maurin and M. Robert, Coord. Chem. Rev., 2017, 334, 184–198 CrossRef CAS.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- G. F. Manbeck and E. Fujita, J. Porphyrins Phthalocyanines, 2015, 19, 45–64 CrossRef CAS.
- K. Kusuda, R. Ishihara and H. Yamaguchi, Electrochim. Acta, 1986, 31, 657–663 CrossRef CAS.
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. F. Jaramillo and M. Robert, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 CrossRef CAS PubMed.
- Z. Jiang, Y. Wang, X. Zhang, H. Zheng, X. Wang and Y. Liang, Nano Res., 2019, 12, 2330–2334 CrossRef CAS.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS.
- N. Corbin, J. Zeng, K. Williams and K. Manthiram, Nano Res., 2019, 12, 2093–2125 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- W. W. Kramer and C. C. L. McCrory, Chem. Sci., 2016, 7, 2506–2515 RSC.
- P. Gotico, Z. Halime and A. Aukauloo, Dalton Trans., 2020, 49, 2381–2396 RSC.
- B. Li, L. Sun, J. Bian, N. Sun, J. Sun, L. Chen, Z. Li and L. Jing, Appl. Catal., B, 2020, 270, 118849 CrossRef CAS.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- S. Gonglach, S. Paul, M. Haas, F. Pillwein, S. S. Sreejith, S. Barman, R. De, S. Müllegger, P. Gerschel, U.-P. Apfel, H. Coskun, A. Aljabour, P. Stadler, W. Schöfberger and S. Roy, Nat. Commun., 2019, 10, 3864 CrossRef PubMed.
- R. De, S. Gonglach, S. Paul, M. Haas, S. S. Sreejith, P. Gerschel, U.-P. Apfel, T. H. Vuong, J. Rabeah, S. Roy and W. Schöfberger, Angew. Chem., Int. Ed., 2020, 59, 10527–10534 CrossRef CAS PubMed.
- Gurudayal, D. Perone, S. Malani, Y. Lum, S. Haussener and J. W. Ager, ACS Appl. Energy Mater., 2019, 2, 4551–4559 CrossRef CAS.
- Y. Liu, A. Deb, K. Yee Leung, W. Nie, W. S. Dean, J. E. Penner-Hahn and C. C. L. McCrory, Dalton Trans., 2020, 49, 16329–16339 RSC.
- T. Zhao, Y. Wang, S. Karuturi, K. Catchpole, Q. Zhang and C. Zhao, Carbon Energy, 2020, 2, 582–613 CrossRef CAS.
- S. Louisia, D. Kim, Y. Li, M. Gao, S. Yu, I. Roh and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2201922119 CrossRef CAS.
- H. Mahmoudi, M. Mahmoudi, O. Doustdar, H. Jahangiri, A. Tsolakis, S. Gu and M. LechWyszynski, Biofuels Engineering, 2017, vol. 2, pp. 11–31 Search PubMed.
- D. Lexa, J.-M. Savéant, J.-P. Battioni, M. Lange and D. Mansuy, Angew. Chem., Int. Ed. Engl., 1981, 20, 578–579 CrossRef.
- I. Artaud, N. Gregoire, P. Leduc and D. Mansuy, J. Am. Chem. Soc., 1990, 112, 6899–6905 CrossRef CAS.
- Y. Liu, W. Xu, J. Zhang, W. Fuller, C. E. Schulz and J. Li, J. Am. Chem. Soc., 2017, 139(14), 5023–5026 CrossRef CAS PubMed.
- H.-H. Liu, Y. Wang, Y.-J. Shu, X.-G. Zhou, J. Wu and S.-Y. Yan, J. Mol. Catal. A: Chem., 2006, 246, 49–52 CrossRef CAS.
- R. D. Lewis, M. Garcia-Borràs, M. J. Chalkley, A. R. Buller, K. N. Houk, S. B. J. Kan and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7308–7313 CrossRef CAS PubMed.
- C. Empel, S. Jana and R. M. Koenigs, Molecules, 2020, 25, 880 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04729b |
| This journal is © The Royal Society of Chemistry 2023 |