
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Luminescent (metallo-supramolecular) cross-linked lanthanide hydrogels from a btp (2,3-bis(1,2,3-triazol-4-yl)picolinamide) monomer give rise to strong Tb(III) and Eu(III) centred emissions†
Isabel N.
Hegarty
a,
Samuel J.
Bradberry
*a,
June I.
Lovitt
a,
Jason M.
Delente
ac,
Niamh
Willis-Fox
b,
Ronan
Daly
b and
Thorfinnur
Gunnlaugsson
 *ac
*ac
aSchool of Chemistry and Trinity Biomedical Sciences Institute, The University of Dublin, Trinity College Dublin, Dublin 2, Ireland. E-mail: gunnlaut@tcd.ie
bInstitute for Manufacturing, Department of Engineering, University of Cambridge, 17 Charles Babbage Road, Cambridge, CB3 0FS, UK
cAMBER (Advanced Materials and Bioengineering Research) Centre, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
First published on 24th January 2023
Abstract
The development of the 2,3-bis(1,2,3- triazol-4-yl)picolinamide (btp) ligand 2, from the amine 1 (which was also structurally characterised) possessing a methyacrylamide unit that facilitated the co-polymerisation of this btp ligand into polymeric hydrogels, using 2-hydroxyethyl methacrylate (HEMA), methylmethacrylate (MMA) and ethylene glycol dimethacrylate (EGDMA), is described, resulting in the formation of two polymers with two different concentrations (cast as polymeric films), F1 (0.03 wt%) and F2 (0.3 wt%), respectively, These polymers were then used to form luminescent soft-materials using lanthanide ions, which can coordinate to the btp ligands; the focus herein on the latter polymer was shown to be more luminescent. The non-polymerisable ligand 3, possessing an acetamide moiety was also developed as a model compound. We also present the formation of a soft material (a supramolecular hydrogel) from the precursor 1, which was shown to be thermoreversibly formed, and its morphological and rheological properties were probed. We also show that upon addition of Tb(III), to this supramolecular gel, we have interrupted the soft-material features upon coordination of the ion to the btp ligands. While the focus herein is on the F2, in both cases the metal ion coordination (which was achieved upon hydration of the polymers in solutions of these ions) results not only in the formation of a luminescent material (Tb(III) is known to give rise to highly luminescent green emission upon coordination of the ion to btp ligands), but also in the instantaneous formation of metal mediated crosslinking within the polymeric matrix. The photophysical properties of both 2 and 3 are presented, both in solution, and as in the case of 2, after the polymerisation within the hydrogel; which in the case of Tb(III) results in the formation of green emitting polymers, clearly visible to the naked eye. Similarly, we demonstrate that upon using Eu(III), red emission is observed from polymer F2.
Introduction
The development of responsive supramolecular systems is currently a highly topical area of research,1–3 with significant developments occurring in the developments of coordination polymers, MOFs, and COFs in recent times.3–6 Self-assembly supramolecular polymers have emerged as highly attractive targets for the formation of hierarchical materials.7–9 The use of low molecular weight gelators (LMWG),10 or more recently, the use of low molecular mass ionic organogelators (LMIOGs),11 as developed by Pfeffer et al., has been particularly studied with this in mind. The incorporation of coordination ligands for such self-assembly formations has also been explored, resulting in the generation of metallo-supramolecular polymers and gels,12–14 some of which have properties beyond that of the ligand or metal-complex alone,15 as we have recently demonstrated using bio-inspired ligand design and Eu(III) as the f-metal ion of choice,16 but the lanthanides have both physical and coordination properties that are highly desirable for the generation of responsive luminescent materials.The lanthanides are strong Lewis-acids, have diverse coordination requirements, are paramagnetic and some also display delayed metal-centred luminescence, which is both line-like, occurring at long wavelengths (visible to NIR) and long lived.17 As such, lanthanide complexes are highly desirable to be employed in the development of higher order supramolecular and responsive structures and materials.18,19 Unfortunately, in most cases, due to their Laporte-forbidden transitions, it is necessary to employ sensitizing antennae to take full advantage of these aforementioned photophysical properties. We have strong interest in harvesting these properties within supramolecular and material chemistries.20–22
Recently we have developed 2,3-bis(1,2,3-triazol-4-yl)picolinamide (btp) derivatives for use in various supramolecular chemistry applications.23 This includes their use in the formation of luminescent coordinating polymers and metallo-gels.24,25 Gratifyingly, the btp based ligands can sensitise the excited states of various lanthanides, including the 5D4 state of Tb(III) (resulting in green emission) resulting in the formation of complexes possessing high Tb-cantered quantum yield (up to 90%), and to a lesser extent, the 5D0 state Eu(III), which results in red emission (but much lower quantum yield) in solution.26 We also recently incorporated btp based lanthanide complexes into polymer matrixes in a non-covalent manner.27 While this work shows promise, the ligand system is found to be susceptible to leaching. To overcome this issue, covalently tethering the btp ligand to the polymer matrix was considered, but we have shown in related work, that ligands and Ln-complexes made from naphthyl-dipicolinic amide (dpa)28 ligands, can be functionalised in two different ways to allow for such incorporation into polymers, and that this modification influences their photophysical properties within the polymers. Consequently, we set out to develop the btp monomer 2, Fig. 1, from amine 1, as well as forming 3, as a model complex for studying the solution properties of the ligand and the corresponding Ln-complexes.
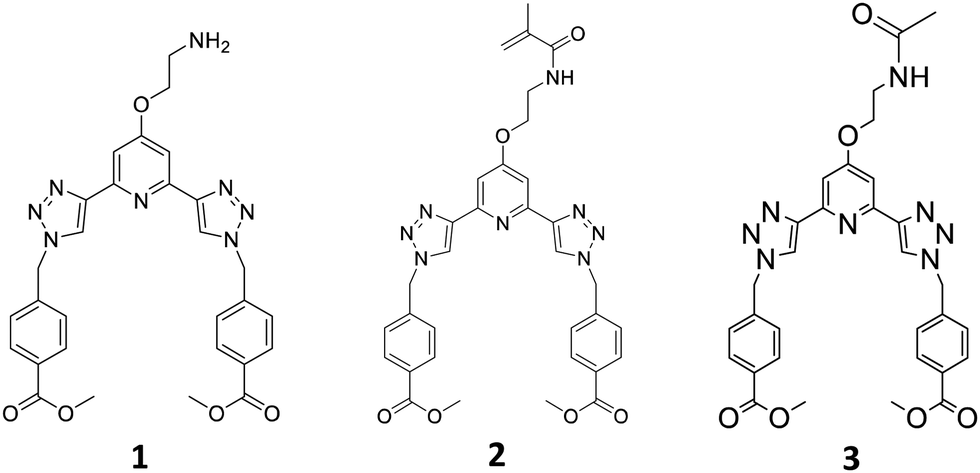 | ||
| Fig. 1 The three main ligands 1(as free base)-3 developed for this project. | ||
In the case of 2, the methylacrylamide unit would facilitate the co-polymerisation of the btp ligand into polymeric hydrogels. This modification would (i) allow for the generation of luminescent responsive systems, and (ii) facilitate the generation of lanthanide cross-linked hydrogels, the functionalization at the 4th position of the pyridine facilitating 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (metal to ligand) complex formation. This second point is very important, as due to the high-coordination requirement of the lanthanides, we would expect three btp ligands to coordinate to a single Ln-ion, thus resulting in the potential to modulate both the ‘hard’ and ‘soft’ nature of the materials. At the same time, the luminescent properties of the lanthanides, would furnish them with rich spectroscopic properties (i.e. point i). Here, we give full account of this work.
:3 (metal to ligand) complex formation. This second point is very important, as due to the high-coordination requirement of the lanthanides, we would expect three btp ligands to coordinate to a single Ln-ion, thus resulting in the potential to modulate both the ‘hard’ and ‘soft’ nature of the materials. At the same time, the luminescent properties of the lanthanides, would furnish them with rich spectroscopic properties (i.e. point i). Here, we give full account of this work.
Results and discussion
Synthesis and characterisation of 1–3 and polymer films F1 an F2
The synthesis of the monomer 2 was achieved from the amine 1, which was formed in a seven-step synthetic procedure (see ESI†), starting from the commercial starting material 2,6-dibromopyridine, using synthetic procedures developed in our laboratory.24–27‡ All the intermediates were fully characterised by NMR spectroscopies, IR spectroscopy and HRMS. The final two steps in the formation of 1 involved the use of click chemistry under CuAAC conditions using CuSO4·5H2O in a DMF:H2O mixture, followed by removal of a Boc protecting group to give the free amine 1, being formed in a moderate yield of 52%, as a brown precipitate, and TFA salt (The structure of 1 was further confirmed by X-ray crystallography as will be discussed further below).
The btp monomer 2 was formed by reacting the amine 1 with methacryloyl chloride for three hours in the presence of triethylamine. After concentrating the reaction mixture, the crude product was extracted into CHCl3, and purified by column chromatography to yield 2 as a white solid in 63% yield. Formation of the desired product was confirmed by 1H NMR spectroscopy (see ESI† for 1, 2 and 3). Two singlet peaks at 5.68 and 5.34 ppm correspond to the alkyne protons, and a peak at 1.85 ppm assigned to the CH3 of the methacrylate moiety, confirm the presence of monomer 2. As for 1, compound 2 was fully characterised as outlined in the Experimental section.
As our main objective was to develop a lanthanide luminescent material, it was essential to photophysically characterise the resulting material, and compare its photophysical properties with a reference complex. As compound 1 has a primary amine at the backbone of this ligand, we anticipated that the luminescent properties of the neutral ligand might be compromised by an active photoinduced electron transfer (PET) from the amine to the excited state of the ligand, resulting in so called PET quenching.29 Because of this it could not be used as a model compound for these studies, and hence, we synthesised compound 3. The acetamide derivative 3 is not PET active, as the amide increases the oxidation potential of the ‘previous amine’, which prevents any PET activity. This structure also provided us with a ‘structural amide model compound’ to investigate the effect of the covalent attachment of 2 to a polymeric backbone, and for studying the photophysical behaviour of this system upon cross-linking by lanthanide ions, such as Tb(III). To achieve this, the primary amine of 1 was simply protected with an acetyl group, by reacting 1 with acetic anhydride in the presence of triethylamine. This generated 3 as a white solid in 83% yield, after workup; 3 being fully characterised by NMR and IR spectroscopy and HRMS (see Experimental).
The general synthesis of the polymeric films F1 and F2 is shown in Scheme 1. This simply involved stirring 2 in a solution of 2-hydroxyethyl methacrylate (HEMA), methylmethacrylate (MMA) and ethylene glycol dimethacrylate (EGDMA,) at room temperature until 2 was completely dissolved (0.3 and 3 mg for F1 and F2, respectively). AIBN was then added and the clear solution was injected into a non-stick mould (see Fig. in ESI†). It was heated in the oven at 80 °C for 6 hours and after cooling to room temperature the material was released from the mould and cut into sections. The material was soaked in H2O overnight, followed by CH3OH overnight to remove any remaining inhibitor or unreacted monomer. As seen in Scheme 1, the obtained film was stiff (and could easily be broken) but upon hydration gave rise to an easily handled soft, flexible film. To ensure that the film remained soft, the films were stored in a deionised aqueous solution. Having these three btp systems at hand as well as F1 and F2, we started exploring their physical and photophysical properties, commencing with 3 and its Tb(III) complex 3.Tb.
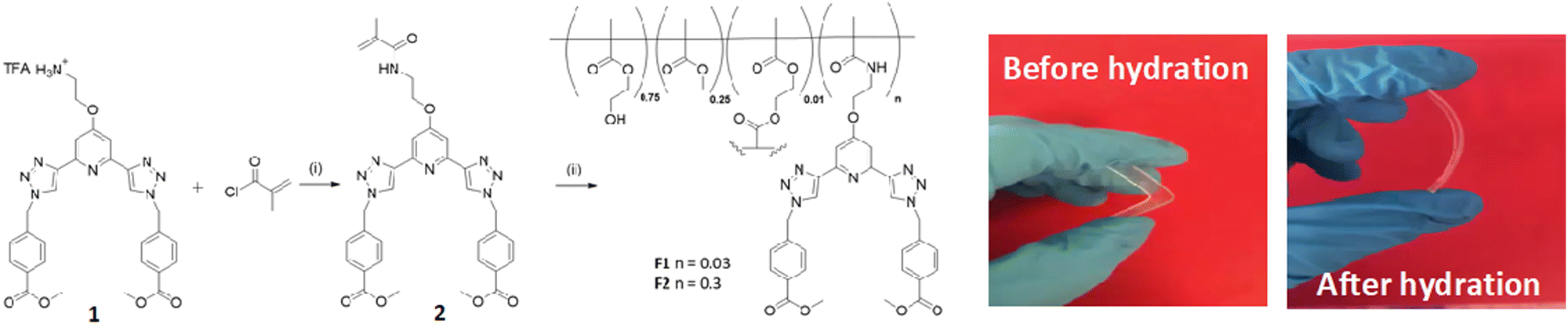 | ||
| Scheme 1 Synthesis and structure of monomer 2, formed from ligand 1, and the two polymeric films F1 and F2. Reagents and conditions: (i) NEt3, CH2Cl2 (ii) 80 °C 6 hours. F1 (0.03 wt%) and F2 (0.3 wt%), respectively. Pictures show the films formed before and after being hydrated. | ||
Synthesis and characterisation of the Tb(III) complex 3.Tb
As mentioned above, we have shown that btp ligands upon complexation with lanthanide ions, give rise to up to 30-fold higher quantum yields for complexes formed from Tb(III) than Eu(III).26a With this in mind, we commenced our work with exploring the photophysical properties of Tb(III), and the Tb(III) complex of 3, 3.Tb, was formed, structurally characterised and photophysically analysed. This was achieved by reacting 3 with Tb(CF3SO3)3 in a 1:3 M:L stoichiometric ratio in CH3OH under microwave irradiation for 15 min at 70 °C. Thereafter, the solvent was concentrated in vacuo, and the desired product isolated by vapour diffusion of diethyl ether into the reaction mixture, to yield a white solid in 79% yield.
The complex was characterised by 1H NMR spectroscopy, IR spectroscopy and HRMS. The 1H NMR spectrum (400 MHz, CD3CN-d3) can be seen in Fig. 2a, and shows both broadening and significant resonances shifts when compared to the free ligand 3, due to the paramagnetic nature of the Tb(III) ion. Elemental analysis was used to confirm the formation of this tris complex, as well as HRMS analysis, though only the characteristic isotopic distribution pattern for the [Tb(3)3(CF3SO3)2]+ complex was observed in the MALDI HRMS with a m/z = 1677.2877 (see ESI†). The photophysical investigation of this ligand (as well as that of the methacryloyl derivative 2) and the resulting Tb(III) complex is outlined in next two sections.
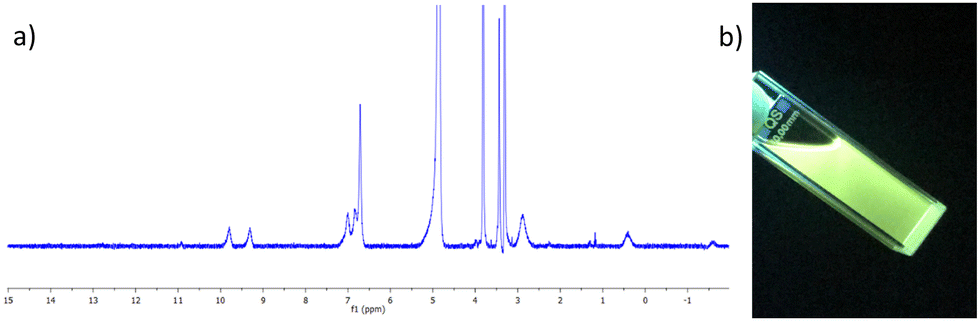 | ||
| Fig. 2 (a) The 1H NMR spectrum (400 MHz, CD3CN-d3) of [Tb(3)3](CF3SO3)3 showing significant shift and broadening of the signals relative to the free ligand 3 (see ESI† for NMR). (b) The bright green Tb(III) emission arising from the 3.Tb complex in CH3CN under UV-light (λex = 254 nm). | ||
Photophysical evaluations of the Tb(III) complex 3.Tb
Having successfully synthesised 3.Tb we next investigated the photophysical properties of the complex in CH3CN, and in MeOH, some key results are shown in Fig. 3. In CH3CN, the UV-visible absorption spectrum of 3.Tb was dominated by a high-energy band at λmax = 236 nm and a lower energy band at λ = 298 nm, with a shoulder at λ = 309 nm (Fig. 3a). Excitation of the complex at λ = 240 nm gave rise to Tb(III)-centred emission with characteristic line-like transitions at λ = 490, 545, 585, 622, 648, 667 and 678 nm, Fig. 3b. These transitions are indicative of Tb(III)-sensitisation by the ligand antenna 3, and can be assigned to the 5D4 → 7FJ transitions (J = 6–0). The complex was found to be stable over 48 hours with no loss of luminescence.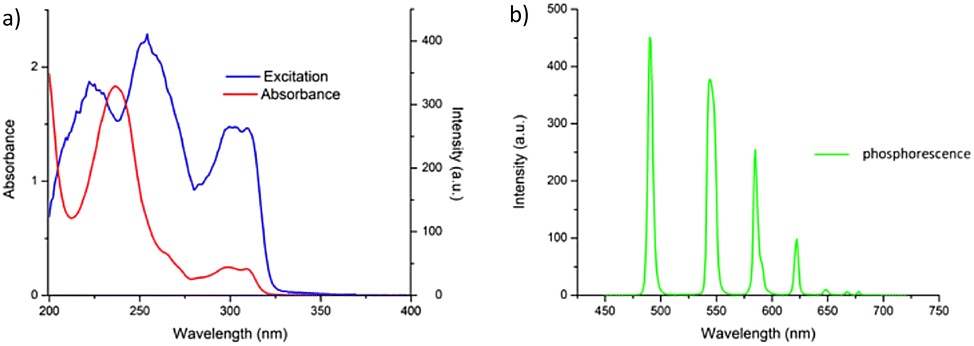 | ||
| Fig. 3 (a) The UV-vis absorption (red) and excitation spectra (λem = 545 nm) of the btp complex 3.Tb in CH3CN showing sensitisation arising from ligand-centred energy transfer (left), and time-gated emission spectrum of 3.Tb showing the characteristic Tb(III) centred-emission. | ||
The excitation spectrum, recorded for the Tb(III)-centred luminescence with λem = 545 nm, also provided evidence for the sensitisation of the Tb(III) ion from the ligand-centred excited states. The structural features of the excitation spectrum closely resemble the absorption spectrum of 3, which indicates indirect excitation of the metal centre from the antenna moiety (Fig. 2a). Furthermore, green emission was also observed from both the isolated solid complex, and from the complex dissolved in solution, such as CH3CN, upon irradiation with UV light (λexc = 254 nm), as demonstrated in Fig. 2b, which was clearly visible to the naked eye.
We also evaluated the hydration state (the so-called q value) of the complex, which indicates the number of associated metal-coordinated solvent molecules, and indicated the coordination number of the complex.30 This was achieved by measuring the Tb(III) excited state lifetimes in CH3OH and CD3OD, respectively. These lifetimes were best fitted to a monoexponential decay function, indicating the presence of a single luminescent species in both solvents, with τ = 1.88 ± 0.1 ms and 1.98 ± 0.2 ms, in CH3OH and CD3OD, respectively. From these measurements a q value of ≈ 0 was determined, demonstrating that the Tb(III) inner coordination sphere was fully saturated by the three terdentate 3 ligands, and that under thermodynamic control, 3.Tb excised as 1:3 (metal to ligand) stoichiometry in solution.
The photoluminescence quantum yield (ΦTot, %) of 3.Tb was also determined in CH3CN as 72% which is very close to relative quantum yields reported for other such complexes as mentioned above.26a,31
Photophysical evaluations of 3 in presence of Tb(III) in situ
Before investigating the photophysical properties of the btp Ln-cross-linked polymers F1 and F2, it was necessary to probe the photophysical properties of the lanthanide complexes formed from 3 under kinetic control. This mimics the condition of the free ligands within the polymers upon their hydration with solutions of lanthanide ions. To achieve this, both the ground and the excited state properties were probed in situ upon titrating the ligand with Tb(III) triflate solutions in CH3CN solution. From this we were able to determine both the stoichiometry and the binding constants for the formation of the Tb(III) complex(es) with 3.32,33The UV-visible absorption spectrum of compound 3 consists of two main bands appearing at λmax = 231 nm and λmax = 300 nm (see ESI†), from which ε = 54487 cm−1 M−1 (λabs = 231 nm) was determined. Significant changes were observed in the UV-visible absorption spectrum upon addition of Tb(III) (see ESI†); the high energy band underwent a hypochromic shift, up to the addition of 0.15 equivalents of Tb(III), which was followed by a redshift to λ = 236 nm and a concomitant hyperchromic shift up to the addition of 0.3 equivalents of Tb(CF3SO3)3, indicating the successful formation of the aforementioned 1:3 stoichiometry (see ESI†). Three isosbestic points at 232, 247 and 286 nm were observed during these titrations. Subsequent additions of Tb(III) did not result in any further changes to the band at 236 nm. The lower energy band at λ = 298 nm also experienced a hyperchromic shift up to the addition of 0.3 equivalents of Tb(III).
Upon excitation of the ligand 3 at λ = 237 nm, ligand centred fluorescence emission was observed, with a band centred at λ = 319 nm (see ESI†). This ligand luminescence was affected by the self-assembly process, which was evident by quenching of the ligand fluorescence upon addition of Tb(III). Upon addition of 0.3 equivalents of Tb(III), a 96% reduction in the emission was observed, which can be attributed to the energy transfer, and hence, sensitisation of the 5D4 excited state of Tb(III) by the ligand 3.
The delayed Tb(III)-centred luminescence was also recorded upon excitation at 237 nm, reflecting the appearance of the Tb(III) emission due to the effective sensitisation by the btp ligand, Fig. 4a. A gradual enhancement in the Tb(III) luminescence was observed from 0 → 0.3 equivalents of Tb(III), with characteristic Tb(III)-centred emission bands appearing at λ = 490, 545, 583, 620, 646, 665, and 675 nm, corresponding to the deactivation of 5D4 → 7FJ states (J = 6–0). The 5D4 → 7F2−0 bands are typically weak and not always seen in such titrations, but they can be clearly observed in the formation of 3.Tbin situ. Subsequent additions of Tb(III), from 0.3→3.0 equivalents, resulted in quenching of the luminescence intensity by approximately 20%.
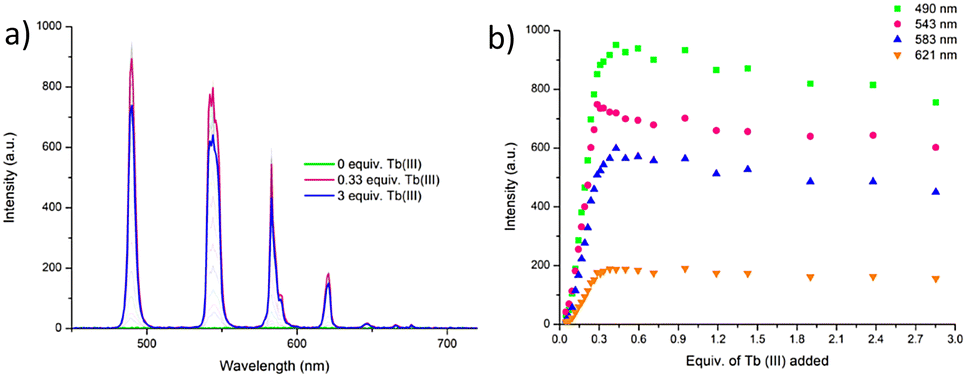 | ||
| Fig. 4 (a) The overall changes to the Tb(III)-centred emission upon titrating 3 (1 × 10−5 M) against Tb(CF3SO3)3 (0 → 3 equiv.) in CH3CN at 22 °C. (b) The corresponding experimental binding isotherms for the Tb(III) emission at λ = 490, 545, 583 and 621 nm. These results were fully reproducible (see enlarged version in ESI†). | ||
As can be seen from Fig. 4a, The most emissive state in solution was seen at approximately 0.3 equivalents of Tb(III), which is consistent with previously reported btp-Tb(III) systems, as at this stoichiometry, the Tb(III) ion is coordinatively saturated, and thus completely shielded from any solvent quenching. Lifetime measurements obtained at the addition of 0.3 equivalents of Tb(III) gave τ = 1.85 ms, which is very close to the lifetimes observed above for the thermodynamically formed 3.Tb. These titrations demonstrate that the 1:3 stoichiometry is the most favourable one formed at low concentration and is quite stable to dissociation upon increasing Tb(III) concentration.
With the view of quantifying the above stability, the changes observed in the ground and the excited state titrations were fitted using non-linear regression analysis program ReactLab EQUILIBRIA®. Analysis of the UV-visible absorption data of 3 titrated with Tb(CF3SO3)3 in CH3CN solution suggests the presence of three main species in solution (3, 3:1 metal:ligand and 1:1 metal:ligand ratios) with the distribution of these species estimated from this analysis (see ESI†); with the ML3 species being the most dominant in solution, reaching 97% abundance at 0.3 equivalents of Tb(III). The 1:3 metal:ligand assembly formed with logβ13 = 23.1 ± 0.6, which is in good agreement with binding constants reported for similar btp systems in the literature with Tb(III).22,23 The second species (1:1 metal:ligand) had a calculated binding constant of logβ11 = 8.3 ± 0.2, which again, is in good agreement with previously reported Tb-btp assemblies.§
Similar analyses of the changes in the Tb(III)-centred emission in Fig. 4a were also carried out and indicated the presence of four main species in solution, (3, 3:1 metal:ligand, 2:1 metal:ligand and 1:1 metal:ligand species). Again, at 0.3 equivalents of Tb(III), the Tb33 species was the most predominant in solution, reaching an abundance of 96%, Fig. 4b. Upon subsequent additions of Tb(III), the 1:2 species was observed, which reaches 20% abundance at the addition of 1.5 equivalents of Tb(III) before reaching a plateau. The evolution of the 1:1 species occurred almost simultaneously with the 1:2 species; however, it is less predominant until the addition of 1.8 equivalents of Tb(III), after which point it continues to increase with a concomitant decreasing of the 1:3 species (see ESI†). From these luminescent changes, the binding constants were determined as logβ1:3 = 22.7, logβ1:2 = 14.5 ± 0.1, and logβ1:1 = 7.0. The stability constants are roughly additive, suggesting that the binding process of each ligand to the metal centre was subject to a similar formation constant.
In addition to these measurements, the self-assembly of compound 2 with Tb(CF3SO3)3 was also investigated in CH3CN solution, in an identical manner to that described above for compound 3. The changes to the UV-visible absorption, fluorescence, and time-gated emission spectra were very similar to those shown for compound 3 with Tb(III). The data was fitted using non-linear regression analysis, and four species were again estimated to be present in solution for the Tb(III) complex formation of 2. The global analysis of these changes was found to be in good agreement with the analysis for compound 3 with Tb(III).
Supramolecular gelation studies of 1
In the past, we have demonstrated that btp ligands possessing carboxylates at both the 4th position of the pyridine, as well as at the two aryl groups on the appended arms (which in the case of 1–3, are protected as methyl esters) are excellent LMWG, and give rise to the formation of transparent or opaque self-healing soft materials, which upon interactions/complexation with lanthanides, can result in the formation of luminescent supramolecular gels.24 Because of this, compound 1 was investigated as well as a potential LMWG.Ligand 1 was suspended in a variety of solvents; CH3CN, CH3OH, CH3OH:H2O, C2H5OH, C2H5OH:H2O, and subject to different gelation conditions including heating and sonication. Of these, the suspension of 1 in CH3CN, followed by sonication, resulted in the formation of an off-white soft material almost immediately. Following this result, the optimum concentration for gelation was determined. Compound 1 (16 mg) was suspended in CH3CN (1 mL) and sonicated for 60 seconds to yield an opaque gel that was robust to the naked eye. The inversion test was performed, and the gel supported its own weight over several hours, Fig. 5c. The thermoreversibility of the gel was investigated by heating the material to up to 60 °C. Upon heating, the gel dissolved and, after standing at room temperature for 15 minutes, the gel reformed, indicating that the material was formed in a thermoreversible manner.
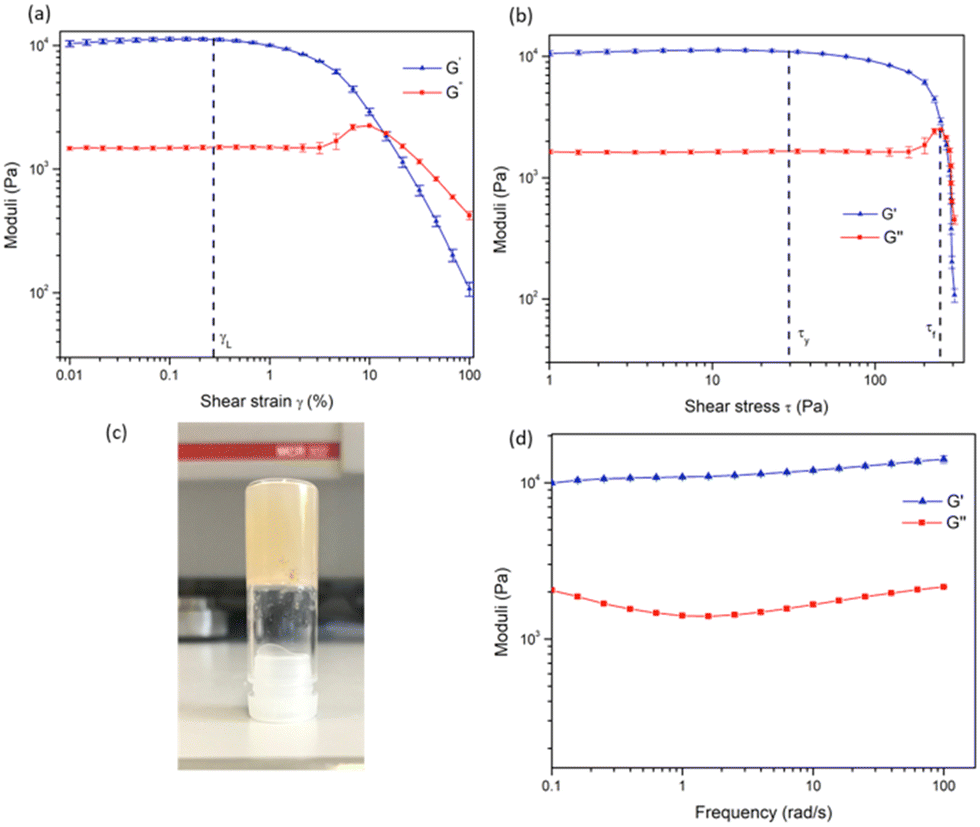 | ||
| Fig. 5 (a) Amplitude (strain) sweep experiment of 1 gel showing the linear viscoelastic region (γL) and confirming the behaviour of the material as a gel. (b) Amplitude (stress) sweep showing yield point (τy) and flow point (τf) of the material. (c) Image of the gel during inversion test. (d) Frequency sweep experiment showing the long-term stability of the gel. | ||
In order to investigate the LMWG wt% at which the gel forms, TGA was carried out on the material. TGA was carried out in the temperature range of 15 to 500 °C, and a mass loss of 96.2% was observed at 100 °C, indicating the gel (as shown in Fig. 5) was formed at 3.8 wt% of 1 (see ESI†). However, as the gel was prepared at 1.6 wt%, this experiment indicates that the gel does not retain all of the solvent in the sample. While inverting the gel upon formation did not result in any loss of residual solvent (i.e the gel did not collapse, as shown in Fig. 5c).
In order to examine the mechanical properties of the gel, rheological experiments were conducted using 3.8 wt% of 1, and an amplitude sweep was conducted in order to confirm the gel-like behaviour of the material. The strain on the sample was increased logarithmically, while the frequency was kept constant. In Fig. 5 it can be seen that the storage modulus, G′ was larger than the loss modulus, G′′, which indicates that the material can be classified as a gel or a viscoelastic solid material (G′ = 1.09 × 104 Pa and G′′ = 1.46 × 103 Pa). The limit of the linear viscoelastic region (LVE, labelled γL in Fig. 5), the range in which further tests could be carried out on the sample without destruction of the sample, was determined to be 0.3% for this sample. After this point, G′ and G′′ begin to converge and the material starts to flow.
From the graph of G′ and G′′ vs. shear stress (τ), in Fig. 5, the yield point and flow point of the material can be determined. The yield point, τy, is value of shear stress at the limit of the LVE region. For this material its value was determined to be 28 Pa, and the flow point, τf, was observed to be 253 Pa. The rise in G′′ to a maximum which begins to drop again prior to the crossover point as seen in Fig. 5a and b suggests the breakdown of the gel microstructure prior to formation of macro cracks leading to material flow. A frequency sweep experiment was also conducted to gain an insight into the long-term stability of the gel. Having determined that the limit of the LVE region was 0.3%, the amplitude was set at 0.1%. As seen in Fig. 5G′ > G′′ over the entire frequency range, which indicates that the material has a cross-linked structure. Rheological tests were repeated three times with three gel samples to ensure reproducibility.
A xerogel of compound 1 (as its salt form) in CH3CN was also generated by drying the sample under vacuum in order to study the morphology of the system by SEM. The SEM images, Fig. 6a, show the fibrous, entangled nature of the gel structure. This morphology is typical of many supramolecular gels in which non-covalent interactions are involved in forming cross-links within the structure immobilising the solvent, such as hydrogen and ionic interactions. At higher wt%, Fig. 6b, a precipitate appeared on the fibres, seen in the SEM images as bright spots. These are very likely due to salt formation, however, the nature of these was not analysed further.
 | ||
| Fig. 6 (a) SEM images of 1 gel (scale bar 200 nm) showing fibrous structure of the material. (b) The formation of salt particles on the surface of the fibres upon gelation at higher concentrations. | ||
Having formed organogels from 1, the possibility of forming a supramolecular metallogel with Tb(III) was next investigated. To do this, a solution of Tb(CF3SO3)3 was added to the pre-formed organogel, by layering a solution of known concentration of the ion on the top of the gel and allowing it to defuse through the gel over period of time. However, unlike previous observations for btp based ligands (cf. above discussion) upon standing overnight, the material lost its soft-material properties and the gel dissolved, giving a solution, which was shown to emit green light under UV-irradiation (λexc = 254 nm) (see ESI†). We have previously seen this behaviour for dpa based gels.32 This strongly indicates the formation of a Tb(III) complex, which likely breaks-down the network of interactions which otherwise generate the supramolecular soft-material upon formation (e.g. such as hydrogen bonding and π–π stacking).
Crystallographic characterisation of [H11+][CF3SO3−]·MeCN
As outlined above, the gelation studies were carried out at different wt% of 1. When the organogel was formed at low, or less than 1.5 wt%, a gel-to-sol transition occurred overnight, and yellow crystals were formed in a colourless solution. These crystals were examined by X-ray diffraction and were found to be crystals of the TFA salt of compound 1, [H11+][CF3SO3−]·MeCN, as illustrated in Fig. 7 (see also ESI†).34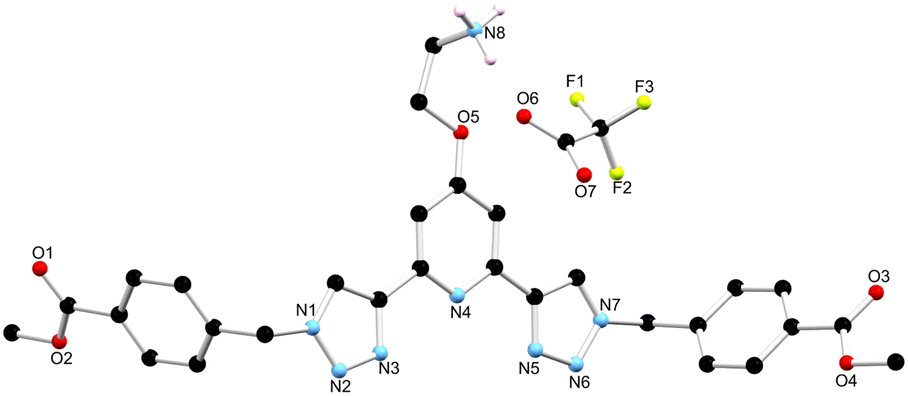 | ||
| Fig. 7 The solid state X-ray structure of compound 1 with heteroatom labelling scheme; lattice acetonitrile molecule and selected hydrogen atoms omitted for clarity. | ||
The structure was solved in the monoclinic C2/c space group, with the asymmetric unit containing one complete protonated molecule of 1, as well as one counterbalancing TFA anion, and a solvent molecule of CH3CN in the lattice. The primary intermolecular interactions in the structure of 1 are hydrogen bonds, involving the oxygen atom of the TFA anion, O6 and the NH3+ of compound 1, with an N⋯O distance of 2.793(5) Å and an N–H⋯O angle of 175.3(3)°. Unexpectedly, the triazoles in ligand 1 adopt a syn–syn conformation. Previous btp ligands have been most commonly reported in the anti–anti conformation, but in the presence of a cation such as a metal ion or a proton, the triazoles can adopt the syn–syn conformation.23,35,36 In this case, the RNH3+ of one ligand molecule, and the triazole N14 and N31 nitrogen atoms of the adjacent ligand molecule, form a hydrogen bonding interaction with an N⋯N distance of 2.998(6) and 2.885(6) Å, respectively, which causes the triazoles to adopt the syn–syn conformation.
Crosslinking polymers F1 and F2 with Ln(III) ions
Having confirmed the incorporation of 2 in the polymer matrix, the ability of the hard dehydrated polymer to uptake Ln(III) ions was investigated. Initially, the dry polymer F2 (which had a higher concentration of the btp ligand) was soaked in an aqueous solution of Tb(CF3SO3)3 and after 10 minutes the resulting soft, flexible polymer was rinsed with water and examined under UV-irradiation at λexc = 254 nm, in water at room temperature. The film exhibited bright green emission under UV-light, which was clearly visible to the naked eye, indicating that crosslinking had occurred with Tb(III) and that the covalently bound btp ligand was sensitising the Tb(III) excited within the polymer film, as is evident from the Fig. 8a. The changes in the Tb(III) centred emission for F1 was similarly enhanced upon soaking in Tb(III) solution over a longer period of time, with all the expected transitions being clearly visible.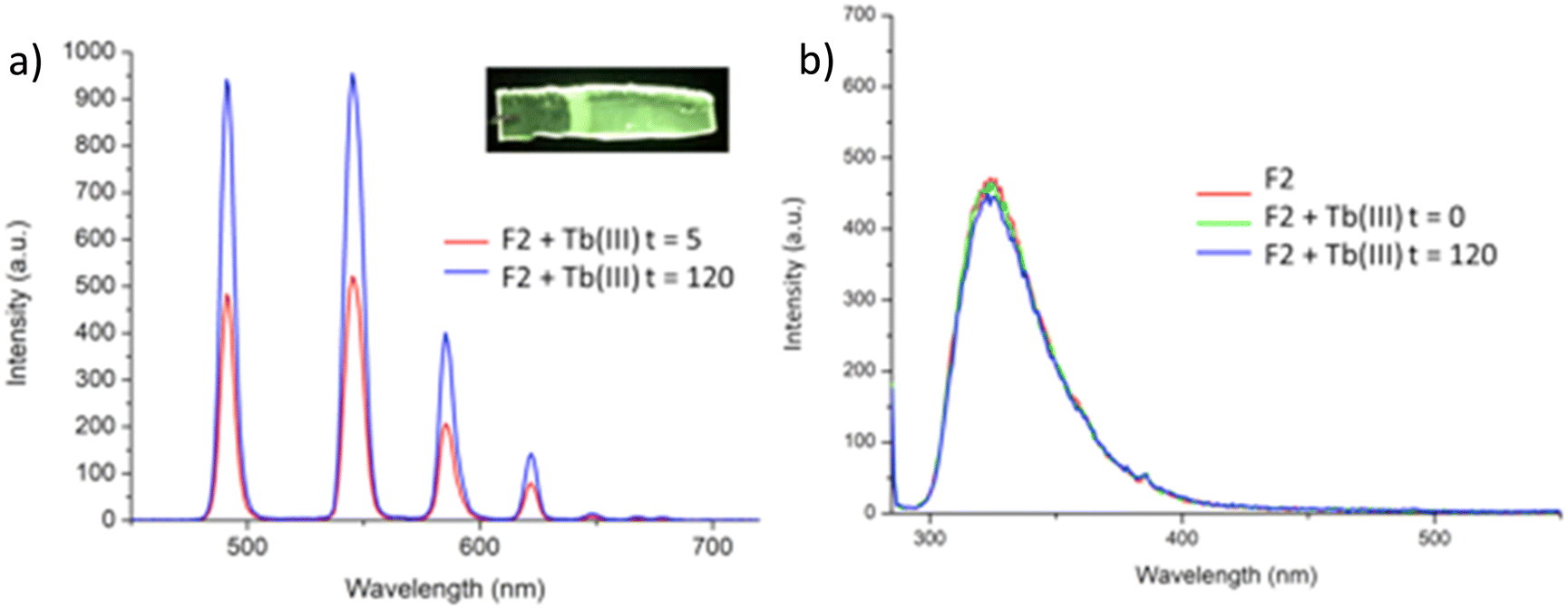 | ||
| Fig. 8 (a) The Tb(III)-centred emission of F2 when measured in H2O after having soaked the polymeric film in a solution of Tb(III) triflate at differ times (given in minutes). Inset (a): Photograph of F2 film following soaked in Tb(CF3SO3)3 under UV-irradiation (λexc = 245 nm), showing the characteristic green Tb(III) luminescence visible to the naked eye. (b) The fluorescence emission spectrum of F2 in H2O at different times. | ||
Examining F2 further, showed that upon excitation of the film (on a quartz slide) at λ = 285 nm in water, the delayed Tb(III)-centred luminescence could be recorded in water and the spectrum can be seen in Fig. 8a. Characteristic Tb(III)-centred luminescence was evident with Tb(III)-centred transitions at λ = 490, 545, 583, 620, 646, 665 and 675 nm, corresponding to deactivation from the 5D4 excited states to the 7FJ states (where J = 6–0). F2 was found to be stable in water; with no evidence of changes in the intensity of the Tb(III)-luminescence spectra seen over several hours. This suggests that the Tb(III) ions underwent diffusion through the polymer film, and did not simply attach to the surface of the film. Thus, it is likely that Tb(III) mediated self-assembly occurs within the polymer matrix, leading to the green luminescence observed, and that the resulting cross-linked btp–Tb complexes within the film are stable to dissociation under such competitive media. This is a very important observation that demonstrates that the grafting of 2 to the backbone of the polymer indeed adds stability to the btp-Ln interaction, which we have not previously seen when btp-Tb complexes were incorporated into such matrixes in a non-covalent manner.22
As was mentioned above, the emission of the Eu(III) is significantly less than for the Tb(III) complexes for the btp based ligands. Nevertheless, to demonstrate that other lanthanides such as Eu(III) could also be used in the formation of luminescent cross-linked polymers was also briefly investigated. As for the Tb(III) above, the polymer film F2 was soaked in a Eu(CF3SO3)3 solution for 10 min at room temperature and the spectra were recorded in H2O resulting in the characteristic red emission of Eu(III) under UV-irradiation, which again, was clearly visible to the naked eye, as shown in Fig. 9a, and demonstrates that Eu(III) can be equally employed in such luminescent material, even though it has (as we have demonstrated in solution on many occasions) much lower Ln-centred quantum yields. As for Tb(III), the delayed Eu(III)-centred luminescence was also recorded upon excitation at λ = 285 nm and the spectrum is shown in Fig. 9a, forF2, Eu(III)-centred transitions appeared at λ = 595, 620, 652 and 695 nm corresponding to the 5D0 → 7FJ (J = 0–3) states characteristic of Eu(III) emission. The fluorescence emission is shown in Fig. 9b, mirroring that seen for Tb(III) above in Fig. 8b for Tb(III).
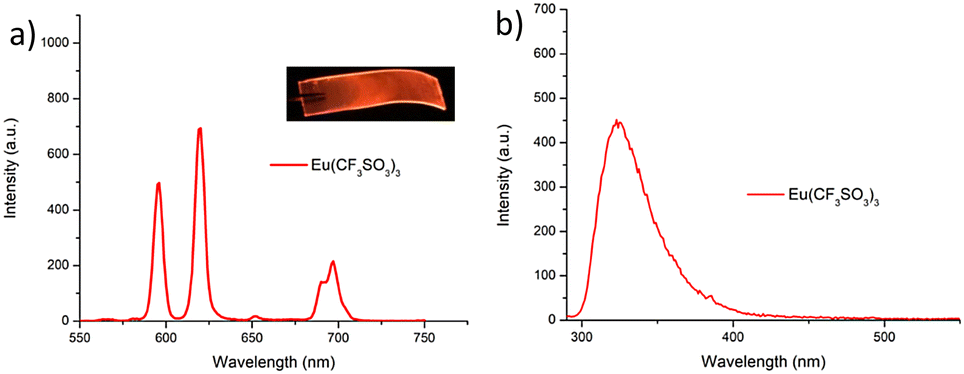 | ||
| Fig. 9 (a) The Eu(III)-centred emission observed for F2 when measured in H2O. (b) The corresponding fluorescence spectrum of F2 measured in H2O. Inset (a) photograph of polymer following soaking for 10 min in Eu(CF3SO3)3 solution under UV-irradiation showing the characteristic red luminescence from Eu(III). | ||
Conclusion
Herein, we have developed the btp ligands 1, 2, and 3 as coordinating ligands for metal ions, and explored the luminescent properties of these ligands upon formation of Tb(III) complexes both in solution and as, in the case of 2, upon co-polymerising the ligand with HEMA, MMA and EGDMA. The model compound 3 was employed to form a Tb(III) complex, which was formed in 1:3 (M:L) stoichiometry. This complex was luminescent, possessing high quantum yield of luminescence, and when formed in situ was produced as the main species in solution at 96%, upon addition of 0.3 equivalents of Tb(III). We also demonstrated that a supramolecular gel with fibrous morphology, can be produced from the precursor 1, which upon interactions with Tb(III) dissolves, as weak supramolecular interactions that produce the gel are replaced with stronger coordination bonds.
The btp based polymers F1 and F2, were formed and both were shown to be luminescent, and metal ion mediated cross-linked, upon interaction with Tb(III) (and Eu(III) in the case of F2). We further demonstrated that unlike the incorporation of btp-based Tb(III) complexes non-covalently into such polymers (such as into a blank HEMA-co-MMA-co-EGDMA polymer), no leakages were observed from the polymeric film upon prolonged soaking in water. Moreover, the Tb(III) centred emission was shown to be stable over a period of time. Furthermore, we show that in competitive media, e.g. water, the polymer is highly luminescent indicating that no dissociation occurs in the cross-linked btp-based Tb(III) complexes within the hydrogel. This is highly important, as it demonstrates significant improvement on the use of non-covalently used btp-based Tb(III) complexes, which indeed showed rapid dissociation and subsequent loss of metal centred luminescence.
The lanthanide cross-linked polymers offer a desirable means of forming functional polymeric systems, where both conventional covalent and coordination metal ion bonds are employed, and we are currently investigating the advantage of such features. This includes the use of ligands that have conjugated receptor sites within the ligand structures of these and related systems, with the view of forming stimulated responding polymers that can be used in luminescent sensing.
Experimental
Synthesis
Please refer to Scheme S1 in ESI.†General experimentals and methods
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the School of Chemistry TCD, Science Foundation Ireland (SFI PI Awards 10/IN.1/B2999 and 13/IA/1865 to TG). TG would also like to thank the SFI funded AMBER Centre for financial support (SFI 12/RC/2278_P2). EPSRC EP/S009000/1 is acknowledged for support of Dr Niamh Willis-Fox.Notes and references
- (a) G. Sinawang, M. Osaki, Y. Takashima, Y. Hiroyasu and A. Harada, Supramolecular self-healing materials from non-covalent cross-linking host–guest interactions, Chem. Commun., 2020, 56, 4381 RSC; (b) X. Yan, F. Wang, B. Zheng and F. Huang, Stimuli-responsive supramolecular polymeric materials, Chem. Soc. Rev., 2012, 41, 6042 RSC.
- (a) A. J. Savyasachi, O. Kotova, S. Shanmugaraju, S. J. Bradberry, G. M. Ó'Máille and T. Gunnlaugsson, Supramolecular Chemistry: A Toolkit for Soft Functional Materials and Organic Particles, Chem, 2017, 3, 764 CrossRef CAS; (b) M. Coste, E. Suárez-Picado and S. Ulrich, Hierarchical self-assembly of aromatic peptide conjugates into supramolecular polymers: it takes two to tango, Chem. Sci., 2022, 13, 909 RSC; (c) G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, New horizons for catalysis disclosed by supramolecular chemistry, Chem. Soc. Rev., 2021, 50, 7681 RSC; (d) C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Biomimetic Self-Healing, Angew. Chem., Int. Ed., 2015, 54, 10428 CrossRef CAS PubMed.
- (a) T. Gorai, W. Schmitt and T. Gunnlaugsson, Highlights of the development and application of luminescent lanthanide based coordination polymers, MOFs and functional nanomaterials, Dalton Trans., 2021, 50, 770 RSC; (b) A. B. Aletti, S. Blasco, S. J. Aramballi, P. E. Kruger and T. Gunnlaugsson, Sulfate-Templated 2D Anion-Layered Supramolecular Self-Assemblies, Chem, 2019, 5, 2617 CrossRef CAS.
- (a) F. Chen, Y.-M. Wang, W. Guo and X.-B. Yin, Color-tunable lanthanide metal–organic framework gels, Chem. Sci., 2019, 10, 1644 RSC; (b) W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Metal–organic frameworks: functional luminescent and photonic materials for sensing applications, Chem. Soc. Rev., 2017, 46, 3242 RSC.
- (a) É. Whelan, F. W. Steuber, T. Gunnlaugsson and W. Schmitt, Tuning photoactive metal–organic frameworks for luminescence and photocatalytic applications, Coord. Chem. Rev., 2021, 437, 213757 CrossRef; (b) T. Gorai, J. I. Lovitt, D. Umadevi, G. McManus and T. Gunnlaugsson, Hierarchical supramolecular co-assembly formation employing multi-component light-harvesting charge transfer interactions giving rise to long-wavelength emitting luminescent microspheres, Chem. Sci., 2022, 13, 7805 RSC.
- J. I. Lovitt, T. Gorai, E. Cappello, J. M. Delente, S. T. Barwich, M. E. Möbius, T. Gunnlaugsson and C. S. Hawes, Supramolecular aggregation properties of 4-(1-morpholino)-1,8-naphthalimide based fluorescent materials, Mater. Chem. Front., 2021, 5, 3458 RSC.
- (a) R. Daly, O. Kotova, M. Boese, T. Gunnlaugsson and J. J. Boland, Chemical Nano-Gardens: Growth of Salt Nanowires from Supramolecular Self-Assembly Gels, ACS Nano, 2013, 7, 4838 CrossRef CAS PubMed; (b) M. Martínez-Calvo, O. Kotova, M. E. Möbius, A. P. Bell, T. McCabe, J. J. Boland and T. Gunnlaugsson, Healable Luminescent Self-Assembly Supramolecular Metallogels Possessing Lanthanide (Eu/Tb) Dependent Rheological and Morphological Properties, J. Am. Chem. Soc., 2015, 137, 1983 CrossRef PubMed.
- (a) E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Supramolecular polymeric hydrogels, Chem. Soc. Rev., 2012, 41, 6195 RSC; (b) Q. Zhang, D. Tang, J. Zhang, R. Ni, L. Xu, T. He, X. Lin, X. Li, H. Qiu, S. Yin and P. J. Stang, Self-Healing Heterometallic Supramolecular Polymers Constructed by Hierarchical Assembly of Triply Orthogonal Interactions with Tunable Photophysical Properties, J. Am. Chem. Soc., 2019, 141, 17909 CrossRef CAS PubMed; (c) J. Uchida, M. Yoshio and T. Kato, Self-healing and shape memory functions exhibited by supramolecular liquid-crystalline networks formed by combination of hydrogen bonding interactions and coordination bonding, Chem. Sci., 2021, 12, 6091 RSC.
- (a) P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Supramolecular Polymerization: A Conceptual Expansion for Innovative Materials, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS; (b) J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, From precision polymers to complex materials and systems, Nat. Rev. Mat, 2016, 1, 16024 CrossRef CAS; (c) Y. Liu, Z. Wang and X. Zhang, Characterization of supramolecular polymers, Chem. Soc. Rev., 2012, 41, 5922 RSC; (d) A. Winter and U. S. Schubert, Synthesis and characterization of metallo-supramolecular polymers, Chem. Soc. Rev., 2016, 45, 5311 RSC; (e) P. Wei, X. Yan and F. Huang, Supramolecular polymers constructed by orthogonal self-assembly based on host–guest and metal–ligand interactions, Chem. Soc. Rev., 2015, 44, 815 RSC; (f) C.-W. Hsu, C. Sauvée, H. Sundén and J. Andréasson, Writing and erasing multicolored information in diarylethene-based supramolecular gels, Chem. Sci., 2018, 9, 8019 RSC.
- (a) X. Du, J. Zhou, J. Shi and B. Xu, Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials, Chem. Rev., 2015, 115, 1316 Search PubMed; (b) E. R. Draper and D. J. Adams, Low-Molecular-Weight Gels: The State of the Art, Chem, 2017, 3, 390 CrossRef CAS; (c) M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Metal- and Anion-Binding Supramolecular Gels, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed; (d) R. G. Weiss and P. Terech, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, Springer, Netherlands, 2006 CrossRef; (e) N. Zweep and J. H. van Esch, Functional Molecular Gels, The Royal Society of Chemistry, 2014, pp. 1–29 Search PubMed; (f) E. R. Draper and D. J. Adams, Controlling the Assembly and Properties of Low-Molecular-Weight Hydrogelators, Langmuir, 2019, 35, 6506 CrossRef CAS PubMed; (g) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Functional π-Gelators and Their Applications, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed.
- J. R. Engstrom, A. J. Savyasachi, M. Parhizkar, A. Sutti, C. S. Hawes, J. M. White, T. Gunnlaugsson and F. M. Pfeffer, Norbornene chaotropic salts as low molecular mass ionic organogelators (LMIOGs), Chem. Sci., 2018, 9, 5233 RSC.
- O. Kotova, S. J. Bradberry, A. J. Savyasachi and T. Gunnlaugsson, Recent advances in the development of luminescent lanthanide-based supramolecular polymers and soft materials, Dalton Trans., 2018, 47, 16377 RSC.
- (a) S. Wei, Z. Li, W. Lu, H. Liu, J. Zhang, T. Chen and B. Z. Tang, Multicolor Fluorescent Polymeric Hydrogels, Angew. Chem., Int. Ed., 2021, 60, 8608 CrossRef CAS PubMed; (b) G. C. Le Goff, R. L. Srinivas, W. A. Hill and P. S. Doyle, Hydrogel microparticles for biosensing, Eur. Polym. J., 2015, 72, 386 CrossRef CAS PubMed; (c) M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Optically healable supramolecular polymers, Nature, 2011, 472, 334 CrossRef CAS PubMed.
- (a) T. Gorai and U. Maitra, Luminescence Resonance Energy Transfer in a Multiple-Component, Self-Assembled Supramolecular Hydrogel, Angew. Chem., Int. Ed., 2017, 56, 10730 CrossRef CAS PubMed; (b) R. Laishram and U. Maitra, Energy transfer in FRET pairs in a supramolecular hydrogel template, Chem. Commun., 2022, 58, 3162 RSC; (c) E. M. Surender, S. J. Bradberry, S. A. Bright, C. P. McCoy, D. C. Williams and T. Gunnlaugsson, Luminescent Lanthanide Cyclen-Based Enzymatic Assay Capable of Diagnosing the Onset of Catheter-Associated Urinary Tract Infections Both in Solution and within Polymeric Hydrogels, J. Am. Chem. Soc., 2017, 139, 381 CrossRef CAS PubMed.
- (a) S. Afzala and U. Maitra, Sensitized Lanthanide Photoluminescence Based Sensors–a Review, Helv. Chim. Acta, 2022, 105, e202100194 Search PubMed; (b) A. Abdollahi, H. Roghani-Mamaqani and B. Razavi, Stimuli-chromism of photoswitches in smart polymers: Recent advances and applications as chemosensors, Prog. Polym. Sci., 2019, 98, 101149 CrossRef CAS; (c) S. Bhowmik, T. Gorai and U. Maitra, Transition metal ion induced hydrogelation by amino-terpyridine ligands, J. Mater. Chem. C, 2014, 2, 1597 RSC; (d) R. Laishram and U. Maitra, Bile Salt-Derived Eu3+ Organogel and Hydrogel: Water-Enhanced Luminescence of Eu3+ in a Gel Matrix, ChemistrySelect, 2018, 3, 519 CrossRef CAS; (e) K. Binnemans, Interpretation of europium(III) spectra, Coord. Chem. Rev., 2015, 295, 1 CrossRef CAS; (f) B. Yang, H. Zhang, H. Peng, Y. Xu, B. Wu, W. Weng and L. Li, Self-healing metallo-supramolecular polymers from a ligand macromolecule synthesized via copper-catalyzed azide–alkyne cycloaddition and thiol–ene double “click” reactions, Polym. Chem., 2014, 5, 1945 RSC; (g) R. J. Wojtecki, M. A. Meador and S. J. Rowan, Using the dynamic bond to access macroscopically responsive structurally dynamic polymers, Nat. Mater., 2010, 10, 14 CrossRef PubMed.
- O. Kotova, C. O’Reilly, S. T. Barwich, L. E. Mackenzie, A. D. Lynes, A. J. Savyasachi, M. Ruether, R. Pal, M. E. Möbius and T. Gunnlaugsson, Lanthanide luminescence from supramolecular hydrogels consisting of bio-conjugated picolinic-acid-based guanosine quadruplexes, Chem, 2022, 8, 1395 CAS.
- D. Parker, J. D. Fradgley and K.-L. Wong, The design of responsive luminescent lanthanide probes and sensors, Chem. Soc. Rev., 2021, 50, 8193 RSC.
- (a) W.-L. Chan, C. Xie, W.-S. Lo, J.-C. G. Bünzli, W.-K. Wong and K.-L. Wong, Lanthanide–tetrapyrrole complexes: synthesis, redox chemistry, photophysical properties, and photonic applications, Chem. Soc. Rev., 2021, 50, 12189 RSC; (b) Y. Zhao and D. Li, Lanthanide-functionalized metal–organic frameworks as ratiometric luminescent sensors, J. Mater. Chem. C, 2020, 8, 12739 RSC.
- (a) P. Sutar, V. M. Suresh and T. K. Maji, Tunable emission in lanthanide coordination polymer gels based on a rationally designed blue emissive gelator, Chem. Commun., 2015, 51, 9876 RSC; (b) P. Sutar and T. K. Maji, Bimodal self-assembly of an amphiphilic gelator into a hydrogel-nanocatalyst and an organogel with different morphologies and photophysical properties, Chem. Commun., 2016, 52, 13136 RSC; (c) S. V. Eliseeva and J.-C. G. Bünzli, Lanthanide luminescence for functional materials and bio-sciences, Chem. Soc. Rev., 2010, 39, 189 RSC; (d) S. E. Bodman and S. J. Butler, Advances in anion binding and sensing using luminescent lanthanide complexes, Chem. Sci., 2021, 12, 2716 RSC; (e) E. Mathieu, A. Sipos, E. Demeyere, D. Phipps, D. Sakaveli and K. E. Borbas, Lanthanide-based tools for the investigation of cellular environments, Chem. Commun., 2018, 54, 10021 RSC.
- (a) E. M. Surender, S. Comby, B. Cavanagh, O. Brennan, T. C. Lee and T. Gunnlaugsson, Two-Photon Luminescent Bone Imaging Using Europium Nanoagents, Chem, 2016, 1, 438 CrossRef CAS; (b) D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Lanthanide-directed synthesis of luminescent self-assembly supramolecular structures and mechanically bonded systems from acyclic coordinating organic ligands, Chem. Soc. Rev., 2016, 45, 3244 RSC.
- D. E. Barry, J. A. Kitchen, K. Pandurangan, A. J. Savyasachi, R. D. Peacock and T. Gunnlaugsson, Formation of Enantiomerically Pure Luminescent Triple-Stranded Dimetallic Europium Helicates and Their Corresponding Hierarchical Self-Assembly Formation in Protic Polar Solutions, Inorg. Chem., 2020, 59, 2646 CrossRef CAS PubMed.
- C. P. McCoy, F. Stomeo, S. E. Plush and T. Gunnlaugsson, Soft Matter pH Sensing: From Luminescent Lanthanide pH Switches in Solution to Sensing in Hydrogels, Chem. Mater., 2006, 18, 4336 CrossRef CAS.
- (a) A. F. Henwood, I. N. Hegarty, E. P. McCarney, J. I. Lovitt, S. Donohoe and T. Gunnlaugsson, Recent advances in the development of the btp motif: A versatile terdentate coordination ligand for applications in supramolecular self-assembly, cation and anion recognition chemistries, Coord. Chem. Rev., 2021, 449, 214206 CrossRef CAS; (b) J. P. Byrne, J. A. Kitchen and T. Gunnlaugsson, The btp [2,6-bis(1,2,3-triazol-4-yl)pyridine] binding motif: a new versatile terdentate ligand for supramolecular and coordination chemistry, Chem. Soc. Rev., 2014, 43, 5302 RSC.
- E. P. McCarney, J. P. Byrne, B. Twamley, M. Martínez-Calvo, G. Ryan, M. E. Mobius and T. Gunnlaugsson, Self-assembly formation of a healable lanthanide luminescent supramolecular metallogel from 2,6-bis(1,2,3-triazol-4-yl)pyridine (btp) ligands, Chem. Commun., 2015, 51, 14123 RSC.
- C. O’Reilly, S. Blasco, B. Parekh, H. Collins, G. Cooke, T. Gunnlaugsson and J. P. Byrne, Ruthenium-centred btp glycoclusters as inhibitors for Pseudomonas aeruginosa biofilm formation, RSC Adv., 2021, 11, 16318 RSC.
- (a) J. P. Byrne, J. A. Kitchen, J. E. O’Brien, R. D. Peacock and T. Gunnlaugsson, Lanthanide Directed Self-Assembly of Highly Luminescent Supramolecular “Peptide” Bundles from α-Amino Acid Functionalized 2,6-Bis(1,2,3-triazol-4-yl)pyridine (btp) Ligands, Inorg. Chem., 2015, 54, 1426 CrossRef CAS PubMed; (b) J. P. Byrne, M. Martínez-Calvo, R. D. Peacock and T. Gunnlaugsson, Chiroptical Probing of Lanthanide-Directed Self-Assembly Formation Using btp Ligands Formed in One-Pot Diazo-Transfer/Deprotection Click Reaction from Chiral Amines, Chem. – Eur. J., 2016, 22, 486 CrossRef CAS PubMed.
- S. J. Bradberry, J. P. Byrne, C. P. McCoy and T. Gunnlaugsson, Lanthanide luminescent logic gate mimics in soft matter: [H+] and [F−] dual-input device in a polymer gel with potential for selective component release, Chem. Commun., 2015, 51, 16565 RSC.
- S. J. Bradberry, G. Dee, O. Kotova, C. P. McCoy and T. Gunnlaugsson, Luminescent lanthanide (Eu(III)) cross-linked supramolecular metallo co-polymeric hydrogels: the effect of ligand symmetry, Chem. Commun., 2019, 55, 1754 RSC.
- M. Martínez-Calvo, S. A. Bright, E. B. Veale, D. C. Williams and T. Gunnlaugsson, 4-Amino-1,8-naphthalimide based fluorescent photoinduced electron transfer (PET) pH sensors as liposomal cellular imaging agents: The effect of substituent patterns on PET directional quenching, Front. Chem. Sci. Eng., 2020, 14, 61 CrossRef.
- A. Beeby, I. Clarkson, R. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, Non-radiative deactivation of the excited states of europium, terbium and ytterbium complexes by proximate energy-matched OH, NH and CH oscillators: an improved luminescence method for establishing solution hydration states, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- A.-S. Chauvin, F. Gumy, D. Imbert and J.-C. Bunzli, Europium and Terbium tris(Dipicolinates) as Secondary Standards for Quantum Yield Determination, Spectrosc. Lett., 2004, 37, 517 CrossRef CAS.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Calculation of Equilibrium Constants from Multiwavelength Spectroscopic Data-IV Model-Free Least-Squares Refinement by Use of Evolving Factor Analysis, Talanta, 1986, 33, 943 CrossRef CAS PubMed.
- S. J. Bradberry, A. J. Savyasachi, R. D. Peacock and T. Gunnlaugsson, Quantifying the Formation of Chiral Luminescent Lanthanide Assemblies in an Aqueous Medium Through Chiroptical Spectroscopy and Generation of Luminescent Hydrogels, Faraday Discuss., 2015, 185, 413 RSC.
- (a) G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed; (b) G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- E. P. McCarney, J. I. Lovitt and T. Gunnlaugsson, Mechanically interlocked chiral self-templated [2]catenanes from 2,6-bis(1,2,3-triazol-4-yl)pyridine (btp) ligands, Chem. – Eur. J., 2021, 27, 12052 CrossRef CAS PubMed.
- J. P. Byrne, S. Blasco, A. B. Aletti, G. Hessman and T. Gunnlaugsson, Formation of self-templated 2,6-bis(1,2,3-triazol-4-yl)pyridine [2]catenanes by triazolyl hydrogen bonding: selective anion hosts for phosphate, Angew. Chem. Int. Ed., 2016, 55, 8938 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray crystallography data table, thermogravimetric analysis data, NMR spectra and additional figures. CCDC 2005056. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qm00998f |
| ‡ One of the intermediates from this synthesis, 2,6-dibromo-4-nitropyridine, crystallised during the workup and the resulting crystals were found to be suitable for X-ray crystallographic analysis. The resulting structure showed the presences of extended intermolecular halogen bonding interactions between the two bromine atoms of the pyridine and that of and the pyridine nitrogen, as well as the nitro group. These interactions are presented in the ESI.† |
| § The changes in the UV-visible absorption spectra were not large and the resulting speciation therefore may not be fully representative of the speciation in solution. No acceptable fit was obtained when the Tb32 species was included in the UV-visible absorption data model. The reliability of this fit should therefore be considered with caution. However, the speciation obtained from the Tb(III)-centred emission spectral data, can be considered a more accurate representation of the likely speciation in solution. These titrations were fully reproducible as was the fitting of the data. |
| This journal is © the Partner Organisations 2023 |