
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTADF-based NIR-II semiconducting polymer dots for in vivo 3D bone imaging†
Keng-Fang
Hsu‡
a,
Shih-Po
Su‡
b,
Hsiu-Feng
Lu‡
cd,
Ming-Ho
Liu
a,
Yuan Jay
Chang
 e,
Yi-Jang
Lee
f,
Huihua Kenny
Chiang
b,
Chao-Ping
Hsu
*cd,
Chin-Wei
Lu
*g and
Yang-Hsiang
Chan
*ahi
e,
Yi-Jang
Lee
f,
Huihua Kenny
Chiang
b,
Chao-Ping
Hsu
*cd,
Chin-Wei
Lu
*g and
Yang-Hsiang
Chan
*ahi
aDepartment of Applied Chemistry, National Yang Ming Chiao Tung University, Hsinchu, Taiwan 30050. E-mail: yhchan@nycu.edu.tw
bInstitute of Biomedical Engineering, National Yang Ming Chiao Tung University, Taipei, Taiwan 11221
cInstitute of Chemistry, Academia Sinica, 128 Section 2, Academia Road, Nankang, Taipei 115, Taiwan. E-mail: cherri@gate.sinica.edu.tw
dNational Center for Theoretical Sciences, 1, Section 4, Roosevelt Road, Taipei 106, Taiwan
eDepartment of Chemistry, Tunghai University, Taichung City 40704, Taiwan
fDepartment of Biomedical Imaging and Radiological Sciences, School of Biomedical Engineering, National Yang Ming Chiao Tung University, Taipei, Taiwan 11221
gDepartment of Applied Chemistry, Providence University, Taichung 43301, Taiwan. E-mail: cwlu@pu.edu.tw
hCenter for Emergent Functional Matter Science, National Yang Ming Chiao Tung University, Hsinchu, Taiwan 30010
iDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung, Taiwan 80708
First published on 18th August 2022
Abstract
Intraoperative fluorescence imaging in the second near-infrared (NIR-II) region heralds a new era in image-guided surgery since the success in the first-in-human liver-tumor surgery guided by NIR-II fluorescence. Limited by the conventional small organic NIR dyes such as FDA-approved indocyanine green with suboptimal NIR-II fluorescence and non-targeting ability, the resulting shallow penetration depth and high false positive diagnostic values have been challenging. Described here is the design of NIR-II emissive semiconducting polymer dots (Pdots) incorporated with thermally activated delayed fluorescence (TADF) moieties to exhibit emission maxima of 1064–1100 nm and fluorescence quantum yields of 0.40–1.58% in aqueous solutions. To further understand how the TADF units affect the molecular packing and the resulting optical properties of Pdots, in-depth and thorough density-functional theory calculations were carried out to better understand the underlying mechanisms. We then applied these Pdots for in vivo 3D bone imaging in mice. This work provides a direction for future designs of NIR-II Pdots and holds promising applications for bone-related diseases.
Introduction
In fundamental studies, fluorescence imaging plays a vital role in visualizing molecular interactions in complicated biological processes. In clinical use, the rapid growth and technological advancement of fluorescence image-guided surgery in humans in these years also highlight the unmet clinical demands of fluorescence imaging techniques in surgery.1–6 The selection of the optimal fluorescent probe is crucial to achieve higher signal-to-background ratios in imaging. Absorption/scattering of light and autofluorescence from tissues of living organisms have been three major obstacles for in vivo imaging in realizing deeper penetration and higher resolution.7 Short-lived autofluorescence from tissues (∼ns) can be effectively eliminated by the use of long-lived inorganic luminescent probes (∼μs to ms) although the potential cytotoxicity of metal ions from these inorganic probes remains a concern.8–11 In terms of absorption and scattering of light by biospecies, fluorescence imaging in the second near-infrared (NIR-II) window (1000–1700 nm) has been recently demonstrated to significantly attenuate photon absorption and scattering in living organisms.6,12–25 More importantly, tissue autofluorescence also reduces drastically in the NIR-II region26,27 so that the necessity for the use of long-lived photoluminescent probes is weakened. To date, a variety of NIR-II fluorophores including carbon-based nanomaterials,28 small organic dyes,29–32 rare-earth materials,33,34 inorganic or metal nanocrystals,35 and polymer-based nanoparticles36–43 have been successfully developed to cover the shortage of commercially available NIR-II dyes.Among the aforementioned fluorophores, semiconducting polymer-based nanoparticles (Pdots) appear to be a promising type of NIR-II fluorescent candidate to be explored due to their strong absorption, extraordinarily high emission brightness, phenomenal photostability, and facile surface functionalization.44–61 However, there are still several unmet challenges for the development of NIR-II emissive Pdots with longer emission wavelengths. Specifically, the main challenge is the difficulty in the synthesis of stable dye molecules possessing a low bandgap. The molecular instability is attributed to the oxidation-labile conjugated system from the raised highest occupied molecular orbital (HOMO) and/or the high electrophilic reactivity from the decreased lowest unoccupied molecular orbital (LUMO).62 The other fundamental constraint is dictated by the energy gap law that radiationless transitions ascend rapidly via vibrational relaxation owing to decreasing energy separation between the excited and ground states.63–65 This is attributed to the fact that, so far, only a few Pdots have been reported to have emission maxima over 1100 nm with acceptable fluorescence quantum yields (>1%) for bioimaging.66,67 Our group recently developed two types of NIR-II emissive Pdots: polymethines68 and donor–acceptor–donor based conjugated polymers,69 trying to overcome the energy gap law and the aggregation-caused quenching (ACQ) issue. However, the unbalance between the longer emission wavelength and the higher fluorescence quantum yield remains a bottleneck for exploring the next generation of NIR-II Pdots.
Inspired by the unique structural and optical properties of thermally activated delayed fluorescence (TADF) materials,70–72 we expect to find the breakthrough point for this dilemma. A TADF moiety usually possesses a highly twisted donor–acceptor molecular framework to separate the HOMO and LUMO in the donor and acceptor segments, respectively, to realize an ultra-narrow singlet-triplet energy splitting.73 We speculate that the highly twisted structure of the TADF molecule could ameliorate the ACQ phenomenon and the same time the interactions of HOMO and LUMO of TADF moeity with that of other segments within a polymer chain might red-shift the resulting emission wavelength. Additionally, the use of long-lived emission of TADF-based fluorophores in bioimaging can effectively preclude the detector from collecting the short-lived autofluorescence of interference species, thereon enhancing the signal-to-background ratios. Algar and Hudson's groups recently developed red-emissive TADF-grafted Pdots for time-gated cellular imaging with high contrast.74 For NIR-II imaging, fortunately, the autofluorescence is less considered because of the significantly reduced autofluorescence in the NIR-II window.
Here, we prepared four types of TADF monomers to copolymerize with donor and acceptor segments through Stille coupling (Fig. 1). The TADF-containing semiconducting polymers exhibited NIR-II fluorescence with different extents of redshift and an increase in fluorescence compared to the control polymer without TADF. Density functional theory (DFT) calculations were performed to investigate the roles of TADF units. NIR-II real-time imaging in mice was then performed to realize through-skull vascular imaging and bone targeting at different time intervals. These results demonstrated the merits of the incorporation of TADF moieties in NIR-II fluorescent Pdots.
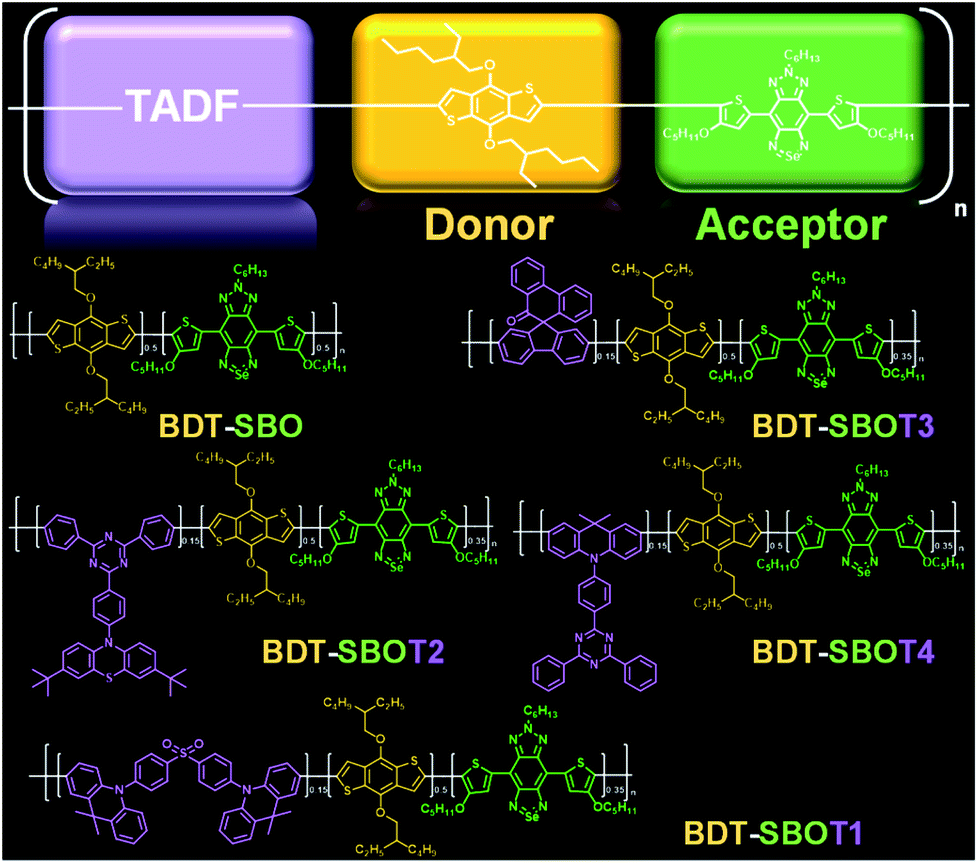 | ||
| Fig. 1 Molecular design of TADF-based conjugated polymers with NIR-II fluorescence. | ||
Results and discussion
Rational design of narrow bandgap donor–acceptor type conjugated polymers incorporated with TADF units
Chalcogena-diazolobenzotriazole (XBTa) derivatives have been shown to possess strong electron affinity with low LUMOs75 and can be used to lower the bandgaps in D–A-type conjugated polymers. The experimental bandgap and LUMO trends of XBTa appear to be Te < Se < S but TeBTa is unstable due to its higher HOMO above −5 eV. Therefore, we selected the SeBTa core (i.e., compound 3 in Fig. 2A) as the acceptor building block in the conjugated polymer. For the selection of the donor, we employed a strong electron donating segment, alkoxyl-substituted benzodithiophene with a rigid coplanar conformation to conjugate with SeBTa-based acceptors. 3-Alkoxy-substituted thiophene was also used as the conjugated linker to enhance the π–π intramolecular interactions. The resulting molecule, compound 4, was then subjected to iodination to obtain compound SBO, followed by the Stille polycondensation reaction to form polymers. In view of the high planarity of the resulting polymers that could induce unwanted ACQ behaviors, we introduced TADF monomers into the polymer backbones to investigate the effect of TADF on the photophysical properties of Pdots. Four different types of TADF monomers (T1–T4 in Fig. 2B) were synthesized for incorporation into the conjugated polymer backbones. Finally, four TADF-based NIR-II fluorescent polymers, BDT-SBOT1, BDT-SBOT2, BDT-SBOT3, and BDT-SBOT4 were synthesized for comparison with the control polymer, BDT-SBO (Fig. 2C).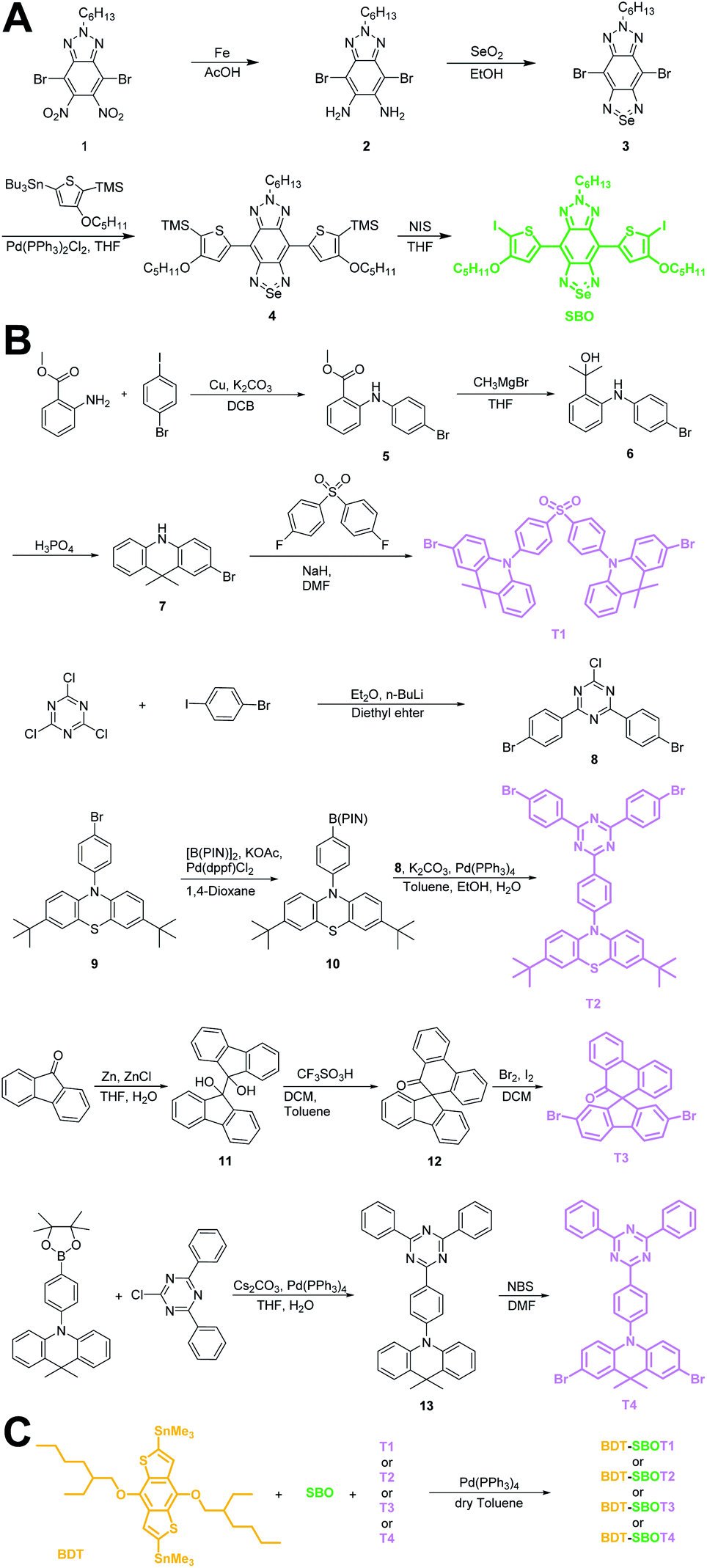 | ||
| Fig. 2 Synthetic procedures of (A) acceptor SBO, (B) TADF monomers T1–T4, and (C) the subsequent polymerization by Stille coupling. | ||
Preparation and characterization of Pdots
The procedures for the preparation of Pdots from polymers are depicted in Fig. 3A. Briefly, conjugated polymers and amphiphilic lipids were dissolved in THF and then co-precipitated in water solution, followed by the evaporation of THF to obtain Pdot solutions. The amphiphilic lipids with hydrophilic polyethylene glycol segments can effectively reduce the non-specific biomolecular adsorption and prolong the blood circulation time of Pdots for efficient in vivo bone targeting. The resulting Pdots can be used to visualize whole-body vasculature and bone in mice at different points of time. The absorption (solid lines) and emission (dashed lines) spectra of these five Pdots are displayed in Fig. 3B and their optical properties are summarized in Table 1. We found that BDT-SBO Pdots appeared to be non-fluorescent with an absorption peak at ∼860 nm. Surprisingly, the TADF moieties incorporated into the copolymers all showed red-shifted absorption/emission with enhanced fluorescence to different extents depending on the chemical structures of TADF monomers. These Pdots exhibited sizes of 23–29 nm in diameter (Fig. 3C and S1 in the ESI†) and negative zeta potentials of −23 to −34 mV. We also measured the fluorescence quantum yields (QYs) of Pdots by using IR-1061 in CH2Cl2 as the reference (Table 1). It was found that the QY of BDT-SBOT2 Pdots was 1.58%, about 4 times higher than that of BDT-SBOT3. To better understand the roles of TADF units in the polymer backbones and the mechanisms underlying their interaction with the donor/acceptor, we conducted in-depth DFT calculations (vide infra). In terms of photostability, a significant factor for long-term imaging, the four types of TADF-based Pdots exhibited superior photostability under continuous 254 nm UV light illumination (Fig. S2†). Their fluorescence intensities remained over 80% of their original intensities even after 15 min of intense UV irradiation (10 W). In contrast, conventional organic dyes including NIR-I emissive indocyanine green (ICG) and NIR-II fluorescent IR-1061 all showed a drastic decrease in emission intensities, especially for IR-1061 which photo-decomposed completely within merely 3 min. To further investigate whether delayed fluorescence was involved, the emission lifetimes of four Pdots were measured, which exhibited only prompt emission (0.52–0.63 ns, Fig. S3†) without the observation of delayed emission for all Pdots. This result implied that thermally activated delayed fluorescence didn't participate in the emission processes of these Pdots. We also measured the QYs of these conjugated polymers under air and inert conditions and no significant difference was observed (Table S1†), further confirming the absence of TADF properties. It is worth mentioning that prolonged emission lifetimes might not be advantageous in the NIR-II window because of the existing low autofluorescence in this region.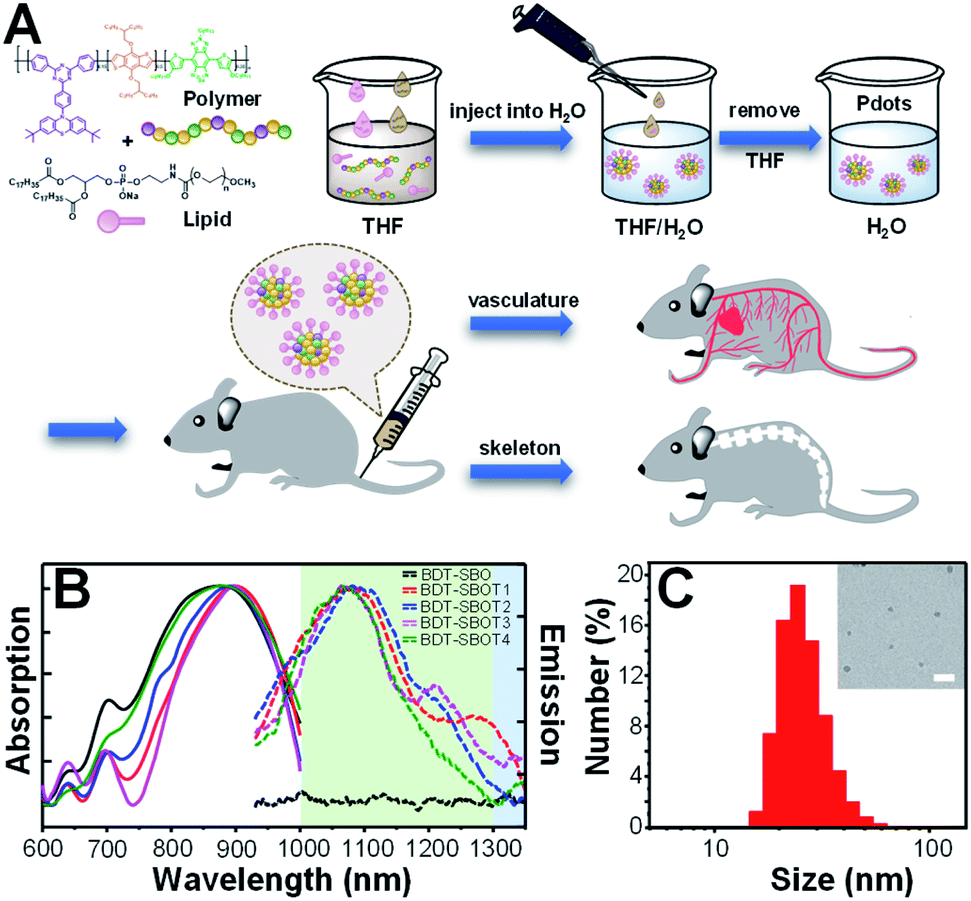 | ||
| Fig. 3 (A) Schematic diagram representing the preparation of lipid-encapsulated Pdots for in vivo vascular and bone imaging. (B) Absorption (solid lines) and emission (dashed lines) spectra of BDT-SBO (black lines), BDT-SBOT1 (red lines), BDT-SBOT2 (blue lines), BDT-SBOT3 (pink lines), and BDT-SBOT4 (green lines). (C) Hydrodynamic diameters of BDT-SBOT2 Pdots obtained by dynamic light scattering with an average size of 27 nm and a zeta potential of −29 mV. The inset shows the transmission electron microscopy image of BDT-SBOT2 Pdots with a scale bar of 100 nm. | ||
| Copolymers | λ absmax (nm) | λ emmax (nm) | Size (nm) | QYc (%) | τ (ns) |
|---|---|---|---|---|---|
| a Absorption maximum. b Fluorescence maximum. c Fluorescence quantum yield in water. d Fluorescence lifetime in CH2Cl2. | |||||
| BDT-SBO | 862 | — | 29 | — | — |
| BDT-SBOT1 | 898 | 1084 | 24 | 1.17 | 0.63 |
| BDT-SBOT2 | 882 | 1100 | 27 | 1.58 | 0.61 |
| BDT-SBOT3 | 900 | 1064 | 28 | 0.40 | 0.54 |
| BDT-SBOT4 | 870 | 1067 | 23 | 0.54 | 0.52 |
Structure–performance relationship of Pdots
To gain insights into the relationship between the structures of polymer chains and their performance in Pdots, we studied the effects of TADF moieties with DFT and time-dependent DFT by using the Tamm-Dancoff approximation76 (TDDFT-TDA) calculation.Model molecules BDT-SBO (D–A and D–A–D) BDT-SBOTn (Tn–D–A and Tn–D–A–D, n = 1–4) were considered in the present work. The structures and basic properties (HOMO, LUMO, excitation energies) are included in the ESI.† When a second D moiety is attached to the oligomer, there is a reduction in the energy gap as well as a red-shift in absorption. In Fig. 4, it is seen that the HOMOs are much more delocalized across the D–A–D region, while the LUMO remains localized to A. However, the lower bandgap is mainly due to a lower LUMO energy. From Table 2, it is seen that the wavelengths of maximum absorption of D–A–D and Tn–D–A–D are red-shifted from their corresponding D–A and Tn–D–A models, which are also close to the experimental values listed in Table S1.† The natural transition orbitals (NTOs)77 for particles and holes for the S1 state are included in Fig. 4, where the case for T2 is shown and those for others are in the ESI.† It is seen that the excitation, as described by the hole and particle orbitals, is very similar to the HOMO and LUMO, respectively.
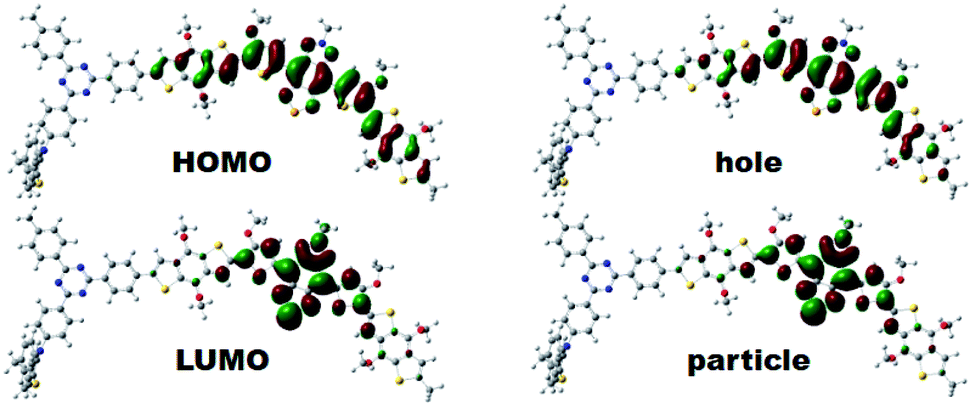 | ||
| Fig. 4 The electron density contours of HOMO and LUMO, and computed natural transition orbital (NTO) pairs for S1 of T2–D–A–D based on DFT calculations. The isosurface value was set at 0.02 a.u. | ||
| E gap | λ cal | E gap | λ cal | ||
|---|---|---|---|---|---|
| D–A | 3.199 | 743 | D–A–D | 2.988 | 814 |
| T1–D–A | 3.153 | 751 | T1–D–A–D | 2.954 | 823 |
| T2–D–A | 3.198 | 744 | T2–D–A–D | 2.985 | 816 |
| T3–D–A | 3.180 | 747 | T3–D–A–D | 2.973 | 819 |
| T4–D–A | 3.139 | 751 | T4–D–A–D | 2.946 | 824 |
In order to understand the reason behind the red shift and increased fluorescence quantum yield in Pdots as compared to those in a solvent, we built simple dimeric models and simulated the stacking of Pdots using the smallest systems, D–A and Tn–D–A (n = 1–4). While Tn–D–A–D is a better model for the optical properties, it is computationally difficult and tedious to optimize and analyze the dimeric structures. Therefore, we aim to gain insights from the spectral changes with Tn–D–A dimers. The full details of the dimer structure and basic properties are included in the ESI.† Briefly, the parallel and antiparallel stacking structure of Tn–D–A was studied by assuming a large overlap in the whole oligomer model to allow the steric effect of TADF on the stacking structure, as shown in Scheme 1. The optimized low-energy structures of D–A dimers are displaced from each other along both the long- or short-molecular axis to various degrees, both in parallel and antiparallel arrangements, as shown in Fig. S10.† The energy of most stable parallel- and antiparallel-stacked dimers is lower by 1∼ kcal mol−1 as compared to other arrangements, and such most-stable structures are used for further studies. The molecular orbital energies of the calculated results, the energy differences between the HOMO and LUMO and LUMO+1, and the absorption wavelength, oscillation strength and occupations of the NTO pair are listed in Tables S2–S4,† and 3, respectively.
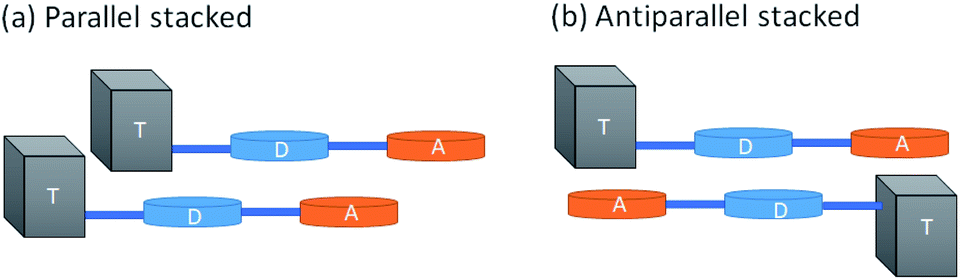 | ||
| Scheme 1 Schematic diagram of the (A) parallel and (B) antiparallel Tn–D–A dimer stacking strategy. | ||
| Monomer | Parallel dimer | |||
|---|---|---|---|---|
| E gap | λ cal | E gap | λ cal (oscillator strength) | |
| D–A | 3.133 | 768 | 2.942 | 895(0.0063), 887(0.0561), 805(0.1716), 728(2.0200) |
| T1–D–A | 3.093 | 774 | 3.020 | 871(0.1711), 843(0.2831), 776(0.1109), 687(1.6088) |
| T2–D–A | 3.134 | 767 | 2.941 | 854(0.9182) |
| T3–D–A | 3.108 | 772 | 2.820 | 975(0.0836), 825(0.2059), 753(0.7187), 715(1.4608) |
| T4–D–A | 3.066 | 778 | 2.871 | 873(0.8813) |
The molecular orbitals and their energy gaps, as well as the low-lying excited states for the stacked dimers are calculated in a nonpolar solvent setting, for modeling the effect of mPEG-DSPE in the Pdot. We note that the vertical transition of monomeric D–A and Tn–D–A is red shifted in this nonpolar setting, and the energy difference between the HOMO and LUMO is also smaller, as seen from the data listed in Table 3, as compared to THF as a polar solvent (Table 2). This implies that the ground states of Tn–D–A are more polarized than its excited state.
In Fig. 5, we include the sketch of optimized stacking structures of Tn–D–A dimers, with TADF groups employed in the search of structure replaced by a methyl group in the graph. It is seen that the parallel D–A dimers are shifted on the short axis (Fig. S10 and S11†). For T1 and T3, the dimers are rotated and twisted, and for T2 and T4, they are shifted along the long axis. In the antiparallel cases, the D–A dimer maintains a good π–π contact, while all other dimers have various degrees of large shift, mainly on the long axis, as shown in Fig. S11.†
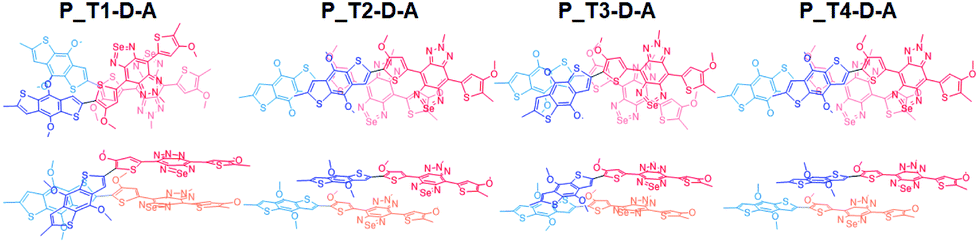 | ||
| Fig. 5 Depicted structures of parallel dimers. For clarity, TADF is replaced by a methyl group in this representation. Donors and accepters are depicted in blue and red respectively, and the corresponding lighter colors are used in the lower layers. | ||
The dimers exhibit a significant red shift in their excitations, for the parallel dimers but not so much for anti-parallel stacked ones, as listed in Tables 3, S3 and S4.† The parallel D–A dimer has a large red shift in excitation (895 and 887 nm) but with a small oscillator strength, and the higher excited state has a large optical activity (728 nm, f = 2.0200), and it is a blue-shifted transition, consistent with the H-aggregate properties.78,79
For P_T2–D–A, the observed strong red-shift and high quantum yield of T2-based Pdots as seen in Table 1 can be supported by the large red-shift excitation with a large oscillator strength. It is seen that the T2–D–A dimer has a large shift on the long axis (Fig. 5), leading to a J-aggregate dimer where the oscillator strength of the lowest excited state of the dimer is the largest. Similar effects can be seen in T4–D–A, but the quantum yield and red shift are not as significant in experimental observation. We note that different TADF moieties result in different structural stackings, as T1 and T3 are the dimers with an angle of rotation in the molecular plane, instead of being shifted. The optical behavior has been closer to that of column aggregation and displays typical absorption and emission features with red-shift properties.80 As shown in Fig. 5, when viewed from the side, the molecule is much twisted, not only in the dihedral angle between D–A itself, and the molecular plane of acceptor (A) is also distorted. Such twisted, atypical dimeric interaction increases the optical activity of the low-energy absorption regions originating from standard H-aggregates, such as the 871 nm (f = 0.1711) and 843 nm (f = 0.2831) excitations seen for P_T1–D–A and 825 nm (f = 0.2059) for P_T3–D–A. Taking T3 as an example, the main transition is found to be HOMO−1 to LUMO, and both intermolecular and intramolecular transitions are involved, as shown in Fig. S12(c).† This is consistent with the experimental observation that P_T3–D–A shows a red shift and decreased activity in Pdots.
Our simple dimer models are a rather limited set of representation for the chromophore Pdots. Nevertheless, there is still a chance for the formation of J-aggregates, with red-shifted, brighter states, as seen in the parallel dimers. Another possibility is forming distorted dimers that are similar H-aggregates, but the low-lying excitonic states are allowed due to the structural distortion. From the theoretical results, it can be inferred that the experimental red shift and QY increase of TADF-containing Pdots are likely due to the interchain aggregated structure, including both J-aggregates and distorted H-aggregate-like structures, which are strongly affected by the structure and interaction in the TADF moieties.
In vivo real-time imaging of vasculature and bone in mice
Before in vivo bioimaging, we first assessed the cytotoxicity of Pdots. As shown in Fig. S13,† MTT assays were performed in HeLa cells with different Pdot concentrations, indicating the high biocompatibility of Pdots in cells. For in vivo bioimaging, we used a 793 nm laser (50 mW cm−2) as the excitation source and a 1200 nm long-pass filter in which BDT-SBOT2 Pdots were taken as an example due to their high QY. We found that BDT-SBOT2 Pdots exhibited much higher fluorescence brightness (∼9 times higher) as compared to ICG under the same optical conditions (Fig. 6A). We then carried out time-lapse whole-blood imaging in living mice by intravenously injecting Pdots through the tail vein. From Fig. 6B, we can find that Pdots flowed throughout the whole body in 5 min and started to accumulate in the excretory system and bones after 2 h post-injection. There have been several bone targeting mechanisms reported using small fluorophores or fluorescent nanomaterials.18,81–84 It was found that the bone targeting efficiency is associated with PEGylation and the modification degree of PEG on the probe.85 Wu's group also demonstrated that small sizes of Pdots (<50 nm) tended to accumulate in endothelium cells of sinusoidal vessels in bone marrow.23 Here we used BDT-SBOT2 Pdots with a diameter of ∼30 nm and these Pdots were capped with PEGylated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) phospholipids on the surface. A high signal to background ratio of the spine was observed, which is in agreement with the reported results.23 To better visualize the precise positions of bones, 3D whole-body mapping was performed at 24 h post-injection (Fig. 6B and video in the ESI†). A high signal-to-background ratio (SBR) of 1.34 with a spatial resolution of 0.52 mm was obtained for the blood vessel around the spinal cord (Fig. 6C and D). Vertical analysis along the yellow line in Fig. 6E for the spine shows the facile discrimination of different vertebrae (Fig. 6F). To better observe the whole skeletal system, the skin and internal organs were removed as shown in Fig. 6G–J. Pdot distribution in major organs was also investigated, showing the large accumulation of Pdots in liver and spleen (Fig. 6K–L). This indicates that the hepatobiliary clearance system is the main metabolic pathway of Pdots. The details of bones at different positions were then imaged, revealing the distinct skeletons of lumbar vertebra, the spinous/transverse process, dorsal rib, thoracic vertebra, tibia, femur and patella (Fig. 6M–O). The above results indicate that these Pdots can be efficiently targeted to bones and can be further used for bone-related disease imaging and study in bone metastasis.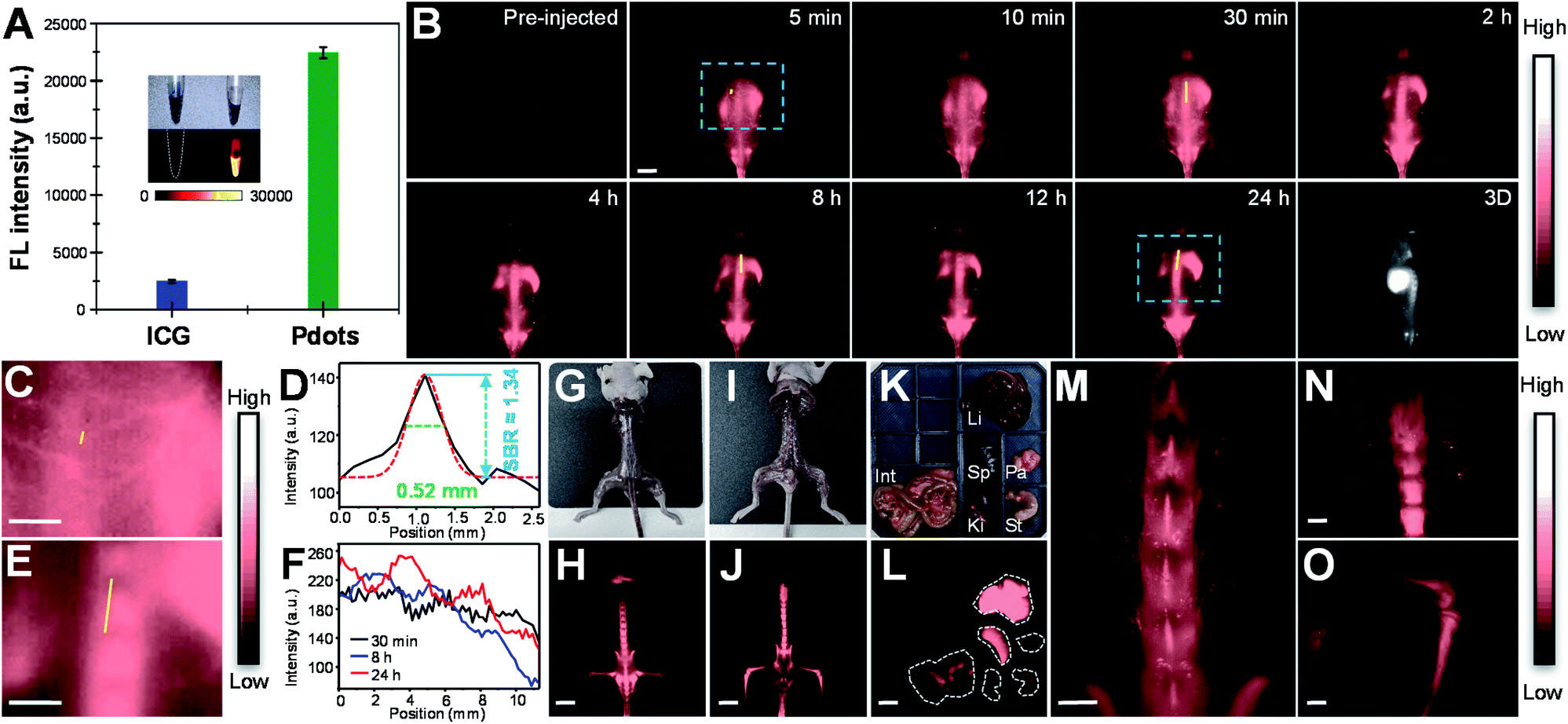 | ||
| Fig. 6 (A) Mean fluorescence intensities of ICG in water and BDT-SBOT2 Pdots in water under a 793 nm laser (50 mW cm−2) with a 1200 nm long-pass filter. The inset represents the corresponding photographs and NIR-II fluorescence images. (B) Real-time whole-body imaging of vascular structures and bones in BALB/c mice (n = 5) intravenously injected with BDT-SBOT2 Pdots in the prone position at certain time intervals from 5 min to 24 h. The bottom-right panel shows reconstructed 3D mapping of a whole-body mouse at 24 h post-injection of BDT-SBOT2 Pdots. (C) The magnified view of vasculature around the spinal cord in the blue square in (B) at 5 min post-injection. (D) Cross-sectional intensity profile along the yellow line in (C). I The enlarged view of spine in the blue square in (B) at 24 h post-injection. (F) Cross-sectional intensity profiles along the yellow lines in mice at 30 min (black line), 8 h (blue line), and 24 h (red line) post-injection of Pdots. (G) Bright-view image and (H) NIR-II fluorescence imaging of the whole skeletal system in mice in the prone position. (I) Bright-view image and (J) NIR-II fluorescence imaging of the whole skeletal system in mice in the supine position. (K) Bright-field image and (L) NIR-II fluorescence imaging of different organs excised from the mouse at 48 h after tail-vein injection of BDT-SBOT2 Pdots. Li: liver; Int: intestinal, Sp: spleen; Pa: pancreas; Ki: kidney; St: stomach. NIR-II fluorescence imaging of (M) lumbar vertebra, the spinous/transverse process, (N) dorsal rib, thoracic vertebra, (O) tibia, femur and patella. The scale bars are 10 mm and 2 mm for (B)/(C)/(E)/(H)/(J)/(L) and (M)/(N)/(O), respectively. | ||
Conclusions
Here we described a series of TADF-incorporated Pdots with NIR-II fluorescence for in vivo imaging. These Pdots show low cytotoxicity in cells and exhibit quantum yields up to 1.58% in pure water. In contrast, bare Pdots without the incorporation of TADF moieties show nonfluorescence under the same conditions. We found that delayed fluorescence was not observed for all Pdots, indicating that the increase in quantum yield was not attributed to the delayed fluorescence. From in-depth DFT calculations, we are surprised to find that the emission red shift and quantum yield increase of TADF-containing Pdots are in fact due to the interchain aggregated structure, including both J-aggregates and distorted H-aggregate-like structures. To demonstrate their in vivo bioimaging ability, whole-body vascular imaging and 3D bond mapping were performed by using these Pdots. This study highlights the unique role of the TADF unit in the Pdot system and thereon opens a new avenue of future molecular design of highly bright NIR-II fluorescent Pdots.Data availability
All data are available in the manuscript and in the ESI.† The raw data directly saved from the instruments of this study are available from the corresponding authors upon reasonable request.Author contributions
K.-F. Hsu and M.-H. Liu conducted synthetic experiments. S.-P. Su and Y.-J. Lee conducted in vivo biological imaging in mice. H. K. Chiang built up NIR-II imaging system. C.-P. Hsu and H.-F. Lu carried out DFT calculations and wrote the manuscript. Y. J. Chang and C.-W. Lu provided DFT molecules. Y.-H. Chan conceived the idea, designed the experiments, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Ethical statement
Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC #1100509) of NYCU.Acknowledgements
We would like to thank the MOST, Taiwan (grant no. 111-2123-M-001-003 and 111-2113-M-A49-027) and the Center for Emergent Functional Matter Science of National Yang Ming Chiao Tung University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. We also thank the Computing Center of Academia Sinica for the computing support.Notes and references
- A. V. DSouza, H. Lin, E. R. Henderson, K. S. Samkoe and B. W. Pogue, J. Biomed. Opt., 2016, 21, 080901 CrossRef PubMed.
- S. L. Troyan, V. Kianzad, S. L. Gibbs-Strauss, S. G. Matsui, R. Oketokoun, L. Ngo, A. Khamene, F. Azar and J. V. Frangioni, Ann. Surg. Oncol., 2009, 16, 2943–2952 CrossRef PubMed.
- J. S. D. Mieog, S. L. Troyan, M. Hutteman, K. J. Donohoe, J. R. v. d. Vorst, A. Stockdale, G.-J. Liefers, H. S. Choi, S. L. Gibbs-Strauss, H. Putter, S. Gioux, P. J. K. Kuppen, Y. Ashitate, C. W. G. M. Löwik, V. T. H. B. M. Smit, R. Oketokoun, L. H. Ngo, C. J. H. v. d. Velde, J. V. Frangioni and A. L. Vahrmeijer, Ann. Surg. Oncol., 2011, 18, 2483–2491 CrossRef PubMed.
- M. Hutteman, J. S. D. Mieog, J. R. v. d. Vorst, G. J. Liefers, H. Putter, C. W. G. M. Löwik, J. V. Frangioni, C. J. H. v. d. Velde and A. L. Vahrmeijer, Breast Cancer Res. Treat., 2011, 127, 163–170 CrossRef CAS PubMed.
- G. M. v. Dam, G. Themelis, L. M. A. Crane, N. J. Harlaar, R. G. Pleijhuis, W. Kelder, A. Sarantopoulos, J. S. d. Jong, H. J. G. Arts, A. G. J. v. d. Zee, J. Bart, P. S. Low and V. Ntziachristos, Nat. Med., 2011, 17, 1315–1319 CrossRef PubMed.
- Z. Hu, C. Fang, B. Li, Z. Zhang, C. Cao, M. Cai, S. Su, X. Sun, X. Shi, C. Li, T. Zhou, Y. Zhang, C. Chi, P. He, X. Xia, Y. Chen, S. S. Gambhir, Z. Cheng and J. Tian, Nat. Biomed. Eng., 2020, 4, 259–271 CrossRef PubMed.
- Y. Tang, F. Pei, X. Lu, Q. Fan and W. Huang, Adv. Opt. Mater., 2019, 7, 1900917 CrossRef CAS.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS PubMed.
- K. M. Tsoi, Q. Dai, B. A. Alman and W. C. W. Chan, Acc. Chem. Res., 2013, 46, 662–671 CrossRef CAS PubMed.
- K. Hanaoka, K. Kikuchi, S. Kobayashi and T. Nagano, J. Am. Chem. Soc., 2007, 129, 13502–13509 CrossRef CAS PubMed.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- Y. Zhong, Z. Ma, F. Wang, X. Wang, Y. Yang, Y. Liu, X. Zhao, J. Li, H. Du, M. Zhang, Q. Cui, S. Zhu, Q. Sun, H. Wan, Y. Tian, Q. Liu, W. Wang, K. C. Garcia and H. Dai, Nat. Biotechnol., 2019, 37, 1322–1331 CrossRef CAS PubMed.
- H. Wan, H. Du and F. W. Dai, Adv. Funct. Mater., 2019, 29, 1900566 CrossRef PubMed.
- C. Yin, X. Lu, Q. Fan and W. Huang, VIEW, 2021, 2, 20200070 CrossRef CAS.
- H. Zhou, X. Zeng, A. Li, W. Zhou, L. Tang, W. Hu, Q. Fan, X. Meng, H. Deng, L. Duan, Y. Li, Z. Deng, X. Hong and Y. Xiao, Nat. Commun., 2020, 11, 6183 CrossRef CAS PubMed.
- Y. Cai, Z. Wei, C. Song, C. Tang, W. Han and X. Dong, Chem. Soc. Rev., 2019, 48, 22–37 RSC.
- Y. Li, Z. Cai, S. Liu, H. Zhang, S. T. H. Wong, J. W. Y. Lam, R. T. K. Kwok, J. Qian and B. Z. Tang, Nat. Commun., 2020, 11, 1255 CrossRef CAS PubMed.
- D. Li, Q. Liu, Q. Qi, H. Shi, E.-C. Hsu, W. Chen, W. Yuan, Y. Wu, S. Lin, Y. Zeng, Z. Xiao, L. Xu, Y. Zhang, T. Stoyanova, W. Jia and Z. Cheng, Small, 2020, 16, 2003851 CrossRef CAS PubMed.
- C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, J. Am. Chem. Soc., 2020, 142, 14789–14804 CrossRef CAS PubMed.
- Q. Zhang, P. Yu, Y. Fan, C. Sun, H. He, X. Liu, L. Lu, M. Zhao, H. Zhang and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 3967–3973 CrossRef CAS PubMed.
- Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16294–16308 CrossRef CAS PubMed.
- B. Li, M. Zhao and F. Zhang, ACS Mater. Lett., 2020, 2, 905–917 CrossRef CAS.
- D. Chen, Y. Liu, Z. Zhang, Z. Liu, X. Fang, S. He and C. Wu, Nano Lett., 2021, 21, 798–805 CrossRef CAS PubMed.
- Z. Zhang, X. Fang, Z. Liu, H. Liu, D. Chen, S. He, J. Zheng, B. Yang, W. Qin, X. Zhang and C. Wu, Angew. Chem., Int. Ed., 2020, 59, 3691–3698 CrossRef CAS PubMed.
- S. Liu, Y. Li, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2021, 12, 3427–3436 RSC.
- H. Dai, Q. Shen, J. Shao, W. Wang, F. Gao and X. Dong, Innovation, 2021, 2, 100082 CAS.
- R.-Q. Yang, K.-L. Lou, P.-Y. Wang, Y.-Y. Gao, Y.-Q. Zhang, M. Chen, W.-H. Huang and G.-J. Zhang, Small Methods, 2021, 5, 2001066 CrossRef CAS PubMed.
- G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816–10906 CrossRef CAS PubMed.
- Y. Liu, Y. Li, S. Koo, Y. Sun, Y. Liu, X. Liu, Y. Pan, Z. Zhang, M. Du, S. Lu, X. Qiao, J. Gao, X. Wang, Z. Deng, X. Meng, Y. Xiao, J. S. Kim and X. Hong, Chem. Rev., 2022, 1, 209–268 CrossRef PubMed.
- J. Huang and K. Pu, Angew. Chem., Int. Ed., 2020, 59, 11717–11731 CrossRef CAS PubMed.
- J. Huang, C. Xie, X. Zhang, Y. Jiang, J. Li, Q. Fan and K. Pu, Angew. Chem., Int. Ed., 2019, 58, 15120–15127 CrossRef CAS PubMed.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC.
- F. Ding, Y. Fan, Y. Sun and F. Zhang, Adv. Healthcare Mater., 2019, 8, 1900260 CrossRef PubMed.
- Y. Zhong and H. Dai, Nano Res., 2020, 13, 1281–1294 CrossRef CAS PubMed.
- F. Yang, Q. Zhang, S. Huang and D. Ma, J. Mater. Chem. B, 2020, 8, 7856–7879 RSC.
- S. Liu, H. Ou, Y. Li, H. Zhang, J. Liu, X. Lu, R. T. K. Kwok, J. W. Y. Lam, D. Ding and B. Z. Tang, J. Am. Chem. Soc., 2020, 35, 15146–15156 CrossRef PubMed.
- G. Hong, Y. Zou, A. L. Antaris, S. Diao, D. Wu, K. Cheng, X. Zhang, C. Chen, B. Liu, Y. He, J. Z. Wu, J. Yuan, B. Zhang, Z. Tao, C. Fukunaga and H. Dai, Nat. Commun., 2014, 5, 4206 CrossRef CAS PubMed.
- K. Shou, Y. Tang, H. Chen, S. Chen, L. Zhang, A. Zhang, Q. Fan, A. Yu and Z. Cheng, Chem. Sci., 2018, 9, 3105–3110 RSC.
- Y. Liu, J. Liu, D. Chen, X. Wang, Z. Liu, H. Liu, L. Jiang, C. Wu and Y. Zou, Macromolecules, 2019, 52, 5735–5740 CrossRef CAS.
- X. Hu, Y. Tang, Y. Hu, F. Lu, X. Lu, Y. Wang, J. Li, Y. Li, Y. Ji, W. Wang, D. Ye, Q. Fan and W. Huang, Theranostics, 2019, 9, 4168–4181 CrossRef CAS PubMed.
- G. Deng, X. Peng, Z. Sun, W. Zheng, J. Yu, L. Du, H. Chen, P. Gong, P. Zhang, L. Cai and B. Z. Tang, ACS Nano, 2020, 14, 11452–11462 CrossRef CAS PubMed.
- Y. Yang, X. Fan, L. Li, Y. Yang, A. Nuernisha, D. Xue, C. He, J. Qian, Q. Hu, H. Chen, J. Liu and W. Huang, ACS Nano, 2020, 14, 2509–2521 CrossRef CAS PubMed.
- Y. Liu, J. Liu, D. Chen, X. Wang, Z. Zhang, Y. Yang, L. Jiang, W. Qi, Z. Ye, S. He, Q. Liu, L. Xi, Y. Zou and C. Wu, Angew. Chem., Int. Ed., 2020, 59, 21049–21057 CrossRef CAS PubMed.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- Y.-H. Chan and P.-J. Wu, Part. Part. Syst. Charact., 2015, 32, 11–28 CrossRef CAS.
- K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570–6597 RSC.
- K. Pu, A. J. Shuhendler, J. V. Jokerst, J. Mei, S. S. Gambhir, Z. Bao and J. Rao, Nat. Nanotechnol., 2014, 9, 233–239 CrossRef CAS PubMed.
- K. Pu, N. Chattopadhyay and J. Rao, J. Controlled Release, 2016, 240, 312–322 CrossRef CAS PubMed.
- X. Lim, Nature, 2016, 531, 26–28 CrossRef CAS PubMed.
- L. Feng, C. Zhu, H. Yuan, L. Liu, F. Lv and S. Wang, Chem. Soc. Rev., 2013, 43, 6620–6633 RSC.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS PubMed.
- J. Yu, Y. Rong, C.-T. Kuo, X.-H. Zhou and D. T. Chiu, Anal. Chem., 2017, 89, 42–56 CrossRef CAS PubMed.
- H.-S. Peng and D. T. Chiu, Chem. Soc. Rev., 2015, 44, 4699–4722 RSC.
- Y. Guo, Y. Li, Y. Yang, S. Tang, Y. Zhang and L. Xiong, ACS Appl. Mater. Interfaces, 2018, 10, 20884–20896 CrossRef CAS PubMed.
- F. Ye, C. Wu, Y. Jin, Y.-H. Chan, X. Zhang and D. T. Chiu, J. Am. Chem. Soc., 2011, 133, 8146–8149 CrossRef CAS PubMed.
- Y. Rong, C. W. J. Yu, X. Zhang, F. Ye, M. Zeigler, M. E. Gallina, I.-C. Wu, Y. Zhang, Y.-H. Chan, W. Sun, K. Uvdal and D. T. Chiu, ACS Nano, 2013, 7, 376–384 CrossRef CAS PubMed.
- S.-Y. Kuo, H.-H. Li, P.-J. Wu, C.-P. Chen, Y.-C. Huang and Y.-H. Chan, Anal. Chem., 2015, 87, 4765–4771 CrossRef CAS PubMed.
- C.-T. Kuo, A. M. Thompson, M. E. Gallina, F. Ye, E. S. Johnson, W. Sun, M. Zhao, J. Yu, I.-C. Wu, B. Fujimoto, C. C. DuFort, M. A. Carlson, S. R. Hingorani, A. L. Paguirigan, J. P. Radich and D. T. Chiu, Nat. Commun., 2016, 7, 11468 CrossRef CAS PubMed.
- K. Sun, Y. Tang, Q. Li, S. Yin, W. Qin, J. Yu, D. T. Chiu, Y. Liu, Z. Yuan, X. Zhang and C. Wu, ACS Nano, 2016, 10, 6769–6781 CrossRef CAS PubMed.
- K. Chang, Z. Liu, X. Fang, H. Chen, X. Men, Y. Yuan, K. Sun, X. Zhang, Z. Yuan and C. Wu, Nano Lett., 2017, 17, 4323–4329 CrossRef CAS PubMed.
- L. Wu, I.-C. Wu, C. C. DuFort, M. A. Carlson, X. Wu, L. Chen, C.-T. Kuo, Y. Qin, J. Yu, S. R. Hingorani and D. T. Chiu, J. Am. Chem. Soc., 2017, 139, 6911–6918 CrossRef CAS PubMed.
- Y. Wang, K. Ogasahara, D. Tomihama, R. Mysliborski, M. Ishida, Y. Hong, Y. Notsuka, Y. Yamaoka, T. Murayama, A. Muranaka, M. Uchiyama, S. Mori, Y. Yasutake, S. Fukatsu, D. Kim and H. Furuta, Angew. Chem., Int. Ed., 2020, 59, 16161–16166 CrossRef CAS PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- W.-C. Chen, P.-T. Chou and Y.-C. Cheng, J. Phys. Chem. C, 2019, 123, 10225–10236 CrossRef CAS.
- E. Thimsen, B. Sadtler and M. Y. Berezin, Nanophotonics, 2017, 6, 1043–1054 CAS.
- J. Mu, M. Xiao, Y. Shi, X. Geng, H. Li, Y. Yin and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202114722 CrossRef CAS PubMed.
- P. Chowdhury and Y.-H. Chan, Mol. Syst. Des. Eng., 2022, 7, 702–719 RSC.
- M.-H. Liu, Z. Zhang, Y.-C. Yang and Y.-H. Chan, Angew. Chem., Int. Ed., 2021, 60, 983–989 CrossRef CAS PubMed.
- Y.-X. Li, S.-P. Su, C.-H. Yang, M.-H. Liu, P.-H. Lo, Y.-C. Chen, C.-P. Hsu, Y.-J. Lee, H. K. Chiang and Y.-H. Chan, Adv. Healthcare Mater., 2021, 10, 2100993 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- X. Yin, Y. He, X. Wang, Z. Wu, E. Pang, J. Xu and J.-A. Wang, Front. Chem., 2020, 8, 725 CrossRef CAS PubMed.
- C. J. Christopherson, N. R. Paisley, Z. Xiao, W. R. Algar and Z. M. Hudson, J. Am. Chem. Soc., 2021, 143, 13342–13349 CrossRef CAS PubMed.
- T. L. Tam, H. Li, Y. M. Lam, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2011, 13, 4612–4615 CrossRef CAS PubMed.
- F. Trani, G. Scalmani, G. Zheng, I. Carnimeo, M. J. Frisch and V. Barone, J. Chem. Theory Comput., 2011, 7, 3304–3313 CrossRef CAS PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- E. E. Jelley, Nature, 1936, 138, 1009–1010 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- Y. J. Wang, Z. Li, J. Tong, X. Y. Shen, A. Qin, J. Z. Sun and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 3559–3568 RSC.
- T. Kowada, J. Kikuta, A. Kubo, M. Ishii, H. Maeda, S. Mizukami and K. Kikuchi, J. Am. Chem. Soc., 2011, 133, 17772–17776 CrossRef CAS PubMed.
- C. Li, Y. Zhang, G. Chen, F. Hu, K. Zhao and Q. Wang, Adv. Mater., 2017, 29, 1605754 CrossRef PubMed.
- S. He, S. Chen, D. Li, Y. Wu, X. Zhang, J. Liu, J. Song, L. Liu, J. Qu and Z. Cheng, Nano Lett., 2019, 19, 2985–2992 CrossRef CAS PubMed.
- Y. Che, S. Feng, J. Guo, J. Hou, X. Zhu, L. Chen, H. Yang, M. Chen, Y. Li, S. Chen, Z. Cheng, Z. Luo and J. Chen, Nanoscale, 2020, 12, 22022 RSC.
- S. Yamashita, H. Katsumi, N. Hibino, Y. Isobe, Y. Yagi, Y. Tanaka, S. Yamada, C. Naito and A. Yamamoto, Biomaterials, 2018, 154, 74–85 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, NMR spectra, and additional information as noted in text. See https://doi.org/10.1039/d2sc03271f |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2022 |