
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBare and ligand protected planar hexacoordinate silicon in SiSb3M3+ (M = Ca, Sr, Ba) clusters†
Chen
Chen
a,
Meng-hui
Wang
a,
Lin-Yan
Feng
b,
Lian-Qing
Zhao
b,
Jin-Chang
Guo
*b,
Hua-Jin
Zhai
 *b,
Zhong-hua
Cui
*a,
Sudip
Pan
*c and
Gabriel
Merino
*d
*b,
Zhong-hua
Cui
*a,
Sudip
Pan
*c and
Gabriel
Merino
*d
aInstitute of Atomic and Molecular Physics, Key Laboratory of Physics and Technology for Advanced Batteries (Ministry of Education), Jilin University, Changchun 130021, China. E-mail: zcui@jlu.edu.cn
bNanocluster Laboratory, Institute of Molecular Science, Shanxi University, Taiyuan 030006, China. E-mail: guojc@sxu.edu.cn; hj.zhai@sxu.edu.cn
cFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Strasse 4, 35032 Marburg, Germany. E-mail: pans@chemie.uni-marburg.de
dDepartamento de Física Aplicada, Centro de Investigación y de Estudios Avanzados, Unidad Mérida, km 6 Antigua carretera a Progreso, Apdo. Postal 73, Cordemex 97310, Mérida, Yuc., Mexico. E-mail: gmerino@cinvestav.mx
First published on 13th June 2022
Abstract
The occurrence of planar hexacoordination is very rare in main group elements. We report here a class of clusters containing a planar hexacoordinate silicon (phSi) atom with the formula SiSb3M3+ (M = Ca, Sr, Ba), which have D3h (1A1′) symmetry in their global minimum structure. The unique ability of heavier alkaline-earth atoms to use their vacant d atomic orbitals in bonding effectively stabilizes the peripheral ring and is responsible for covalent interaction with the Si center. Although the interaction between Si and Sb is significantly stronger than the Si–M one, sizable stabilization energies (−27.4 to −35.4 kcal mol−1) also originated from the combined electrostatic and covalent attraction between Si and M centers. The lighter homologues, SiE3M3+ (E = N, P, As; M = Ca, Sr, Ba) clusters, also possess similar D3h symmetric structures as the global minima. However, the repulsive electrostatic interaction between Si and M dominates over covalent attraction making the Si–M contacts repulsive in nature. Most interestingly, the planarity of the phSi core and the attractive nature of all the six contacts of phSi are maintained in N-heterocyclic carbene (NHC) and benzene (Bz) bound SiSb3M3(NHC)6+ and SiSb3M3(Bz)6+ (M = Ca, Sr, Ba) complexes. Therefore, bare and ligand-protected SiSb3M3+ clusters are suitable candidates for gas-phase detection and large-scale synthesis, respectively.
Exploring the bonding capacity of main-group elements (such as carbon or silicon) beyond the traditional tetrahedral concept has been a fascinating subject in chemistry for five decades. The 1970 pioneering work of Hoffmann and coworkers1 initiated the field of planar tetracoordinate carbons (ptCs), or more generally, planar hypercoordinate carbons. The past 50 years have witnessed the design and characterization of an array of ptC and planar pentacoordinate carbon (ppC) species.2–14 However, it turned out to be rather challenging to go beyond ptC and ppC systems. The celebrated CB62− cluster and relevant species15,16 were merely model systems because C avoids planar hypercoordination in such systems.17,18 In 2012, the first genuine global minimum D3h CO3Li3+ cluster was reported to have six interactions with carbon in planar form, although electrostatic repulsion between positively charged phC and Li centers and the absence of any significant orbital interaction between them make this hexacoordinate assignment questionable.19 It was only very recently that a series of planar hexacoordinate carbon (phC) species, CE3M3+ (E = S–Te; M = Li–Cs), were designed computationally by the groups of Tiznado and Merino (Fig. 1; left panel),20 in which there exist pure electrostatic interactions between the negative Cδ− center and positive Mδ+ ligands. These phC clusters were achieved following the so-called “proper polarization of ligand” strategy.
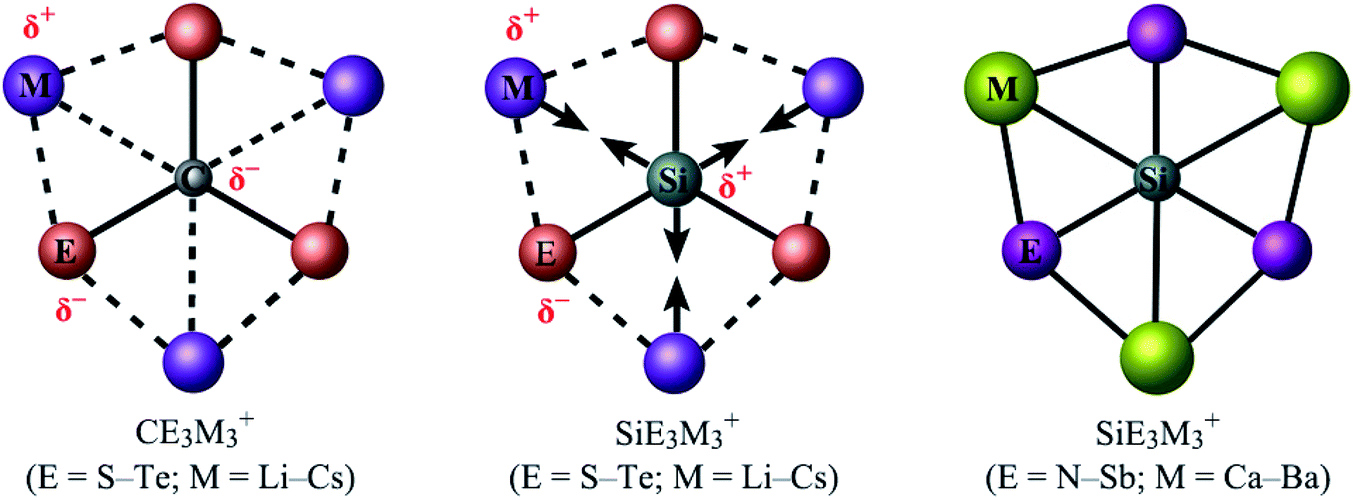 | ||
| Fig. 1 The pictorial depiction of previously reported phC CE3M3+ (E = S–Te; M = Li–Cs) clusters and the present SiE3M3+ (E = S–Te and N–Sb; M = Li–Cs and Ca–Ba) clusters. Herein the solid and dashed lines represent covalent and ionic bonding, respectively. The opposite double arrows illustrate electrostatic repulsion. | ||
The concept of planar hypercoordinate carbons has been naturally extended to their next heavier congener, silicon-based systems. Although the steric repulsion between ligands decreases due to the larger size, the strength of π- and σ-bonding between the central atom and peripheral ligands dramatically decreases, which is crucial for stability. Planar tetracoordinate silicon (ptSi) was first experimentally observed in a pentaatomic C2v SiAl4− cluster by Wang and coworkers in 2000.21 Very recently, this topic got a huge boost by the room-temperature, large-scale syntheses of complexes containing a ptSi unit.22 A recent computational study also predicted the global minimum of SiMg4Y− (Y = In, Tl) and SiMg3In2 to have unprecendented planar pentacoordinate Si (ppSi) units.23 Planar hexacoordinate Si (phSi) systems seem to be even more difficult to stabilize. Previously, a C2v symmetric Cu6H6Si cluster was predicted as the true minimum,24 albeit its potential energy surface was not fully explored. A kinetically viable phSi SiAl3Mg3H2+ cluster cation was also predicted.25 However, these phSi systems24,25 are only local minima and not likely to be observed experimentally. In 2018, the group of Chen identified the Ca4Si22− building block containing a ppSi center and constructed an infinite CaSi monolayer, which is essentially a two-dimensional lattice of the Ca4Si2 motif.26 Thus, it is still an open question to achieve a phSi atom to date.
Herein we have tried to find the correct combination towards a phSi system as the most stable isomer. Gratifyingly, we found a series of clusters, SiE3M3+ (E = N, P, As, Sb; M = Ca, Sr, Ba), having planar D3h symmetry with Si at the center of the six membered ring, as true global minimum forms. Si–E bonds are very strong in all the clusters, and alkaline-earth metals interact with the Si center by employing their d orbitals. However, electrostatic repulsion originated from the positively charged Si and M centers for E = N, P, and As dominates over attractive covalent interaction, making individual Si–M contacts repulsive in nature. This makes the assignment of SiE3M3+ (E = N, P, As; M = Ca, Sr, Ba) as genuine phSi somewhat skeptical. SiSb3M3+ (M = Ca, Sr, Ba) clusters are the sole candidates which possess genuine phSi centers as both electrostatic and covalent interactions in Si–M bonds are attractive. The d orbitals of M ligands play a crucial role in stabilizing the ligand framework and forming covalent bonds with phSi. Such planar hypercoordinate atoms are, in general, susceptible to external perturbations. However, the present title clusters maintain the planarity and the attractive nature of the bonds even after multiple ligand binding at M centers in SiSb3M3(NHC)6+ and SiSb3M3(Bz)6+. This would open the door for large-scale synthesis of phSi as well.
Two major computational efforts were made before reaching our title phSi clusters. The first one is to examine SiE3M3+ (E = S–Po; M = Li–Cs) clusters, which adopt D3h or C3v structures as true minima (see Table S1 in ESI†), being isoelectronic to the previous phC CE3M3+ (E = S–Po; M = Li–Cs) clusters. In the SiE3M3+ (E = S–Po; M = Li–Cs) clusters, the Si center always carries a positive charge ranging from 0.01 to +1.03|e|, in contrast to the corresponding phC species (see Fig. 1). Thus, electrostatic interactions between the Siδ+ and Mδ+ centers would be repulsive (Fig. 1). Given that the possibility of covalent interaction with an alkali metal is minimal, it would be a matter of debate whether they could be called true coordination. A second effort is to tune the electronegativity difference between Si and M centers so that the covalent contribution in Si–M bonding becomes substantial. Along this line, we consider the combinations of SiE3M3+ (E = N, P, As, Sb; M = Be, Mg, Ca, Sr, Ba). The results in Fig. S1† show that for E = Be and Mg, the phSi geometry has a large out-of-plane imaginary frequency mode, which indicates a size mismatch between the Si center and peripheral E3M3 (E = N–Bi; M = Be, Mg) ring. On the other hand, the use of larger M = Ca, Sr, Ba atoms effectively expands the size of the cavity and eventually leads to perfect planar geometry with Si atoms at the center as minima. In the case of SiBi3M3+, the planar isomer possesses a small imaginary frequency for M = Ca. Although planar SiBi3Sr3+ and SiBi3Ba3+ are true minima, they are 2.2 and 2.5 kcal mol−1 higher in energy than the lowest energy isomer, respectively (Fig. S2†). Fig. 2 displays some selected low-lying isomers of SiE3M3+ (E = N, P, As, Sb; M = Ca, Sr, Ba) clusters (see Fig. S3–S6† for additional isomers). The global minimum structure is a D3h symmetric phSi with an 1A1′ electronic state for all the twelve cases. The second lowest energy isomer, a ppSi, is located more than 49 kcal mol−1 above phSi for E = N. This relative energy between the most stable and nearest energy isomer gradually decreases upon moving from N to Sb. In the case of SiSb3M3+ clusters, the second-lowest energy isomer is 4.6–6.1 kcal mol−1 higher in energy than phSi. The nearest triplet state isomer is very high in energy (by 36–53 kcal mol−1, Fig. S3–S6†) with respect to the global minimum.
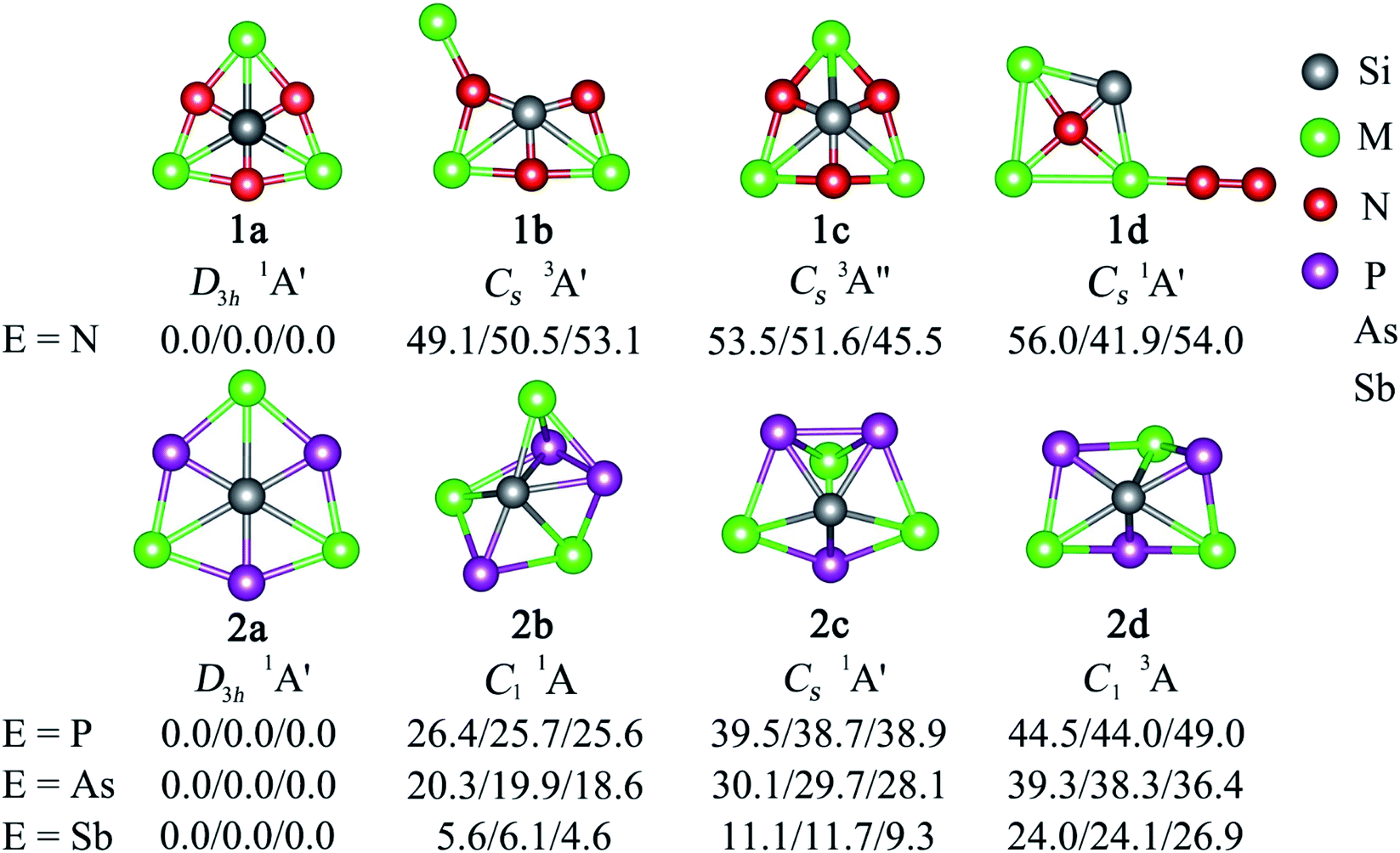 | ||
| Fig. 2 The structures of low-lying isomers of SiE3M3+ (E = N, P, As, Sb; M = Ca, Sr, Ba) clusters. Relative energies (in kcal mol−1) are shown at the single-point CCSD(T)/def2-TZVP//PBE0/def2-TZVP level, followed by a zero-energy correction at PBE0. The values from left to right refer to Ca, Sr, and Ba in sequence. The group symmetries and electronic states are also given. | ||
Born–Oppenheimer molecular dynamics (BOMD) simulations at room temperature (298 K), taking SiE3Ca3+ clusters as case studies, were also performed. The results are displayed in Fig. S7.† All trajectories show no isomerization or other structural alterations during the simulation time, as indicated by the small root mean square deviation (RMSD) values. The BOMD data suggest that the global minimum also has reasonable kinetic stability against isomerization and decomposition.
The bond distances, natural atomic charges, and bond indices for SiE3Ca3+ clusters are given in Table 1 (see also Tables S2–S5† for M = Sr, Ba). The Si–E bond distances are shorter than the typical Si–E single bond distance computed using the self-consistent covalent radii proposed by Pyykkö.27 In contrast, the Si–M bond distance is almost equal to the single bond distance. This gives the first hint of the presence of covalent bonding therein. However, the Wiberg bond indices (WBIs) for the Si–M links are surprisingly low (0.02–0.04). We then checked the Mayer bond order (MBO), which can be seen as a generalization of WBIs and is more acceptable since the approach of WBI calculations assumes orthonormal conditions of basis functions while the MBO considers an overlap matrix. The MBO values for the Si–M links are now sizable (0.13–0.18). These values are reasonable considering the large difference in electronegativity between Si and M, and, therefore, only a very polar bond is expected between them. In fact, the calculations of WBIs after orthogonalization of basis functions by the Löwdin method gives significantly large bond orders (0.48–0.55), which is known to overestimate the bond orders somewhat. The above results indicate that the presence of covalent bonding cannot be ruled out only by looking at WBI values.
| r Si–E | r Si–Ca | r E–Ca | q Si | q E | q Ca | |
|---|---|---|---|---|---|---|
| E = N | 1.669 | 2.555 | 2.246 | 1.57 | −1.93 | 1.74 |
| (1.14) {1.23} [1.84] | (0.02) {0.13} [0.51] | (0.22) {0.67} [0.84] | ||||
| E = P | 2.180 | 2.935 | 2.640 | 0.25 | −1.42 | 1.67 |
| (1.34) {1.11} [1.52] | (0.03) {0.14} [0.54] | (0.27) {0.74} [1.05] | ||||
| E = As | 2.301 | 3.004 | 2.721 | 0.07 | −1.34 | 1.65 |
| (1.33) {1.10} [1.45] | (0.03) {0.15} [0.55] | (0.29) {0.71} [1.12] | ||||
| E = Sb | 2.538 | 3.155 | 2.896 | −0.39 | −1.16 | 1.62 |
| (1.29) {1.01} [1.33] | (0.04) {0.18} [0.48] | (0.30) {0.78} [1.14] |
Our following argument regarding the presence of covalent Si–M bonding is based on energy decomposition analysis (EDA) in combination with natural orbital for chemical valence (NOCV) theory. We first performed EDA by taking Ca and SiE3Ca2 in different charge and electronic states as interacting fragments to get the optimum fragmentation scheme that suits the best to describe the bonding situation (see Tables S6–S9†). The size of orbital interaction (ΔEorb) is used as a probe.28 For all cases, Ca+ (D, 4s1) and SiE3Ca2 (D) in their doublet spin states turn out to be the best schemes, which give the lowest ΔEorb value. Table 2 shows the numerical results of EDA-NOCV calculations. The relative contribution of electrostatic and orbital terms shows that the interaction between Ca+ and SiE3Ca2 is a bit more covalent in nature than electrostatic. The intrinsic interaction energy (ΔEint) between the two fragments gradually diminishes upon moving from E = N to its heavier homologues. Both decreased electrostatic and orbital interactions are responsible for such a reduction in ΔEint.
| Energy term | Interaction | Ca+ (D, 4s1) + SiN3Ca2 (D) | Ca+ (D, 4s1) + SiP3Ca2 (D) | Ca+ (D, 4s1) + SiAs3Ca2 (D) | Ca+ (D, 4s1) + SiSb3Ca2 (D) |
|---|---|---|---|---|---|
| a The values in parentheses are the percentage contributions to total attractive interactions (ΔEelstat + ΔEorb). b The values in parentheses are the percentage contributions to the total orbital interaction ΔEorb. | |||||
| ΔEint | −192.9 | −153.0 | −144.9 | −129.9 | |
| ΔEPauli | 139.8 | 115.2 | 115.7 | 110.9 | |
| ΔEelstata | −162.0 (48.7%) | −116.4 (43.4%) | −113.0 (43.4%) | −100.9 (41.9%) | |
| ΔEorba | −170.7 (51.3%) | −151.8 (56.6%) | −147.6 (56.6%) | −140.0 (58.1%) | |
| ΔEorb(1)b | SiE3Ca2–Ca+(s) electron-sharing σ-bond | −89.2 (52.3%) | −79.4 (52.3%) | −74.3 (50.3%) | −66.9 (47.8%) |
| ΔEorb(2)b | SiE3Ca2 → Ca+(d) π‖-donation | −32.9 (19.3%) | −32.0 (21.1%) | −31.8 (21.5%) | −30.8 (22.0%) |
| ΔEorb(3)b | SiE3Ca2 → Ca+(d) σ-donation | −13.1 (7.7%) | −11.9 (7.8%) | −12.0 (8.1%) | −11.9 (8.5%) |
| ΔEorb(4)b | SiE3Ca2 → Ca+(d) π⊥-donation | −12.3 (7.2%) | −12.2 (8.0%) | −12.5 (8.5%) | −12.5 (8.9%) |
| ΔEorb(5)b | SiE3Ca2 → Ca+(d) δ-donation | −8.1 (4.7%) | −9.9 (6.5%) | −10.9 (7.4%) | −11.8 (8.4%) |
| ΔEorb(rest)b | −15.1 (8.8%) | −6.4 (4.2%) | −6.1 (4.1%) | −6.1 (4.4%) | |
The decomposition of ΔEorb into pair-wise orbital interaction ΔEorb(n) in Table 2 and the corresponding deformation densities Δρ(n) provide us with the most important information about bonding. The strongest orbital contribution, ΔEorb(1), which accounts for 48–52% of the total covalent interaction, originates from the coupling of unpaired electrons of Ca+ and SiE3Ca2. The remaining orbital terms, ΔEorb(2)–(5), mainly come from the donation of electron density from SiE3Ca2 to vacant atomic orbitals (AOs) of Ca+. The inspection of the shape of Δρ(n) and the related orbitals of the fragments (Fig. 3) helps us to identify the Si–Ca covalent bond and the orbitals involved in the pairwise interactions. The s orbital of Ca+ takes part in the electron-sharing σ-bond formation with SiE3Ca2, whereas vacant d AOs of Ca+ act as acceptor orbitals in the dative interactions, ΔEorb(2)–(5). Therefore, d AOs of Ca+ are responsible for 39–48% of the total orbital interaction. The present results further strengthen the proposal29–33 that heavier alkaline-earth elements (Ca, Sr, and Ba) should be classified as transition metals rather than main-group elements. Furthermore, a careful look at the Δρ(n) plots shows that in ΔEorb(1) and ΔEorb(2) only peripheral atoms are involved, but in ΔEorb(3)–(5) there is direct covalent interaction between Si and Ca centers. To correlate with the molecular orbitals (MOs) of the SiE3Ca3+ cluster, the related MOs for 24 valence electrons are given in Fig. S8.† Δρ(3)–(5) can be correlated with HOMO-4, the HOMO and the HOMO′, respectively. Therefore, although the MO coefficient of Ca centers is small, they should not be neglected as the energy stabilization coming from them is significant. Si and M centers are only connected through delocalized bonds which is the reason for not having any gradient path between them as is indicated in the electron density analysis. Instead, there is a ring critical point at the center of the SiE2M ring (see Fig. S9†). The results of adaptive natural density partitioning (AdNDP) analysis also corroborate this, where M centers are connected with the Si center through 7c–2e π-bonds (see Fig. S10†).
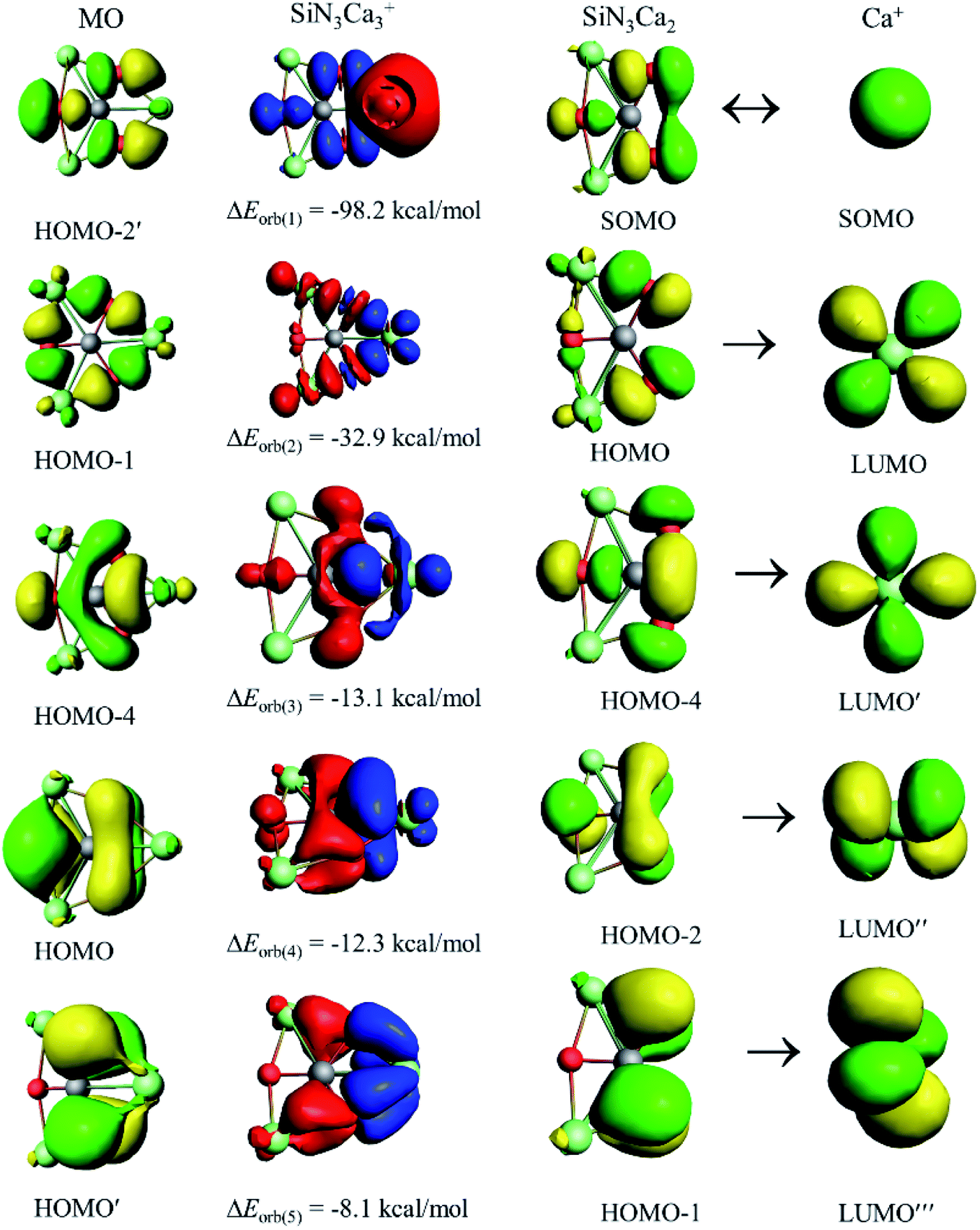 | ||
| Fig. 3 Plot of the deformation densities, Δρ(1)–(5) corresponding to ΔEorb(1)–(5) and the related interacting orbitals of the fragments in the SiN3Ca3+ cluster at the PBE0/TZ2P-ZORA//PBE0/def2-TZVP level. The orbital energy values are in kcal mol−1. The charge flow of the deformation densities is from red to blue. The isovalue for Δρ(1) is 0.001 au and for the rest is 0.0005 au. | ||
Another aspect is to check the nature of electrostatic interaction between Si and M. The natural charges in Table 1 shows significant electron transfer from M to E centers that imposes a positive partial charge of 1.7|e|. The E centers possess large negative charges (ranging from −1.2 to −1.9|e|), which decrease with the reduction in electronegativity of E. For a given M, with the variation of E, a drastic change in partial charge on Si is noted. For E = N, the Si center carries a high positive charge (1.6|e|), which is greatly reduced upon moving to E = P (0.3|e|). For E = As, the Si center becomes almost neutral, and eventually, it possesses a negative charge for E = Sb. Therefore, the electrostatic repulsion between Si and M should be largely reduced from N to As, and finally for SiSb3M3+, the electrostatic interaction between Si and M should be attractive. Note that the energy partitioning scheme in EDA-NOCV taking Ca+ + SiE3Ca2 provides combined energy contributions for one M–Si contact and two M–E contacts. This argument based on point charges is further verified by the energy components obtained in interacting quantum atom (IQA) analysis, which shows that the covalent (VCoval) part of the Si–Ca interaction is attractive (ranging between −4.6 and −11.6 kcal mol−1) in all cases. However, the electrostatic (ionic, VIonic) part is highly repulsive for E = N, P, As, making the overall interaction repulsive. Such electrostatic repulsion is sharply reduced in moving from N to As, and finally, for E = Sb, it becomes attractive (see Tables S10–S12†). Thus, the SiSb3M3+ cluster presents a case in which covalent bonding is robust and ionic interaction between Si and M centers is attractive in nature. If we look at the inter-atomic interaction energies (VTotal) for Si–M bonds and M–E bonds, it can be understood that the repulsive energy in Si–M bonds is largely overcompensated by two M–E bonds, even for E = N. This is the reason why electrostatic repulsion between Si and M centers does not result in a very large Si–M bond distance. Nevertheless, repulsive Si–M contacts in SiE3M3+ (E = N, P, As) make hexacoordination assignment skeptical. SiSb3M3+ clusters should be considered to possess phSi convincingly. Note that the IUPAC definition of coordination number only demands “the number of other atoms directly linked to that specified atom”,34 but does not say about the overall nature of interaction between them. In SiSb3M3+, phSi is linked to three Sb atoms through strong covalent bonds and is bound to three M atoms through ionic interaction in combination with a weaker covalent interaction. These clusters are only weakly aromatic because of such polar electronic distribution (see Fig. S11†).
The next challenge is to protect the reactive centers of phSi clusters with bulky ligands, which is required for large scale synthesis. This is not an easy task since slight external perturbation of most of the planar hypercoordinate atom species could result in a loss in planarity. Few years ago, the groups of Ding and Merino35 reported CAl4MX2 (M = Zr, Hf; X = F–I, C5H5) where ppC is sandwiched and protected by a metallocene framework. Therefore, the presence of X groups is mandatory to provide the electronic stabilization in ppC. In the present cases, surprisingly, SiSb3M3+ clusters are found to maintain the planarity around hexagons even after the coordination of M centers with six N-heterocyclic carbene (NHC) and benzene (Bz) ligands forming SiSb3M3(NHC)6+ and SiSb3M3(Bz)6+ (M = Ca, Sr, Ba) complexes, respectively (see Fig. 4). These complexes are highly stable against ligand dissociation as reflected by the high bond dissociation energy (De = 236.1 (Ca), 203.9 (Sr) and 171.3 (Ba) kcal mol−1) for SiSb3M3(NHC)6+ → SiSb3M3+ + 6NHC and De = 153.8 (Ca), 128.0 (Sr) and 114.0 (Ba) kcal mol−1 for SiSb3M3(Bz)6+ → SiSb3M3+ + 6Bz. The Si–M bond distances are slightly elongated because of coordination with the ligands. But the results of IQA given in Table S13† show that Si–M bonds have attractive interaction energies ranging between −20.0 and −32.4 kcal mol−1. Therefore, the planarity of the phSi core and the attractive nature of all the six contacts of phSi are maintained in ligand-bound SiSb3M3(NHC)6+ and SiSb3M3(Bz)6+ (M = Ca, Sr, Ba) complexes.
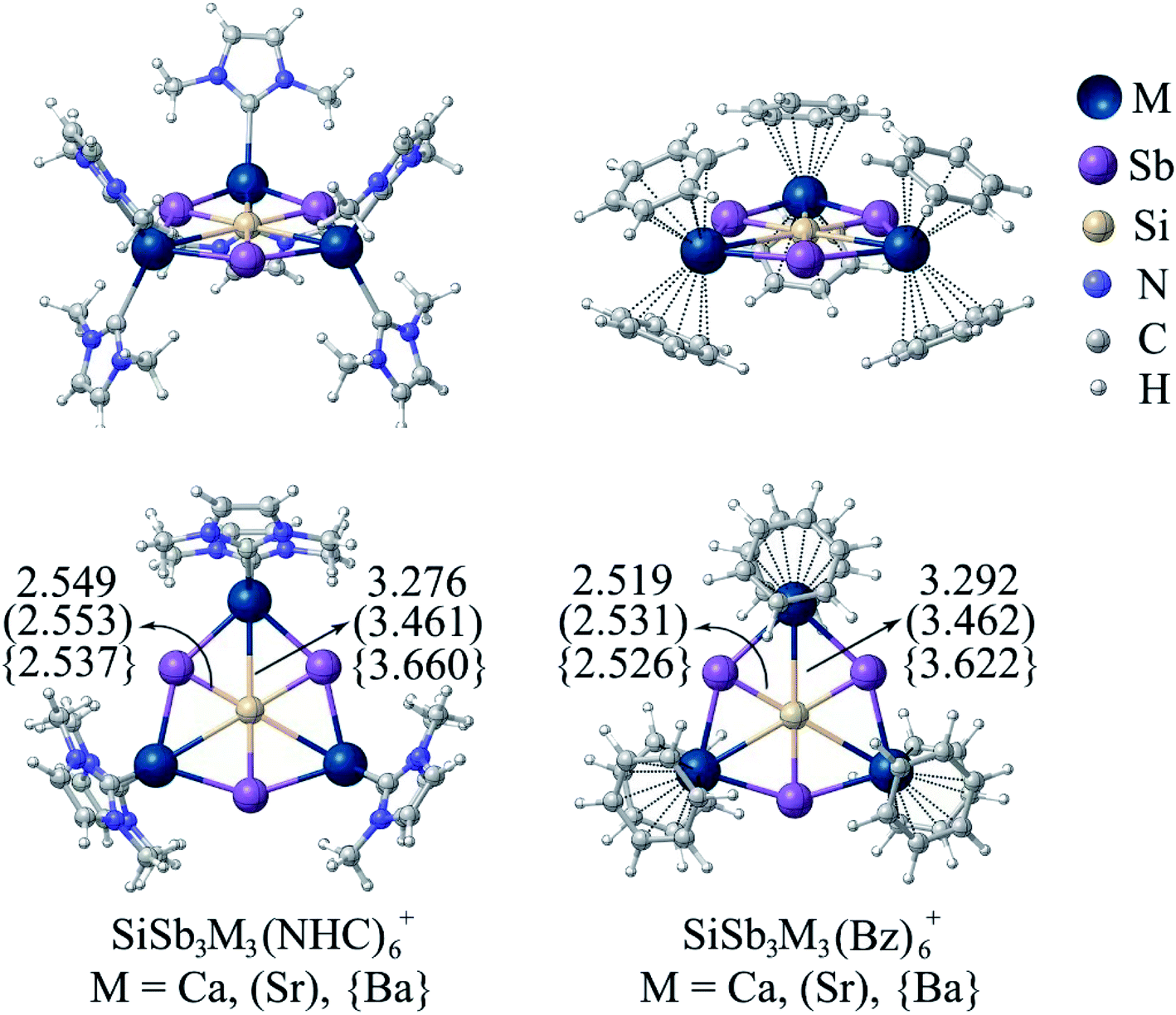 | ||
| Fig. 4 The minimum energy geometries of SiSb3M3(NHC)6+ and SiSb3M3(Bz)6+ (M = Ca, Sr, Ba) complexes at the PBE0-D3(BJ)/def2-TZVP level. | ||
In summary, we have theoretically achieved the first series of planar hexacoordinate silicon (phSi) clusters, SiSb3M3+ (M = Ca, Sr, Ba), by exploring their potential energy surfaces. These phSi systems are both thermodynamically and kinetically stable. The global minimum structures of SiE3M3+ (E = N, P, As, Sb) clusters have a D3h symmetry with the 1A1′ electronic state. The ability of the heavier alkaline-earth metals (Ca–Ba) to utilize their d orbitals in chemical bonding is a key factor that underlies the stability of these systems. The Ca–Ba ligands form weak covalent bonding with Si centers through their d orbitals, mimicking transition metals. The electronic charge distribution and IQA analysis show that electrostatic interaction in the Si–Ca links is essentially repulsive in SiN3M3+, but it sharply reduces with the decrease in electronegativity of E. Eventually, a sizable electrostatic attractive interaction exists between Si and M centers in SiSb3M3+, leading to a truly unprecedented phSi bonding motif that is held together by both covalent bonding and attractive ionic interaction. For SiE3M3+ (E = N, P, As) clusters, the electrostatic repulsion between Si and M dominates over covalent interaction, making Si–M contacts repulsive in nature. Most interestingly, the planarity of the phSi core and the attractive nature of all the six contacts of phSi are maintained in N-heterocyclic carbene (NHC) and benzene (Bz) bound SiSb3M3(NHC)6+ and SiSb3M3(Bz)6+ (M = Ca, Sr, Ba) complexes. Therefore, such clusters protected by bulky ligands would be suitable candidates for large scale synthesis in the presence of bulky counter-ions. Recent experimental reports on ptSi systems have already stimulated much curiosity within the community, and the present results would undoubtedly act as a stimulus to it.
Data availability
Computational details, extra data and the Cartesian coordinates for all compounds are provided in the ESI† accompanying this paper.Author contributions
JCG, H-JZ, Z-hC, SP, and GM designed the works and concepts, analyzed the data, wrote the draft, and finalized it. CC and M-hW performed the global minima searching. L-YF and L-QZ performed NBO and IQA. SP performed EDA-NOCV. All authors took part in the discussions and approved the final version.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 11922405, 11874178, and 91961204). H. J. Z. acknowledges support from the National Natural Science Foundation of China (22173053 and 21873058). Part of the calculations in this work were supported by the High-Performance Computing Center of Jilin University, China.References
- R. Hoffmann, R. W. Alder and C. F. Wilcox, J. Am. Chem. Soc., 1970, 92, 4992–4993 CrossRef CAS.
- J. B. Collins, J. D. Dill, E. D. Jemmis, Y. Apeloig, P. v. R. Schleyer, R. Seeger and J. A. Pople, J. Am. Chem. Soc., 1976, 98, 5419–5427 CrossRef CAS.
- P. v. R. Schleyer and A. I. Boldyrev, J. Chem. Soc., Chem. Commun., 1991, 21, 1536–1538 RSC.
- X. Li, L. S. Wang, A. I. Boldyrev and J. Simons, J. Am. Chem. Soc., 1999, 121, 6033–6038 CrossRef CAS.
- L. S. Wang, A. I. Boldyrev, X. Li and J. Simons, J. Am. Chem. Soc., 2000, 122, 7681–7687 CrossRef CAS.
- X. Li, H. F. Zhang, L. S. Wang, G. D. Geske and A. I. Boldyrev, Angew. Chem., Int. Ed., 2000, 39, 3630–3632 CrossRef CAS PubMed.
- J. Xu, X. Zhang, S. Yu, Y. H. Ding, K. H. Bowen and J. Phys, Chem. Lett., 2017, 8, 2263–2267 CAS.
- W. Siebert and A. Gunale, Chem. Soc. Rev., 1999, 28, 367–371 RSC.
- R. Keese, Chem. Rev., 2006, 106, 4787–4808 CrossRef CAS PubMed.
- G. Merino, M. A. Mendez-Rojas, A. Vela and T. Heine, J. Comput. Chem., 2007, 28, 362–372 CrossRef CAS PubMed.
- L. M. Yang, E. Ganz, Z. Chen, Z. X. Wang and P. v. R. Schleyer, Angew. Chem., Int. Ed., 2015, 54, 9468–9501 CrossRef CAS PubMed.
- Y. Pei, W. An, K. Ito, P. v. R. Schleyer and X. C. Zeng, J. Am. Chem. Soc., 2008, 130, 10394–10400 CrossRef CAS PubMed.
- V. Vassilev-Galindo, S. Pan, K. J. Donald and G. Merino, Nat. Rev. Chem., 2018, 2, 1–10 CrossRef.
- J. C. Guo, L. Y. Feng, J. Barroso, G. Merino and H. J. Zhai, Chem. Commun., 2020, 56, 8305–8308 RSC.
- K. Exner and P. v. R. Schleyer, Science, 2000, 290, 1937–1940 CrossRef CAS PubMed.
- (a) K. Ito, Z. Chen, C. Corminboeuf, C. S. Wannere, X. H. Zhang, Q. S. Li and P. v. R. Schleyer, J. Am. Chem. Soc., 2007, 129, 1510–1511 CrossRef CAS PubMed; (b) R. Islas, T. Heine, K. Ito, P. v. R. Schleyer and G. Merino, J. Am. Chem. Soc., 2007, 129, 14767–14774 CrossRef CAS PubMed.
- L. M. Wang, W. Huang, B. B. Averkiev, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2007, 46, 4550–4553 CrossRef CAS PubMed.
- B. B. Averkiev, D. Y. Zubarev, L. M. Wang, W. Huang, L. S. Wang and A. I. Boldyrev, J. Am. Chem. Soc., 2008, 130, 9248–9250 CrossRef CAS PubMed.
- Y. B. Wu, Y. Duan, G. Lu, H. G. Lu, P. Yang, P. V. R. Schleyer, G. Merino, R. Islas and Z. X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 14760–14763 RSC.
- L. Leyva-Parra, L. Diego, O. Yanez, D. Inostroza, J. Barroso, A. Vasquez-Espinal, G. Merino and W. Tiznado, Angew. Chem., Int. Ed., 2021, 60, 8700–8704 CrossRef CAS PubMed.
- A. I. Boldyrev, X. Li and L. S. Wang, Angew. Chem., Int. Ed., 2000, 39, 3307–3310 CrossRef CAS PubMed.
- (a) P. Ghana, J. Rump, G. Schnakenburg, M. I. Arz and A. C. Filippou, J. Am. Chem. Soc., 2021, 143, 420–432 CrossRef CAS PubMed; (b) F. Ebner and L. Greb, Chem, 2021, 7, 2151–2159 CrossRef CAS PubMed.
- M. H. Wang, X. Dong, Z. H. Cui, M. Orozco-Ic, Y. H. Ding, J. Barroso and G. Merino, Chem. Commun., 2020, 56, 13772–13775 RSC.
- S. D. Li, G. M. Ren and C. Q. Miao, Inorg. Chem., 2004, 43, 6331–6333 CrossRef CAS PubMed.
- A. J. Kalita, S. S. Rohman, C. Kashyap, S. S. Ullah, I. Baruah, L. J. Mazumder and A. K. Guha, Int. J. Quantum Chem., 2021, 121, e2664 CrossRef CAS.
- Y. Wang, M. Qiao, Y. Li and Z. Chen, Nanoscale Horiz., 2018, 3, 327–334 RSC.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- F. A. Bickelhaupt, C. Fonseca Guerra, M. P. Mitoraj, F. Sagan, A. Michalak, S. Pan and G. Frenking, Phys. Chem. Chem. Phys., 2022 10.1039/D2CP02153F.
- X. Wu, L. L. Zhao, J. Y. Jin, S. Pan, W. Li, X. Y. Jin, G. J. Wang, M. F. Zhou and G. Frenking, Science, 2018, 361, 912–916 CrossRef CAS PubMed.
- Q. Wang, S. Pan, Y. B. Wu, G. H. Deng, J. H. Bian, G. J. Wang, L. L. Zhao, M. F. Zhou and G. Frenking, Angew. Chem., Int. Ed., 2019, 58, 17365–17374 CrossRef CAS PubMed.
- Q. Wang, S. Pan, S. J. Lei, J. Y. Jin, G. H. Deng, G. J. Wang, L. L. Zhao, M. F. Zhou and G. Frenking, Nat. Commun., 2019, 10, 3375 CrossRef PubMed.
- (a) I. Fernandez, N. Holzmann and G. Frenking, Chem.–Eur. J., 2020, 26, 14194–14210 CrossRef CAS PubMed; (b) T. Bettens, S. Pan, F. De Proft, G. Frenking and P. Geerlings, Chem.–Eur. J., 2020, 26, 12785–12793 CrossRef CAS PubMed; (c) G. Frenking, I. Fernández, N. Holzmann, S. Pan, I. Krossing and M. Zhou, JACS Au, 2021, 1, 623–645 CrossRef CAS PubMed; (d) P. Stegner, C. Färber, J. Oetzel, U. Siemeling, M. Wiesinger, J. Langer, S. Pan, N. Holzmann, G. Frenking, U. Albold, B. Sarkar and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 14615–14620 CrossRef CAS PubMed; (e) Y. Zhou, S. Pan, X. Dong, L. Wang, M. Zhou and G. Frenking, J. Am. Chem. Soc., 2022, 144, 8355–8361 CrossRef CAS PubMed.
- M. F. Zhou and G. Frenking, Acc. Chem. Res., 2021, 54, 3071–3082 CrossRef CAS PubMed.
- P. Muller, Pure Appl. Chem., 1994, 66, 1077–1184 CrossRef.
- Z.-H. Cui, V. Vassilev-Galindo, J. L. Cabellos, E. Osorio, M. Orozco, S. Pan, Y.-H. Ding and G. Merino, Chem. Commun., 2017, 53, 138–141 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01761j |
| This journal is © The Royal Society of Chemistry 2022 |