
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular mechanisms of amyloid formation in living systems
Tessa
Sinnige

Bijvoet Centre for Biomolecular Research, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands. E-mail: t.sinnige1@uu.nl
First published on 17th May 2022
Abstract
Fibrillar protein aggregation is a hallmark of a variety of human diseases. Examples include the deposition of amyloid-β and tau in Alzheimer's disease, and that of α-synuclein in Parkinson's disease. The molecular mechanisms by which soluble proteins form amyloid fibrils have been extensively studied in the test tube. These investigations have revealed the microscopic steps underlying amyloid formation, and the role of factors such as chaperones that modulate these processes. This perspective explores the question to what extent the mechanisms of amyloid formation elucidated in vitro apply to human disease. The answer is not yet clear, and may differ depending on the protein and the associated disease. Nevertheless, there are striking qualitative similarities between the aggregation behaviour of proteins in vitro and the development of the related diseases. Limited quantitative data obtained in model organisms such as Caenorhabditis elegans support the notion that aggregation mechanisms in vivo can be interpreted using the same biophysical principles established in vitro. These results may however be biased by the high overexpression levels typically used in animal models of protein aggregation diseases. Molecular chaperones have been found to suppress protein aggregation in animal models, but their mechanisms of action have not yet been quantitatively analysed. Several mechanisms are proposed by which the decline of protein quality control with organismal age, but also the intrinsic nature of the aggregation process may contribute to the kinetics of protein aggregation observed in human disease.
1. Introduction
More than fifty human diseases are characterised by the deposition of fibrillar protein aggregates.1,2 These include neurodegenerative disorders such as Alzheimer's, Parkinson's, and Huntington's diseases. Proteins also aggregate in organs other than the brain, for example in systemic amyloidosis and type II diabetes. Early investigations into the nature of the deposits found in disease tissue showed that they could be stained with iodine, leading to the name ‘amyloid’ derived from the Greek word for starch.3 The different proteins that aggregate in human diseases are generally unrelated in terms of sequence or native structure (see Table 1 for the main proteins discussed in this article). However, they have in common that in disease they fold into a fibrillar structure with a cross-β architecture, referred to as ‘amyloid fibril’.| Protein | Disease | Isoforms and post-translational modifications | Aggregation behaviour in vitro | Ref. |
|---|---|---|---|---|
| a 9 polyQ expansion diseases are known, which are each associated with polyQ stretches in a different protein. | ||||
| Amyloid-β | Alzheimer's disease | Cleavage product of APP, different lengths including 1–38, 1–40, 1–42, 1–43 | Spontaneous under physiological conditions; dominated by secondary nucleation | 15 and 16 |
| Tau | Alzheimer's disease and other tauopathies | 6 isoforms, heavily phosphorylated in disease | Promoted by the addition of polyanions; dominated by secondary nucleation and/or fragmentation | 19 and 38 |
| α-Synuclein | Parkinson's disease and other synucleinopathies | C-terminal cleavage and phosphorylation of S129 and other sites in disease | Slow intrinsic aggregation; promoted by the addition of negatively charged lipid bilayers (primary nucleation), low pH (secondary nucleation) or shaking (fragmentation) | 17, 18 and 20–22 |
| Huntingtin (htt) fragments with expanded polyQ | Huntington's diseasea | Aberrant splicing and/or cleavage give rise to N-terminal fragments including exon 1 | Simple polyQ stretches and htt exon 1 undergo spontaneous aggregation under physiological conditions; dominated by secondary nucleation; strong dependence on polyQ length | 23–25 |
| Islet amyloid polypeptide (IAPP) | Type II diabetes | Mature IAPP is produced from a precursor protein and secreted | Spontaneous aggregation; accelerated by membranes containing anionic lipids | 26–30 |
| Superoxide dismutase 1 (SOD1) | Amyotrophic lateral sclerosis (ALS) | Mature protein is a disulfide-crosslinked dimer with copper and zinc ions bound | The apoSOD1 monomer forms fibrils via an unfolded intermediate; fragmentation-driven mechanism under agitation | 31–33 |
| Prion protein (PrP) | Creutzfeldt-Jakob disease | Glycosylated and lipid-anchored | Natively folded PrPC is converted into PrPSc through a fragmentation-based mechanism | 22 |
The relationship between amyloid formation and toxicity is still unclear, leading some to question whether protein aggregation plays a causal role in human disease.4 Amyloids can also have functional roles, for example in bacterial biofilm formation5 and the storage of peptide hormones.6 However, a large body of evidence suggests that the process of pathogenic fibril formation contributes to cellular dysfunction and death. Toxicity associated with amyloid formation has been attributed to intermediate or off-pathway oligomeric species, which may engage in a variety of aberrant interactions and thereby disrupt cellular pathways.7–10 The molecular features of oligomers remain largely elusive, given their heterogeneous and – in the case of intermediate species – transient nature. The process of fibril growth may in addition cause significant perturbation to cellular structures such as membranes.11,12 Mature fibrils are considered to be relatively inert, but they have also been shown to engage in non-physiological interactions and to sequester essential cellular components.13,14
The precise relationship between amyloid formation and toxicity is difficult to establish without knowledge of the molecular mechanisms of protein aggregation in a cellular context. Great progress has been made in elucidating the molecular mechanisms of fibril formation using test tube experiments with purified proteins and peptides, where detailed kinetic analysis has quantitatively revealed the aggregation processes of e.g. amyloid-β15,16 and α-synuclein.17,18 However, it is not clear to what extent these mechanisms are relevant for the situation in vivo. Even though the biophysical properties of a protein remain fundamentally the same, its aggregation behaviour may be modulated in living cells and organisms, for example by the presence of molecular chaperones. It is not clear whether these factors merely change the rates of the different aggregation steps, or whether the aggregation mechanisms are more extensively remodelled. This perspective explores the question to what extent the mechanisms elucidated in vitro apply to human disease, by combining our knowledge on the biophysics of amyloid formation with the genetics of human disease and information from model organisms.
2. Lessons learnt from in vitro studies
In vitro studies of amyloid formation using isolated peptides and proteins have the advantage that the intrinsic aggregation behaviour is observed, and that the conditions can be carefully controlled. Even so, the process of amyloid formation in itself is rather complex, comprising several microscopic steps (Fig. 1A) that are not immediately evident from the macroscopic aggregation data. The nature and contributions of the microscopic steps can be elucidated by means of kinetic analysis, based on global fitting of experimental data to mathematical models describing the aggregation processes.34,35 The experimental data are typically based on Thioflavin T fluorescence or other measurements that provide a read-out for the fibril mass, monitored for a range of protein concentrations over time.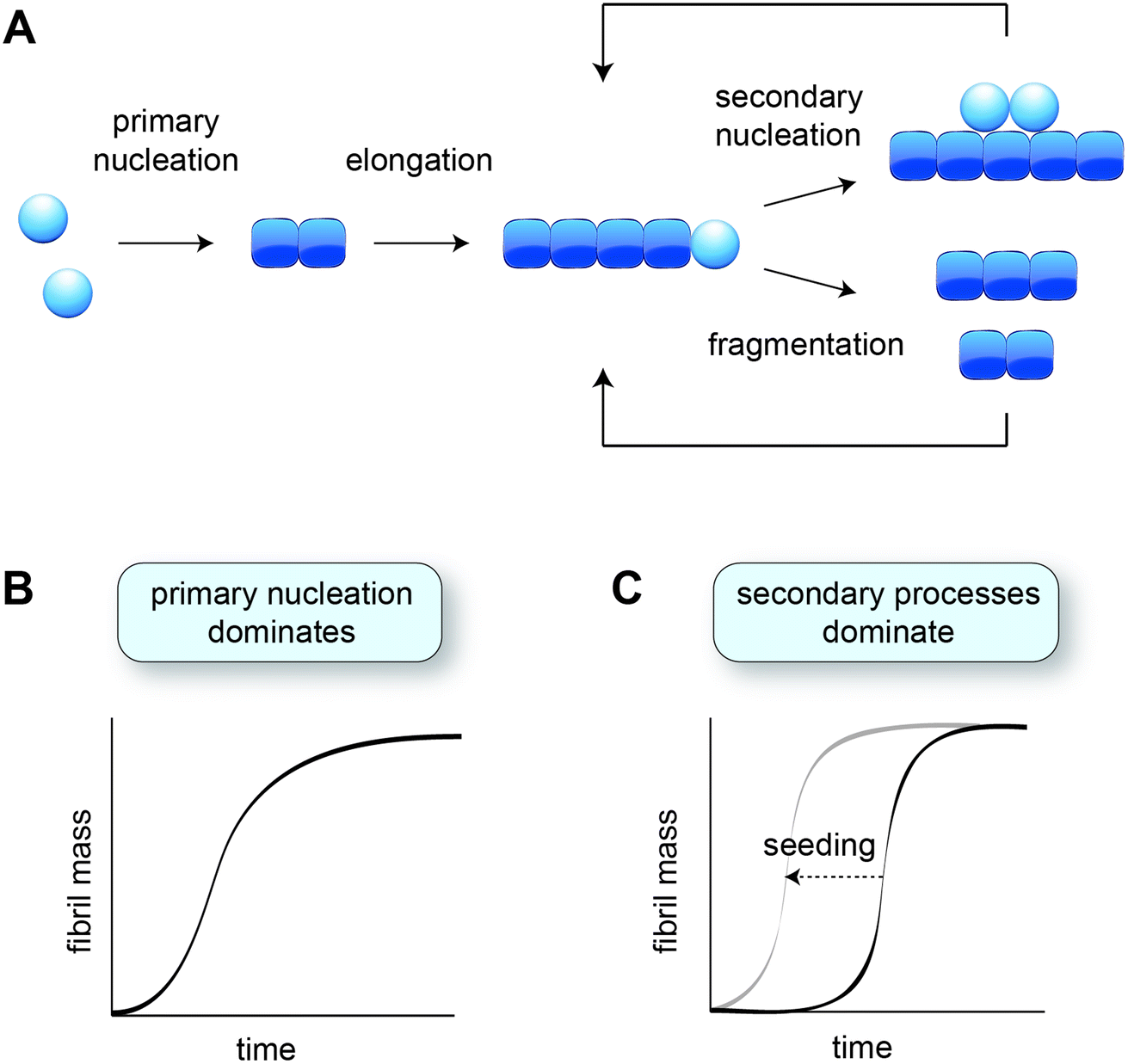 | ||
| Fig. 1 Aggregation kinetics in vitro. (A) Schematic of the different microscopic aggregation steps. Spheres indicate monomeric protein, and cubes the monomeric subunits in the fibril. The nucleus is formed by two monomers in the case of amyloid-β, but may have a different size for other proteins. Secondary nucleation and fragmentation cause a positive feedback loop, leading to the generation of more fibrils. (B) Cartoon of an aggregation profile characterised by primary nucleation. (C) Cartoon of an aggregation profile characterised by secondary processes, evident from the long lag phase. Adding pre-formed fibril seeds shortens the lag phase by bypassing primary nucleation. | ||
2.1 Mechanisms of amyloid formation established in vitro
The formation of fibrils from purely monomeric protein depends on an initial conformational change, referred to as primary nucleation. The critical nucleus can be seen as a transition state onto which the addition of monomers is energetically favourable. Nucleation combined with elongation represents the simplest mechanism of amyloid formation starting from monomeric protein, and is characterised by a sigmoidal aggregation curve (Fig. 1B). A more strongly sigmoidal curve with a long lag phase and a rapid increase towards the plateau is indicative of secondary processes, which accelerate aggregation as a function of the fibril mass already present (Fig. 1C). Secondary processes include fibril fragmentation, leading to the generation of new elongation-competent fibril ends, and secondary nucleation, the catalysis of nucleation on the surface of existing fibrils (Fig. 1A). The lag phase can be shortened by adding pre-formed fibril seeds, bypassing the need for primary nucleation (Fig. 1C).36Secondary nucleation has been found to dominate the overall process of fibril formation for many disease-associated peptides and proteins in vitro, including amyloid-β,15 a fragment of tau,37 huntingtin exon 1 with expanded polyglutamine,25 and islet amyloid polypeptide (IAPP)30,38 (Table 1). Single-molecule studies on a full length tau isoform, α-synuclein and mouse prion protein highlighted fragmentation as the dominant mechanism producing new fibrils (Table 1).19,22 Given that fragmentation is independent of the monomer concentration, it may play a more significant role relative to secondary nucleation at the low protein concentrations expected in vivo. Secondary processes can explain aggregate spreading across tissues by the transfer of fibril seeds, and may contribute to the long lag phase of aggregation observed in human disease, as further discussed in Section 6.
2.2 Experimental conditions affect aggregation behaviour
It is likely that the biological environment changes the rates of the different aggregation steps to varying extents, thereby shifting the mechanism to a different dominant process. Even in vitro, experimental conditions greatly affect the aggregation mechanism. A good example is presented by α-synuclein (Table 1), which accumulates in Lewy Bodies in Parkinson's disease.39 Under quiescent conditions and physiological pH, α-synuclein fibrillization occurs very slowly, taking weeks until completion at 37 °C.20 The aggregation process can be accelerated by vigorous shaking,21 leading to fragmentation being the main driver of fibril formation. The addition of purely anionic lipid vesicles strongly promotes aggregation by accelerating primary nucleation.18 Other lipid compositions and higher lipid ratios were found to have the opposite effect, however.40 At pH values below 6.0, α-synuclein aggregation occurs readily due to a major increase in the rate of secondary nucleation.17 α-Synuclein engages in functional interactions with synaptic vesicles in vivo,41,42 and is thought to traffic via acidic cellular organelles such as endosomes.43–45 Whether aggregation is promoted and which of the mechanisms is dominant may thus strongly depend on the intracellular localisation of the protein.The confinement of proteins inside the cell by itself has major implications for the aggregation mechanisms, and these small volume effects have also been explored in vitro. Nucleation is a stochastic process, yet in bulk experiments comprising many molecules this behaviour is not observable. Encapsulation of insulin into nanodroplets, however, has revealed highly stochastic behaviour, whereby aggregation is initiated in different droplets at different points in time.46 Once aggregation has started, it spreads rapidly across the droplet, suggesting that stochastic nucleation events are followed by secondary processes that lead to the propagation of fibril formation within the droplet. The volume range in which stochastic nucleation is noticeable (fL-pL) encompasses that of most living cells,47 and this behaviour has indeed been observed in cultured cells48 as well as a multicellular animal model49 as further discussed in Section 4.
Liquid–liquid phase separation has recently emerged as a way to further control protein localisation inside cells.50–52 Given that the local protein concentration within the phase-separated compartment is orders of magnitude higher than in the dilute surroundings, this phenomenon can greatly increase aggregation rates. Liquid–liquid phase separation has functional roles, but has also been shown to lead to the aggregation of a range of disease-associated proteins in vitro, including FUS,53 α-synuclein,54,55 and tau.56 In the case of α-synuclein, the aggregation of phase-separated protein was found to be independent of the bulk monomer concentration, showing that nucleation is initiated within the droplets in which the concentration remains constant.57 Translating this behaviour to the situation in vivo, phase-separation may thus induce aggregation even when the overall concentration of a particular protein would be too low to lead to significant aggregation in a dispersed state.
3. Biophysical principles of aggregation play a role in human disease
Despite the multitude of factors that may affect aggregation pathways and rates in a biological environment, key aspects of the behaviour of aggregation-prone proteins as established in vitro appear to be preserved in vivo. Genetic forms of protein aggregation diseases are often associated with increased protein levels or the production of more aggregation-prone variants, as detailed below. When comparing different diseases, the trend emerges that proteins that are more intrinsically aggregation-prone in vitro (Table 1) cause a more robust pathology in vivo. Altogether, aggregation propensity appears to be an important factor for the onset and severity of human disease, even though the precise relationship is not yet clear and there are notable exceptions.3.1 Concentration dependence of amyloid formation in human disease
The aggregation kinetics of amyloid formation in vitro are dependent on the starting concentration of monomeric protein. The identification of gene multiplications in familial forms of Alzheimer's and Parkinson's diseases suggests that this may also be the case in humans. An early-onset familial form of Parkinson's disease with dementia was found to arise from a triplication of the SNCA gene encoding α-synuclein,58 resulting in an approximate doubling of mRNA and soluble protein levels.59 Gene duplication of SNCA is associated with a less aggressive, late-onset form of the disease, suggesting a direct level between α-synuclein levels and disease progression.60The APP gene encoding the amyloid precursor protein is located on chromosome 21, and people with Down syndrome that carry an additional copy of this chromosome are at high risk to develop early-onset Alzheimer's disease.61 Duplication of the APP gene itself has also been associated with a rare early-onset form of the disease.62 Other familial forms of Alzheimer's disease are linked to the cleavage of APP by the γ-secretase complex, releasing amyloid-β. Typically, these mutations result in higher levels of the total pool of amyloid-β peptides, or specifically of the more aggregation-prone 1–42 isoform compared to 1–40.63 Sporadic Alzheimer's disease may in fact also be linked to increased amyloid-β levels, given that the main genetic risk factor, APOE, is involved in its clearance.64
3.2 Disease mutations affect aggregation propensity
Disease mutations in the part of APP that encodes amyloid-β generally enhance the aggregation kinetics of the peptide in vitro, in particular due to an increased contribution of secondary nucleation.65,66 Mutations in tau are typically not found in Alzheimer's disease, but are associated with the development of frontotemporal dementia and other tauopathies. Certain variants such as the P301L mutation have been shown to aggregate faster than wild-type under a range of experimental conditions,67–72 and to induce a strong phenotype in animal models.73–75 Other mutations seem to have a stronger effect on microtubule binding, which is the physiological role of tau.70,71 In a cellular context, this may lead to an increased concentration of free tau, which also has the effect of promoting protein aggregation. Yet other mutations have been found to affect the relative abundance of the different tau isoforms,76 but their respective aggregation propensities remain to be determined.When looking at amino acid substitutions in α-synuclein that underly familial forms of Parkinson's disease, the picture is rather diverse. Kinetic analysis in vitro revealed that the A53T mutant protein has increased rates of both anionic lipid-catalysed aggregation and secondary processes occurring at mildly acidic pH.77 For A30P only the latter were accelerated, whereas the E46K, G51D and H50Q mutations had either unaffected or lower aggregation rates compared to wild-type α-synuclein. These mutants might display different aggregation behaviour in the human brain compared to the test tube. For example, A30P has a reduced affinity for neutral membranes,78 which may lead to an increase in free protein thereby promoting aggregation, similarly as described above for tau. It is also conceivable that α-synuclein fibril formation is not the factor driving these familial disease forms. In support of this notion, certain genetic forms of Parkinson's disease with mutations in parkin or LRKK2 present cases without Lewy Body pathology.79,80
Perhaps the most direct evidence for a correlation between protein aggregation and disease is provided by Huntington's and other polyglutamine (polyQ) expansion diseases, which all have a genetic cause. The expansion of CAG repeats leads to mutant proteins with an extended polyQ tract, causing intracellular protein inclusions and disease symptoms when the pathogenic threshold is crossed. The longer the polyQ stretch, the faster the aggregation kinetics in vitro,24 and the earlier disease symptoms occur.81–84 In the case of Huntington's disease, the typical polyQ threshold of ∼40 residues causes disease in mid-life, whereas expansions of over 60 residues are associated with juvenile Huntington's disease affecting patients younger than 21 years old.82,83
The peptides and proteins considered above are largely intrinsically disordered, although structural segments may be formed upon physiological interactions, e.g. in the case of tau binding to microtubules. Proteins with a globular fold are also capable of forming amyloid fibrils, which occurs via partial unfolding. In these cases, destabilisation of the folded state by mutations typically promotes amyloid formation and disease. This has been shown for SOD1, which aggregates in familial forms of ALS (Table 1),85 but also for proteins associated with systemic amyloidosis including lysozyme,86 β-2 microglobulin,87 and transthyretin.88 Drugs that stabilise the native transthyretin tetramer slow the progression of disease forms associated with mutant as well as wild-type proteins, providing strong evidence for a causal link between protein aggregation and the disease.89
3.3 Correlation between aggregation propensities in vitro and in disease
It is difficult to compare the aggregation kinetics of different proteins in vitro, for which the experiments have been carried out at varying conditions, and even more so in vivo, where limited quantitative information is available. Still, it is interesting to qualitatively consider how the aggregation propensities in vitro correlate with the onset and extent of aggregation in different diseases (Table 1). For example, amyloid-β 1–42 is so aggregation-prone that it is difficult to handle in test tube experiments. Amyloid plaques appear decades before the onset of symptoms in Alzheimer's disease, and are even found in cognitively normal aged individuals.90,91 Thus, it is tempting to speculate that amyloid-β aggregation in vivo happens spontaneously once sufficiently high concentrations are reached (see Sections 3.1 and 6.2).A similar argument can be made for polyQ diseases. Simple peptides comprising as few as 15 glutamine residues spontaneously aggregate into fibrils in the test tube.92 A physical explanation for the higher threshold for polyQ aggregation observed in human disease is the transition from a tetrameric or dimeric nucleus for shorter polyQ lengths, towards a monomeric nucleus at longer polyQ lengths.24 The longer polyQ peptides are able to form a β-hairpin structure within a single chain, accelerating fibril formation by orders of magnitude. The aggregation kinetics of polyQ proteins are modulated by the flanking regions,93,94 and consistently the threshold is somewhat different between the different polyQ diseases.95 The diseases are not fully penetrant at intermediate polyQ lengths (e.g. 35–39 for Huntington's disease), suggesting that in this regime the cellular environment has a major influence on the aggregation process. At polyQ lengths above the pathogenic threshold (40 for Huntington's disease), the disease will however invariably manifest, accompanied by the characteristic protein inclusions.
Contrastingly, wild-type tau is much less prone to spontaneous aggregation in vitro, and most assays include negatively charged molecules such as heparin to drive tau self-association. In humans, the protein occurs in six alternatively spliced isoforms, and in Alzheimer's brain it is heavily phosphorylated.96,97 The effects of the amino acid sequence and posttranslational modifications on the aggregation propensity of tau are not yet clear, given that most in vitro studies have been performed using short and unmodified tau fragments. However, it appears that the aggregation of wild-type tau in Alzheimer's disease requires certain biological triggers, as evidenced by the fact that it occurs downstream of amyloid-β deposition.90 Most of the mouse models for Alzheimer's disease based on APP and γ-secretase mutations fail to develop tau tangles in spite of extensive amyloid plaque formation, and the relationship between the aggregation of the two proteins remains to be determined.98
Wild-type α-synuclein is also less intrinsically aggregation-prone than for example amyloid-β, and in vitro aggregation assays typically rely on the addition of seeds, anionic lipid vesicles or a shift to acidic pH as discussed in Section 2.2. In cultured cells, α-synuclein is highly soluble and found as a disordered monomer99 or an α-helical tetramer.100 Intriguingly, Lewy Bodies in human brain tissue have been suggested to be a medley of cellular organelles and membranes, in the apparent absence of α-synuclein fibrils.101 However, α-synuclein aggregation and neuronal dysfunction can be robustly induced by introducing pre-formed fibrils in cellular and mouse models,102–107 leading to the hypothesis that α-synuclein pathology in the human brain develops through the spreading of fibril seeds. Seeded aggregation is highly efficient in vitro by bypassing the need for primary nucleation (Fig. 1C), and in this way aggregation could proceed in vivo even for proteins with very low intrinsic aggregation propensity. The hypothesis of aggregation via the cell-to-cell transfer of seeds in combination with secondary processes will be further discussed in Section 6.3.
4. In vivo studies on the kinetics of amyloid formation
A more quantitative and mechanistic comparison of protein aggregation in vitro and in vivo is more challenging to achieve, although some progress has been made in analysing protein aggregation in humans and animal models. The development of PET tracers has made it possible to investigate protein aggregation in the human brain in a longitudinal fashion,108,109 although at limited resolution. High-resolution cryoEM structures of fibrils extracted from the human brain,110–115 on the other hand, are based on the post-mortem state and lack kinetic information. As such, mechanistic studies largely depend on the use of animal models, and even simple models can be appropriate to address the fundamental aspects of protein aggregation in vivo. A limited number of studies on the kinetics of amyloid formation in living organisms are available, as discussed in the following.4.1 Aggregation kinetics in the nematode C. elegans
The nematode Caenorhabditis elegans is a relatively simple multicellular animal model, composed of 959 somatic cells for the hermaphrodite adult. These cells comprise different tissues including the muscles, the intestine, and a basic nervous system. The main advantages of using C. elegans to study protein aggregation mechanisms are its fast reproductive cycle of ca. 3 days and short lifespan of 2–3 weeks, and its optical transparency, which enables the visualisation of protein aggregation in live animals.In C. elegans models that were developed to identify genetic modifiers of aggregation and toxicity, the disease-associated proteins are typically fluorescently labelled and overexpressed in the large muscle cells for clear visualisation and correlation with a motility phenotype.116,117 As such, these models are relatively artificial, yet they provide the opportunity to characterise the behaviour of disease-related proteins in a living and ageing animal. For example, using fluorescence lifetime imaging (FLIM), the aggregation kinetics and the nature of the inclusions formed by polyQ and α-synuclein were shown to be clearly distinct (Fig. 2). Whereas polyQ aggregates rapidly into amyloid-like inclusions, α-synuclein forms a long-lived intermediate state that appears liquid-like.55,118 Given that the proteins were studied in the same biological environment, these differences can be attributed to their intrinsic biophysical properties. The ability of α-synuclein to undergo liquid–liquid phase separation, after which the droplets mature into amyloid-like aggregates, has indeed also been shown in vitro.55,57
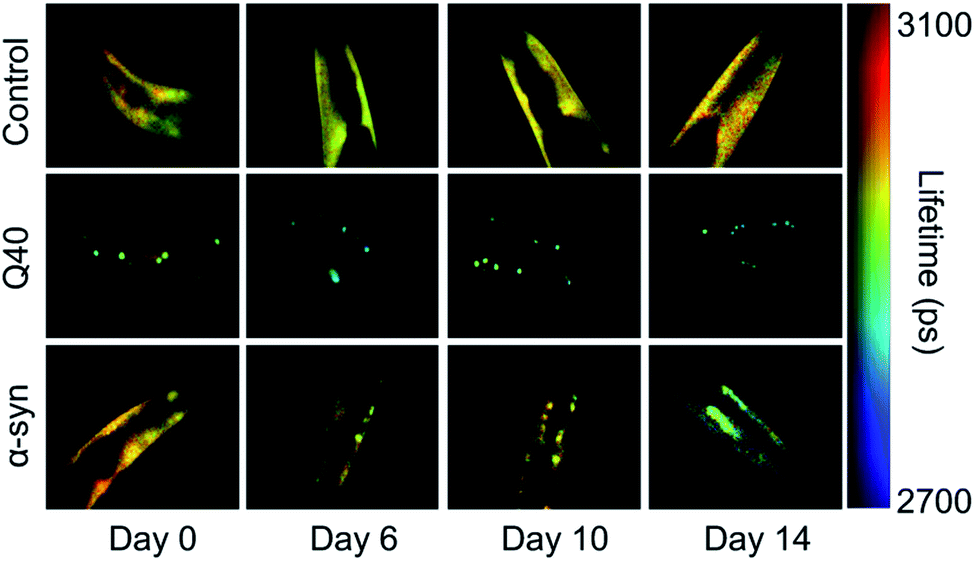 | ||
| Fig. 2 FLIM shows the difference in kinetics and the nature of the inclusions formed by polyQ versus α-synuclein. Shown are the head regions of C. elegans with expression in the muscle cells of YFP (control), 40 glutamines tagged with YFP (Q40) and α-synuclein tagged with YFP (α-syn). Amyloid formation is accompanied by a reduction in the fluorescence lifetime (blue-green colours). Q40 forms amyloid-like inclusions directly, whereas α-synuclein accumulates in inclusions that are not amyloid-like until the worms have reached old age. Based on ref. 118. | ||
In the case of polyQ aggregation, the experimental tractability of the C. elegans system allowed for a quantitative analysis similar as performed in vitro (Fig. 3).49 Fitting the kinetics for multiple strains expressing the protein at different levels in the muscle cells revealed that aggregation is governed by independent nucleation events in individual cells (Fig. 3B). Cell-to-cell spreading or other non-cell-autonomous effects do not noticeably contribute to the aggregation kinetics in this C. elegans model (Fig. 3C). Instead, the muscle cells essentially operate as ‘individual test tubes’, similarly as found in a kinetic study using cultured neuronal cells.48 Moreover, the nucleation rate is effectively constant across the investigated timespan, and contributions arising from ageing processes are not detectable. Future studies are required to confirm whether this spontaneous aggregation behaviour holds true for more disease-relevant protein constructs, cell types and expression levels.
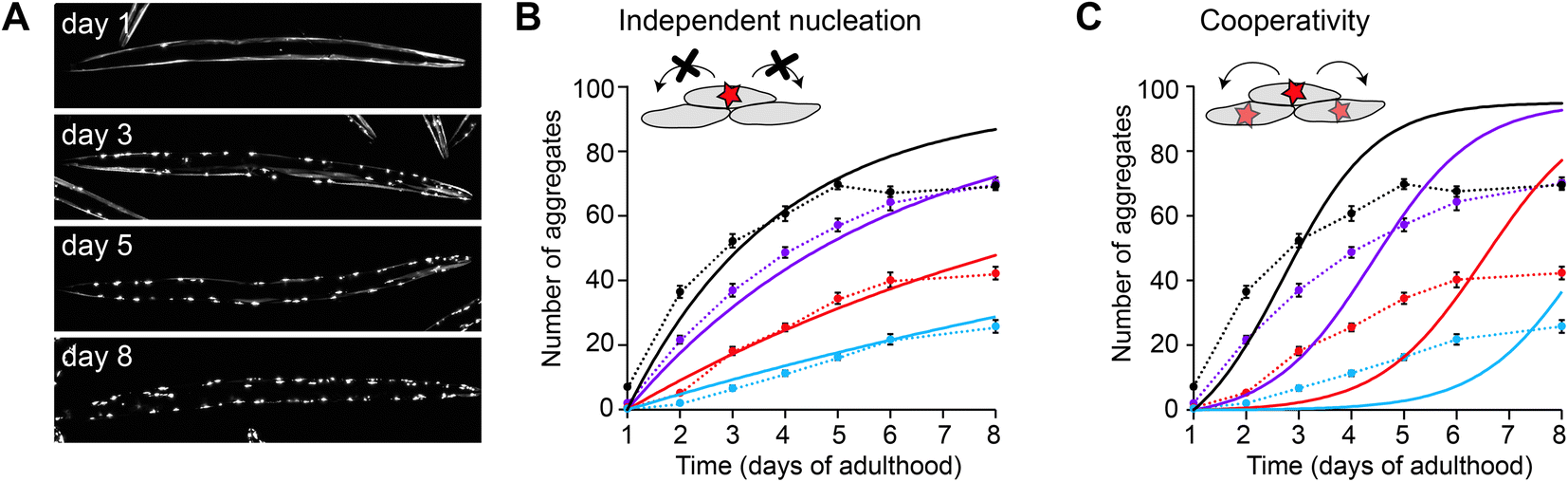 | ||
| Fig. 3 Aggregation kinetics of Q40-YFP in C. elegans muscle cells. (A) Fluorescence images of individual animals showing the increase in aggregates over different days of adulthood. Note that aggregation is only initiated once the animals are adults, in contrast to previous work using a strain with higher overexpression levels (Fig. 2). (B) Experimental data for four strains expressing the protein at different levels, fitted to a model based on independent nucleation events in individual cells. (C) The same dataset as in B, fitted to a model including cooperativity. In this model, which does not recapitulate the data well, the aggregation rate increases as a function of the aggregate mass already present, e.g. due to cell-to-cell spreading or a proteostasis decline. Based on ref. 49. | ||
4.2 Aggregation kinetics in flies, mice, and the human brain
It is more challenging to obtain high-quality experimental data in more complex organisms such as fruit flies and mice, yet a number of noteworthy studies have provided mechanistic insights into aggregation pathways and rates in these systems. In Drosophila, a striking correlation was found between the aggregation propensity of amyloid-β mutants and the neurotoxicity assessed by longevity and motor function.119 A study on mice expressing SOD1 mutants found in ALS reported that survival times were directly correlated with the levels of unfolded protein.120 In the case of this globular protein, unfolding is required for amyloid formation to occur, and decreased protein stability is expected to lead to faster aggregation. The kinetic profiles obtained in the mice were similar to those in vitro and consistent with a fragmentation-based mechanism.Also in the case of a mouse model of prion disease, the scaling of the aggregation kinetics with the protein levels in different strains pointed towards a fragmentation-dominated mechanism.121 The aggregation rates were found to be orders of magnitude lower than determined for the prion protein (PrP) in vitro. It is perhaps expected that aggregation rates are lower in vivo given the presence of molecular chaperones, as discussed in Section 5. However, analysis of tau P301S aggregation in mice yielded very similar rates as in vitro.122 The overexpression as well as the highly aggregation-prone mutation may favour the spontaneous aggregation of this protein in mice, although chaperones should remain effective against protein aggregation under these circumstances, as detailed in the next section.
The experimental possibilities to elucidate protein aggregation mechanisms in the human brain are evidently more limited compared to model organisms. Nevertheless, the progression of tau aggregation in Alzheimer's brain was recently quantitatively analysed based on different datasets, including antibody staining, tau seeding assays and longitudinal PET data.122 The determined rates for tau aggregation in the human brain were orders of magnitude lower than those in vitro, which could again be attributed to the presence of molecular chaperones.
Notably, the recent cryo-EM structures of tau, α-synuclein and amyloid-β fibrils extracted from patient brain material do not typically match those formed by the recombinant proteins in vitro.114,115,123 Although these structures represent kinetic end points, it can be inferred that the aggregation processes must differ. The structure of amyloid-β fibrils purified from a mouse model does correspond to that of the main fibril type identified in familial Alzheimer's disease.115 Thus, further studies into the aggregation mechanisms in animal models have the potential to bridge the gap with in vitro experiments and provide insights relevant to amyloid formation in human disease.
5. Role of molecular chaperones in controlling amyloid formation
Although many proteins spontaneously fold into their native structure when diluted into buffer from a denatured state, efficient protein folding inside the cell is dependent on the presence of molecular chaperones. Various classes of chaperones and co-chaperones exist (see e.g. ref. 124–128 for reviews) and many of these have been shown to inhibit amyloid formation in test tube reactions (Fig. 4). Different microscopic steps of the aggregation process may be affected, depending on the species the molecular chaperone interacts with. Affinity for the monomeric protein reduces several aggregation steps including primary nucleation, whereas binding to the fibril surface is a potent way to block secondary nucleation, and binding to fibril ends specifically interferes with elongation.129 | ||
| Fig. 4 Molecular chaperones preventing amyloid formation. (A) Representative structures of different classes of chaperones: Hsp70 (Escherichia coli DnaK, PDB 2KHO), Hsp90 (human Hsp90β, ref. 153), J-protein (Thermus Thermophilus DnaJ dimer, PDB 4J80), chaperonin (human TRiC/CCT complex, PDB 6NRA), small heat shock protein (sHsp; human Hsp27 oligomer, PDB 6DV5). (B) Schematic of the aggregation pathway with the action of molecular chaperones, derived from a combination of in vitro and in vivo data as described in the main text. | ||
5.1 Molecular chaperones inhibit amyloid formation in model organisms
Many studies have shown that chaperones also inhibit protein aggregation in cultured cells and animal models (see ref. 130 for a more extensive review), although mechanistic investigations in vivo are limited. A notable exception is presented by a study on the J-protein DNAJB6, which inhibits polyQ aggregation in vitro, in cells and in mouse models.25 Kinetic analysis of the in vitro data revealed that DNAJB6 predominately inhibits primary nucleation. Consistently with this mechanism, the chaperone prevented polyQ aggregation in cultured cells, but failed to do so in the presence of added fibril seeds. Aggregation kinetics are difficult to monitor in mouse models (see Section 4), but at the examined time points the numbers of aggregates in mice overexpressing DNAJB6 were lowered, and the onset of symptoms was delayed. These data suggest that indeed the early aggregation events are inhibited by DNAJB6 in vivo.The importance of the chaperone network in preventing protein aggregation and toxicity is evident from the results of genetic screens in C. elegans models, which have implicated major chaperone classes such as Hsp70, Hsp90, TRiC (the eukaryotic chaperonin), and several co-chaperones (Fig. 4).131,132 It is striking that these screens are based on strains overexpressing aggregation-prone proteins such as polyQ, which might be expected to overwhelm the available chaperone capacity. However, the knock-down of the identified chaperones exacerbates protein aggregation compared to control conditions, meaning that these chaperones are effective even at low stoichiometric ratios. The recently developed kinetic analysis of protein aggregation in C. elegans discussed above may prove useful to dissect and quantify the contributions of individual chaperones in this model system.49
Chaperones typically act to prevent protein aggregation and not to promote it. The knock-down of a number of co-chaperones was found, perhaps surprisingly, to decrease polyQ aggregation in a genetic screen in C. elegans.133 This result was suggested to arise from negative regulation of chaperone function by these co-chaperones, so that their knock-down actually improves protein folding capacity. The Hsp90 co-chaperone Aha1 was shown to promote tau aggregation when the two components were added to in vitro assays, and the accumulation of insoluble tau was found to be increased in mice overexpressing Aha1.134 These data indeed provide evidence that co-chaperones may modulate the interaction with chaperones in a manner that reduces their anti-aggregation effects both in vitro and in vivo.
5.2 Disaggregation, remodelling and degradation
Another intriguing way in which chaperones may effectively promote aggregation, is by disassembling existing fibrils into seeding-competent species (Fig. 4B). A metazoan disaggregation machinery consisting of a Hsp70, a Hsp110 nucleotide exchange factor and a specific J-protein co-chaperone was shown to break down preformed α-synuclein fibrils in vitro via a combination of fragmentation and depolymerisation, finally releasing non-toxic monomers.135,136 Other studies came to slightly different conclusions, suggesting an “all-or-none” mechanism of protofilament unzipping followed by depolymerisation,137 or even simpler the direct release of monomers from fibril ends.138 However, knocking down the disaggregase component Hsp110 in C. elegans reduced foci formation of both α-synuclein and polyQ, suggesting that in vivo the disaggregation machinery promotes aggregation by creating seeding-competent species.139 The spreading of α-synuclein to neighbouring tissues was also reduced by the knock-down of Hsp110 in C. elegans,139 implying that the disaggregation machinery may drive an aggregation mechanism of fragmentation coupled to the cell-to-cell transmission of seeds. It will be interesting to also analyse the effects of the disaggregation machinery in a quantitative manner in vivo to establish its contribution to the overall aggregation process.Altogether, chaperones are likely to affect the rates of individual aggregation processes in vivo and may thus shift the dominant mechanism by which fibrils arise, compared to the situation in vitro. Another way of remodelling the aggregation pathway is by changing the morphology of the final aggregate species (Fig. 4B). This possibility has not yet been widely investigated, but studies in vitro have shown that a small heat shock protein reroutes the aggregation pathway of amyloid-β 1–40 towards amorphous, rather than fibrillar, aggregates.140 Along the same lines, overexpression of the co-chaperone Sis1 in yeast induces the cloud-like condensation of polyQ, rather than the typical compact fibrillar inclusions, also suggesting a change in aggregate morphology.141 Studies on the remodelling of aggregate structures in more complex organisms are lacking thus far, although small heat shock proteins have been suggested to promote aggregation into less toxic species in ageing C. elegans.142 Whether chaperones contribute to the diversity of fibril morphologies as observed e.g. across different tauopathies110–115 remains to be determined. Recent advances in cryo-electron tomography will be particularly helpful to reveal the molecular nature of aggregates under the influence of different chaperone levels in single cells,12,107,143,144 or potentially in small model organisms such as the nematode.145,146
Perhaps the most effective way for chaperones to deal with aggregation-prone proteins is to target them for degradation (Fig. 4B). Hsp70 and Hsp90 cooperate with the co-chaperone CHIP, which is an E3 ubiquitin ligase that mediates proteasomal degradation.147 Increasing the levels of Hsp70 in a tauopathy mouse model decreases tau aggregation by reducing its levels, which was shown to be accomplished via CHIP.148 Blocking the folding capacity of Hsp90 similarly reroutes phosphorylated tau for degradation in cultured cells and in mice.149 The C. elegans homolog of CHIP was not identified in the genetic screens mentioned above, which were carried out in polyQ and amyloid-β expressing animals, and it will be interesting to shed more light on the substrate specificity of this system. Chaperones are also connected with the other main degradation pathway next to the ubiquitin-proteasome system, which is autophagy. The Hsp70 co-chaperone BAG3 promotes the autophagy of ubiquitinated substrates when proteasomal degradation is impaired, and together with the small heat shock protein HSPB8 reduces the levels of expanded huntingtin in cultured cells.150 Promoting degradation may in general be a good strategy to prevent protein aggregation in disease by lowering the levels of the soluble protein, as well as clearing aggregates (see e.g. ref. 151 and 152 for further reviews on this topic).
6. What determines the kinetics of aggregation in human disease?
The late onset of sporadic forms of protein aggregation diseases such as Alzheimer's and Parkinson's is often attributed to the decline in protein homeostasis (proteostasis) with age. However, the accumulation of fibril mass also intrinsically takes time to develop, as obvious from in vitro experiments (Fig. 1B and C). The question thus emerges to what extent the time course of amyloid-like protein aggregation in vivo is determined by the decline in proteostasis during ageing, versus the kinetics of spontaneous amyloid formation as observed in vitro. In other words, is protein aggregation a function of biological ageing, or of age as simply “the passing of time”? The typical course of human protein aggregation diseases with a long lag phase and an onset of aggregation and symptoms in mid-to late-life can be explained by several reasons, both from the point of a decline in proteostasis as well as from the intrinsic kinetics of amyloid formation. Some possible scenarios will be discussed below. These are by no means exhaustive, and different combinations may be at play depending on the protein and the associated disease.6.1 Proteostasis decline during ageing
Proteostasis encompasses the pathways that collectively ensure that proteins are functional, including biogenesis, folding, translocation and degradation.154 The use of C. elegans has been instrumental in demonstrating the fact that proteostasis capacity is limited. In a hallmark study, it was shown that the expression of metastable proteins alongside polyQ results in premature aggregation and loss of function.155 Metastable proteins furthermore start to aggregate and lose their function over the course of adulthood of the animal, suggesting that proteostasis capacity declines with age.156,157 Similar discoveries were recently made in the mouse brain.158 Widespread changes in the proteome have also been found to take place during the reproductive phase of C. elegans, followed by the aggregation of many endogenous proteins at a later stage.142 The ability to mount robust stress responses such as the heat shock response deteriorates even earlier in the life cycle of the nematode, at the onset of the reproductive phase.159 Although the decline of proteostasis with age encompasses changes in a variety of pathways, the focus here is on chaperone function and degradation, since these most notably modulate the propensity for amyloid formation.It is important to note that amyloid formation and amorphous aggregation are two distinct processes. The metastable proteins that have been used as proteostasis sensors in animal models155–158 are natively globular proteins that precipitate when they fail to reach their native state, due to the exposure of hydrophobic patches and loss of solubility. Many amyloid-forming proteins such as tau and α-synuclein are however intrinsically disordered, and relatively soluble in their unfolded native states. In contrast to amorphous aggregation, amyloid formation is a highly ordered process, which is limited by an energetically unfavourable nucleation event. As for every phase transition, amyloid-like aggregation only occurs at levels above the protein's solubility, here defined as the critical protein concentration.160 Beyond this threshold, fibril elongation by monomer addition takes place spontaneously, so that aggregation will invariably proceed once nucleation has occurred. It should be noted that once the critical concentration has been surpassed, it still takes time for the fibril mass to build up via the mechanisms described in Section 2 (Fig. 1), in contrast to amorphous precipitation which takes place almost immediately.161
6.2 Aggregation mechanisms driven by a decline in proteostasis
Considering this framework, a first possibility explaining late-onset protein aggregation is that a decline in chaperone capacity lowers the critical concentration for amyloid formation, causing the disease-associated protein to cross this barrier (Fig. 5A). A reduction of the levels of (active) chaperones that transiently interact with amyloid-prone proteins would increase the levels of protein free to undergo amyloid aggregation, effectively lowering its critical concentration. Changes in chaperone levels during ageing in C. elegans are relatively modest, but some J-proteins are notably reduced.142 In brain tissue of Alzheimer's patients, most chaperones also remain unchanged, but Hsp90 paralogs are selectively downregulated.162 For specific client proteins, such disturbances may be sufficient to exceed their solubility limits. Computational predictions have revealed that many disease-associated proteins are expressed at levels close to their solubility,163 meaning that subtle perturbations may be sufficient to cross this threshold.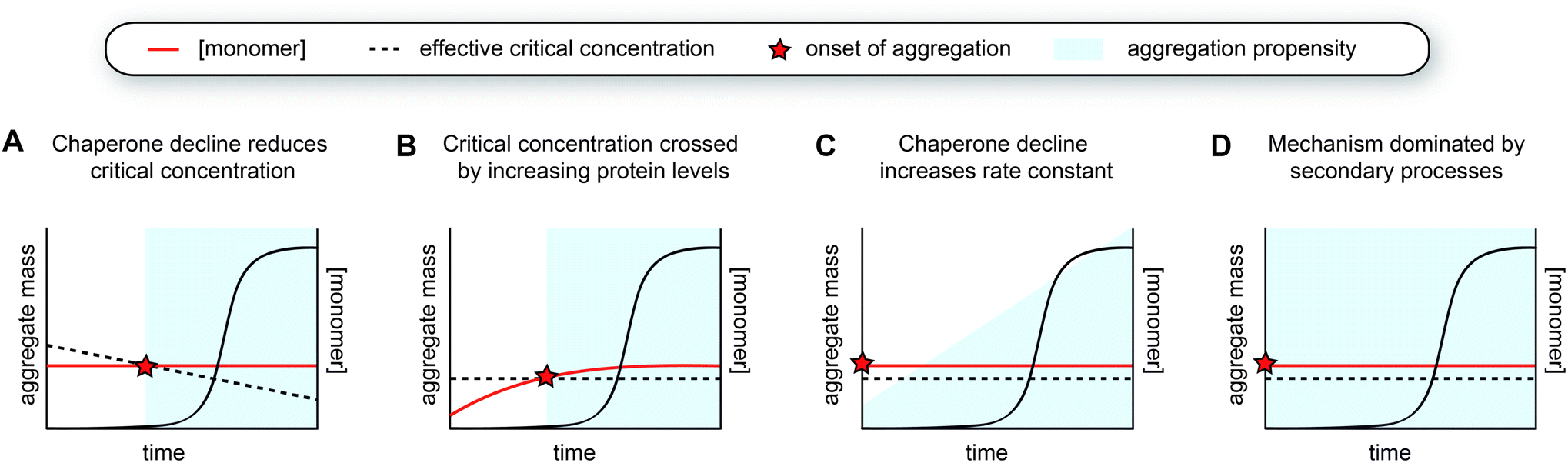 | ||
| Fig. 5 Models for the aggregation time course observed in human disease. (A) The decline of chaperone capacity during ageing leads to a decrease in the effective critical protein concentration. Once the monomer concentration exceeds this threshold, aggregation is initiated. (B) Similar as model A, but the critical concentration remains constant, whereas the monomer concentration increases. The concentration curves in A and B can have any shape, as long as the two curves intersect. (C) The monomer is already expressed beyond the critical threshold, yet the aggregation propensity (i.e., the rate constant for nucleation or other microscopic steps) increases due to a decline in chaperone capacity. The accumulation of a co-factor would cause a similar scenario. (D) The aggregation mechanism is strongly dominated by secondary processes, which lead to cooperative kinetics with a long lag phase. | ||
Another way to cross the critical concentration is an increase in the level of the amyloid-prone protein (Fig. 5B). This may be either due to decreased clearance or increased production, the latter also including the generation of cleavage products or aggregation-prone post-translationally modified species. The genetics behind Alzheimer's disease support several of these points, given that familial mutants lead to the generation of more and/or longer amyloid-β peptides, and the main genetic risk factor for sporadic disease is associated with impaired clearance. Proteins may also accumulate in a particular subcellular compartment, as exemplified by FUS and TDP-43 for which the mislocalisation from the nucleus to the cytoplasm is thought to be one of the triggers for aggregation and disease.164 In the cytoplasm, these proteins undergo liquid–liquid phase separation and subsequently aggregate, whereas the high RNA concentrations in the nucleus are thought to prevent their phase separation and aggregation.165
A decline in proteostasis may also contribute to the kinetic profile observed in human disease in a more subtle manner. Perhaps, certain proteins are already present above their critical concentrations, but the aggregation rates are too low to lead to significant problems and/or can be dealt with by the degradation machinery. However, changes in the chaperone network may lead to increased aggregation propensities, for example by promoting nucleation or by enhancing fibril fragmentation (Fig. 5C). Such effects may in particular lead to strongly sigmoidal curves in the case of a positive feedback loop, whereby protein aggregation induces further disruption of proteostasis. The critical concentration hypothesis is somewhat more appealing because it precludes any incidence of aggregation prior to crossing the barrier (Fig. 5A and B). Given that amyloid formation can proceed quickly once nucleation has occurred, especially when secondary processes are at play, the possibility of the cell to degrade these aggregates may be limited.
As a variation to the mechanism described above, small molecules such as metabolites or lipids that can act as co-factors for nucleation may also accumulate during ageing and hence effectively increase the nucleation rate constant (Fig. 5C). Unfortunately, not much is known about factors that directly promote aggregation in vivo, although it is likely that nucleation is heterogeneous in the complex biological environment and involves interplay with other molecules and cellular structures. Interestingly, a cryoEM structure of tau fibrils extracted from brain tissue of chronic traumatic encephalopathy (CTE) patients contains additional density in a hydrophobic cavity, which may correspond to a sterol or a fatty acid.112 The build-up of a co-factor or a catalyst may be a particularly important trigger for the aggregation of proteins that do not have a high intrinsic aggregation propensity, such as tau, which also requires additives to undergo aggregation in vitro. Whereas CTE has a clear external cause, namely repetitive head impact, other neurodegenerative diseases are also associated with lifestyle factors and changes in metabolism,166 and it will be interesting to further explore this possibility.
6.3 Aggregation mechanisms driven by secondary processes
Alternatively, the long lag phase observed in human disease can also be attributed solely to an aggregation mechanism that is dominated by secondary processes, which by itself leads to a strongly sigmoidal aggregation curve (Fig. 5D, see Fig. 1C and 3C). Both the rates of fragmentation and secondary nucleation increase with increasing fibril mass, and in particular when the monomer concentrations are low and/or primary nucleation is very slow compared to the secondary process, these mechanisms induce a long lag phase followed by a rapid increase in fibril formation. In the case of intracellular aggregation as observed for example in the case of tau, α-synuclein and polyQ proteins, these mechanisms would have to involve cell-to-cell spreading of seeds for aggregation to propagate across the tissue.The spreading of protein aggregation following neuronal connectivity was already suggested from the Braak stages of pathology in Alzheimer's and Parkinson's diseases.167,168 Over the past years, a wide range of evidence for aggregate spreading of tau, α-synuclein and other proteins has been gathered in a range of experimental systems (see ref. 169–173 for reviews). A mechanism dominated by secondary processes combined with spreading is an attractive explanation for the kinetics observed in human disease, although much remains to be uncovered. In particular, it is not clear to what extent fragmentation and secondary nucleation occur in vivo, with respect to in vitro measurements. The fact that disease brains typically contain only one or two structural polymorphs suggests that the fibril structure is preserved throughout the process of spreading and replication, yet this can be the consequence of either one of the secondary processes. The rate of fragmentation is fundamentally independent of the concentration of monomeric protein, perhaps rendering this process somewhat more likely as the main mechanism to generate seeds compared to secondary nucleation at low physiological protein concentrations. The disaggregase machinery can further promote the breakage of fibrils (see Section 5.2), although the contribution of this activity to the overall aggregation process is not yet clear.
In the case of PrP as well as SOD1, kinetic analysis in mice (Section 4.2) pointed towards fragmentation as the dominant mechanism.120,121 Direct evidence for the occurrence of secondary nucleation in vivo is not yet available. The fibril structures of e.g. tau from patient brains have revealed a fuzzy coat formed by disordered regions of the protein surrounding the amyloid core,110 but it is not clear whether this would interfere with or enhance the absorption of monomers followed by their nucleation. Other interactors, including molecular chaperones, may also decorate the surface of fibrils making them unavailable for monomer interactions. Fibrils are moreover typically found in dense clusters in vivo. This may limit secondary nucleation on the one hand, but on the other hand may also be a consequence of exactly this process. Further quantitative analysis of (seeded) aggregation in model organisms will be critical to resolve these outstanding questions.
7. Conclusions and outlook
The biophysical principles driving amyloid formation in vitro fundamentally hold true in vivo, despite the complexity of the cellular and organismal environment. However, each of the microscopic processes, for example primary nucleation or fibril elongation, is modulated by the surroundings. Chaperones have been shown to affect the rates of several microscopic aggregation steps in vitro, but it is not yet clear to what extent these mechanisms remodel the aggregation pathways in vivo. Physical constraints such as cellular confinement and phase separation furthermore lead to different processes being dominant, compared to bulk in vitro studies. Intracellular protein aggregation is strongly limited by primary nucleation, which needs to occur in every individual cell in the absence of aggregate spreading. The spreading of seeds combined with secondary nucleation or fragmentation is likely to dominate the overall aggregation process for many cells in a tissue to be affected when primary nucleation rates are low. Phase separation, on the other hand, efficiently promotes primary nucleation due to the high local protein concentration within the phase-separated state.The answer to what extent the in vitro mechanisms are relevant to human disorders depends on the specifics of the protein and the associated disease. For highly aggregation-prone protein variants, such as long polyQ proteins or aggressive point mutants, the aggregation process may be more ‘spontaneous’ and resemble the in vitro mechanism. The case of juvenile Huntington's diseases exemplifies the fact that protein aggregation diseases are not necessarily age-related, and can manifest at an age where protein quality control is expected to be intact. In these cases, the lag phase observed in disease could be due to the fact that secondary processes dominate the aggregation mechanism, but an increase in protein levels over time may also contribute. In contrast, for proteins that are less intrinsically aggregation-prone, such as wild-type α-synuclein or tau, cellular handling including localisation and post-translational modifications may trigger the onset of aggregation.
A problem with elucidating the contributions of spontaneous aggregation versus changes in the biological environment is that most animal models used to study protein aggregation diseases, including the C. elegans models discussed in this perspective as well as most mouse models, are based on high levels of overexpression of the disease-associated protein. These models typically display aggregation starting from early adulthood or even during development (reviewed in ref. 174). It will be important to investigate aggregation mechanisms in animal models expressing more modest protein levels. In such models, it will be possible to quantify the critical concentration for aggregation and to elucidate which aspects of ageing may trigger amyloid formation. In addition, obtaining more quantitative data about the concentrations of disease-associated proteins in humans at different time points would be extremely helpful. Altogether, bridging the gap between biophysics and the biology of ageing and disease is an exciting avenue that will hopefully contribute to a molecular understanding of the mechanisms underlying protein aggregation diseases, and the development of novel therapeutics.
Author contributions
T. S. wrote the article.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
I would like to thank Stefan Rüdiger and the members of my lab for critical reading of the manuscript, and Utrecht University for a start-up grant.References
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, The amyloid state and its association with protein misfolding diseases, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- R. A. Kyle, Amyloidosis: a convoluted story, Br. J. Haematol., 2001, 114, 529–538 CrossRef CAS PubMed.
- K. Herrup, The case for rejecting the amyloid cascade hypothesis, Nat. Neurosci., 2015, 18, 794–799 CrossRef CAS PubMed.
- M. R. Chapman, L. S. Robinson, J. S. Pinkner, R. Robyn, H. John, H. Mårten, N. Staffan and S. J. Hultgren, Role of Escherichia coli Curli Operons in Directing Amyloid Fiber Formation, Science, 2002, 295, 851–855 CrossRef CAS PubMed.
- S. K. Maji, M. H. Perrin, M. R. Sawaya, S. Jessberger, K. Vadodaria, R. A. Rissman, P. S. Singru, K. P. R. Nilsson, R. Simon, D. Schubert, D. Eisenberg, J. Rivier, P. Sawchenko, W. Vale and R. Riek, Functional amyloids as natural storage of peptide hormones in pituitary secretory granules, Science, 2009, 325, 328–332 CrossRef CAS PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- S. Campioni, B. Mannini, M. Zampagni, A. Pensalfini, C. Parrini, E. Evangelisti, A. Relini, M. Stefani, C. M. Dobson, C. Cecchi and F. Chiti, A causative link between the structure of aberrant protein oligomers and their toxicity, Nat. Chem. Biol., 2010, 6, 140–147 CrossRef CAS PubMed.
- G. Fusco, S. W. Chen, P. T. F. Williamson, R. Cascella, M. Perni, J. A. Jarvis, C. Cecchi, M. Vendruscolo, F. Chiti, N. Cremades and L. Ying, Structural basis of membrane disruption and cellular toxicity by a-synuclein oligomers, Science, 2017, 358, 1440–1443 CrossRef CAS PubMed.
- A. Yu, S. G. Fox, A. Cavallini, C. Kerridge, M. J. O’Neill, J. Wolak, S. Bose and R. I. Morimoto, Tau protein aggregates inhibit the protein-folding and vesicular trafficking arms of the cellular proteostasis network, J. Biol. Chem., 2019, 294(19), 7917–7930 CrossRef CAS PubMed.
- M. F. M. Engel, L. Khemtémourian, C. C. Kleijer, H. J. D. Meeldijk, J. Jacobs, A. J. Verkleij, B. De Kruijff, J. A. Killian and J. W. M. Höppener, Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6033–6038 CrossRef CAS PubMed.
- F. J. B. Bäuerlein, I. Saha, A. Mishra, M. Kalemanov, A. Martínez-Sánchez, R. Klein, I. Dudanova, M. S. Hipp, F. U. Hartl, W. Baumeister and R. Fernández-Busnadiego, In situ architecture and cellular interactions of PolyQ inclusions, Cell, 2017, 171, 179–187 CrossRef PubMed.
- L. Ferrari, R. Stucchi, K. Konstantoulea, G. van de Kamp, R. Kos, W. J. C. Geerts, L. S. van Bezouwen, F. G. Förster, M. Altelaar, C. C. Hoogenraad and S. G. D. Rüdiger, Arginine π-stacking drives binding to fibrils of the Alzheimer protein Tau, Nat. Commun., 2020, 11, 571 CrossRef CAS PubMed.
- H. Olzscha, S. M. Schermann, A. C. Woerner, S. Pinkert, M. H. Hecht, G. G. Tartaglia, M. Vendruscolo, M. Hayer-Hartl, F. U. Hartl and R. M. Vabulas, Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions, Cell, 2011, 144, 67–78 CrossRef CAS PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS PubMed.
- G. Meisl, X. Yang, E. Hellstrand, B. Frohm, J. B. Kirkegaard, S. I. A. Cohen, C. M. Dobson, S. Linse and T. P. J. Knowles, Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9384–9389 CrossRef CAS PubMed.
- A. K. Buell, C. Galvagnion, R. Gaspar, E. Sparr, M. Vendruscolo, T. P. J. Knowles, S. Linse and C. M. Dobson, Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7671–7676 CrossRef CAS PubMed.
- C. Galvagnion, A. K. Buell, G. Meisl, T. C. T. Michaels, M. Vendruscolo, T. P. J. Knowles and C. M. Dobson, Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation, Nat. Chem. Biol., 2015, 11, 229–234 CrossRef CAS PubMed.
- F. Kundel, L. Hong, B. Falcon, W. A. McEwan, T. C. T. Michaels, G. Meisl, N. Esteras, A. Y. Abramov, T. J. P. Knowles, M. Goedert and D. Klenerman, Measurement of Tau Filament Fragmentation Provides Insights into Prion-like Spreading, ACS Chem. Neurosci., 2018, 9, 1276–1282 CrossRef CAS PubMed.
- K. A. Conway, J. D. Harper and P. T. Lansbury, Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease, Nat. Med., 1998, 4, 1318–1320 CrossRef CAS PubMed.
- L. Giehm and D. E. Otzen, Strategies to increase the reproducibility of protein fibrillization in plate reader assays, Anal. Biochem., 2010, 400, 270–281 CrossRef CAS PubMed.
- J. C. Sang, G. Meisl, A. M. Thackray, L. Hong, A. Ponjavic, T. P. J. Knowles, R. Bujdoso and D. Klenerman, Direct Observation of Murine Prion Protein Replication in Vitro, J. Am. Chem. Soc., 2018, 140, 14789–14798 CrossRef CAS PubMed.
- K. Kar, C. L. Hoop, K. W. Drombosky, M. A. Baker, R. Kodali, I. Arduini, P. C. A. Van Der Wel, W. S. Horne and R. Wetzel, β-Hairpin-mediated nucleation of polyglutamine amyloid formation, J. Mol. Biol., 2013, 425, 1183–1197 CrossRef CAS PubMed.
- K. Kar, M. Jayaraman, B. Sahoo, R. Kodali and R. Wetzel, Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent, Nat. Struct. Mol. Biol., 2011, 18, 328–336 CrossRef CAS PubMed.
- V. Kakkar, et al., The S/T-Rich Motif in the DNAJB6 Chaperone Delays Polyglutamine Aggregation and the Onset of Disease in a Mouse Model, Mol. Cell, 2016, 62, 272–283 CrossRef CAS PubMed.
- J. D. Knight and A. D. Miranker, Phospholipid catalysis of diabetic amyloid assembly, J. Mol. Biol., 2004, 341, 1175–1187 CrossRef CAS PubMed.
- S. A. Jayasinghe and R. Langen, Lipid membranes modulate the structure of Islet Amyloid Polypeptide, Biochemistry, 2005, 44, 12113–12119 CrossRef CAS PubMed.
- J. R. Brender, J. Krishnamoorthy, M. F. M. Sciacca, S. Vivekanandan, L. D'Urso, J. Chen, C. La Rosa and A. Ramamoorthy, Probing the Sources of the Apparent Irreproducibility of Amyloid Formation: Drastic Changes in Kinetics and a Switch in Mechanism Due to Micellelike Oligomer Formation at Critical Concentrations of IAPP, J. Phys. Chem. B, 2015, 119, 2886–2896 CrossRef CAS PubMed.
- X. Zhang, J. R. St Clair, E. London and D. P. Raleigh, Islet amyloid polypeptide membrane interactions: Effects of membrane composition, Biochemistry, 2017, 56, 376–390 CrossRef CAS PubMed.
- B. O. W. Elenbaas, L. Khemtemourian, J. A. Killian and T. Sinnige, Membrane-catalyzed IAPP aggregation is dominated by secondary nucleation, bioRxiv, 2022 DOI:10.1101/2022.02.04.479144.
- S. D. Khare, M. Caplow and N. V. Dokholyan, The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15094–15099 CrossRef CAS PubMed.
- Y. Furukawa, K. Kaneko, K. Yamanaka, T. V. O'Halloran and N. Nukina, Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis, J. Biol. Chem., 2008, 283, 24167–24176 CrossRef CAS PubMed.
- L. Lang, M. Kurnik, J. Danielsson and M. Oliveberg, Fibrillation precursor of superoxide dismutase 1 revealed by gradual tuning of the protein-folding equilibrium, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17868–17873 CrossRef CAS PubMed.
- G. Meisl, J. B. Kirkegaard, P. Arosio, T. C. T. T. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. J. J. Knowles, Molecular mechanisms of protein aggregation from global fitting of kinetic models, Nat. Protoc., 2016, 11, 252–272 CrossRef CAS PubMed.
- T. C. T. Michaels, A. Šarić, J. Habchi, S. Chia, G. Meisl, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Chemical kinetics for bridging molecular mechanisms and macroscopic measurements of amyloid fibril formation, Annu. Rev. Phys. Chem., 2018, 69, 273–298 CrossRef CAS PubMed.
- S. I. A. Cohen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, From macroscopic measurements to microscopic mechanisms of protein aggregation, J. Mol. Biol., 2012, 421, 160–171 CrossRef CAS PubMed.
- D. C. Rodriguez Camargo, E. Sileikis, S. Chia, E. Axell, K. Bernfur, R. L. Cataldi, S. I. A. Cohen, G. Meisl, J. Habchi, T. P. J. Knowles, M. Vendruscolo and S. Linse, Proliferation of Tau 304–380 Fragment Aggregates through Autocatalytic Secondary Nucleation, ACS Chem. Neurosci., 2021, 12(23), 4406–4415 CrossRef CAS PubMed.
- D. C. Rodriguez Camargo, S. Chia, J. Menzies, B. Mannini, G. Meisl, M. Lundqvist, C. Pohl, K. Bernfur, V. Lattanzi, J. Habchi, S. I. Cohen, T. P. J. Knowles, M. Vendruscolo and S. Linse, Surface-Catalyzed Secondary Nucleation Dominates the Generation of Toxic IAPP Aggregates, Front. Mol. Biosci., 2021, 8, 757425 CrossRef PubMed.
- G. M. Spillantini, M. L. Schmidt, V. M.-Y. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Alpha-Synuclein in Lewy bodies, Nature, 1997, 388, 839–840 CrossRef PubMed.
- A. S. Kurochka, D. A. Yushchenko, P. Bouř and V. V. Shvadchak, Influence of lipid membranes on α-synuclein aggregation, ACS Chem. Neurosci., 2021, 12, 825–830 CrossRef CAS PubMed.
- G. Fusco, T. Pape, A. D. Stephens, P. Mahou, A. R. Costa, C. F. Kaminski, G. S. Kaminski Schierle, M. Vendruscolo, G. Veglia, C. M. Dobson and A. De Simone, Structural basis of synaptic vesicle assembly promoted by α-synuclein, Nat. Commun., 2016, 7, 12563 CrossRef PubMed.
- J. Lautenschläger, et al., C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction, Nat. Commun., 2018, 9, 712 CrossRef PubMed.
- J. Liu, J.-P. Zhang, M. Shi, T. Quinn, J. Bradner, R. Beyer, S. Chen and J. Zhang, Rab11a and HSP90 Regulate Recycling of Extracellular α-Synuclein, J. Neurosci., 2009, 29, 1480–1485 CrossRef CAS PubMed.
- M. M. Apetri, R. Harkes, V. Subramaniam, G. W. Canters, T. Schmidt and T. J. Aartsma, Direct Observation of α-Synuclein Amyloid Aggregates in Endocytic Vesicles of Neuroblastoma Cells, PLoS One, 2016, 11, e0153020 CrossRef PubMed.
- M. A. A. Fakhree, I. B. M. Konings, J. Kole, A. Cambi, C. Blum and M. M. A. E. Claessens, The Localization of Alpha-synuclein in the Endocytic Pathway, Neuroscience, 2021, 457, 186–195 CrossRef CAS PubMed.
- T. P. J. Knowles, D. A. White, A. R. Abate, J. J. Agresti, S. I. A. Cohen, R. A. Sperling, E. J. De Genst, C. M. Dobson and D. A. Weitz, Observation of spatial propagation of amyloid assembly from single nuclei, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14746–14751 CrossRef CAS PubMed.
- T. C. T. Michaels, A. J. Dear and T. P. J. Knowles, Stochastic calculus of protein filament formation under spatial confinement, New J. Phys., 2018, 20, 055007 CrossRef.
- D. W. Colby, J. P. Cassady, G. C. Lin, V. M. Ingram and K. D. Wittrup, Stochastic kinetics of intracellular huntingtin aggregate formation, Nat. Chem. Biol., 2006, 2, 319–323 CrossRef CAS PubMed.
- T. Sinnige, G. Meisl, T. C. T. Michaels, M. Vendruscolo, T. P. J. Knowles and R. I. Morimoto, Kinetic analysis reveals that independent nucleation events determine the progression of polyglutamine aggregation in C. elegans, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2021888118 CrossRef CAS PubMed.
- C. P. Brangwynne, P. Tompa and R. V. Pappu, Polymer physics of intracellular phase transitions, Nat. Phys., 2015, 11, 899–904 Search PubMed.
- Y. Shin and C. P. Brangwynne, Liquid phase condensation in cell physiology and disease, Science, 2017, 357, eaaf4382 CrossRef PubMed.
- S. Boeynaems, S. Alberti, N. L. Fawzi, T. Mittag, M. Polymenidou, F. Rousseau, J. Schymkowitz, J. Shorter, B. Wolozin, L. Van Den Bosch, P. Tompa and M. Fuxreiter, Protein Phase Separation: A New Phase in Cell Biology, Trends Cell Biol., 2018, 28(6), 420–435 CrossRef CAS PubMed.
- A. Patel, et al., A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation, Cell, 2015, 162, 1066–1077 CrossRef CAS PubMed.
- S. Ray, et al., α-Synuclein aggregation nucleates through liquid–liquid phase separation, Nat. Chem., 2020, 12, 705–716 CrossRef CAS PubMed.
- M. C. Hardenberg, T. Sinnige, S. Casford, S. T. Dada, C. Poudel, E. A. Robinson, M. Fuxreiter, C. F. Kaminksi, G. S. Kaminski Schierle, E. A. A. Nollen, C. M. Dobson and M. Vendruscolo, Observation of an α-synuclein liquid droplet state and its maturation into Lewy body-like assemblies, J. Mol. Cell Biol., 2021, 13, 282–294 CAS.
- S. Wegmann, et al., Tau protein liquid–liquid phase separation can initiate tau aggregation, EMBO J., 2018, e98049 Search PubMed.
- S. T. Dada, M. C. Hardenberg, L. K. Mrugalla and M. O. Mckeon, Spontaneous nucleation and fast aggregate-dependent proliferation of α-synuclein aggregates within liquid condensates at physiological pH, bioRxiv, 2021 DOI:10.1101/2021.09.26.461836.
- A. B. Singleton, et al., α-Synuclein Locus Triplication Causes Parkinson's Disease, Science, 2003, 302, 841 CrossRef CAS PubMed.
- M. Farrer, J. Kachergus, L. Forno, S. Lincoln, D. S. Wang, M. Hulihan, D. Maraganore, K. Gwinn-Hardy, Z. Wszolek, D. Dickson and J. W. Langston, Comparison of Kindreds with Parkinsonism and α-Synuclein Genomic Multiplications, Ann. Neurol., 2004, 55, 174–179 CrossRef CAS PubMed.
- M.-C. C. Chartier-Harlin, J. Kachergus, C. Roumier, V. Mouroux, X. Douay, S. Lincoln, C. Levecque, L. Larvor, J. Andrieux, M. Hulihan, N. Waucquier, L. Defebvre, P. Amouyel, M. Farrer and A. Destée, α-synuclein locus duplication as a cause of familial Parkinson's disease, Lancet, 2004, 364, 1167–1169 CrossRef CAS.
- F. K. Wiseman, T. Al-Janabi, J. Hardy, A. Karmiloff-Smith, D. Nizetic, V. L. J. Tybulewicz, E. M. C. Fisher and A. Strydom, A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome, Nat. Rev. Neurosci., 2015, 16, 564–574 CrossRef CAS PubMed.
- B. V. Hooli, G. Mohapatra, M. Mattheisen, A. R. Parrado, J. T. Roehr, Y. Shen, J. F. Gusella, R. Moir, A. J. Saunders, C. Lange, R. E. Tanzi and L. Bertram, Role of common and rare APP DNA sequence variants in Alzheimer disease, Neurology, 2012, 78, 1250–1257 CrossRef CAS PubMed.
- Alzforum, at http://www.alzforum.org/mutations on 28 February 2022.
- K. R. Wildsmith, M. Holley, J. C. Savage, R. Skerrett and G. E. Landreth, Evidence for impaired amyloid β clearance in Alzheimer's disease, Alzheimers. Res. Ther., 2013, 5, 33 CrossRef CAS PubMed.
- G. Meisl, X. Yang, B. Frohm, T. P. J. Knowles and S. Linse, Quantitative analysis of intrinsic and extrinsic factors in the aggregation mechanism of Alzheimer-associated Aβ-peptide, Sci. Rep., 2016, 6, 18728 CrossRef CAS PubMed.
- X. Yang, G. Meisl, B. Frohm, E. Thulin, T. P. J. Knowles and S. Linse, On the role of sidechain size and charge in the aggregation of A β 42 with familial mutations, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5849–E5858 CrossRef CAS PubMed.
- M. Goedert, R. Jakes and R. A. Crowther, Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments, FEBS Lett., 1999, 450, 306–311 CrossRef CAS PubMed.
- P. Nacharaju, J. Lewis, C. Easson, S. Yen, J. Hackett, M. Hutton and S.-H. Yen, Accelerated filament formation from tau protein with specific FTDP-17 missense mutations, FEBS Lett., 1999, 447, 195–199 CrossRef CAS PubMed.
- T. C. Gamblin, M. E. King, H. Dawson, M. P. Vitek, J. Kuret, R. W. Berry and L. I. Binder, Vitro Polymerization of Tau Protein Monitored by Laser Light Scattering: Method and Application to the Study of FTDP-17 Mutants, Biochemistry, 2000, 39, 6136–6144 CrossRef CAS PubMed.
- S. Barghorn, Q. Zheng-Fischhöfer, M. Ackmann, J. Biernat, M. von Bergen, E.-M. Mandelkow and E. Mandelkow, Structure, Microtubule Interactions, and Paired Helical Filament Aggregation by Tau Mutants of Frontotemporal Dementias, Biochemistry, 2000, 39, 11714–11721 CrossRef CAS PubMed.
- B. Combs and T. C. Gamblin, FTDP-17 Tau Mutations Induce Distinct Effects on Aggregation and Microtubule Interactions, Biochemistry, 2012, 51, 8597–8607 CrossRef CAS PubMed.
- Q. Q. Yao, L. Hong, S. Wu and S. Perrett, Distinct microscopic mechanisms for the accelerated aggregation of pathogenic Tau mutants revealed by kinetic analysis, Phys. Chem. Chem. Phys., 2020, 22, 7241–7249 RSC.
- J. Götz, F. Chen, R. Barmettler and R. M. Nitsch, Tau filament formation in transgenic mice expressing P301L tau, J. Biol. Chem., 2001, 276, 529–534 CrossRef PubMed.
- J. Lewis, E. McGowan, J. Rockwood, H. Melrose, P. Nacharaju, M. Van Slegtenhorst, K. Gwinn-Hardy, M. Paul Murphy, M. Baker, X. Yu, K. Duff, J. Hardy, A. Corral, W.-L. Lin, S.-H. Yen, D. W. Dickson, P. Davies and M. Hutton, Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein, Nat. Genet., 2000, 25, 402–405 CrossRef CAS PubMed.
- Y. Yoshiyama, M. Higuchi, B. Zhang, S. M. Huang, N. Iwata, T. C. C. Saido, J. Maeda, T. Suhara, J. Q. Trojanowski and V. M. Y. Lee, Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model, Neuron, 2007, 53, 337–351 CrossRef CAS PubMed.
- M. Hutton, et al., Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17, Nature, 1998, 393, 702–705 CrossRef CAS PubMed.
- P. Flagmeier, G. Meisl, M. Vendruscolo, T. P. J. Knowles, C. M. Dobson, A. K. Buell and C. Galvagnion, Mutations associated with familial Parkinson's disease alter the initiation and amplification steps of α-synuclein aggregation, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 201604645 CrossRef PubMed.
- V. C. Ruf, G. S. Nübling, S. Willikens, S. Shi, F. Schmidt, J. Levin, K. Bötzel, F. Kamp and A. Giese, Different Effects of α-Synuclein Mutants on Lipid Binding and Aggregation Detected by Single Molecule Fluorescence Spectroscopy and ThT Fluorescence-Based Measurements, ACS Chem. Neurosci., 2019, 10, 1649–1659 CrossRef CAS PubMed.
- K. K. Johansen, S. H. Torp, M. J. Farrer, E. K. Gustavsson and J. O. Aasly, A Case of Parkinson's Disease with No Lewy Body Pathology due to a Homozygous Exon Deletion in Parkin, Case Rep. Neurol. Med., 2018, 2018, 6838965 Search PubMed.
- C. Gaig, M. J. Martí, M. Ezquerra, M. J. Rey, A. Cardozo and E. Tolosa, G2019S LRRK2 mutation causing Parkinson's disease without Lewy bodies, J. Neurol. Neurosurg. Psychiatry, 2007, 78, 626–628 CrossRef PubMed.
- L. P. Ranum, M. Y. Chung, S. Banfi, A. Bryer, L. J. Schut, R. Ramesar, L. A. Duvick, A. McCall, S. H. Subramony and L. Goldfarb, et al., Molecular and clinical correlations in spinocerebellar ataxia type I: evidence for familial effects on the age at onset, Am. J. Hum. Genet., 1994, 55, 244–252 CAS.
- S. N. Illarioshkin, S. Igarashi, O. Onodera, E. D. Markova, N. N. Nikolskaya, H. Tanaka, T. Z. Chabrashwili, N. G. Insarova, K. Endo, I. A. Ivanova-Smolenskaya and S. Tsjui, Trinucleotide Repeat Length and Rate of Progression of Huntington's Disease, Ann. Neurol., 1994, 36, 630–635 CrossRef CAS PubMed.
- N. S. Wexler, Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3498–3503 CrossRef CAS PubMed.
- B. P. C. Van De Warrenburg, H. Hendriks, A. Dürr, M. C. A. Van Zuijlen, G. Stevanin, A. Camuzat, R. J. Sinke, A. Brice and B. P. H. Kremer, Age at onset variance analysis in spinocerebellar ataxias: A study in a Dutch-French cohort, Ann. Neurol., 2005, 57, 505–512 CrossRef PubMed.
- M. J. Lindberg, R. Byström, N. Boknäs, P. M. Andersen and M. Oliveberg, Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9754–9759 CrossRef CAS PubMed.
- M. Ahn, C. L. Hagan, A. Bernardo-Gancedo, E. De Genst, F. N. Newby, J. Christodoulou, A. Dhulesia, M. Dumoulin, C. V. Robinson, C. M. Dobson and J. R. Kumita, The Significance of the Location of Mutations for the Native-State Dynamics of Human Lysozyme, Biophys. J., 2016, 111, 2358–2367 CrossRef CAS PubMed.
- S. Valleix, et al., Hereditary Systemic Amyloidosis Due to Asp76Asn Variant β 2 -Microglobulin, N. Engl. J. Med., 2012, 366, 2276–2283 CrossRef CAS PubMed.
- Y. Sekijima, R. L. Wiseman, J. Matteson, P. Hammarström, S. R. Miller, A. R. Sawkar, W. E. Balch and J. W. Kelly, The Biological and Chemical Basis for Tissue-Selective Amyloid Disease, Cell, 2005, 121, 73–85 CrossRef CAS PubMed.
- J. W. Kelly, Does protein aggregation drive postmitotic tissue degeneration?, Sci. Transl. Med., 2021, 13, eaax0914 CrossRef CAS PubMed.
- C. R. Jack, D. S. Knopman, W. J. Jagust, R. C. Petersen, M. W. Weiner, P. S. Aisen, L. M. Shaw, P. Vemuri, H. J. Wiste, S. D. Weigand, T. G. Lesnick, V. S. Pankratz, M. C. Donohue and J. Q. Trojanowski, Tracking pathophysiological processes in Alzheimer's disease: An updated hypothetical model of dynamic biomarkers, Lancet Neurol, 2013, 12, 207–216 CrossRef CAS PubMed.
- V. L. Villemagne, S. Burnham, P. Bourgeat, B. Brown, K. A. Ellis, O. Salvado, C. Szoeke, S. L. Macaulay, R. Martins, P. Maruff, D. Ames, C. C. Rowe and C. L. Masters, Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: A prospective cohort study, Lancet Neurol, 2013, 12, 357–367 CrossRef CAS.
- S. Chen, V. Berthelier, J. B. Hamilton, B. O'Nuallai and R. Wetzel, Amyloid-like features of polyglutamine aggregates and their assembly kinetics, Biochemistry, 2002, 41, 7391–7399 CrossRef CAS PubMed.
- A. K. Thakur, M. Jayaraman, R. Mishra, M. Thakur, V. M. Chellgren, I.-J. L. Byeon, D. H. Anjum, R. Kodali, T. P. Creamer, J. F. Conway, A. M. Gronenborn and R. Wetzel, Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism, Nat. Struct. Mol. Biol., 2009, 16, 380–389 CrossRef CAS PubMed.
- S. L. Crick, K. M. Ruff, K. Garai, C. Frieden and R. V. Pappu, Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20075–20080 CrossRef CAS PubMed.
- A. P. Lieberman, V. G. Shakkottai and R. L. Albin, Polyglutamine repeats in neurodegenerative diseases, Annu. Rev. Pathol. Mech. Dis., 2019, 14, 1–27 CrossRef CAS PubMed.
- Y. Wang and E. Mandelkow, Tau in physiology and pathology, Nat. Rev. Neurosci., 2016, 17, 5–21 CrossRef PubMed.
- D. P. Hanger, B. H. Anderton and W. Noble, Tau phosphorylation: the therapeutic challenge for neurodegenerative disease, Trends Mol. Med., 2009, 15, 112–119 CrossRef CAS PubMed.
- H. Sasaguri, P. Nilsson, S. Hashimoto, K. Nagata, T. Saito, B. De Strooper, J. Hardy, R. Vassar, B. Winblad and T. C. Saido, APP mouse models for Alzheimer's disease preclinical studies, EMBO J., 2017, 36, 2473–2487 CrossRef CAS PubMed.
- F.-X. Theillet, A. Binolfi, B. Bekei, A. Martorana, H. M. Rose, M. Stuiver, S. Verzini, D. Lorenz, M. van Rossum, D. Goldfarb and P. Selenko, Structural disorder of monomeric α-synuclein persists in mammalian cells, Nature, 2016, 530(7588), 45–50 CrossRef CAS PubMed.
- T. Bartels, J. G. Choi and D. J. Selkoe, α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation, Nature, 2011, 477, 107–110 CrossRef CAS PubMed.
- S. H. Shahmoradian, et al., Lewy pathology in Parkinson's disease consists of crowded organelles and lipid membranes, Nat. Neurosci., 2019, 22, 1099–1109 CrossRef CAS PubMed.
- L. A. Volpicelli-Daley, K. C. Luk, T. P. Patel, S. A. Tanik, D. M. Riddle, A. Stieber, D. F. Meaney, J. Q. Trojanowski and V. M.-Y. Lee, Exogenous α-Synuclein Fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death, Neuron, 2011, 72, 57–71 CrossRef CAS PubMed.
- L. Bousset, L. Pieri, G. Ruiz-Arlandis, J. Gath, P. H. Jensen, B. Habenstein, K. Madiona, V. Olieric, A. Böckmann, B. H. Meier and R. Melki, Structural and functional characterization of two alpha-synuclein strains, Nat. Commun., 2013, 4, 2575 CrossRef PubMed.
- W. Peelaerts, L. Bousset, A. Van der Perren, A. Moskalyuk, R. Pulizzi, M. Giugliano, C. Van den Haute, R. Melki and V. Baekelandt, α-Synuclein strains cause distinct synucleinopathies after local and systemic administration, Nature, 2015, 522, 340 CrossRef CAS PubMed.
- A. Lau, et al., α-Synuclein strains target distinct brain regions and cell types, Nat. Neurosci., 2020, 23, 21–31 CrossRef CAS PubMed.
- A.-L. Mahul-Mellier, J. Burtscher, N. Maharjan, L. Weerens, M. Croisier, F. Kuttler, M. Leleu, G. W. Knott and H. A. Lashuel, The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4971–4982 CrossRef CAS PubMed.
- V. A. Trinkaus, I. Riera-Tur, A. Martínez-Sánchez, F. J. B. Bäuerlein, Q. Guo, T. Arzberger, W. Baumeister, I. Dudanova, M. S. Hipp, F. U. Hartl and R. Fernández-Busnadiego, In situ architecture of neuronal α-Synuclein inclusions, Nat. Commun., 2021, 12, 2110 CrossRef CAS PubMed.
- C. R. Jack Jr, H. J. Wiste, C. G. Schwarz, V. J. Lowe, M. L. Senjem, P. Vemuri, S. D. Weigand, T. M. Therneau, D. S. Knopman, J. L. Gunter, D. T. Jones, J. Graff-Radford, K. Kantarci, R. O. Roberts, M. M. Mielke, M. M. Machulda and R. C. Petersen, Longitudinal tau PET in ageing and Alzheimer's disease, Brain, 2018, 141, 1517–1528 CrossRef PubMed.
- L. J. Renaud, et al., Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau-PET, Sci. Transl. Med., 2020, 12, eaau5732 CrossRef PubMed.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. W. Scheres, Cryo-EM structures of tau filaments from Alzheimer's disease, Nature, 2017, 547, 185–190 CrossRef CAS PubMed.
- B. Falcon, W. Zhang, A. G. Murzin, G. Murshudov, H. J. Garringer, R. Vidal, R. A. Crowther, B. Ghetti, S. H. W. W. Scheres and M. Goedert, Structures of filaments from Pick's disease reveal a novel tau protein fold, Nature, 2018, 561, 137–140 CrossRef CAS PubMed.
- B. Falcon, J. Zivanov, W. Zhang, A. G. Murzin, H. J. Garringer, R. Vidal, R. A. Crowther, K. L. Newell, B. Ghetti, M. Goedert and S. H. W. Scheres, Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules, Nature, 2019, 568, 420–423 CrossRef CAS PubMed.
- T. Arakhamia, C. E. Lee, Y. Carlomagno, D. M. Duong, S. R. Kundinger, K. Wang, D. Williams, M. DeTure, D. W. Dickson, C. N. Cook, N. T. Seyfried, L. Petrucelli and A. W. P. Fitzpatrick, Posttranslational modifications mediate the structural diversity of tauopathy strains, Cell, 2020, 180, 633–644 CrossRef CAS PubMed.
- M. Schweighauser, Y. Shi, A. Tarutani, F. Kametani, A. G. Murzin, B. Ghetti, T. Matsubara, T. Tomita, T. Ando, K. Hasegawa, S. Murayama, M. Yoshida, M. Hasegawa, S. H. W. Scheres and M. Goedert, Structures of α-synuclein filaments from multiple system atrophy, Nature, 2020, 585, 464–469 CrossRef CAS PubMed.
- Y. Yang, et al., Cryo-EM Structures of Amyloid-β 42 Filaments from Human Brains, Science, 2021, 375, 167–172 CrossRef PubMed.
- J. F. Morley, H. R. Brignull, J. J. Weyers and R. I. Morimoto, The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10417–10422 CrossRef PubMed.
- T. J. van Ham, K. L. Thijssen, R. Breitling, R. M. W. W. Hofstra, R. H. A. A. Plasterk and E. A. A. A. Nollen, C. elegans Model Identifies Genetic Modifiers of α-Synuclein Inclusion Formation During Aging, PLoS Genet., 2008, 4, e1000027 CrossRef PubMed.
- R. F. Laine, T. Sinnige, K. Y. Ma, A. J. Haack, C. Poudel, P. Gaida, N. Curry, M. Perni, E. A. A. A. Nollen, C. M. Dobson, M. Vendruscolo, G. S. K. Schierle, C. F. Kaminski, G. S. Kaminski Schierle and C. F. Kaminski, Fast fluorescence lifetime imaging reveals the aggregation processes of α-synuclein and polyglutamine in aging Caenorhabditis elegans, ACS Chem. Biol., 2019, 414714, 1628–1636 CrossRef PubMed.
- L. M. Luheshi, G. G. Tartaglia, A.-C. Brorsson, A. P. Pawar, I. E. Watson, F. Chiti, M. Vendruscolo, D. A. Lomas, C. M. Dobson and D. C. Crowther, Systematic In Vivo Analysis of the Intrinsic Determinants of Amyloid β Pathogenicity, PLoS Biol., 2007, 5, e290 CrossRef PubMed.
- L. Lang, P. Zetterström, T. Brännström, S. L. Marklund, J. Danielsson and M. Oliveberg, SOD1 aggregation in ALS mice shows simplistic test tube behavior, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9878–9883 CrossRef CAS PubMed.
- G. Meisl, T. Kurt, I. Condado-Morales, C. Bett, S. Sorce, M. Nuvolone, T. C. T. Michaels, D. Heinzer, M. Avar, S. I. A. Cohen, S. Hornemann, A. Aguzzi, C. M. Dobson, C. J. Sigurdson and T. P. J. Knowles, Scaling analysis reveals the mechanism and rates of prion replication in vivo, Nat. Struct. Mol. Biol., 2021, 28, 365–372 CrossRef CAS PubMed.
- G. Meisl, E. Hidari, K. Allinson, T. Rittman, S. L. Devos, J. S. Sanchez, C. K. Xu, K. E. Duff, K. A. Johnson, J. B. Rowe, B. T. Hyman, T. P. J. Knowles and D. Klenerman, In vivo rate-determining steps of tau seed accumulation in Alzheimer's disease, Sci. Adv., 2021, 7, eabh1448 CrossRef CAS PubMed.
- W. Zhang, B. Falcon, A. G. Murzin, J. Fan, R. A. Crowther, M. Goedert and S. H. W. Scheres, Heparin-induced tau filaments are polymorphic and differ from those in alzheimer’s and pick’s diseases, Elife, 2019, 8, e43584 CrossRef PubMed.
- Y. E. Kim, M. S. Hipp, A. Bracher, M. Hayer-Hartl and F. Ulrich Hartl, Molecular Chaperone Functions in Protein Folding and Proteostasis, Annu. Rev. Biochem., 2013, 82, 323–355 CrossRef CAS PubMed.
- H. H. Kampinga and E. A. Craig, The HSP70 chaperone machinery: J proteins as drivers of functional specificity, Nat. Rev. Mol. Cell Biol., 2010, 11, 579–592 CrossRef CAS PubMed.
- T. Morán Luengo, M. P. Mayer and S. G. D. Rüdiger, The Hsp70–Hsp90 Chaperone Cascade in Protein Folding, Trends Cell Biol., 2019, 29, 164–177 CrossRef PubMed.
- R. Rosenzweig, N. B. Nillegoda, M. P. Mayer and B. Bukau, The Hsp70 chaperone network, Nat. Rev. Mol. Cell Biol., 2019, 20, 665–680 CrossRef CAS PubMed.
- A. Mogk, C. Ruger-Herreros and B. Bukau, Cellular functions and mechanisms of action of small heat shock proteins, Annu. Rev. Microbiol., 2019, 73, 89–110 CrossRef CAS PubMed.
- P. Arosio, T. C. T. Michaels, S. Linse, C. Månsson, C. Emanuelsson, J. Presto, J. Johansson, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation, Nat. Commun., 2016, 7, 10948 CrossRef CAS PubMed.
- T. Sinnige, A. Yu and R. I. Morimoto, Challenging Proteostasis: Role of the Chaperone Network to Control Aggregation-Prone Proteins in Human Disease, HSF1 and Molecular Chaperones in Biology and Cancer, 2020, pp. 53–68 Search PubMed.
- E. A. A. Nollen, S. M. Garcia, G. van Haaften, S. Kim, A. Chavez, R. I. Morimoto and R. H. A. Plasterk, Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6403–6408 CrossRef CAS PubMed.
- M. Brehme, C. Voisine, T. Rolland, S. Wachi, J. H. Soper, Y. Zhu, K. Orton, A. Villella, D. Garza, M. Vidal, H. Ge and R. I. Morimoto, A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease, Cell Rep., 2014, 9(3), 1135–1150 CrossRef CAS PubMed.
- M. C. Silva, S. Fox, M. Beam, H. Thakkar, M. D. Amaral and R. I. Morimoto, A genetic screening strategy identifies novel regulators of the proteostasis network, PLoS Genet., 2011, 7, e1002438 CrossRef CAS PubMed.
- L. B. Shelton, J. D. Baker, D. Zheng, L. E. Sullivan, P. K. Solanki, J. M. Webster, Z. Sun, J. J. Sabbagh, B. A. Nordhues, J. Koren, S. Ghosh, B. S. J. Blagg, L. J. Blair and C. A. Dickey, Hsp90 activator Aha1 drives production of pathological tau aggregates, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 201707039 CrossRef PubMed.
- X. Gao, M. Carroni, C. Nussbaum-Krammer, A. Mogk, N. B. B. Nillegoda, A. Szlachcic, D. L. L. Guilbride, H. R. R. Saibil, M. P. P. Mayer and B. Bukau, Human Hsp70 disaggregase reverses Parkinson’s-linked α-synuclein amyloid fibrils, Mol. Cell, 2015, 59, 781–793 CrossRef CAS PubMed.
- A. S. Wentink, N. B. Nillegoda, J. Feufel, G. Ubartaitė, C. P. Schneider, P. De Los Rios, J. Hennig, A. Barducci and B. Bukau, Molecular dissection of amyloid disaggregation by human HSP70, Nature, 2020, 587, 483–488 CrossRef CAS PubMed.
- A. Franco, P. Gracia, A. Colom, J. D. Camino, J. Á. Fernández-Higuero, N. Orozco, A. Dulebo, L. Saiz, N. Cremades, J. M. G. Vilar, A. Prado and A. Muga, All-or-none amyloid disassembly via chaperone-triggered fibril unzipping favors clearance of α-synuclein toxic species, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2105548118 CrossRef CAS PubMed.
- M. M. Schneider, S. Gautam, T. W. Herling, E. Andrzejewska, G. Krainer, A. M. Miller, Q. A. E. Peter, F. S. Ruggeri, M. Vendruscolo, A. Bracher, C. M. Dobson, F. U. Hartl and T. P. Knowles, The Hsc70 Disaggregation Machinery Removes Monomer Units Directly from α -Synuclein Fibril Ends, bioRxiv, 2020 DOI:10.1101/2020.11.02.365825.
- J. Tittelmeier, C. A. Sandhof, H. M. Ries, S. Druffel-Augustin, A. Mogk, B. Bukau and C. Nussbaum-Krammer, The HSP110/HSP70 disaggregation system generates spreading-competent toxic α-synuclein species, EMBO J., 2020, 39, 1–16 CrossRef PubMed.
- H. Müller, D. M. Dias, A. V. D. Zalm and A. J. Baldwin, αB-crystallin affects the morphology of Aβ(1-40) aggregates, bioRxiv, 2021 DOI:10.1101/2021.03.07.433908.
- C. L. Klaips, M. H. M. Gropp, M. S. Hipp and F. U. Hartl, Sis1 potentiates the stress response to protein aggregation and elevated temperature, Nat. Commun., 2020, 11, 6271 CrossRef CAS PubMed.
- D. M. Walther, P. Kasturi, M. Zheng, S. Pinkert, G. Vecchi, P. Ciryam, R. I. Morimoto, C. M. Dobson, M. Vendruscolo, M. Mann and F. U. Hartl, Widespread proteome remodeling and aggregation in aging C. elegans, Cell, 2015, 161, 919–932 CrossRef CAS PubMed.
- Q. Guo, C. Lehmer, A. Martínez-Sánchez, T. Rudack, F. Beck, H. Hartmann, M. Pérez-Berlanga, F. Frottin, M. S. Hipp, F. U. Hartl, D. Edbauer, W. Baumeister and R. Fernández-Busnadiego, In situ structure of neuronal C9orf72 poly-GA aggregates reveals proteasome recruitment, Cell, 2018, 172, 696–705 CrossRef CAS PubMed.
- A. Gruber, D. Hornburg, M. Antonin, N. Krahmer, J. Collado, M. Schaffer, G. Zubaite, C. Lüchtenborg, T. Sachsenheimer, B. Brügger, M. Mann, W. Baumeister, F. U. Hartl, M. S. Hipp and R. Fernández-Busnadiego, Molecular and structural architecture of polyQ aggregates in yeast, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3446–E3453 CrossRef CAS PubMed.
- J. Harapin, M. Börmel, K. T. Sapra, D. Brunner, A. Kaech and O. Medalia, Structural analysis of multicellular organisms with cryo-electron tomography, Nat. Methods, 2015, 12, 634–636 CrossRef CAS PubMed.
- M. Schaffer, S. Pfeffer, J. Mahamid, S. Kleindiek, T. Laugks, S. Albert, B. D. Engel, A. Rummel, A. J. Smith, W. Baumeister and J. M. Plitzko, A cryo-FIB lift-out technique enables molecular-resolution cryo-ET within native Caenorhabditis elegans tissue, Nat. Methods, 2019, 16, 757–762 CrossRef CAS PubMed.
- P. Connell, C. A. Ballinger, J. Jiang, Y. Wu, L. J. Thompson, J. Höhfeld and C. Patterson, The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins, Nat. Cell Biol., 2001, 3, 93–96 CrossRef CAS PubMed.
- L. Petrucelli, et al., CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation, Hum. Mol. Genet., 2004, 13, 703–714 CrossRef CAS PubMed.
- C. A. Dickey, A. Kamal, K. Lundgren, N. Klosak, R. M. Bailey, J. Dunmore, P. Ash, S. Shoraka, J. Zlatkovic, C. B. Eckman, C. Patterson, D. W. Dickson, N. S. Nahman, M. Hutton, F. Burrows and L. Petrucelli, The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins, J. Clin. Invest., 2007, 117, 648–658 CrossRef CAS PubMed.
- S. Carra, S. J. Seguin, H. Lambert and J. Landry, HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy, J. Biol. Chem., 2008, 283, 1437–1444 CrossRef CAS PubMed.
- A. Ciechanover and Y. T. Kwon, Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies, Exp. Mol. Med., 2015, 47, e147 CrossRef CAS PubMed.
- A. Djajadikerta, S. Keshri, M. Pavel, R. Prestil, L. Ryan and D. C. Rubinsztein, Autophagy Induction as a Therapeutic Strategy for Neurodegenerative Diseases, J. Mol. Biol., 2020, 432, 2799–2821 CrossRef CAS PubMed.
- G. E. Karagöz, A. M. S. Duarte, E. Akoury, H. Ippel, J. Biernat, T. Morán Luengo, M. Radli, T. Didenko, B. A. Nordhues, D. B. Veprintsev, C. A. Dickey, E. Mandelkow, M. Zweckstetter, R. Boelens, T. Madl and S. G. D. Rüdiger, Hsp90-tau complex reveals molecular basis for specificity in chaperone action, Cell, 2014, 156, 963–974 CrossRef PubMed.
- J. Labbadia and R. I. Morimoto, The biology of proteostasis in aging and disease, Annu. Rev. Biochem., 2015, 84, 435–464 CrossRef CAS PubMed.
- T. Gidalevitz, A. Ben-Zvi, K. K. H. Ho, H. R. H. Brignull and R. I. Morimoto, Progressive disruption of cellular protein folding in models of polyglutamine diseases, Science, 2006, 311, 1471–1474 CrossRef CAS PubMed.
- A. Ben-Zvi, E. A. Miller and R. I. Morimoto, Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14914–14919 CrossRef CAS PubMed.
- R. Gupta, P. Kasturi, A. Bracher, C. Loew, M. Zheng, A. Villella, D. Garza, F. U. Hartl and S. Raychaudhuri, Firefly luciferase mutants as sensors of proteome stress, Nat. Methods, 2011, 8, 879–884 CrossRef CAS PubMed.
- S. Blumenstock, E. K. Schulz-Trieglaff, K. Voelkl, A. Bolender, P. Lapios, J. Lindner, M. S. Hipp, F. U. Hartl, R. Klein and I. Dudanova, Fluc-EGFP reporter mice reveal differential alterations of neuronal proteostasis in aging and disease, EMBO J., 2021, 40, e107260 CrossRef CAS PubMed.
- J. Labbadia and R. I. Morimoto, Repression of the heat shock response is a programmed event at the onset of reproduction, Mol. Cell, 2015, 59, 639–650 CrossRef CAS PubMed.
- M. So, D. Hall and Y. Goto, Revisiting supersaturation as a factor determining amyloid fibrillation, Curr. Opin. Struct. Biol., 2016, 36, 32–39 CrossRef CAS PubMed.
- M. Adachi, M. So, K. Sakurai, J. Kardos and Y. Goto, Supersaturation-limited and Unlimited Phase Transitions Compete to Produce the Pathway Complexity in Amyloid Fibrillation, J. Biol. Chem., 2015, 290, 18134–18145 CrossRef CAS PubMed.
- M. B. Koopman and S. G. D. Rüdiger, Alzheimer cells on their way to derailment show selective changes in protein quality control network, Front. Mol. Biosci., 2020, 7, 214 CrossRef CAS PubMed.
- P. Ciryam, G. G. Tartaglia, R. I. Morimoto, C. M. Dobson and M. Vendruscolo, Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins, Cell Rep., 2013, 5, 781–790 CrossRef CAS PubMed.
- H. Ederle and D. Dormann, TDP-43 and FUS en route from the nucleus to the cytoplasm, FEBS Lett., 2017, 591, 1489–1507 CrossRef CAS PubMed.
- S. Maharana, J. Wang, D. K. Papadopoulos, D. Richter, A. Pozniakovsky, I. Poser, M. Bickle, S. Rizk, J. Guillén-Boixet, T. M. Franzmann, M. Jahnel, L. Marrone, Y. T. Chang, J. Sterneckert, P. Tomancak, A. A. Hyman and S. Alberti, RNA buffers the phase separation behavior of prion-like RNA binding proteins, Science, 2018, 360, 918–921 CrossRef CAS PubMed.
- C. Procaccini, M. Santopaolo, D. Faicchia, A. Colamatteo, L. Formisano, P. de Candia, M. Galgani, V. De Rosa and G. Matarese, Role of metabolism in neurodegenerative disorders, Metabolism, 2016, 65, 1376–1390 CrossRef CAS PubMed.
- H. Braak and E. Braak, Neuropathological stageing of Alzheimer-related changes, Acta Neuropathol., 1991, 82, 239–259 CrossRef CAS PubMed.
- H. Braak, K. Tredici, U. Del Rüb, R. A. I. de Vos, E. N. H. Jansen Steur and E. Braak, Staging of brain pathology related to sporadic Parkinson's disease, Neurobiol. Aging, 2003, 24, 197–211 CrossRef PubMed.
- C. Peng, J. Q. Trojanowski and V. M. Y. Lee, Protein transmission in neurodegenerative disease, Nat. Rev. Neurol., 2020, 16, 199–212 CrossRef CAS PubMed.
- M. Jucker and L. C. Walker, Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases, Nat. Neurosci., 2018, 21, 1341–1349 CrossRef CAS PubMed.
- G. Meisl, T. P. Knowles and D. Klenerman, The molecular processes underpinning prion-like spreading and seed amplification in protein aggregation, Curr. Opin. Neurobiol., 2020, 61, 58–64 CrossRef CAS PubMed.
- J. Vaquer-Alicea and M. I. Diamond, Propagation of protein aggregation in neurodegenerative diseases, Annu. Rev. Biochem., 2019, 88, 785–810 CrossRef CAS PubMed.
- M. Goedert, D. S. Eisenberg and R. A. Crowther, Propagation of tau aggregates and neurodegeneration, Annu. Rev. Neurosci., 2017, 40, 189–210 CrossRef CAS PubMed.
- T. Sinnige, K. Stroobants, C. M. Dobson and M. Vendruscolo, Biophysical studies of protein misfolding and aggregation in in vivo models of Alzheimer's and Parkinson's diseases, Q. Rev. Biophys., 2020, 49, e22 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |