
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bioorthogonal chemical reporter for the detection and identification of protein lactylation†
Yanan
Sun‡
a,
Yanchi
Chen‡
a and
Tao
Peng
 *ab
*ab
aState Key Laboratory of Chemical Oncogenomics, Guangdong Provincial Key Laboratory of Chemical Genomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China. E-mail: tpeng@pku.edu.cn
bInstitute of Chemical Biology, Shenzhen Bay Laboratory, Shenzhen 518132, China
First published on 26th April 2022
Abstract
L-Lactylation is a recently discovered post-translational modification occurring on histone lysine residues to regulate gene expression. However, the substrate scope of lactylation, especially that in non-histone proteins, remains unknown, largely due to the limitations of current methods for analyzing lactylated proteins. Herein, we report an alkynyl-functionalized bioorthogonal chemical reporter, YnLac, for the detection and identification of protein lactylation in mammalian cells. Our in-gel fluorescence and chemical proteomic analyses show that YnLac is metabolically incorporated into lactylated proteins and directly labels known lactylated lysines of histones. We further apply YnLac to the proteome-wide profiling of lactylation, revealing many novel modification sites in non-histone proteins for the first time. Moreover, we demonstrate that lactylation of a newly identified substrate protein PARP1 regulates its ADP-ribosylation activity. Our study thus provides a powerful chemical tool for characterizing protein lactylation and greatly expands our understanding of substrate proteins and functions of this new modification.
Introduction
L-Lactate, the end product of glycolysis generated from the reduction of pyruvate by lactate dehydrogenase, is an essential metabolite in mammals.1 Although historically considered to be a metabolic waste product, L-lactate has since been recognized to serve as a major circulating energy source2 and, additionally, act as a multifunctional signaling molecule involved in diverse physiological and pathological processes, including angiogenesis, inflammation, neural activity, neoplasia, and autoimmune diseases.3–6 Recently, a seminal study by Zhao laboratory reported that L-lactate is a precursor to drive direct modifications of histones.7 This novel type of post-translational modification (PTM), named L-lactylation8 and referred to as lactylation hereinafter, occurs on the side-chains of lysine residues in core histones with covalent attachment of L-lactyl groups (Fig. 1A). The study suggested that histone lactylation is physiologically relevant and contributes to epigenetic gene regulation. For instance, in pro-inflammatory macrophages, intracellular lactate can stimulate histone H3 lactylation on the promoters of homeostatic genes, which activates their expression.7 Later studies have shown that histone lactylation plays key regulatory roles under a variety of biological conditions, such as cell state transition,9 cell reprogramming,10 neuronal excitation,11 tumorigenesis,12,13 glucose metabolism,14 and so on.15–18 The discovery of lactylation greatly extends our understanding of the functions of lactate and implies broad involvement of this modification in diverse biological processes,19,20 given the widespread existence and high concentrations of L-lactate in mammals.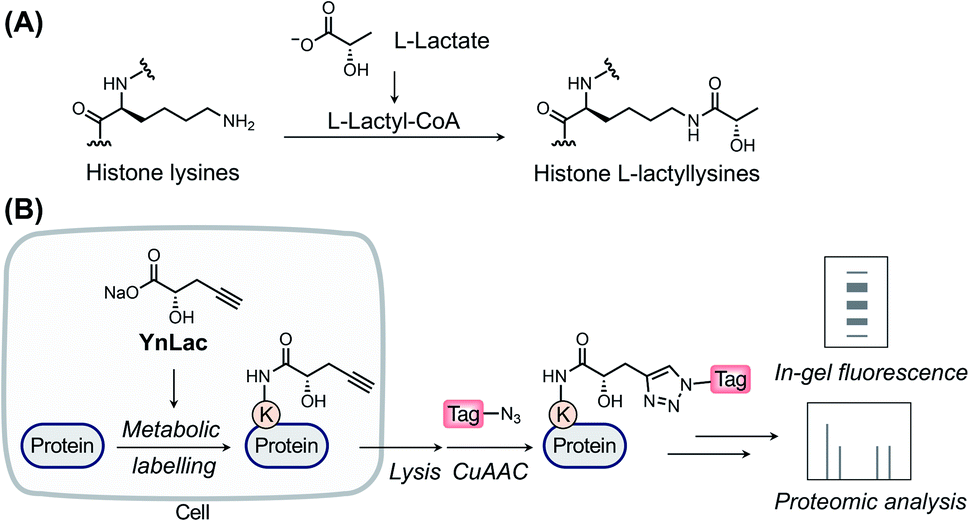 | ||
| Fig. 1 The chemical reporter strategy for studying protein lactylation. (A) Chemical structure of histone lysine lactylation. (B) Schematic for the detection and identification of lactylated proteins using the bioorthogonal chemical reporter. The alkynyl-functionalized L-lactate analogue (i.e., YnLac) is metabolically incorporated into lactylated proteins in live cells. Then alkynyl-labelled proteins are conjugated with fluorescent or affinity tags for in-gel fluorescence detection or proteomic identification, respectively. | ||
As a newly identified PTM, the biological roles and regulatory elements of lactylation remain undiscovered. Identification of lactylated substrate proteins is an essential step required for dissecting the biological functions of lactylation and demands a robust method for the detection and enrichment of lactylated proteins. Current studies mainly rely on isotopic L-lactate and pan anti-lactyllysine antibodies.7 However, the isotopic labelling method requires autoradiography for detection that is time-consuming and hazardous to implement. What's more, this method cannot be used for protein or peptide enrichment. While amenable for immunopurification, the antibody-based method is limited by the cost, availability, and specificity of antibodies and, moreover, is not ideal for monitoring the turnover of lactylation. Due to these technical limitations, the substrate proteins and modification sites of lactylation remain largely unexplored, especially in mammalian cells. In particular, initially discovered in histone proteins, lactylation in non-histone proteins has only been identified in microbial pathogens and plants.21–23 While antibody-based immunoprecipitation coupled with mass spectrometry (MS) analysis was attempted to identify potential lactylated proteins at the protein level in mouse brain tissues,11 proteome-wide identification of lactylation sites in mammalian cells has not been reported yet. Consequently, histone proteins remain to be the only well-characterized lactylated proteins in mammalian cells. Lactylation of mammalian non-histone proteins and its substrate scope are still not defined, which has seriously impeded its biological characterization. To overcome the limitations of current methods and characterize lactylated substrate proteins, a new approach is imperatively needed for efficient detection and comprehensive profiling of protein lactylation in mammalian cells.
Over the past two decades, the bioorthogonal chemical reporter strategy has emerged as a powerful alternative method for studying diverse protein PTMs,24–27 including glycosylation,28–30 acylation,31–33 lipidation,34–37 and so on.25,27 Specifically, chemical reporters, i.e., the alkynyl or azido derivatives of PTM donor precursors, can be metabolically incorporated into PTM-bearing proteins and allow for labelling of these proteins of interest with bioorthogonal groups. The labelled proteins are subsequently conjugated with fluorophores or affinity tags through bioorthogonal reactions38 for fluorescence detection or proteomic identification, respectively. Herein, we report an alkynyl-functionalized L-lactate analogue as a bioorthogonal chemical reporter for metabolic labelling, fluorescence detection, and chemical proteomic profiling of protein lactylation in mammalian cells.
Results and discussion
Design of lactylation chemical reporters
In a previous study by Zhao laboratory, exogenous sodium L-lactate was supplemented into cell culture media to enhance histone lactylation levels.7 It has also been shown that isotopic sodium L-lactate was metabolically incorporated into lactylated histones. Moreover, an analogue of L-lactate appended with a fluorescent tag has been utilized as an L-lactate mimic to study its transport and metabolism in live cells.39 Inspired by these studies, we envisioned that alkynyl analogues of L-lactate could be metabolized and incorporated into lactylated proteins in live mammalian cells. We thus designed and synthesized a sodium L-lactate derivative, i.e., sodium (S)-2-hydroxypent-4-ynoate (YnLac), that contains a terminal alkynyl on the original methyl group (Fig. 1B and Scheme S1 in the ESI†), as a potential chemical reporter for the detection and profiling of protein lactylation (Fig. 1B). According to previous studies on chemical reporters for protein acetylation and malonylation,31,32 we expected that the structural alteration stemming from the introduction of a small alkynyl functionality could be tolerated by the promiscuous cellular lactylation machinery.We first examined whether YnLac could be metabolically incorporated into proteins in live mammalian cells. For this purpose, HEK293T cells were incubated with YnLac and lysed with highly denatured lysis buffer to maximize the extraction of proteins from various cellular compartments. The resulting whole cell lysates were then reacted with azido-rhodamine (az-rho) via Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction40 to conjugate YnLac-labelled proteins with the rhodamine dye for in-gel fluorescence analysis (Fig. 1B). The results showed that a wide range of proteins was metabolically labelled by YnLac in a dose-dependent manner with strong fluorescence signals at the concentrations of 25–50 mM (Fig. S1 in the ESI†). It is worth mentioning that this concentration range is consistent with the dose of exogenous sodium L-lactate (i.e., 25 mM) utilized to boost histone lactylation levels in a previous study.7 In addition to YnLac, we also synthesized and examined additional L-lactate analogues, i.e., sodium (S)-2-hydroxyhex-5-ynoate (YnLac-2) and methyl (S)-2-hydroxypent-4-ynoate (YnLac-OMe), for metabolic labelling of proteins in live cells (Fig. S2A and Scheme S1 in the ESI†). The in-gel fluorescence results demonstrated that YnLac-2 containing a bulkier side-chain was less efficient for protein labelling than YnLac (Fig. S2B in the ESI†). The YnLac-OMe was designed to increase the cellular delivery of YnLac through reversible masking of the negatively charged carboxylate group with an ester, which was previously demonstrated as the pro-metabolite strategy for short-chain fatty acylation chemical reporters.41 However, our results showed that YnLac-OMe did not enhance the protein labelling signals compared to YnLac (Fig. S2C in the ESI†). As YnLac resembles the carboxylate structure of L-lactate and exhibits relatively high protein labelling efficiency in live cells, we, therefore, focused on YnLac for subsequent studies.
In-gel fluorescence characterization of the lactylation chemical reporter YnLac
We then investigated whether YnLac could metabolically label known lactylated proteins. Histone proteins (i.e., H3, H4, H2A, and H2B) have been well established to be lactylated.7 We thus separated the nuclear fractions from YnLac-labelled HEK293T cells and extracted the core histones, followed by click reactions with az-rho and in-gel fluorescence analysis. As a result, dose- and time-dependent profiling demonstrated that core histones with characteristic molecular weights were clearly labelled by YnLac at concentrations of 5–50 mM upon incubation for 1–8 hours (Fig. 2A and S3 in the ESI†). Notably, the YnLac concentrations are in the range of pathophysiological levels of L-lactate from 1.5–3 mM in healthy tissues42 to 10–40 mM in cancer tissues.42,43 In addition, comparable concentrations (e.g., 10–25 mM) of sodium L-lactate were previously used in cell culture media for stimulation and isotopic labelling of protein lactylation.7 We also examined whether protein labelling by YnLac was sensitive to competition from natural L-lactate. Importantly, we observed that pre-treatment and co-incubation with L-lactate attenuated the labelling of core histones by YnLac in a dose-dependent manner (Fig. 2B), probably because L-lactate competes with YnLac for lactylation enzymes and sites. These results thus suggest that YnLac can be metabolically incorporated into known lactylated core histones.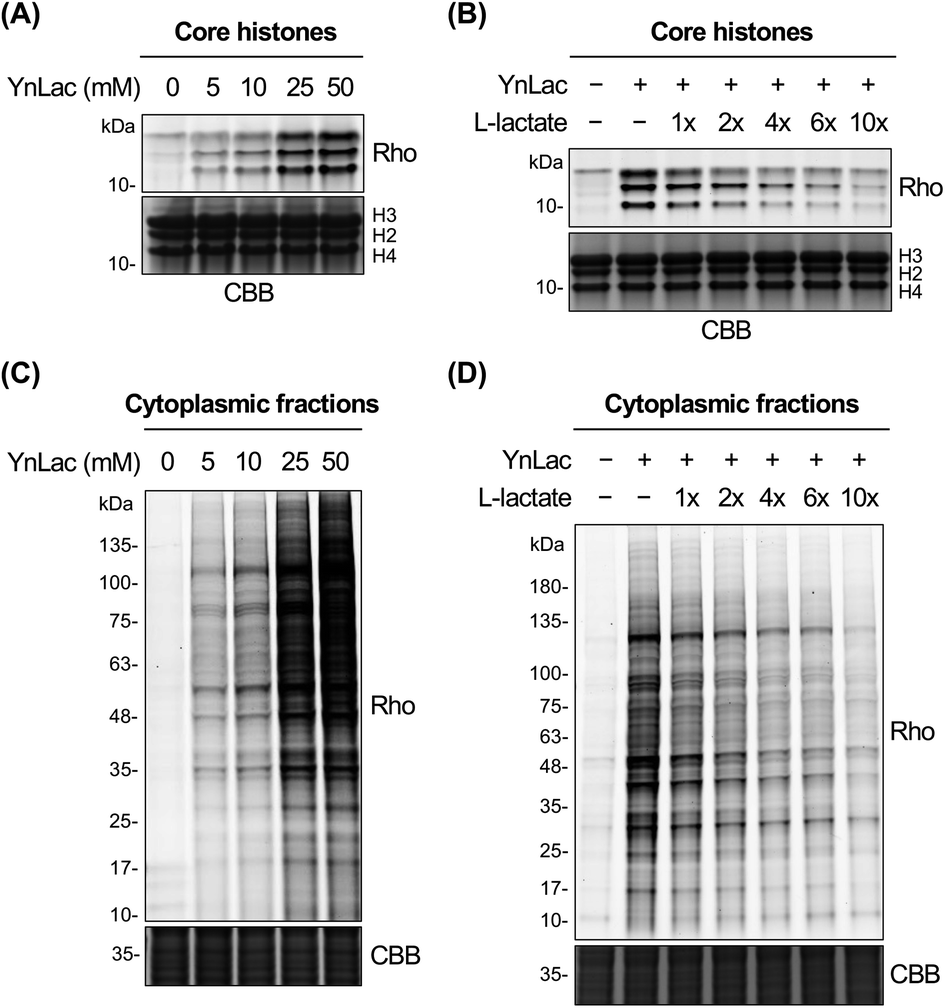 | ||
| Fig. 2 In-gel fluorescence detection of lactylated proteins with YnLac. (A) Concentration-dependent metabolic labelling of core histones by YnLac. (B) Dose-dependent competition of core histone labelling by YnLac with sodium L-lactate. (C) Concentration-dependent metabolic labelling of cytoplasmic proteins by YnLac. (D) Dose-dependent competition of cytoplasmic protein labelling by YnLac with sodium L-lactate. HEK293T cells were incubated with YnLac at varying concentrations for 8 hours. The core histones and cytoplasmic proteins were separated for click conjugation with az-rho and in-gel fluorescence analysis. For L-lactate competition, HEK293T cells were pre-treated with sodium L-lactate for 4 hours and co-incubated with YnLac (20 mM) and sodium L-lactate at indicated doses (shown as fold of YnLac concentration) for 8 hours. “Rho” represents the rhodamine fluorescence channel. Coomassie brilliant blue (CBB) staining is included as the protein loading control. | ||
We proceeded to explore the metabolic labelling of non-histone proteins by YnLac. Notably, whether lactylation occurs in non-histone proteins in mammalian cells remains unknown. Our in-gel fluorescence analysis on whole cell lysates has suggested that, in addition to histones, many non-histone proteins were also labelled by YnLac (Fig. S1 in the ESI†). In line with these results, the fluorescence imaging analysis on HeLa cells that were metabolically labelled with YnLac revealed that YnLac-labelled proteins were indeed localized in both the nucleus and cytoplasm (Fig. S4 in the ESI†). To focus on non-histone proteins in the cytoplasm, we prepared cytoplasmic extracts from YnLac-labelled HEK293T cells for click conjugation with az-rho and in-gel fluorescence detection. Like core histones, proteins in cytoplasmic fractions were also metabolically labelled by YnLac in a dose- and time-dependent manner (Fig. 2C and S5 in the ESI†). More importantly, we observed decreased labelling of cytoplasmic proteins by YnLac upon pre-addition and coadministration of L-lactate (Fig. 2D), indicating that YnLac most likely was metabolically incorporated into lactylated non-histone proteins.
To further characterize YnLac as a chemical reporter for protein lactylation, we conducted additional in-gel fluorescence profiling experiments. We evaluated the role of histone acetyltransferase p300 in protein labelling by YnLac. Inhibition of p300 by curcumin44 generally reduced the labelling of core histones by YnLac (Fig. S6 in the ESI†), suggesting that p300 is a potential lactylation writer protein. This is notably consistent with previous results in a cell-free assay.7 Many types of protein short-chain lysine acylations such as acetylation, propionylation, and malonylation are reversible and regulated by two major families of deacylases, i.e., histone deacetylases (HDACs) and sirtuins.45,46 Therefore, lactylation is likely also reversible.19 To assess the turnover and regulation of lactylation, we performed a pulse-chase labelling experiment47 using YnLac. Specifically, HEK293T cells were pulse-labelled with YnLac and chased with YnLac-deficient media for different periods, followed by cell lysis, click conjugation, and in-gel fluorescence detection. The results showed that the fluorescence signals of YnLac-labelled histones decreased gradually (Fig. S7 in the ESI†). Interestingly, we observed that the turnover of YnLac labelling was delayed when cells were treated with a pan-HDAC inhibitor trichostatin A (TSA),48 but not when treated with a pan-sirtuin inhibitor nicotinamide (NAM)49 (Fig. S7 in the ESI†). These results therefore suggest that protein lactylation is reversible and likely regulated by the HDAC family, consistent with a very recent report.8 Furthermore, we examined the applicability and generality of YnLac for the metabolic labelling of proteins in various cell lines. Robust protein labelling by YnLac was observed for all cell types tested (Fig. S8 in the ESI†), highlighting the general utility of YnLac. Notably, different cell types demonstrated overlapping, but discrete, profiles of protein labelling, indicating that YnLac labels a specific set of proteins dependent on the function and protein expression of individual cell lines. Together, these results collectively demonstrate that YnLac can be metabolically incorporated into lactylated proteins in mammalian cells and enables robust fluorescence detection of protein lactylation.
Chemical proteomic profiling of lactylation using YnLac
We next sought to verify that YnLac modifies lysine residues of proteins by MS analysis. We initially focused on the nuclear fractions that contain a large number of histones with known lactylation sites. Specifically, the nuclear fractions of YnLac-labelled HEK293T cells were prepared, conjugated with the acid-cleavable azido-DADPS-biotin tag50 (Fig. S9A in the ESI†), and subjected to affinity purification with streptavidin beads. After on-bead digestion, YnLac-modified peptides were eluted with acid for MS analysis (Fig. 3A). The acquired MS/MS spectra were analyzed using MaxQuant software51 by setting the fragment left on YnLac-labelled lysine residues as a variable modification (Fig. S9B in the ESI†). To enhance the reliability and coverage of the site identification, we processed four replicates of YnLac-labelled samples, in parallel with L-lactate-treated samples serving as the controls. Two modification sites that were identified in both YnLac-labelled samples and L-lactate-treated controls were assigned as false positives and excluded. The MS/MS analysis demonstrated that YnLac was indeed incorporated into multiple known lactylated lysine residues in histone proteins, such as H3K23, H3K27, H4K8, H4K12, H2BK5, H2BK11, and H2BK20 (Tables S1 and S2 in the ESI†). For example, as shown in Fig. 3B, an H3 peptide, i.e., 18KQLATKYnLacAAR,26 containing the YnLac modification at K23, was identified by MS/MS analysis (Fig. S9C in the ESI†). Notably, we also identified two previously unidentified histone lactylation sites including H2AK4 and H2AK9 (Table S1 in the ESI†). More importantly, in addition to sites in histones, four novel lactylation sites in non-histone proteins, including HMGN1, NPM1, PDCD4, and PHF3, were also retrieved in the proteomic analysis (Tables S1 and S2 in the ESI†). Overall, 19 YnLac-modified sites were identified in the nuclear fractions, including 13 known and two unreported lactylation sites in histones and four novel sites in non-histone proteins (Fig. 3C and D). Taken together, these data demonstrate that YnLac directly targets lysine residues on proteins and, more importantly, can be used to identify new lactylation sites at a proteome-wide scale.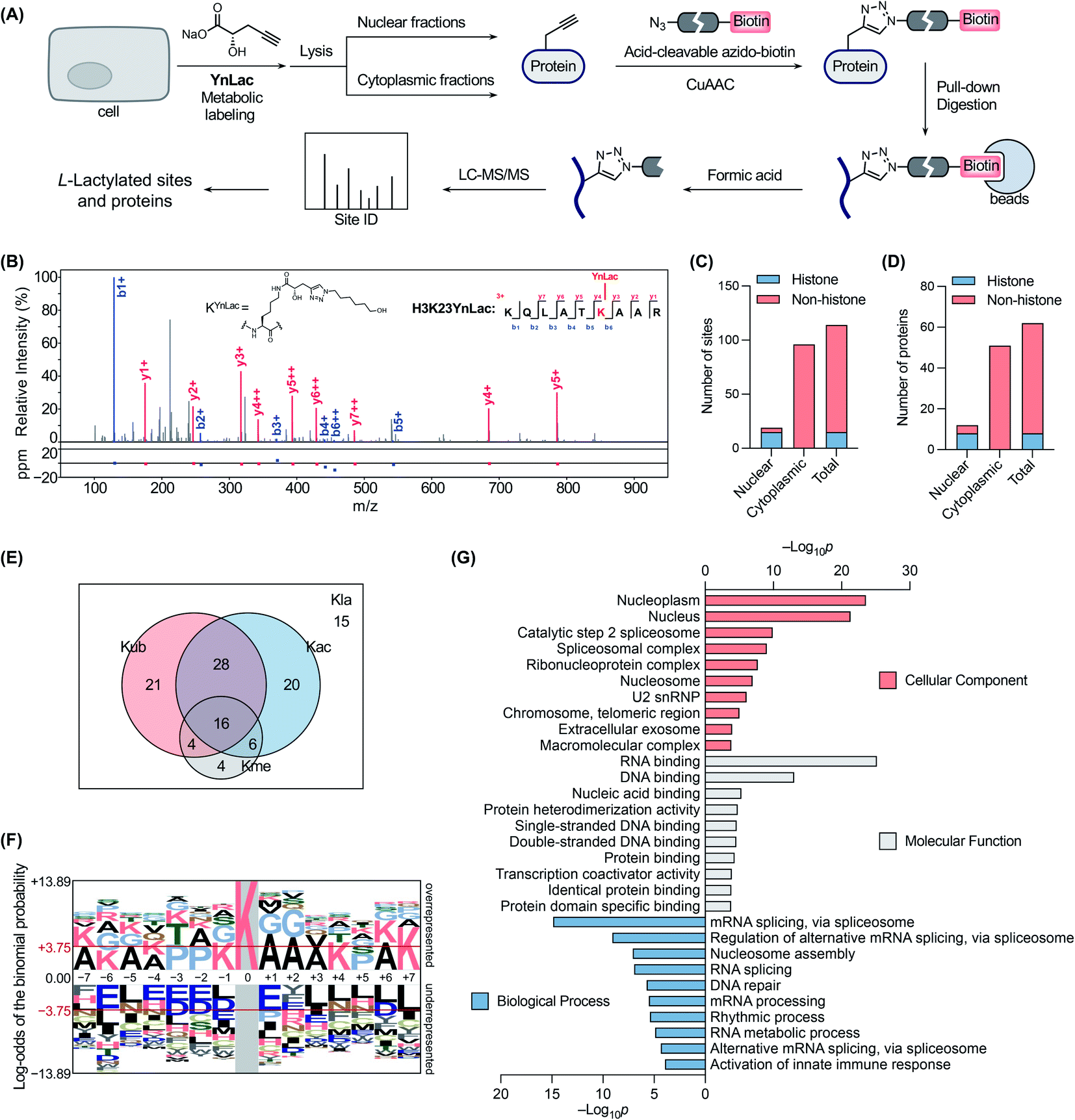 | ||
| Fig. 3 Chemical proteomic profiling of protein lactylation with YnLac. (A) Workflow for the chemical proteomic profiling of lactylation sites using YnLac. HEK293T cells were metabolically labelled with YnLac and the nuclear and cytoplasmic fractions were separated for click conjugation with acid-cleavable azido-biotin and affinity pull-down. After on-bead digestion, the peptides were eluted with acid and analyzed by MS. (B) A representative MS/MS spectrum shows the identification of YnLac modification on the K23 residue of histone H3. (C) Summary of identified lactylation sites in the nuclear and cytoplasmic fractions. (D) Summary of identified lactylated proteins in the nuclear and cytoplasmic fractions. (E) The Venn diagram shows the overlap of identified lactylation sites (Kla) with three abundant types of lysine PTMs, i.e., acetylation (Kac), ubiquitination (Kub), and methylation (Kme). (F) Sequence motif analysis shows the preference of amino acid residues around the identified lactylation sites. The red horizontal lines denote thresholds of p < 0.05. (G) GO term enrichment analysis of identified lactylated proteins. | ||
Encouraged by these results, we continued to apply YnLac to globally identify additional lactylation sites in non-histone proteins. To this end, the cytoplasmic fractions of YnLac-labelled cells were processed as aforementioned for affinity purification and on-bead digestion, followed by acid elution and MS analysis (Fig. 3A). We again processed L-lactate-treated controls in parallel with YnLac-labelled samples for MS analysis. And no YnLac-modified peptides were found in L-lactate-treated controls, suggesting the accuracy and reliability of our proteomic analysis. In particular, 96 lysine residues in 51 non-histone proteins from the cytoplasmic fractions were identified to contain the YnLac modification (Fig. 3C and D), representing novel lactylation sites (Tables S3 and S4 in the ESI†). For example, the modification sites of YnLac in representative non-histone proteins, such as NCL, HMGB1, PARP1, NOLC1, NUCKS1, DNAJC8, and PCNP, were identified by the MS/MS analysis as shown in Fig. S10, S11, S12, S13, S14, S15, and S16, respectively, in the ESI.† Notably, there is only one common modification site, i.e., NPM1K202 (Tables S1 and S3 in the ESI†), that was also retrieved in the nuclear fractions, indicating that both nuclear and cytoplasmic fractions are valuable and complementary sources for lactylation identification. In total, we identified 114 lactylation sites across 62 proteins (Fig. 3C and D), among which 63% contain single lactylation sites (Fig. S17A in the ESI†).
With these newly identified lactylation sites, we searched the PTM databases to determine whether the corresponding lysine residues were also subjected to three abundant types of lysine PTMs, such as acetylation, ubiquitination, and methylation. While most of the lactylation sites bear at least one or more other PTMs, 13% of them are exclusively lactylated (Fig. 3E), suggesting unique functions of lactylation and potential crosstalk between lactylation and other types of lysine PTMs. To explore the possible motifs for lactylation, we analyzed the amino acid sequences around the identified lactylation sites using pLogo.52 We found that positively charged lysines were preferred at many positions (e.g., −7, −6, −5, −4, −1, +4, +6, and +7), while negatively charged glutamic acid and aspartic acid residues were generally underrepresented in the sequences, especially at the −6 and +1 positions (Fig. 3F). In addition, alanine and proline were highly enriched at the +1/+2 and −3/−2 positions, respectively. Interestingly, this consensus sequence showed a high similarity to that recently reported in a computational study for lactylation site prediction.53 For example, lysine residues were enriched at positions −4 and +4 in both consensus sequences. Moreover, small amino acid residues such as alanine and glycine were preferred at positions near the lactylation site (e.g., −2, −1, +1, +2, and +3) in both sequences. As a comparison, sequence analysis surrounding the acetylation sites54 revealed that glutamic acid and aspartic acid residues were enriched at the −1 position, while bulky aromatic residues (i.e., tyrosine and phenylalanine) and aspartic acid were preferred at the +1 position (Fig. S17B in the ESI†). Therefore, the sequence motif for lactylation is distinct from that of acetylation.
To better understand the newly identified lactylated substrate proteins, we performed a series of bioinformatics analyses at the protein level. PTM enrichment analysis showed that the majority of lactylated proteins contain other types of PTMs with a high enrichment in acetylation and ubiquitination (Fig. S18A and Table S5 in the ESI†), consistent with the above analysis on lactylation sites (Fig. 3E). We also carried out a protein class enrichment analysis to describe the classification of lactylated proteins (Fig. S18B and Table S6 in the ESI†). Notably, nucleic acid metabolism proteins, RNA metabolism proteins, histones, chromatin/chromatin-binding proteins, and RNA splicing/processing factors are highly represented. Cellular component analysis using the Gene Ontology (GO) database55 revealed that lactylated proteins are more represented in the nucleoplasm, nucleus, and spliceosome (Fig. 3G and Table S7 in the ESI†). It is notable that lactylated proteins were mainly identified in the cytoplasmic fractions (Fig. 3D), indicating that the majority of them may have dual subcellular localizations. Molecular function analysis demonstrated that many of the lactylated proteins possess RNA, DNA, and protein binding activities (Fig. 3G and Table S7 in the ESI†). In addition, GO analysis on biological processes indicated a significant enrichment of lactylated proteins in processes such as RNA splicing, nucleosome assembly, and DNA repair (Fig. 3G and Table S7 in the ESI†). Furthermore, the Kyoto Encyclopedia of Genes and Genomes (KEGG)56 pathway enrichment analysis demonstrated that lactylated proteins are largely clustered in spliceosome and DNA repair-related pathways like base excision repair (Fig. S18C and Table S8 in the ESI†). Consistently, the REACTOME57 pathway analysis also indicated an enrichment in pathways such as RNA splicing, base excision repair, and chromosome maintenance (Fig. S18C and Table S8 in the ESI†). Lastly, we compared the list of lactylated substrate proteins with that of proteins bearing lysine lactoylation (or D-lactylation), a new type of PTM formed via a nonenzymatic reaction between lysine residues and S-D-lactylglutathione.58 We found that there is a limited overlap between these two sets of proteins (Fig. S19 in the ESI†). While lactylated proteins were predominately annotated to localize in the nucleus and nucleoplasm, D-lactylation is more enriched in cytosolic proteins,58 indicating the differential nature of these two PTM types. Collectively, these results suggest that lactylation mainly occurs on proteins that are associated with diverse nuclear biological processes such as RNA processing/metabolism, chromosome organization, and DNA repair.
Validation of new lactylated substrate proteins
To corroborate the proteomic profiling data, we experimentally validated the identification of lactylation on several non-histone proteins, including NCL, HMGB1, PARP1, NOLC1, NUCKS1, DNAJC8, and PCNP (Table S3 in the ESI†). For this purpose, HEK293T cells expressing individual target proteins were labelled with YnLac. The whole cell lysates were conjugated with azido-biotin (az-biotin) and subjected to pull-down with streptavidin beads, followed by elution and western blot analysis. The results demonstrated that these candidate lactylated proteins were indeed specifically labelled and enriched by YnLac (Fig. 4A and S20 in the ESI†). Moreover, we selected NOLC1 and DNAJC8 to examine the YnLac modification sites, as they were identified with two and one sites, respectively, in the proteomic analysis. The results validated that mutation of the identified YnLac-modified sites on NOLC1 and DNAJC8 decreased their enrichment by YnLac, to different extents, in the pull-down samples (Fig. S21 in the ESI†).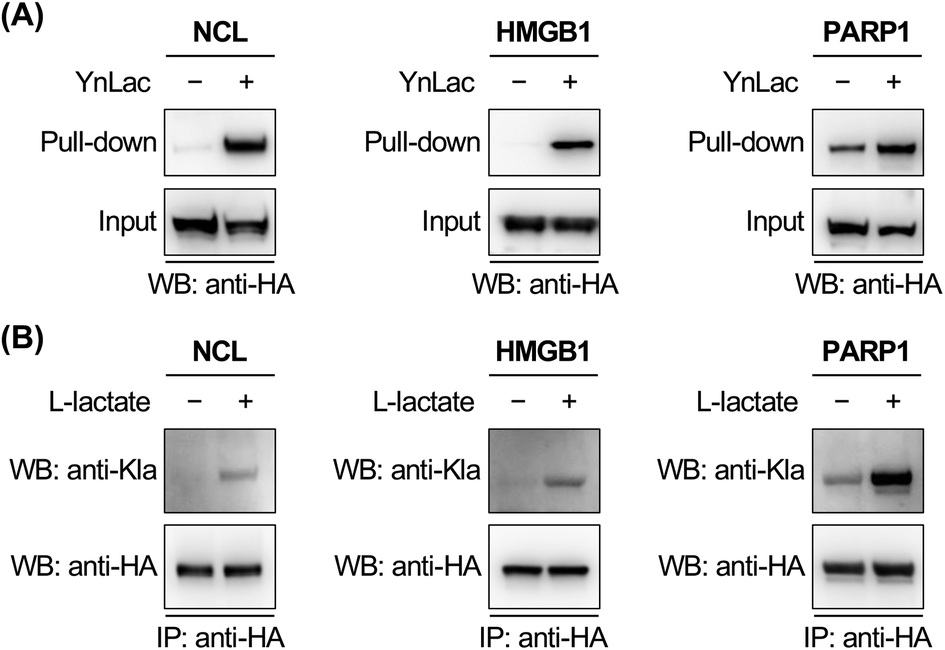 | ||
| Fig. 4 Validation of the identification of selected lactylated non-histone proteins. (A) Verification of the enrichment of newly identified lactylated non-histone proteins. HEK293T cells expressing HA-tagged candidate lactylated proteins were labelled with YnLac (20 mM) for 8 hours and lysed for click conjugation with az-biotin. After pull-down with streptavidin beads, the enriched proteins were eluted for western blot analysis. (B) Verification of the lactylation of newly identified non-histone proteins. HEK293T cells expressing HA-tagged candidate lactylated proteins were treated with or without sodium L-lactate (25 mM) and lysed for immunoprecipitation. The immunoprecipitates were analyzed by western blot using a pan anti-lactyllysine antibody (anti-Kla). Anti-HA blotting shows the protein loading for each lane. | ||
We further confirmed the lactylation on three selected non-histone proteins, i.e., NCL, HMGB1, and PARP1, using a pan anti-lactyllysine antibody. In this regard, HEK293T cells expressing HA-tagged target proteins were treated with or without L-lactate. Western blot analysis of the immunoprecipitated target proteins showed enhanced levels of lactylation on NCL, HMGB1, and PARP1 upon L-lactate treatment (Fig. 4B), verifying that they are bona fide lactylated proteins. Notably, lactylation of mouse HMGB1 in macrophages was very recently reported.59 Using YnLac labelling and streptavidin pull-down, we determined the YnLac modification sites on HMGB1. Although single mutation of the identified K177 and additional mutation of two nearby lysines (i.e., K172 and K173) that are located upstream of the second nuclear localization signal (NLS; residues 178–184)60 of HMGB1 hardly reduced the YnLac labeling (Fig. S22 in the ESI†), triple mutation of the conserved N-terminal K7, K8, and K12 located before the first NLS (residues 27–43)60 attenuated the enrichment by YnLac. Moreover, mutation of all of these six lysines resulted in a significant loss of the YnLac labeling (Fig. S22 in the ESI†), indicating that HMGB1 is likely lactylated on multiple lysine residues upstream of the two NLS domains.
Functional investigation of PARP1 lactylation
Finally, we focused on PARP1 to further explore its lactylation. PARP1, i.e., poly(ADP-ribose) polymerase 1, is an abundant, ubiquitously expressed ADP-ribosyl transferase that catalyzes the attachment of negatively charged ADP-ribose polymers to itself and other substrate proteins.61 The enzyme consists of three functional domains: an N-terminal DNA-binding domain, a central auto-modification domain, and a C-terminal catalytic domain.61 PARP1 is activated by DNA damage and stimulates ADP-ribosylation of multiple proteins including itself in the auto-modification domain and others, resulting in recruitment of DNA repair factors and chromatin remodelling.61 Therefore, PARP1 plays an important role in DNA repair and maintenance of genome stability.To determine the lactylation sites of PARP1, the three YnLac-modified lysine residues, i.e., K498, K505, and K506, identified in our proteomic analysis (Table S3 in the ESI†) were mutated singly or in combination to arginines (Fig. S23A in the ESI†). The mutants were evaluated by immunoprecipitation and western blot analysis using the anti-lactyllysine antibody. While single-residue mutation (i.e., K498R, K505R, and K506R) did not alter lactylation levels of PARP1, the triple mutant (i.e., 3KR; K498/505/506R) provided a decreased lactylation signal (Fig. S23B in the ESI†). However, the residual lactylation level of PARP1-3KR suggested additional lactylation sites in the protein. We noticed that K498, K505, and K506 fall within an intrinsically disordered segment of the PARP1 auto-modification domain (residues 494–524),62 which is also a hyperacetylated region.63 We therefore mutated surrounding lysines in this segment to arginines and constructed a lysine-less 7KR mutant of PARP1 (i.e., K498/505/506/508/518/521/524R) (Fig. S23A in the ESI†). Western blot analysis of immunoprecipitated PARP1 and mutants showed that lactylation of the 7KR mutant was almost abolished (Fig. 5A), suggesting that these lysine residues are the main acceptor sites for lactylation in cells.
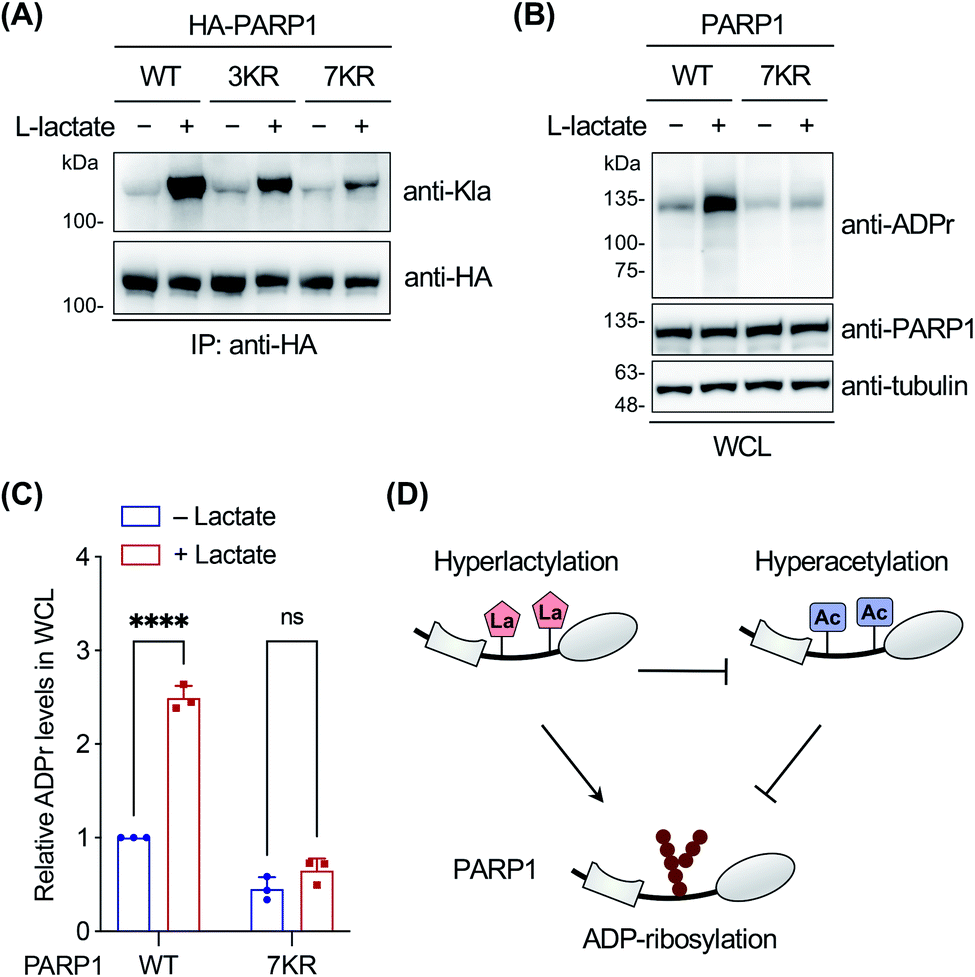 | ||
| Fig. 5 Investigation on PARP1 lactylation. (A) Analysis of lactylation of PARP1, 3KR (K498/505/506R), and 7KR (K498/505/506/508/518/521/524R) mutants. HEK293T cells expressing HA-tagged PARP1 or mutants were treated with or without sodium L-lactate (25 mM) and lysed for immunoprecipitation. The immunoprecipitates were analyzed by western blot using a pan anti-lactyllysine antibody (anti-Kla). Anti-HA blotting shows the protein loading for each lane. (B) Analysis of ADP-ribosylation activity of wild-type (WT) PARP1 and the 7KR (K498/505/506/508/518/521/524R) mutant. PARP1 knockout HEK293T cells were transfected with PARP1 or the 7KR mutant, treated with or without sodium L-lactate (10 mM), and stimulated with 2 mM H2O2 for 10 min. Whole cell lysates (WCL) were prepared for western blot analysis using an anti-ADP-ribosylation antibody (anti-ADPr). (C) Quantification of overall ADP-ribosylation levels relative to anti-PARP1 signals shown in (B). Data are represented as mean ± s.d., n = 3. **** indicates a p-value < 0.0001, and ns indicates a p-value > 0.05, calculated by two-way ANOVA test. (D) Model for differential regulation of PARP1 ADP-ribosylation by hyperlactylation and hyperacetylation in the auto-modification domain. | ||
As the lactylation sites are within the PARP1 auto-modification domain (residues 373–524), we explored the effect of lactylation on PARP1 ADP-ribosylation activity in the oxidative DNA damage response. To eliminate the interference from abundant endogenous PARP1, we utilized a PARP1 knockout HEK293T cell line, which was verified to be deficient in PARP1 expression and oxidative stress-induced ADP-ribosylation (Fig. S24 in the ESI†).62 The cells were transfected to express wild-type PARP1 or the 7KR mutant, treated with or without L-lactate, and then stimulated with H2O2 to induce oxidative DNA damage. Western blot analysis on whole cell lysates showed that L-lactate boosted the overall ADP-ribosylation levels in PARP1-expressing cells, but not in 7KR-expressing cells (Fig. 5B and C). In addition, we confirmed that both the lactylation and auto-ADP-ribosylation levels of immunoprecipitated wild-type PARP1 were increased with L-lactate treatment (Fig. S25 in the ESI†). By contrast, the 7KR mutant was minimally lactylated in PARP1 knockout cells and, more importantly, its auto-modification level was marginally changed upon L-lactate treatment (Fig. S25 in the ESI†). These results demonstrate that L-lactate induces PARP1 lactylation and that lactylated PARP1 exhibits increased overall and auto-ADP-ribosylation activities.
Notably, a hyperacetylation mimic of PARP1, 7KQ (Fig. S23A in the ESI†), bearing lysine-to-glutamine mutations was reported to oppositely display a diminished auto-ADP-ribosylation activity,62 demonstrating the inhibitory effect of hyperacetylation. As hyperlactylation and hyperacetylation occur on the same residues in the auto-modification domain, it is likely that lactylation competes with and thereby suppresses PARP1 acetylation, resulting in recovery of its ADP-ribosylation activity (Fig. 5D). These results together indicate that hyperlactylation and hyperacetylation in the auto-modification domain differentially regulate the ADP-ribosylation activity of PARP1 (Fig. 5D). As the ADP-ribosylation activity of PARP1 contributes to most of its known functions in DNA repair,61 these results also suggest a potential regulatory role of lactylation in the DNA repair process. Further studies remain to be performed to investigate how lactylation modulates and competes with acetylation to regulate the ADP-ribosylation activity of PARP1, as well as to examine the mechanism of crosstalk among these three PTMs.
Conclusions
To summarize, we have developed a bioorthogonal chemical reporter, YnLac, for studying protein lactylation in mammalian cells. YnLac has been thoroughly characterized for general metabolic labelling and efficient fluorescent detection of known lactylated histones as well as uncharacterized lactylated non-histone proteins. Our in-gel fluorescence analyses indicate that protein lactylation is reversible and likely regulated by p300 and the HDAC family. It is also worth noting that further investigations are still needed to identify all regulatory enzymes for lactylation, as chemical inhibition of p300 and HDAC does not completely block YnLac labelling and its turnover, respectively. In combination with chemical proteomics, we show that YnLac directly targets lysine residues and enables the identification of lactylation sites. We further apply YnLac to global proteomic profiling in the mammalian proteome, which identifies many new lactylation sites in non-histone proteins for the first time and thus greatly expands the substrate scope of protein lactylation. To the best of our knowledge, this is the first proteome-wide analysis on the identification of lactylation sites in mammalian cells. Moreover, we experimentally validated the lactylation modifications on several selected non-histone proteins. Furthermore, we demonstrate that hyperlactylation of PARP1 regulates its ADP-ribosylation activity and may contribute to DNA repair. Overall, our study not only provides a powerful and readily accessible chemical tool for the general detection and identification of protein lactylation in different biological contexts, but also suggests novel substrate proteins and regulatory roles of lactylation. The bioorthogonal lactylation chemical reporter described herein should open up a new avenue for the analytical and functional studies of this newly identified PTM in physiology and disease.Data availability
Primary data for the synthesis of YnLac, in-gel fluorescence characterization, proteomic analysis, and bioinformatics analysis are provided in the ESI.† The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE64 partner repository with the dataset identifier PXD033454.Author contributions
Y. Sun performed the in-gel fluorescence, proteomic, and bioinformatics analyses on YnLac. Y. Chen synthesized YnLac and assisted in the in-gel fluorescence analysis. T. Peng conceived and supervised the project, and wrote the manuscript with input from all authors.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
We thank Dr Liyan Zhou for assistance in mass spectrometry analysis. We also thank Prof. Wenbiao Gan at Peking University Shenzhen Graduate School and Peng lab members for helpful discussion. T. P. acknowledges support from Shenzhen Science and Technology Innovation Committee (GXWD20201231165807007-20200814103057002) and Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2019SHIBS0004).Notes and references
- M. Adeva-Andany, M. López-Ojén, R. Funcasta-Calderón, E. Ameneiros-Rodríguez, C. Donapetry-García, M. Vila-Altesor and J. Rodríguez-Seijas, Mitochondrion, 2014, 17, 76–100 CrossRef CAS PubMed.
- J. D. Rabinowitz and S. Enerbäck, Nat. Metab., 2020, 2, 566–571 CrossRef CAS PubMed.
- Z. Chen, M. Liu, L. Li and L. Chen, J. Cell. Physiol., 2018, 233, 2839–2849 CrossRef CAS PubMed.
- P. J. Magistretti and I. Allaman, Nat. Rev. Neurosci., 2018, 19, 235–249 CrossRef CAS PubMed.
- F. Baltazar, J. Afonso, M. Costa and S. Granja, Front. Oncol., 2020, 10, 231 CrossRef PubMed.
- T.-Y. Lee, J. Yeungnam Med. Sci., 2021, 38, 183–193 CrossRef CAS PubMed.
- D. Zhang, Z. Tang, H. Huang, G. Zhou, C. Cui, Y. Weng, W. Liu, S. Kim, S. Lee, M. Perez-Neut, J. Ding, D. Czyz, R. Hu, Z. Ye, M. He, Y. G. Zheng, H. A. Shuman, L. Dai, B. Ren, R. G. Roeder, L. Becker and Y. Zhao, Nature, 2019, 574, 575–580 CrossRef CAS PubMed.
- C. Moreno-Yruela, D. Zhang, W. Wei, M. Bæk, W. Liu, J. Gao, D. Danková, A. L. Nielsen, J. E. Bolding, L. Yang, S. T. Jameson, J. Wong, C. A. Olsen and Y. Zhao, Sci. Adv., 2022, 8, eabi6696 CrossRef CAS PubMed.
- R. A. Irizarry-Caro, M. M. McDaniel, G. R. Overcast, V. G. Jain, T. D. Troutman and C. Pasare, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30628–30638 CrossRef CAS PubMed.
- L. Li, K. Chen, T. Wang, Y. Wu, G. Xing, M. Chen, Z. Hao, C. Zhang, J. Zhang, B. Ma, Z. Liu, H. Yuan, Z. Liu, Q. Long, Y. Zhou, J. Qi, D. Zhao, M. Gao, D. Pei, J. Nie, D. Ye, G. Pan and X. Liu, Nat. Metab., 2020, 2, 882–892 CrossRef CAS PubMed.
- H. Hagihara, H. Shoji, H. Otabi, A. Toyoda, K. Katoh, M. Namihira and T. Miyakawa, Cell Rep., 2021, 37, 109820 CrossRef CAS PubMed.
- J. Yu, P. Chai, M. Xie, S. Ge, J. Ruan, X. Fan and R. Jia, Genome Biol., 2021, 22, 85 CrossRef CAS PubMed.
- J. Xiong, J. He, J. Zhu, J. Pan, W. Liao, H. Ye, H. Wang, Y. Song, Y. Du, B. Cui, M. Xue, W. Zheng, X. Kong, K. Jiang, K. Ding, L. Lai and Q. Wang, Mol. Cell, 2022 DOI:10.1016/j.molcel.2022.02.033.
- R.-Y. Pan, L. He, J. Zhang, X. Liu, Y. Liao, J. Gao, Y. Liao, Y. Yan, Q. Li, X. Zhou, J. Cheng, Q. Xing, F. Guan, J. Zhang, L. Sun and Z. Yuan, Cell Metab., 2022, 34, 634–648 CrossRef CAS PubMed .e636..
- H. Cui, N. Xie, S. Banerjee, J. Ge, D. Jiang, T. Dey, Q. L. Matthews, R.-M. Liu and G. Liu, Am. J. Respir. Cell Mol. Biol., 2021, 64, 115–125 CrossRef CAS PubMed.
- W. Yang, P. Wang, P. Cao, S. Wang, Y. Yang, H. Su and B. Nashun, Epigenet. Chromatin, 2021, 14, 57 CrossRef CAS PubMed.
- Q. Yang, J. Liu, Y. Wang, W. Zhao, W. Wang, J. Cui, J. Yang, Y. Yue, S. Zhang, M. Chu, Q. Lyu, L. Ma, Y. Tang, Y. Hu, K. Miao, H. Zhao, J. Tian and L. An, J. Biol. Chem., 2022, 298, 101456 CrossRef CAS PubMed.
- X. Chu, C. Di, P. Chang, L. Li, Z. Feng, S. Xiao, X. Yan, X. Xu, H. Li, R. Qi, H. Gong, Y. Zhao, F. Xiao and Z. Chang, Front. Immunol., 2022, 12, 786666 CrossRef PubMed.
- L. T. Izzo and K. E. Wellen, Nature, 2019, 574, 492–493 CrossRef CAS PubMed.
- A.-N. Chen, Y. Luo, Y.-H. Yang, J.-T. Fu, X.-M. Geng, J.-P. Shi and J. Yang, Front. Immunol., 2021, 12, 688910 CrossRef CAS PubMed.
- M. Gao, N. Zhang and W. Liang, Front. Microbiol., 2020, 11, 594743 CrossRef PubMed.
- N. Zhang, N. Jiang, L. Yu, T. Guan, X. Sang, Y. Feng, R. Chen and Q. Chen, Front. Cell Dev. Biol., 2021, 9, 719720 CrossRef PubMed.
- X. Meng, J. M. Baine, T. Yan and S. Wang, J. Agric. Food Chem., 2021, 69, 8287–8297 CrossRef CAS PubMed.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- M. Grammel and H. C. Hang, Nat. Chem. Biol., 2013, 9, 475–484 CrossRef CAS PubMed.
- C. G. Parker and M. R. Pratt, Cell, 2020, 180, 605–632 CrossRef CAS PubMed.
- F. Yang and C. Wang, Chem. Commun., 2020, 56, 13506–13519 RSC.
- D. Rabuka, S. C. Hubbard, S. T. Laughlin, S. P. Argade and C. R. Bertozzi, J. Am. Chem. Soc., 2006, 128, 12078–12079 CrossRef CAS PubMed.
- B. Cheng, Q. Tang, C. Zhang and X. Chen, Annu. Rev. Anal. Chem., 2021, 14, 363–387 CrossRef CAS PubMed.
- N. J. Pedowitz and M. R. Pratt, RSC Chem. Biol., 2021, 2, 306–321 RSC.
- Y.-Y. Yang, J. M. Ascano and H. C. Hang, J. Am. Chem. Soc., 2010, 132, 3640–3641 CrossRef CAS PubMed.
- X. Bao, Q. Zhao, T. Yang, Y. M. E. Fung and X. D. Li, Angew. Chem., Int. Ed., 2013, 52, 4883–4886 CrossRef CAS PubMed.
- X. Bao, Y. Xiong, X. Li and X. D. Li, Chem. Sci., 2018, 9, 7797–7801 RSC.
- B. R. Martin and B. F. Cravatt, Nat. Methods, 2009, 6, 135–138 CrossRef CAS PubMed.
- G. Charron, M. M. Zhang, J. S. Yount, J. Wilson, A. S. Raghavan, E. Shamir and H. C. Hang, J. Am. Chem. Soc., 2009, 131, 4967–4975 CrossRef CAS PubMed.
- E. Thinon, R. A. Serwa, M. Broncel, J. A. Brannigan, U. Brassat, M. H. Wright, W. P. Heal, A. J. Wilkinson, D. J. Mann and E. W. Tate, Nat. Commun., 2014, 5, 4919 CrossRef CAS PubMed.
- T. Peng, E. Thinon and H. C. Hang, Curr. Opin. Chem. Biol., 2016, 30, 77–86 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- S. Benson, A. Fernandez, N. D. Barth, F. de Moliner, M. H. Horrocks, C. S. Herrington, J. L. Abad, A. Delgado, L. Kelly, Z. Chang, Y. Feng, M. Nishiura, Y. Hori, K. Kikuchi and M. Vendrell, Angew. Chem., Int. Ed., 2019, 58, 6911–6915 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- W. R. Sinclair, J. H. Shrimp, T. T. Zengeya, R. A. Kulkarni, J. M. Garlick, H. Luecke, A. J. Worth, I. A. Blair, N. W. Snyder and J. L. Meier, Chem. Sci., 2018, 9, 1236–1241 RSC.
- S. Walenta, M. Wetterling, M. Lehrke, G. Schwickert, K. Sundfør, E. K. Rofstad and W. Mueller-Klieser, Cancer Res., 2000, 60, 916–921 CAS.
- D. M. Brizel, T. Schroeder, R. L. Scher, S. Walenta, R. W. Clough, M. W. Dewhirst and W. Mueller-Klieser, Int. J. Radiat. Oncol., Biol., Phys., 2001, 51, 349–353 CrossRef CAS.
- K. Balasubramanyam, R. A. Varier, M. Altaf, V. Swaminathan, N. B. Siddappa, U. Ranga and T. K. Kundu, J. Biol. Chem., 2004, 279, 51163–51171 CrossRef CAS PubMed.
- C. Choudhary, B. T. Weinert, Y. Nishida, E. Verdin and M. Mann, Nat. Rev. Mol. Cell Biol., 2014, 15, 536–550 CrossRef CAS PubMed.
- B. R. Sabari, D. Zhang, C. D. Allis and Y. Zhao, Nat. Rev. Mol. Cell Biol., 2017, 18, 90–101 CrossRef CAS PubMed.
- M. M. Zhang, L. K. Tsou, G. Charron, A. S. Raghavan and H. C. Hang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8627–8632 CrossRef CAS PubMed.
- D. M. Vigushin, S. Ali, P. E. Pace, N. Mirsaidi, K. Ito, I. Adcock and R. C. Coombes, Clin. Cancer Res., 2001, 7, 971–976 CAS.
- A. A. Sauve, C. Wolberger, V. L. Schramm and J. D. Boeke, Annu. Rev. Biochem., 2006, 75, 435–465 CrossRef CAS PubMed.
- J. Szychowski, A. Mahdavi, J. J. L. Hodas, J. D. Bagert, J. T. Ngo, P. Landgraf, D. C. Dieterich, E. M. Schuman and D. A. Tirrell, J. Am. Chem. Soc., 2010, 132, 18351–18360 CrossRef CAS PubMed.
- J. Cox and M. Mann, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef CAS PubMed.
- J. P. O'Shea, M. F. Chou, S. A. Quader, J. K. Ryan, G. M. Church and D. Schwartz, Nat. Methods, 2013, 10, 1211–1212 CrossRef PubMed.
- P. Jiang, W. Ning, Y. Shi, C. Liu, S. Mo, H. Zhou, K. Liu and Y. Guo, Comput. Struct. Biotechnol. J., 2021, 19, 4497–4509 CrossRef CAS PubMed.
- H. Huang, S. Tang, M. Ji, Z. Tang, M. Shimada, X. Liu, S. Qi, J. W. Locasale, R. G. Roeder, Y. Zhao and X. Li, Mol. Cell, 2018, 70, 663–678 CrossRef CAS PubMed .e666..
- T. G. O. Consortium, Nucleic Acids Res., 2020, 49, D325–D334 CrossRef PubMed.
- M. Kanehisa and S. Goto, Nucleic Acids Res., 2000, 28, 27–30 CrossRef CAS PubMed.
- M. Gillespie, B. Jassal, R. Stephan, M. Milacic, K. Rothfels, A. Senff-Ribeiro, J. Griss, C. Sevilla, L. Matthews, C. Gong, C. Deng, T. Varusai, E. Ragueneau, Y. Haider, B. May, V. Shamovsky, J. Weiser, T. Brunson, N. Sanati, L. Beckman, X. Shao, A. Fabregat, K. Sidiropoulos, J. Murillo, G. Viteri, J. Cook, S. Shorser, G. Bader, E. Demir, C. Sander, R. Haw, G. Wu, L. Stein, H. Hermjakob and P. D'Eustachio, Nucleic Acids Res., 2021, 50, D687–D692 CrossRef PubMed.
- D. O. Gaffney, E. Q. Jennings, C. C. Anderson, J. O. Marentette, T. Shi, A.-M. Schou Oxvig, M. D. Streeter, M. Johannsen, D. A. Spiegel, E. Chapman, J. R. Roede and J. J. Galligan, Cell Chem. Biol., 2020, 27, 206–213 CrossRef CAS PubMed .e206..
- K. Yang, M. Fan, X. Wang, J. Xu, Y. Wang, F. Tu, P. S. Gill, T. Ha, L. Liu, D. L. Williams and C. Li, Cell Death Differ., 2022, 29, 133–146 CrossRef CAS PubMed.
- T. Bonaldi, F. Talamo, P. Scaffidi, D. Ferrera, A. Porto, A. Bachi, A. Rubartelli, A. Agresti and M. E. Bianchi, EMBO J., 2003, 22, 5551–5560 CrossRef CAS PubMed.
- A. Ray Chaudhuri and A. Nussenzweig, Nat. Rev. Mol. Cell Biol., 2017, 18, 610–621 CrossRef CAS PubMed.
- G. Liszczak, K. L. Diehl, G. P. Dann and T. W. Muir, Nat. Chem. Biol., 2018, 14, 837–840 CrossRef CAS PubMed.
- P. O. Hassa, S. S. Haenni, C. Buerki, N. I. Meier, W. S. Lane, H. Owen, M. Gersbach, R. Imhof and M. O. Hottiger, J. Biol. Chem., 2005, 280, 40450–40464 CrossRef CAS PubMed.
- Y. Perez-Riverol, J. Bai, C. Bandla, D. García-Seisdedos, S. Hewapathirana, S. Kamatchinathan, D. J. Kundu, A. Prakash, A. Frericks-Zipper, M. Eisenacher, M. Walzer, S. Wang, A. Brazma and J. A. Vizcaíno, Nucleic Acids Res., 2021, 50, D543–D552 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures and tables, and detailed experimental methods. See https://doi.org/10.1039/d2sc00918h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |