
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn unprecedented azobenzene-based organic single-component ferroelectric†
Hang
Peng‡
,
Jun-Chao
Qi‡
,
Xian-Jiang
Song
,
Ren-Gen
Xiong
 * and
Wei-Qiang
Liao
*
* and
Wei-Qiang
Liao
*
Ordered Matter Science Research Center, Nanchang University, 330031, P. R. China. E-mail: liaowq@ncu.edu.cn; xiongrg@seu.edu.cn
First published on 5th April 2022
Abstract
Organic single-component ferroelectrics, as an important class of metal-free ferroelectrics, are highly desirable because of their easy processing, mechanical flexibility, and biocompatibility. However, although nearly 50 years have passed since the discovery of photochromism in azobenzene-doped cholesteric liquid crystals, ferroelectricity has never been found in azobenzene-based crystals. Here, we use an amino group to substitute a fluorine atom of 2,2′,4,4′,6,6′-hexafluoroazobenzene, which successfully introduces ferroelectricity into 2-amino-2′,4,4′,6,6′-pentafluoroazobenzene (APFA). APFA shows an extremely high Curie temperature (Tc) of 443 K, which is outstanding among single-component ferroelectrics. It also exhibits an indirect optical band gap of 2.27 eV as well as photoisomerization behavior between the trans-form and the cis-form triggered by pedal motion. To our knowledge, APFA is the first azobenzene-based ferroelectric crystal. This work opens an avenue to design excellent single-component ferroelectrics and will inspire the exploration of azobenzene-based ferroelectrics for promising applications in biofriendly ferroelectric devices.
Introduction
Over the past century, ferroelectrics, as an indispensable class of functional materials, have been endowed with abundant physical properties including ferroelectricity, pyroelectricity, piezoelectricity and so on.1–3 Since ferroelectricity was discovered in Rochelle salt by Valasek in 1920, they have become key components in manifold areas as ferroelectric random access memory (FeRAM), capacitors, sensors, infrared detectors, semiconductors, photonic switches, actuators, and surface acoustic wave devices.4–6 Because of their unique superiority such as light weight, good degradability, mechanical flexibility, homochirality, and structural tunability, molecular ferroelectrics have continued to generate new functional materials and intriguing phenomena.7,8 Among them, organic single-component ferroelectrics have attracted wide attention by virtue of their satisfactory biocompatibility, low acoustic impedance (matching with human bodies), and environmental friendliness.9,10 Specifically, croconic acid, an ingredient of black dyes, is a hydrogen-bonded single-component ferroelectric with the absence of phase transition, possessing a large spontaneous polarization (Ps) of 20 μC cm−2 and a wide ferroelectric temperature window.9 Despite extensive interest, however, organic single-component ferroelectrics have rarely been reported until now.Azobenzene and its derivatives, as a class of classical single-component organic photochromic compounds, are promising for optical memories, coloring fiber, organic photoconductors, printing systems, and liquid crystal displays.11–13 Their photochromic phenomenon was first discovered in an azobenzene-doped cholesteric liquid crystal by Sackmann et al. in the early 1970s.14 Photochromism is defined as the reversible structural transformation of a chemical species between two isomers showing different absorption spectra induced by photoirradiation.15–19 Given this, could photochromic molecules be allowed to enrich the structural diversity of organic ferroelectrics under an elaborate design? If possible, the combination of photoisomerization and ferroelectricity will bring about extraordinary scientific and technical significance. Although azobenzene and its derivatives are desirable for the construction of organic photoswitchable ferroelectrics, azobenzene-based ferroelectric crystals have not been found to date.
In this work, we successfully designed a single-component azobenzene-based crystal, 2-amino-2′,4,4′,6,6′-pentafluoroazobenzene (APFA), which crystallizes in the monoclinic polar space group P21 at room temperature. Based on the original crystal 2,2′,4,4′,6,6′-hexafluoroazobenzene (HFA) with the centrosymmetric space group of P21/n at room temperature, we use an amino group to substitute the F atom at the 2-position of HFA (Fig. 1a). The F atom and the amino group have similar steric parameters but totally different electronegativity, which may lead to a drastic change in their molecular symmetry and thereby import a molecular dipole moment as well as further ferroelectricity. The ferroelectricity of APFA is confirmed by the piezoresponse force microscopy (PFM) technique and polarization-electric field (P–E) hysteresis loop measurement. It is worth noting that APFA exhibits an extremely high Tc of 443 K, which is higher than the Tc of most organic single-component ferroelectrics. Meanwhile, solid-state ultraviolet-visible (UV-vis) absorption spectra indicate that APFA shows an indirect optical band gap of 2.27 eV and undergoes a photo-induced structural phase transition between the trans- and cis-conformation through pedal motion. To the best of our knowledge, APFA is the first single-component azobenzene-based ferroelectric crystal. This finding inspires the exploration of azobenzene-based organic ferroelectric crystals and holds promise for future photo-controllable smart materials and biofriendly optoelectronic devices such as wearable medical devices.
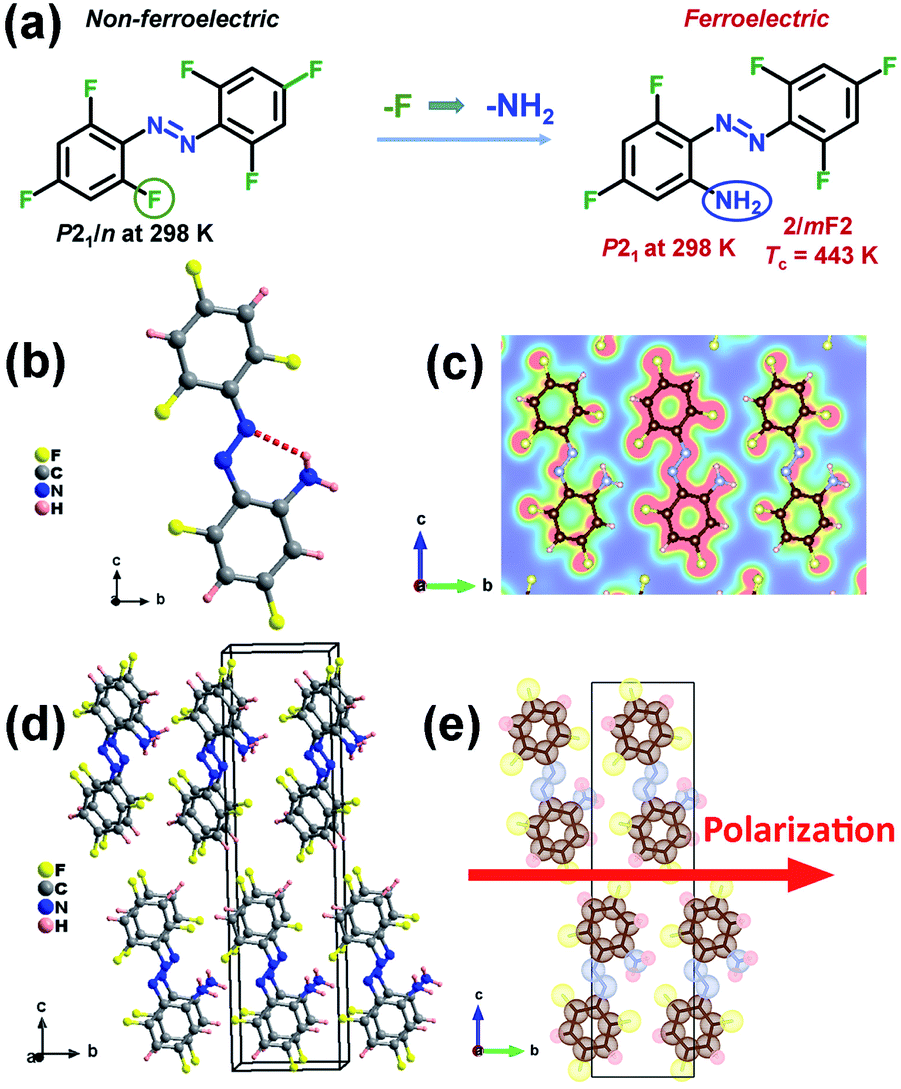 | ||
| Fig. 1 (a) Design strategy of ferroelectric APFA by replacing the F atom with an amino group. (b) Crystal structure of APFA. The dashed line denotes the intramolecular N–H–N hydrogen bonding interaction. (c) Electron density distribution on the selected plane built with the three N atoms of APFA at 298 K. (d) Packing views of the structure of APFA at 298 K. (e) Packing views of the structure of APFA along the a axis represented by the dot-surface model at 298 K. The direction of spontaneous polarization in the ferroelectric phase is shown with a red arrow. | ||
Results and discussion
The synthesis of compound APFA is shown in Fig. S1.†20 The precursor compound HFA was synthesized according to literature procedures.21 The phase purity of APFA was confirmed by powder X-ray diffraction (PXRD) (Fig. S2†) and infrared (IR) analysis (Fig. S3†). The IR characteristic peaks of the C–F, –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– (trans), and C–N bonds of APFA are shown at around 1333, 1405, and 1049 cm−1, respectively.22 The crystal structure determination reveals that both HFA and APFA adopt the trans-form at room temperature (Fig. S4† and 1b). HFA crystallizes in the centrosymmetric nonpolar space group P21/n at room temperature, which does not meet the necessary conditions of becoming a ferroelectric (Table S1†). Meanwhile, it does not undergo a thermodynamic structural phase transition (Fig. S5†). Thus, based on the chemical design strategy of lowering molecular symmetry for inducing ferroelectricity, we made a subtle modification, namely, a substitution of an amino group for the F atom at the 2-position on the compound HFA to achieve a lower-symmetry ferroelectric phase by introducing a molecular dipole moment. The resultant APFA molecule is a push–pull molecule, similar to pseudostilbene-type molecules, which contains an electron-acceptor (A) component and an electron-donor (D) unit.23 They connected with each other by a π-conjugated linkage. Such a structure (D–π–A) enables good transport of charge from the D to the A part, showing a high value of dipole moment.23 Crystal APFA adopts a polar space group P21 at 298 K (Table S1†), and its bond lengths and angles are listed in Tables S2 and S3.† The structural asymmetric unit is composed of one APFA molecule. It should also be noted that the introduction of an amino group supports the formation of the intramolecular N–H⋯N hydrogen bonding interaction, whose bond length and angle are 2.696(6) Å and 121.8°, respectively (Fig. 1b and Table S4†). The electron density distribution (slice) pictures show that these intramolecular hydrogen bonding interactions make the trans-isomer more stable (Fig. 1c). Due to the similar steric parameters of the F atom and amino group, HFA and APFA present similar packing structures (Fig. S6† and 1d). It has been found that both the HFA and APFA molecules were parallelly aligned along the a-direction through the typical π–π stacking once found in some biological macromolecular systems (Fig. S7†), which contributes greatly to the stabilization of the neighbor base pair stacking in duplex DNA and RNA, the recognition of protein–ligand interactions, etc.24,25 In the structure of HFA, the π–π distance between neighboring molecules is 4.487 Å (Fig. S7†). After substitution with an amino group, in the structure of APFA, the π–π distance between neighboring molecules decreases to 3.767 Å, indicating the enhancement of π–π interactions in APFA (Fig. S7†). This π–π stacking not only aids the self-assembly process but significantly ensures the oriented alignment of APFA molecules. This contributes to a striking structural feature that all of the C–N bonds in APFA align along the b-axis direction, resulting in spontaneous polarization (Fig. 1e). As shown in Fig. 1e, the iso-surfaces of each atom of the APFA crystal are shown as dotted surfaces, which is useful for understanding how atoms are combined with each other in a molecule. However, the molecules in the adjacent columns present a cross arrangement from the structural perspective view along the c-axis (Fig. S8†). It is noted that in the azobenzene-based host–guest compound [(4-phenylazoanilinium)([18]crown-6)]BF4,26 although π–π interactions between two adjacent 4-phenylazoanilinium cations exist, the molecules show no parallel stacking along one direction through the π–π interactions, different from the case of APFA, which may be because the existence of host molecules hinders the stacking of guest phenylazoanilinium cations. This host–guest compound was found to adopt the centrosymmetric pace group P21/n at room temperature, showing no ferroelectricity.26
N– (trans), and C–N bonds of APFA are shown at around 1333, 1405, and 1049 cm−1, respectively.22 The crystal structure determination reveals that both HFA and APFA adopt the trans-form at room temperature (Fig. S4† and 1b). HFA crystallizes in the centrosymmetric nonpolar space group P21/n at room temperature, which does not meet the necessary conditions of becoming a ferroelectric (Table S1†). Meanwhile, it does not undergo a thermodynamic structural phase transition (Fig. S5†). Thus, based on the chemical design strategy of lowering molecular symmetry for inducing ferroelectricity, we made a subtle modification, namely, a substitution of an amino group for the F atom at the 2-position on the compound HFA to achieve a lower-symmetry ferroelectric phase by introducing a molecular dipole moment. The resultant APFA molecule is a push–pull molecule, similar to pseudostilbene-type molecules, which contains an electron-acceptor (A) component and an electron-donor (D) unit.23 They connected with each other by a π-conjugated linkage. Such a structure (D–π–A) enables good transport of charge from the D to the A part, showing a high value of dipole moment.23 Crystal APFA adopts a polar space group P21 at 298 K (Table S1†), and its bond lengths and angles are listed in Tables S2 and S3.† The structural asymmetric unit is composed of one APFA molecule. It should also be noted that the introduction of an amino group supports the formation of the intramolecular N–H⋯N hydrogen bonding interaction, whose bond length and angle are 2.696(6) Å and 121.8°, respectively (Fig. 1b and Table S4†). The electron density distribution (slice) pictures show that these intramolecular hydrogen bonding interactions make the trans-isomer more stable (Fig. 1c). Due to the similar steric parameters of the F atom and amino group, HFA and APFA present similar packing structures (Fig. S6† and 1d). It has been found that both the HFA and APFA molecules were parallelly aligned along the a-direction through the typical π–π stacking once found in some biological macromolecular systems (Fig. S7†), which contributes greatly to the stabilization of the neighbor base pair stacking in duplex DNA and RNA, the recognition of protein–ligand interactions, etc.24,25 In the structure of HFA, the π–π distance between neighboring molecules is 4.487 Å (Fig. S7†). After substitution with an amino group, in the structure of APFA, the π–π distance between neighboring molecules decreases to 3.767 Å, indicating the enhancement of π–π interactions in APFA (Fig. S7†). This π–π stacking not only aids the self-assembly process but significantly ensures the oriented alignment of APFA molecules. This contributes to a striking structural feature that all of the C–N bonds in APFA align along the b-axis direction, resulting in spontaneous polarization (Fig. 1e). As shown in Fig. 1e, the iso-surfaces of each atom of the APFA crystal are shown as dotted surfaces, which is useful for understanding how atoms are combined with each other in a molecule. However, the molecules in the adjacent columns present a cross arrangement from the structural perspective view along the c-axis (Fig. S8†). It is noted that in the azobenzene-based host–guest compound [(4-phenylazoanilinium)([18]crown-6)]BF4,26 although π–π interactions between two adjacent 4-phenylazoanilinium cations exist, the molecules show no parallel stacking along one direction through the π–π interactions, different from the case of APFA, which may be because the existence of host molecules hinders the stacking of guest phenylazoanilinium cations. This host–guest compound was found to adopt the centrosymmetric pace group P21/n at room temperature, showing no ferroelectricity.26
To investigate the phase transition properties of APFA, we firstly carried out differential scanning calorimetry (DSC) measurement. In the DSC curves of APFA, one pair of large heat anomalies in the heating–cooling run with a hysteresis of approximately 13 K can be clearly observed, indicating a reversible first-order phase transition at a Curie temperature (Tc) of 443 K (Fig. 2a). Such a high Tc is beyond that of most organic single-component molecular ferroelectrics such as thiourea (169 K),8 2,2,6,6-tetramethyl-1-piperidinyloxy (287 K),8 trichloroacetamide (355 K),8 cyclohexan-1,1′-diacetic acid (397 K),8 (R)-3-quinuclidinol (400 K),10 (−)-camphanic acid (414 K),27 and R-10-camphorsulfonylimine (429 K),28 and is even greater than that of the inorganic ferroelectric BaTiO3 (393 K).29 This high Tc of APFA could be mainly attributed to the intramolecular hydrogen bonding interaction as well as the interlaminar π–π stacking interaction. From the polar ferroelectric phase to the centrosymmetric paraelectric phase (see below), the APFA molecules show orientational change to make the spontaneous polarization in the ferroelectric phase disappear in the paraelectric phase. During the ferroelectric-to-paraelectric phase transition, the presence of intramolecular N–H⋯N hydrogen bonding interaction and interlaminar π–π stacking interaction in the structure increases the potential energy barrier for triggering the orientational change of molecules, which would greatly contribute to high Tc in APFA, as found in the high-Tc molecular ferroelectric [3-oxo-quinuclidinium]ClO4 with intermolecular N–H⋯OC hydrogen-bonding interactions.30 The entropy change (ΔS) in the heating process is about 56.98 J K−1 mol−1. It should be noted that APFA having a large entropy change of 56.98 J K−1 mol−1 and a thermal hysteresis of 13 K is a promising candidate for electrocaloric applications.31 The real part (ε′) of the complex dielectric constant of APFA displays a step-like dielectric anomaly near Tc (Fig. 2b), which further confirms the phase transition. The thermogravimetric analysis (TGA) measurement of APFA indicates that APFA shows good thermal stability up to 499 K (Fig. S9†), which is higher than the Tc of 443 K.
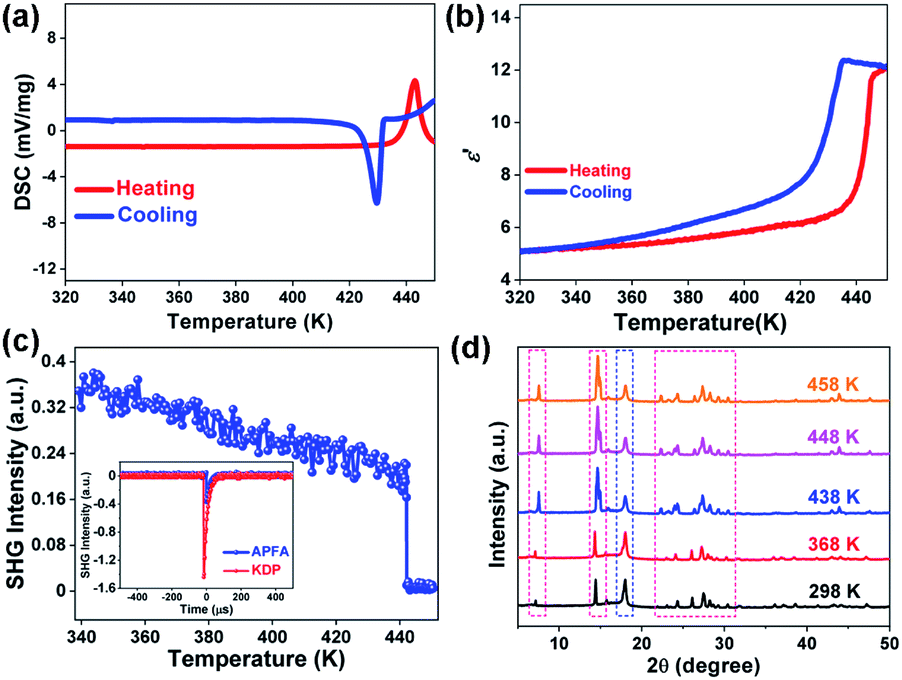 | ||
| Fig. 2 Phase transition of APFA. (a) DSC curve. (b) Temperature-dependent ε′ at 1 MHz. (c) Temperature-dependent SHG response. Inset: comparison of the SHG intensity of APFA and KDP. (d) Variable-temperature PXRD patterns. | ||
Ferroelectric phase transitions are always accompanied by symmetry breaking. As a second-order nonlinear optical effect, the SHG effect could be used to detect non-centrosymmetric crystals, because SHG response is only allowed in crystals that are devoid of inversion symmetry.32,33 APFA shows a clear SHG signal at room temperature with intensity being about 1/4 of that of the KDP standard, in accordance with the non-centrosymmetric polar P21 space group (the inset of Fig. 2c). In the vicinity of Tc, the SHG intensity displays a sharp decrease, and then keeps an inactive state in the high-temperature phase (HTP, above Tc), indicating that APFA should crystallize in a centrosymmetric space group in HTP (Fig. 2c).
Due to the severe sublimation of crystal APFA, we failed to obtain its crystal structure in the paraelectric phase. To end this, we carried out variable-temperature PXRD measurement on APFA from 298 to 458 K. We used a high temperature resistant polymeric perfluoroalkoxy film covered on the sample tank in order to avoid the sublimation of APFA. As plotted in Fig. S10,† the PXRD pattern of this polymeric film was obtained at 298 K with an obvious diffraction peak located at 18°. Thus, we obtained the PXRD patterns of the APFA sample with the polymeric film covered or uncovered (Fig. S10†). The remarkably consistent PXRD patterns under these two conditions suggest that the usage of this polymeric film will not influence the variable-temperature PXRD results of APFA. Upon heating from 298 to 368 K, as recorded in Fig. 2d, the PXRD patterns show no obvious change. With the temperature increasing above Tc, some of the diffraction peaks observed in the ferroelectric phase were marked with red dotted rectangles split or merged in the HTP. Through the Pawley refinements of the PXRD data at 458 K (Fig. S11†), a monoclinic crystal system was suggested. Considering the absence of the SHG signal in the HTP, we inferred that APFA adopts a centrosymmetric monoclinic point group 2/m above Tc. Thus, the ferroelectric phase transition of APFA could be described as 2/mF2 according to the Aizu rule, accompanied by the loss of macroscopic symmetry elements from four (E, C2, i, and σh) in the paraelectric point group 2/m (C2h) to two (E and C2) in ferroelectric 2 (C2).
Because of the possible ferroelectric phase transition with an Aizu notation of 2/mF2, APFA may possess the ferroelectricity. The PFM technique can provide non-destructive visualization of the statics and dynamics of ferroelectric domains as well as the manipulation of the domain switching process at the nanometer scale, and it has long been regarded as a powerful tool to characterize ferroelectric materials.34–37 Each PFM image is composed of amplitude and phase parameters, providing information about the value of the piezoelectric coefficient (proportional to the local polarization) and the orientation of the domain polarization, respectively. To comprehensively investigate the ferroelectric properties for APFA, we employed the PFM technique to study the ferroelectric domain structures and the local domain switching behavior on its single-crystalline thin film. Firstly, we observed the striking 180° phase contrast and the 180° domain wall in the vertical PFM image on the single-crystalline thin film of APFA without any thermal treatment, which has no obvious correlation between the piezoelectric signals and the topography (Fig. 3a–c). This provides direct proof for the existence of ferroelectric domains, corresponding to a uniaxial 2/mF2-type molecular ferroelectric.
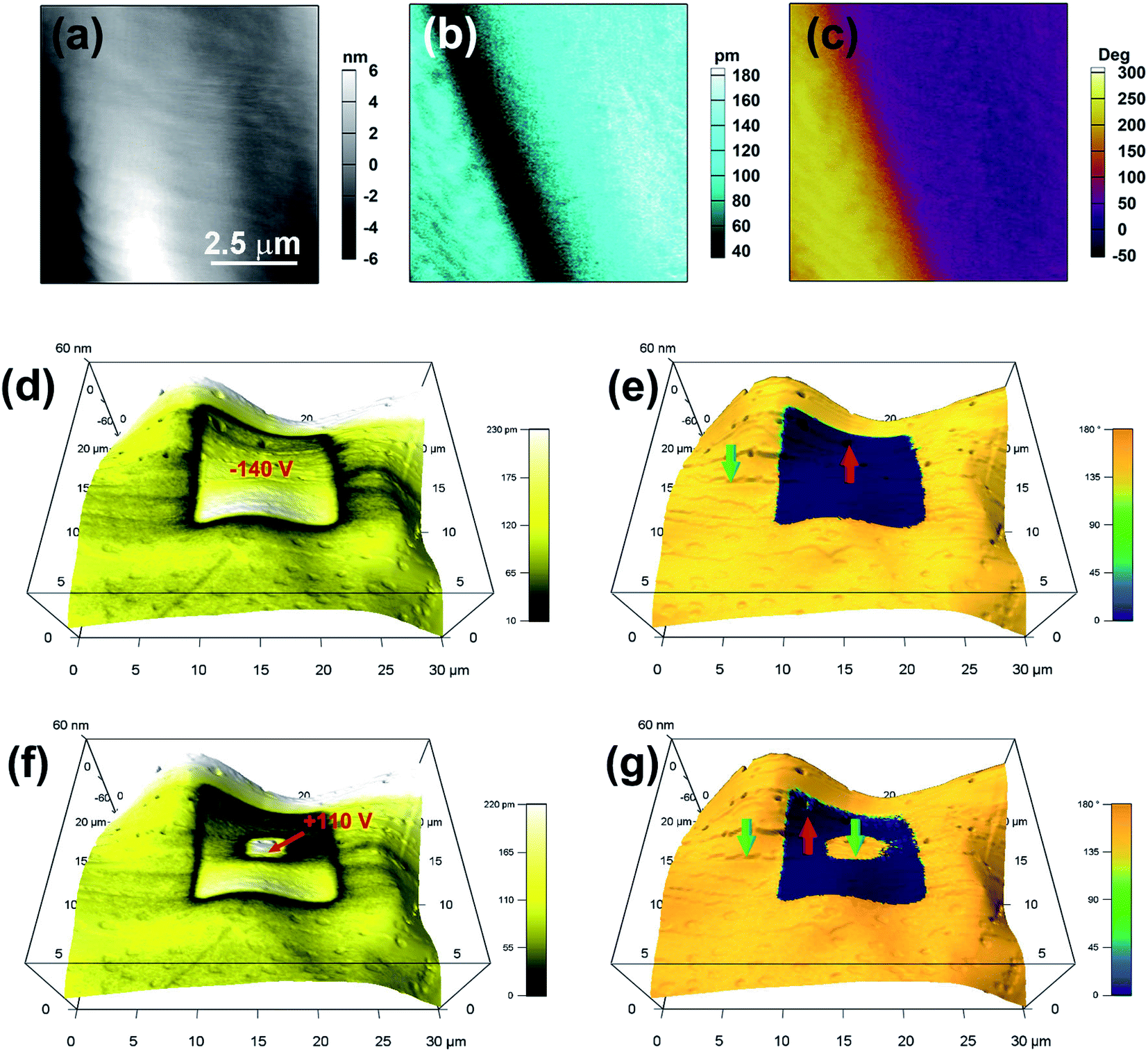 | ||
| Fig. 3 Domain switching for the APFA thin film. Topography images (a), PFM amplitude images (b), and PFM phase images (c) of an 8 × 8 μm2 region. Vertical PFM amplitude (d) and (f) and phase (e) and (g) images superimposed on the topographic image, which were recorded after writing a square area with a voltage of −140 V (d) and (e) followed by a smaller central square with a voltage of +110 V (f) and (g) using a biased conductive tip. The blue and yellow regions in the phase images indicate the regions with out-of-plane polarization oriented upward and downward, respectively. | ||
The salient feature of ferroelectric materials is the possession of spontaneous polarization that can be reversed via an external electric field. Here, the ferroelectric characteristics of APFA were investigated at the nanoscale by PFM in its thin film sample with a thickness of about 300 nm. In PFM, the switching of the polarization can be realized by using an electrically biased tip that scans over a selected area and visualized it by the subsequent PFM imaging. Electrical writing was performed by scanning a square region with a tip voltage of −140 V, followed by vertical PFM imaging. Fig. 3d and e show the resulting domain pattern: the electrically poled area has been fully polarized upward while downward polarization outside this area remains intact. Domain walls appear as narrow dark lines with a typically weak signal that can be discerned in the amplitude image. Fig. 3f and g further show that the poled area can be switched back simply by the application of an opposite voltage of +110 V. This robust microscopic ferroelectricity demonstrated by PFM is an inherent feature of APFA. Besides, we also applied PFM switching spectroscopy measurements for the thin film of APFA. The amplitude loops show a distinct hysteresis and a butterfly shape, while the phase loops present two polarization states with a difference of 180°, which suggests that polarization reversal occurs locally (Fig. S12†). The PFM results undoubtedly provide solid evidence for the switchable polarization of APFA, which is an intrinsic characteristic of ferroelectrics.
For further confirmation, we also investigated macroscopic ferroelectric polarization reversal through P–E hysteresis loop measurement on a single-crystalline thin film of APFA at 298 K (Fig. 4a). Using the double-wave method, a typical current density–electric filed (J–E) curve with two opposite peaks is observed, corresponding to two stable states with opposite polarity (Fig. 4a).27 Then, by integrating the polarization switching current based on the J–E curve, a well-shaped P–E hysteresis loop can be obtained, providing convincing evidence for the ferroelectricity of APFA (Fig. 4a). Specifically, the measured saturated polarization (Ps) value of APFA is about 1.83 μC cm−2 on its thin-film form. To estimate the ferroelectric polarization of the crystal APFA, we first calculated the vector sum of the molecular dipole moments in the unit cell. The dipole moment of APFA is 2.6421 D, and the direction is depicted with a blue arrow (Fig. S13†). Considering the arrangement and dipole orientation of the molecules in the unit cell, we could estimate the total polarization value to be about 3.09 μC cm−2 along the b axis, in good accordance with the symmetry requirement of the space group P21. Furthermore, we also employed the Berry phase method to evaluate this ferroelectric polarization of 4.98 μC cm−2 along the b axis. The Berry phase method provides a more accurate value because the non-negligible intermolecular interactions and stacking effects from the former are taken into consideration. The measured Ps value of APFA is somewhat smaller than that of the calculated one, which is reasonable because of the uncertainty of the polarization direction on the single-crystalline thin film of APFA. The possible ferroelectric mechanism of APFA could be ascribed to the flipping motion of the APFA molecules. As shown in Fig. S13 and S14,† the ferroelectric polarization of APFA originates from the directional alignment of APFA molecular dipoles, resulting in polarization along the crystallographic b-axis. If the ferroelectric polarization flips under an applied electric field, all molecular dipoles also need to be reoriented under the electric field. The molecular dipoles of APFA are mainly derived from polar carbon–fluorine and carbon–nitrogen bonds (Fig. S13†). The inversion of a single molecular dipole can be achieved by the in-plane rotation of the benzene ring involving the amino group and the pedal motion of the nitrogen–nitrogen double bond. During the reversal of the dipole, another benzene ring involving three fluorine atoms can act as a stator, because the high symmetry of this part does not contribute to the total dipole reversal.
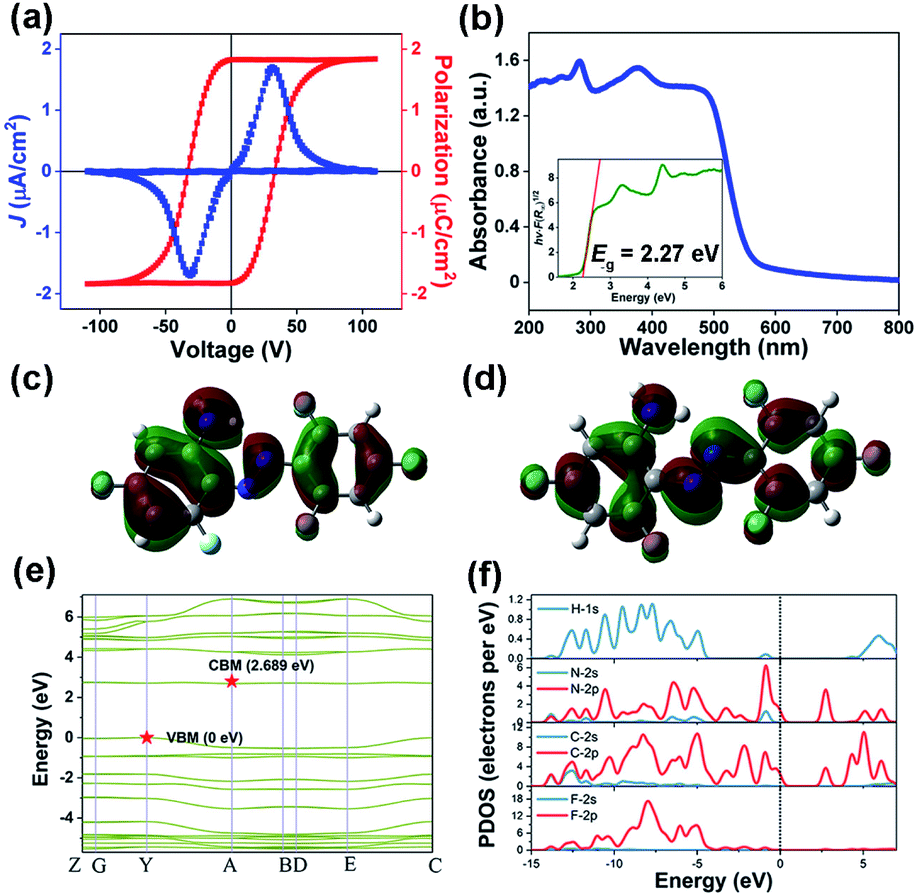 | ||
| Fig. 4 (a) P–E hysteresis loops of the APFA thin film via a double-wave method at room temperature. (b) UV-vis absorption spectra of APFA. Inset: the Tauc plot for APFA. (c) HOMO and (d) LUMO of the APFA molecule. (e) The calculated band structure of APFA and (f) the corresponding PDOS. | ||
Azobenzene and its derivatives generally possess the ability of configuration transition between trans- and cis-configuration under light irradiation or heating. Such a photoisomerization behaviour is an important property for azobenzene-based materials. It was found that the optical properties of azobenzene materials strongly depend on the type of substitution in the azobenzene moiety.38 In order to investigate the optical properties of APFA, UV-vis absorption spectra were recorded in the solid state at room temperature. As shown in Fig. 4b, the UV-vis absorption spectrum of APFA without any treatment displays an intense absorption at the band edge onset of 557 nm, corresponding to an energy for absorption onset of 2.226 eV. According to the Tauc equation, the optical band gap of APFA is determined to be 2.27 eV (Fig. 4b, inset). There are two strong absorption bands at approximately 466 nm and 282 nm, which can be attributed to the n → π* and the π → π* transition of APFA in its trans-configuration, respectively. Notably, after the irradiation of 405 nm for 60 s on the powder sample of APFA, a slight decrease and an increase in the intensity of the π → π* and n → π* transition bands occurred respectively, indicating the occurrence of the transforming tendency from the trans- to cis-isomers (Fig. S15a†).39 Furthermore, under the irradiation of 450 nm for 60 s, the cis-isomers could be transformed into the initial trans-form, indicating the reversibility of photoisomerization behavior (Fig. S15b†). Meanwhile, we recorded the solid-state UV-vis adsorption spectra of APFA before and after open-air exposure under sunlight from 1 h to 6 h (Fig. S16†). The UV-vis absorption spectra show no obvious change after open-air exposure under sunlight for 6 h, reflecting good photostability. The transformation from the trans- to the cis-isomer in APFA might occur via pedal motion which is commonly found in the thermal motion of azobenzene crystals.40,41 It should be noted that the F atoms on the benzene ring of APFA have superelectronegativity, which makes the electron cloud on the benzene ring and the lone pair on the N atoms deviate to the side of F atoms and finally weakens the intramolecular N–H⋯N hydrogen bonding interaction. With the weakening of the hydrogen-bonding interaction, the energy barrier for the trans–cis transition of APFA is lowered, which makes the photoisomerization of APFA possible. We also calculated the conversion rate of the trans-to-cis photoisomerization of APFA based on the relative intensity of the absorption band of π → π* transition in the solid-state UV-vis spectra shown in Fig. S15a.†42,43 After trans-to-cis photoisomerization, the maximum relative intensity of the absorption band of π → π* transition decreases slightly due to the production of cis-isomer APFA. Assuming that the APFA was all in the trans-isomer at the initial stage, based on the change of the maximum relative intensity of the absorption band of π → π* transition, the conversion rate of trans-to-cis photoisomerization can be estimated to be about 4%. The low conversion rate may be related to the fact that the photoisomerization of APFA can only occur on the surface of the solid-state sample because the light can hardly penetrate into the bulk of the solid-state sample. Additionally, the existence of intramolecular N–H⋯N hydrogen bonding interactions and interlaminar π–π stacking interactions in the structure is not beneficial for pedal motion for inducing the trans-to-cis isomerization, which may also contribute to the low conversion rate of trans-to-cis photoisomerization in APFA.
From the perspective of the molecule, we first calculated the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the APFA molecule to investigate its electronic structure (Fig. 4c and d). The obtained HOMO–LUMO gap of the APFA molecule is 0.12996 hartree, corresponding to an energy of 3.54 eV, which corresponds to absorption at a wavelength of 350 nm. It should be noted that the electron density of the APFA molecule is mainly distributed in the π-conjugated region, and there is a significant change between the HOMO and the LUMO.
To gain a deep insight into the electronic structure, we then calculated the band structure of APFA based on density functional theory (DFT). As depicted in Fig. 4e, the calculated band structure of APFA indicates an indirect bandgap characteristic, where the conduction band (CB) minimum and the valence band (VB) maximum are localized at different points in the Brillouin zone, in good agreement with the gradual UV-vis absorption edge. The calculated band gap is 2.689 eV, slightly larger than the optical value of 2.27 eV, considering the limitation of DFT methods. Besides, the density of states (DOS) is a powerful solution to assign the bands and study the bonding properties among the atoms in the structure.44 Obviously, from the PDOS plot of APFA (Fig. 4f), we found that H 1s states match well with those of C-2s2p and N-2s2p states over nearly the whole energy region, indicative of strong covalent interactions of the C–H and N–H bonds. With regard to the C panel, the electronic states around −4.1 and 6.0 eV could be attributed to the σ bonding and σ* antibonding states of C–C bonds, respectively, and those of −2.5–0 eV and 1.8–4.0 eV are the π bonding and π* antibonding states of C–C bonds, respectively. For APFA, the highest valence bands and the bottommost conduction bands are mainly dominated by the electronic states of C-2p and N-2p.
Conclusions
In summary, we report a single-component azobenzene-based ferroelectric crystal APFA, which experiences a paraelectric–ferroelectric transition at an exceptionally high Tc of 443 K, higher than those of most single-component ferroelectrics. In comparison with its prototype, the substitution of an amino group for the F atom on the HFA molecule not only forms intramolecular N–H⋯N hydrogen bonding interaction stabilizing the trans-isomer but also lowers the molecular symmetry and further introduces a molecular dipole moment into the APFA compound, which is of great importance for the ultimate achievement of ferroelectricity. As far as we are aware, this is the first case of single-component azobenzene-based ferroelectric crystals. The satisfactory biocompatibility as well as the environmental friendliness give the metal-free single-component ferroelectric APFA the possibility for application in medical and wearable optoelectronic sensor devices.Experimental procedures
Materials
The compound HFA was synthesized according to literature procedures.21 The synthesis procedure is shown in Fig. S1.† KMnO4 (15 g, 95 mmol) and FeSO4·7H2O (15 g, 54 mmol) were thoroughly mixed and ground into a dark red powder. 2,4,6-Trifluoroaniline was dissolved in 120 mL dichloromethane in a single-neck flask, and the powder obtained above was added to the solution in batches over roughly an hour. The reaction solution was heated to reflux overnight, and then filtered through Celite, and concentrated under reduced pressure. The crude product was then purified by column chromatography (petroleum ether) to obtain an orange solid (1.65 g, 18.58%).The compound APFA was synthesized according to literature procedures.20 The HFA (1.45 g, 5 mmol) obtained above, ammonia water (wt. 25–28%, 50 mL), and tetrahydrofuran (100 mL) were transferred to a closed pressure-resistant bottle, and the mixture was stirred at room temperature overnight. When the reaction was completed, the organic layer was separated, the aqueous layer was extracted with ethyl acetate (30 mL × 3), and the organic phases were combined, fully dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. The crude product was then purified by column chromatography (ethyl acetate/petroleum ether = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40) to obtain a yellow solid. Orange needle-like crystals were obtained by recrystallization of the synthesized products in ethyl acetate. The 1H NMR (nuclear magnetic resonance) and 19F NMR spectra of HFA and APFA are shown in Fig. S17–S20.† NMR results for HFA: 1H NMR (300 MHz, DMSO-d6) δ 7.65–7.31 (m, 4H); 19F NMR (282 MHz, DMSO-d6) δ −102.03 (t, J = 8.8 Hz), −117.33 (d, J = 8.8 Hz). NMR results for APFA: 1H NMR (300 MHz, DMSO-d6) δ 8.24 (s, 2H), 7.64–7.27 (m, 2H), 6.60 (m, 1H), 6.51 (m, 1H). 19F NMR (282 MHz, DMSO-d6) δ −102.42 (d, J = 11.9 Hz), −106.55 (t, J = 6.6 Hz), −114.87 (d, J = 11.9 Hz), −119.31 (d, J = 6.6 Hz). The NMR results combined with single-crystal X-ray diffraction analysis well confirm the structures of HFA and APFA.
:40) to obtain a yellow solid. Orange needle-like crystals were obtained by recrystallization of the synthesized products in ethyl acetate. The 1H NMR (nuclear magnetic resonance) and 19F NMR spectra of HFA and APFA are shown in Fig. S17–S20.† NMR results for HFA: 1H NMR (300 MHz, DMSO-d6) δ 7.65–7.31 (m, 4H); 19F NMR (282 MHz, DMSO-d6) δ −102.03 (t, J = 8.8 Hz), −117.33 (d, J = 8.8 Hz). NMR results for APFA: 1H NMR (300 MHz, DMSO-d6) δ 8.24 (s, 2H), 7.64–7.27 (m, 2H), 6.60 (m, 1H), 6.51 (m, 1H). 19F NMR (282 MHz, DMSO-d6) δ −102.42 (d, J = 11.9 Hz), −106.55 (t, J = 6.6 Hz), −114.87 (d, J = 11.9 Hz), −119.31 (d, J = 6.6 Hz). The NMR results combined with single-crystal X-ray diffraction analysis well confirm the structures of HFA and APFA.
Thin-film preparation
The precursor solution of APFA was prepared by dissolving 50 mg of the as-grown crystal in 1 mL ethyl acetate. Thin films of APFA were deposited by drop-coating the precursor solution of 20 μL onto a cleaned indium-tin-oxide-coated glass and then dried at 30 °C for 20 min.Single-crystal X-ray crystallography
Single-crystal X-ray diffraction data at different temperatures were measured using a Rigaku Saturn 924 diffractometer with Cu-Kα radiation (λ = 1.54184 Å). Data collection and structural refinement were performed using Rigaku CrystalClear and SHELXTL software packages. Powder X-ray diffraction (PXRD) data were collected by using a Rigaku D/MAX 2000 PC X-ray diffraction system with Cu Kα radiation with a step size of 0.02°.DSC, dielectric and SHG measurements
Differential scanning calorimetry (DSC) measurements were performed on a PerkinElmer Diamond DSC under a nitrogen atmosphere in aluminum crucibles with a heating or cooling rate of 20 K min−1. The complex permittivity was measured on a Tonghui TH2828A. Silver conduction paste deposited on plate surfaces was used as the electrodes. For second harmonic generation (SHG) experiments, an unexpanded laser beam with low divergence (pulsed Nd:YAG at a wavelength of 1064 nm, 5 ns pulse duration, 1.6 MW peak power, and 10 Hz repetition rate) was used. The instrument model is an Ins 1210058, INSTEC Instruments, while the laser is a Vibrant 355 II, OPOTEK.UV-vis absorbance spectra
UV-vis absorbance spectroscopy was performed on powder samples by using a Shimadzu (Tokyo, Japan) UV-3600 Plus spectrophotometer equipped with an ISR-603 integrating sphere at room temperature, respectively. BaSO4 was used as a 100% reflectance reference.P–E hysteresis loop measurements
A thin-film crystal capacitor was fabricated for the P–E hysteresis loop measurement. For the film, GaIn eutectic was used as the top electrode. Ferroelectric hysteresis measurements were conducted with this capacitor architecture (GaIn/sample film/ITO) by the double wave method.PFM measurements
PFM visualization of the ferroelectric domain structures was carried out using a commercial atomic force microscope system (MFP-3D, Asylum Research) on the polycrystalline thin-film sample. Conductive Pt/Ir-coated silicon probes (EFM-50, Nanoworld) were used for domain imaging and polarization switching studies.Calculation conditions
The dipole moment was calculated at the b3lyp/6-31G(d) level with Gaussian 16 software. We constructed a molecular conformation based on the experimentally measured single crystal X-ray diffraction structure. We carried out density functional calculations based on the Berry phase method developed by King-Smith and Vanderbilt.45,46 First-principles calculations were performed within the framework of density functional theory implemented in the Vienna ab initio simulation package (VASP; 5.4.4).47,48 The energy cut-off for the expansion of the wave functions was fixed at 550 eV and the exchange–correlation interactions were treated within the generalized gradient approximation of the Perdew–Burke–Ernzerhof type.49 Firstly, geometrical optimization was performed by fixing the lattice constant based on the experimentally measured X-ray crystal structure. Then, Berry phase calculation was performed on the optimized geometry.Electronic structure calculations, including the band structure and PDOS, were performed using a plane-wave pseudopotential method within DFT in the CASTEP program. We chose the Heyd–Scuseria–Ernzerhof-06 (HSE-06) and the norm-conserving pseudopotential with the following valence-electron configurations: N-2s22p3, F-2s22p5, C-2s22p2, and H-1s1. A Monkhorst–Pack k-point sampling of 4 × 2 × 1 and an energy cutoff of 850 eV were set.
NMR measurements
1H NMR (300 MHz) and 19F NMR (282 MHz) spectra were recorded on Bruker Ascend TM 300 instruments. Chemical shifts for 1H-NMR spectra are reported as δ (parts per million) relative to the residual proton signal of DMSO-d6 at 2.51 ppm (m).Data availability
The ESI footnote details the data available as part of the ESI.†Author contributions
R.-G. X. and W.-Q. L. devised and developed the project. H. P and J.-C. Q. contributed equally to this work. H. P. carried out X-ray, thermodynamic and SHG characterization. J.-C. Q. carried out UV-vis and NMR measurements. X.-J. S. carried out other characterization. All the authors analyzed the data, discussed the results and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Jun-Chao Liu (Nanchang University) for his synthesis work. This work was supported by the National Natural Science Foundation of China (21991142, 21831004, and 22175082).Notes and references
- C. R. Qiu, B. Wang, N. Zhang, S. J. Zhang, J. F. Liu, D. Walker, Y. Wang, H. Tian, T. R. Shrout, Z. Xu, L. Q. Chen and F. Li, Nature, 2020, 577, 350–354 CrossRef CAS PubMed.
- Y. Zhang, H. D. Lu, X. X. Yong, X. X. Cheng, L. Xie, T. Aoki, L. Z. Li, C. Heikes, S. P. Lau, D. C. Schlom, L. Q. Chen, A. Gruverman and X. Q. Pan, Adv. Mater., 2019, 31, 1902099 CrossRef PubMed.
- C.-L. Jia, K. W. Urban, M. Alexe, D. Hesse and I. Vrejoiu, Science, 2011, 331, 1420–1423 CrossRef CAS PubMed.
- J. Valasek, Phys. Rev., 1921, 17, 475–481 CrossRef CAS.
- J. F. Scott and C. A. Paz de Araujo, Science, 1989, 246, 1400–1405 CrossRef CAS PubMed.
- R. W. Whatmore, Y. M. You, R. G. Xiong and C. B. Eom, APL Mater., 2021, 9, 070401 CrossRef CAS.
- W.-Q. Liao, D. Zhao, Y.-Y. Tang, Y. Zhang, P.-F. Li, P.-P. Shi, X.-G. Chen, Y.-M. You and R.-G. Xiong, Science, 2019, 363, 1206–1210 CrossRef CAS PubMed.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357–366 CrossRef CAS PubMed.
- S. Horiuchi, Y. Tokunaga, G. Giovannetti, S. Picozzi, H. Itoh, R. Shimano, R. Kumai and Y. Tokura, Nature, 2010, 463, 789–792 CrossRef CAS PubMed.
- P. F. Li, W. Q. Liao, Y. Y. Tang, W. C. Qiao, D. W. Zhao, Y. Ai, Y. F. Yao and R. G. Xiong, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5878–5885 CrossRef CAS PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- W. Szymanski, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef CAS PubMed.
- A. Fihey, A. Perrier, W. R. Browne and D. Jacquemin, Chem. Soc. Rev., 2015, 44, 3719–3759 RSC.
- E. Sackmann,, J. Am. Chem. Soc., 1971, 93, 7088–7090 CrossRef.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2016, 116, 15089–15166 CrossRef CAS PubMed.
- A. M. Rice, C. R. Martin, V. A. Galitskiy, A. A. Berseneva, G. A. Leith and N. B. Shustova, Chem. Rev., 2020, 120, 8790–8813 CrossRef CAS PubMed.
- E. Hadjoudis and I. M. Mavridis, Chem. Soc. Rev., 2004, 33, 579–588 CAS.
- Y. Y. Tang, J. C. Liu, Y. L. Zeng, H. Peng, X. Q. Huang, M. J. Yang and R. G. Xiong, J. Am. Chem. Soc., 2021, 143, 13816–13823 CrossRef CAS PubMed.
- W. Q. Liao, Y. L. Zeng, Y. Y. Tang, H. Peng, J. C. Liu and R. G. Xiong, J. Am. Chem. Soc., 2021, 143, 21685–21693 CrossRef CAS PubMed.
- M. Matsui, N. Tanaka, N. Andoh, K. Funabiki, K. Shibata, H. Muramatsu, Y. Ishigure, E. N. Kohyama, Y. Abe and M. Kaneko, Chem. Mater., 1998, 10, 1921–1930 CrossRef CAS.
- D. Bleger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef CAS PubMed.
- L. Sharma and T. Kimura, Polym. Adv. Technol., 2003, 14, 392–399 CrossRef CAS.
- B. Derkowska-Zielinska, L. Skowronski, M. Sypniewska, D. Chomicki, V. Smokal, O. Kharchenko, M. Naparty and O. Krupka, Opt. Mater., 2018, 85, 391–398 CrossRef CAS.
- B. J. Klein, K. R. Vann, F. H. Andrews, W. W. Wang, J. Zhang, Y. Zhang, A. A. Beloglazkina, W. Mi, Y. Li, H. Li, X. Shi, A. G. Kutateladze, B. D. Strahl, W. R. Liu and T. G. Kutateladze, Nat. Commun., 2018, 9, 4574 CrossRef PubMed.
- V. Sivasakthi, A. Anbarasu and S. Ramaiah, Cell Biochem. Biophys., 2013, 67, 853–863 CrossRef CAS PubMed.
- R. K. Huang, Z. F. Xiao, D. X. Liu, W. X. Zhang and X. M. Chen, Chem. Commun., 2019, 55, 7159–7162 RSC.
- Y. Ai, P.-F. Li, M.-J. Yang, Y.-Q. Xu, M.-Z. Li and R.-G. Xiong, Chem. Sci., 2022, 13, 748–753 RSC.
- P.-F. Li, Y. Ai, Y.-L. Zeng, J.-C. Liu, Z.-K. Xu, Z.-K. Xu and Z.-X. Wang, Chem. Sci., 2022, 13, 657–664 RSC.
- Y.-M. You, W.-Q. Liao, D. Zhao, H.-Y. Ye, Y. Zhang, Q. Zhou, X. Niu, J. Wang, P.-F. Li, D.-W. Fu, Z. Wang, S. Gao, K. Yang, J.-M. Liu, J. Li, Y. Yan and R.-G. Xiong, Science, 2017, 357, 306–309 CrossRef CAS PubMed.
- C.-K. Yang, W.-N. Chen, Y.-T. Ding, J. Wang, Y. Rao, W.-Q. Liao, Y. Xie, W. Zou and R.-G. Xiong, J. Am. Chem. Soc., 2019, 141, 1781–1787 CrossRef CAS PubMed.
- J.-J. Wang, D. Fortino, B. Wang, X. Zhao and L.-Q. Chen, Adv. Mater., 2020, 32, 1906224 CrossRef CAS PubMed.
- X.-X. Chen, X.-Y. Zhang, D.-X. Liu, R.-K. Huang, S.-S. Wang, L.-Q. Xiong, W.-X. Zhang and X.-M. Chen, Chem. Sci., 2021, 12, 8713–8721 RSC.
- Y. Ai, R. Sun, Y.-L. Zeng, J.-C. Liu, Y.-Y. Tang, B.-W. Wang, Z.-M. Wang, S. Gao and R.-G. Xiong, Chem. Sci., 2021, 12, 9742–9747 RSC.
- H.-Y. Zhang, X.-G. Chen, Y.-Y. Tang, W.-Q. Liao, F.-F. Di, X. Mu, H. Peng and R.-G. Xiong, Chem. Soc. Rev., 2021, 50, 8248–8278 RSC.
- H. Uršič and U. Prah, Proc. R. Soc. A, 2019, 475, 20180782 CrossRef PubMed.
- O. Kwon, D. Seol, H. Qiao and Y. Kim, Adv. Sci., 2020, 7, 1901391 CrossRef CAS PubMed.
- H. Ma, W. X. Gao, J. L. Wang, T. Wu, G. L. Yuan, J. M. Liu and Z. G. Liu, Adv. Electron. Mater., 2016, 2, 1600038 CrossRef.
- Q. Tang, L.-H. He, Y.-H. Yang, J.-F. Long, X.-K. Fu and C.-B. Gong, Org. Electron., 2016, 30, 200–206 CrossRef CAS.
- R. Dong, B. Zhu, Y. Zhou, D. Yan and X. Zhu, Polym. Chem., 2013, 4, 912–915 RSC.
- J. Harada and K. Ogawa, J. Am. Chem. Soc., 2001, 123, 10884–10888 CrossRef CAS PubMed.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Nather, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed.
- M. Suzuki, J. C. Lim and T. Saegusa, Macromolecules, 2000, 33, 5347–5352 CrossRef.
- H.-Y. Zhang, C.-L. Hu, Z.-B. Hu, J.-G. Mao, Y. Song and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 3240–3245 CrossRef CAS PubMed.
- R. King-Smith and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 1651 CrossRef CAS PubMed.
- D. Vanderbilt and R. King-Smith, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 4442 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2144165 and 2144166. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00689h |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |