
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoregular functionalized polysaccharides via cationic ring-opening polymerization of biomass-derived levoglucosan†
Mayuri K.
Porwal
 a,
Yernaidu
Reddi‡
b,
Derek J.
Saxon‡
b,
Christopher J.
Cramer
bc,
Christopher J.
Ellison
*a and
Theresa M.
Reineke
*b
a,
Yernaidu
Reddi‡
b,
Derek J.
Saxon‡
b,
Christopher J.
Cramer
bc,
Christopher J.
Ellison
*a and
Theresa M.
Reineke
*b
aDepartment of Chemical Engineering and Materials Science, University of Minnesota, Minneapolis, Minnesota 55455, USA. E-mail: porwa001@umn.edu; cellison@umn.edu
bDepartment of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, USA. E-mail: treineke@umn.edu
cUnderwriters Laboratories Inc., 333 Pfingsten Rd., Northbrook, Illinois 60620, USA
First published on 17th March 2022
Abstract
We report the facile synthesis and characterization of 1,6-α linked functional stereoregular polysaccharides from biomass-derived levoglucosan via cationic ring-opening polymerization (cROP). Levoglucosan is a bicyclic acetal with rich hydroxyl functionality, which can be synthetically modified to install a variety of pendant groups for tailored properties. We have employed biocompatible and recyclable metal triflate catalysts – scandium and bismuth triflate – for green cROP of levoglucosan derivatives, even at very low catalyst loadings of 0.5 mol%. Combined experimental and computational studies provided key kinetic, thermodynamic, and mechanistic insights into the cROP of these derivatives with metal triflates. Computational studies reveal that ring-opening of levoglucosan derivatives is preferred at the 1,6 anhydro linkage and cROP proceeds in a regio- and stereo-specific manner to form 1,6-α glycosidic linkages. DFT calculations also show that biocompatible metal triflates efficiently coordinate with levoglucosan derivatives as compared to the highly toxic PF5 used previously. Post-polymerization modification of levoglucosan-based polysaccharides is readily performed via UV-initiated thiol–ene click reactions. The reported levoglucosan based polymers exhibit good thermal stability (Td > 250 °C) and a wide glass transition temperature (Tg) window (<−150 °C to 32 °C) that is accessible with thioglycerol and lauryl mercaptan pendant groups. This work demonstrates the utility of levoglucosan as a renewably-derived scaffold, enabling facile access to tailored polysaccharides that could be important in many applications ranging from sustainable materials to biologically active polymers.
Introduction
Polymers are indispensable in modern life and the global economy; however, more than 90% of current synthetic polymers are derived from fossil fuels and 87% of these produced polymers contribute to plastic waste, usually after a single use.1–4 Lignocellulosic biomass is one of the most promising renewable feedstocks for sustainable polymers due to its worldwide abundance and availability.5–10 The major component of lignocellulosic biomass is cellulose, and thermochemical conversion processes—such as fast pyrolysis—provide an efficient route to convert biomass cellulose into renewable chemicals.5–8 The major product of cellulose fast pyrolysis is levoglucosan (Fig. 1A), which can be obtained with yields as high as 80%.11–14 Not surprisingly, levoglucosan has been identified as a building block for high value-added chemicals, pharmaceuticals, and surfactants.13–15 As a result, there has been increasing interest in the commercial production of levoglucosan from biomass. Techno-economic analyses indicate that levoglucosan could be produced by thermochemical conversion at a low cost of $1.33 per kg to $3.0 per kg, which is competitive with many petroleum-derived monomers.12,15–17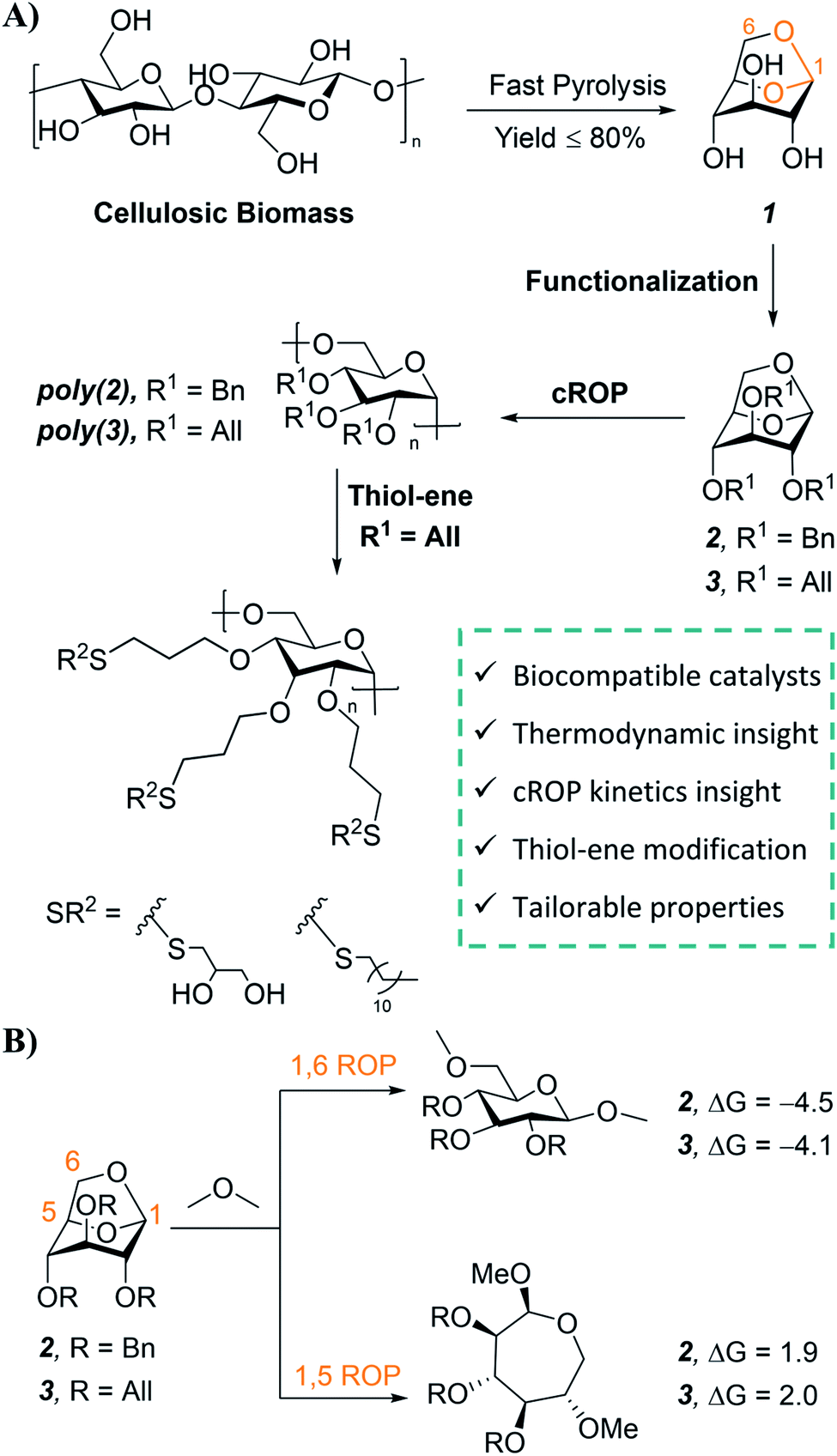 | ||
| Fig. 1 (A) Schematic depicting synthesis of levoglucosan (1) from cellulosic biomass via fast pyrolysis. In this work, tribenzyl (2) and triallyl levoglucosan monomers (3) are subjected to metal triflate catalyzed cROP to synthesize poly(2) and poly(3) respectively. UV-initiated thiol–ene click reaction is utilized for further modification of poly(3). (B) Isodesmic reaction for ring-opening of 2 and 3 with dimethyl ether and the corresponding ring strain free energies (kcal mol−1) at the SMD(DCM)/M06-2X/6-311++G(d,p)//SMD(DCM)/M06-L/6-31+G(d,p) level. | ||
Structurally, levoglucosan is an attractive feedstock for the synthesis of sustainable polymers. This anhydrosugar contains a bicyclic acetal linkage (Fig. 1A, highlighted in orange) that is amenable to cationic ring-opening polymerization (cROP).18 Levoglucosan also offers rich functionality due to its three hydroxyl groups, which can be synthetically modified prior to cROP to install a variety of pendant groups for tailored properties. Furthermore, the incorporation of the rigid glucopyranose ring in the polymer backbone via cROP can increase the thermal stability of levoglucosan based polysaccharides.19 Stereoregular polysaccharides are also of interest in biological and biomedical applications such as drug delivery and blood anti-coagulation due to their biocompatibility, multiple functionalities, and stereochemistry dependent properties.20–24 In addition, stereoselective cROP of levoglucosan to obtain polysaccharides with 1,6-α-glycosidic linkages is attractive from a sustainability standpoint since these linkages have demonstrated susceptibility to enzymatic and acidic hydrolysis.25–27 Despite these advantages, levoglucosan has received considerably less attention in the sustainable polymers field as compared to levoglucosenone,28–37 another anhydrosugar than can be obtained in small quantities from cellulose pyrolysis.13
The cROP of protected levoglucosan derivatives was first reported more than 50 years ago, however the synthetic routes employed had some major drawbacks.38 These derivatives are generally synthesized from levoglucosan by reaction with alkyl/aryl bromide in the presence of sodium hydride in dimethylformamide, which is hazardous due to its thermal instability.39–43 Benzyl and allyl derivatives of levoglucosan have been two of the most widely studied monomers with cROP proceeding at low temperatures in the presence of PF5 or TMSOTf.44–46 The resulting polymers were de-benzylated/de-allylated for use as polysaccharide mimetics and HPLC stationary phases, respectively.38,44,47 These polymerization conditions can afford high molecular weight polymers (Mn of 50 kDa), however reaction times were generally long (80 h) and initiators (PF5 & TMSOTf) that are both highly toxic and difficult-to-handle were used.38,48 Additionally, in these previous studies stereoselective cROP of levoglucosan required rigorous conditions such as high vacuum and very low temperature (−60 °C for the tribenzyl monomer, 0 °C for the triallyl monomer).38,41,44,45 Overall, identification of alternative low toxicity catalysts for cROP of levoglucosan under mild conditions has remained a challenge, thereby hindering their large scale and sustainable synthesis. This has potentially limited the use of levoglucosan in the sustainable polymers field as compared to other biomass-derived sugars such as levoglucosenone.
Despite the initial investigations in cROP of levoglucosan, the material properties of the resulting polysaccharides have not been characterized and computational insight into the thermodynamics and mechanism of levoglucosan cROP has not been provided. Additionally, very limited work has been done to utilize the rich hydroxyl functionality of levoglucosan for the synthesis of functional polysaccharides. Fu et al. synthesized amphiphilic polysaccharides by ring-opening copolymerization of tripropargyl levoglucosan, followed by azide–alkyne cycloaddition modification of the copolymer.49 However to the best of our knowledge this is the only example of reactive levoglucosan-based materials, providing ample space for the development and characterization of functional levoglucosan polymers. Taken together, these factors have led us to explore methods to improve levoglucosan polymerization conditions while preserving reactive functionalities for polymer modification.
Herein, we report a synthetic platform for stereoregular 1,6-α linked levoglucosan-based polysaccharides with different pendant functional groups. An array of catalysts was screened for the cROP of levoglucosan-based benzyl (Bn) 2 and allyl (All) 3 functional monomers (Fig. 1A) to identify green alternatives. Scandium and bismuth triflate—which are biocompatible and recyclable catalysts—were identified as promising candidates and used for further studies to understand the kinetics, thermodynamics, and mechanism of ring-opening for 2 and 3. Post-polymerization modification of poly(3) was performed with UV-mediated thiol–ene click chemistry to afford functional polymers with a range of solubility and thermal properties. This work paves the way for future development of this renewable platform, enabling facile access to tailored polysaccharides for sustainable polymers to biomaterials applications.
Results and discussion
Monomer synthesis
Monomer 2 was purchased from a commercial supplier and monomer 3 was synthesized on a multi-gram scale using a modified method for the etherification of starch (75% yield, ESI Sections 2.1 & 9.1†).50 This synthetic route is safe with easy to handle reagents, employing NaOH as a mild base in dimethyl sulfoxide. We also explored a “greener” synthetic pathway for 3 using NaOH, a phase-transfer catalyst, and water as the reaction medium;51 the monomer was successfully prepared with this method, albeit in lower yield (36% yield; ESI Section 2.2†).Screening of cROP conditions
Combined molecular mechanics and density functional theory (DFT) calculations were performed to determine the ring-strain free energies for isodesmic reaction of 2 and 3 with dimethyl ether. Unsurprisingly, both the monomers have comparable ring-strain values, and for both 2 and 3 ring-opening is energetically favored at the 1,6-anhydro linkage over the 1,5-linkage (Fig. 1B), supporting polymerization of 2 and 3.To identify less toxic catalysts to promote cROP of 2 and 3, and to understand the effect of solvent and catalyst loading on cROP of 2 and 3, a library of polymerization experiments was performed. We initially screened a range of metal and organic catalysts due to their commercial availability, low toxicity, and ability to ring open cyclic ethers for the polymerization of 2 (Fig. S3 and Table S1†).48,52–58 Cationic initiators BF3OEt2 and MeOTf that are conventionally employed for cROP of cyclic acetals were also screened in addition to the array of catalysts for direct comparison.57,58 All screening reactions took place at room temperature for 72 h in dichloromethane (initial monomer concentration [M]0 = 1.0 mol L−1). Successful catalysts were then used to screen cROP of 3; two metal triflates [Sc(OTf)3 and Bi(OTf)3] were identified for cROP of both 2 and 3 (Table 1, entries 1, 2, 7, 8). Both metal triflates provided comparable conversion to the BF3OEt2 and MeOTf controls (Table S2†) and provide the additional benefit of being recyclable (via a simple aqueous extraction) while not releasing corrosive byproducts such as triflic acid commonly released by alkyl triflates.48 However, these metal triflates have limited solubility in dichloromethane and hence further studies were performed to understand solvent effects.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Monomer | Cat. | Solvent CH2C12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN :MeCN |
[M]0 (mol L−1) | [M]:[cat] |
Conva (%) | DPb | M w (g mol−1) | Đ |
| a Monomer conversion determined by 1H NMR spectroscopy. b Molecular weight and dispersity determined by SEC-MALS in DMF. | |||||||||
| 1 | 2 | Sc(OTf)3 | 100:0 |
1 | 50:1 |
74 | 16 | 11030 |
1.6 |
| 2 | 2 | Bi(OTf)3 | 100:0 |
1 | 50:1 |
84 | 8 | 4400 | 1.3 |
| 3 | 2 | Sc(OTf)3 | 100:0 |
2 | 100:1 |
82 | 17 | 12200 |
1.6 |
| 4 | 2 | Sc(OTf)3 | 100:0 |
2 | 200:1 |
71 | 27 | 16500 |
1.4 |
| 5 | 2 | Sc(OTf)3 | 99:1 |
2 | 100:1 |
57 | 31 | 18600 |
1.4 |
| 6 | 2 | Sc(OTf)3 | 99:1 |
2 | 200:1 |
34 | 22 | 13400 |
1.4 |
| 7 | 3 | Bi(OTf)3 | 100:0 |
1 | 20:1 |
86 | 13 | 6100 | 1.7 |
| 8 | 3 | Sc(OTf)3 | 100:0 |
1 | 20:1 |
77 | 12 | 7900 | 2.3 |
| 9 | 3 | Bi(OTf)3 | 100:0 |
Bulk | 100:1 |
75 | 24 | 10500 |
1.5 |
| 10 | 3 | Bi(OTf)3 | 100:0 |
Bulk | 200:1 |
71 | 21 | 9500 | 1.7 |
| 11 | 3 | Bi(OTf)3 | 99:1 |
2 | 100:1 |
68 | 16 | 6600 | 1.5 |
| 12 | 3 | Bi(OTf)3 | 99:1 |
Bulk | 100:1 |
97 | 24 | 12500 |
1.9 |
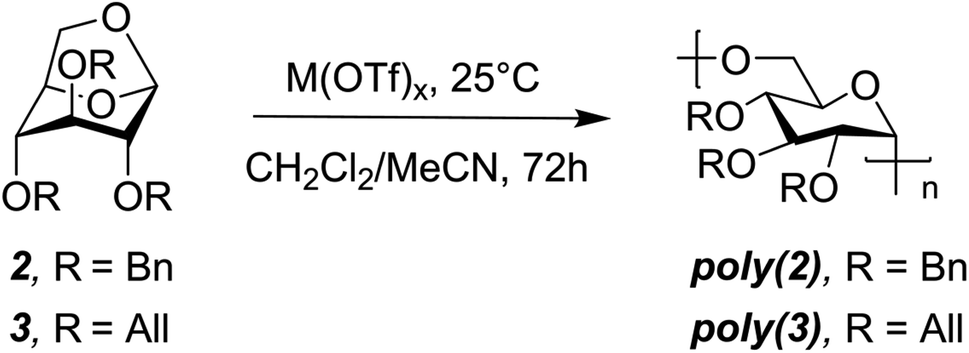
We investigated the addition of varying quantities of acetonitrile (MeCN), which is a better solvent for the catalyst, in the polymerization mixture. To our surprise, no polymerization occurred when >10% MeCN by volume was present in the reaction medium. Moreover, monomer conversion generally decreased with increasing amount of MeCN from 0–10% (Tables S3 and S4†) and we hypothesize this is due to the better solvation of the ionic active species by MeCN, as reported in the literature.59,60 We then studied the effect of varying catalyst loading on cROP of 2 and 3 and found that polymerization of both monomers could be conducted at M(OTf)3 loadings as low as 0.5 mol% (Table 1, entries 4, 6, 10). Notably, the pendant groups do not seem to drastically impact conversion, as similar values were observed for both the monomers likely due to their comparable ring-strain (Fig. 1B). With the identified metal triflates, Mw values up to 18.6 kDa for poly(2) (DP = 31) and 12.5 kDa for poly(3) (DP = 24) could be achieved with moderate dispersities (Table 1, entries 5 and 12). These moderate molecular weights are most likely caused by intermolecular chain transfer and back-biting reactions which are common features in cROP of cyclic acetals.57 Chain transfer reactions are favored in the cROP of cyclic acetals due to the higher basicity of oxygen atoms in the polymer chain as compared to the monomer.57 Moreover, it is hypothesized that metal triflate mediated cROP of levoglucosan derivatives follows a catalytic approach as reported in the literature.54 In a catalytic approach, one metal triflate molecule catalytically produces a large number of polymer molecules, thereby leading to shorter polymer chains.54 Since the metal triflate mediated cROP of levoglucosan follows a catalytic approach, calculating a desired or target molecular weight for a given set of polymerization conditions is difficult.
Thermodynamics of polymerization
DFT calculations were employed to provide insights into the mechanism of levoglucosan cROP catalyzed by Bi(OTf)3, Sc(OTf)3, and PF5 (as a control for comparison to previous studies), as well as to understand catalyst efficiency. Various mechanistic pathways were considered (higher energy pathways are provided in the ESI Section 11†), and the lower energy pathway is depicted in Fig. 2A. First, monomer 2 or 3 is activated by the catalyst to form a thermodynamically stable complex (I1). When PF5 is utilized as the catalyst, activation of 2 or 3 is unfavorable relative to the metal triflates due to poor coordination of PF5. This suggests that PF5 exhibits less efficient initiation, leading to the high Mn observed in previous studies.44,46 After the first step, I1 undergoes ring opening of 1,6-anhydro linkage to generate the carbenium intermediate I2 through a TS structure TS1, and the activation free energies are similar for monomer 2 or 3 with their respective catalysts, Bi(OTf)3 and Sc(OTf)3 (Fig. 2A, B and S45†). Next, the nucleophilic addition of another molecule of monomer to the electrophilic carbon of intermediate I2 generates intermediate I3via a pseudoaxial approach through TS2. In this step, the ΔG‡ values for nucleophilic addition of 2 and 3 are similar in all cases (Fig. 2B). Interestingly, it is evident that computed energetics of the nucleophilic addition of monomer to I2 to form the alternative intermediate structure I4 (Fig. 2A) indicate it as energetically disfavored (possibly due to the steric hindrance between the incoming monomer and catalyst), thereby leading to stereospecific cROP of 2 and 3 (Fig. S46, ESI Section 11.3†). Further, Fig. 2C shows that the monomer addition at C6 of 2 and 3 is energetically disfavored compared to the anomeric carbon. This indicates that cROP proceeds in a regiospecific manner with attack on the anomeric carbon to form 1,6-glycosidic linkages. Finally, computational mechanistic insights also provide some understanding of the moderate molecular weights observed experimentally. Specifically, higher energy barrier pathways may contribute to slower propagation leading to shorter polymer chains (Fig. S48†). Finally, the increase in ΔG‡ and the increase in charge separation between the active ionic species with increased chain length may hinder propagation to high molecular weight polymer.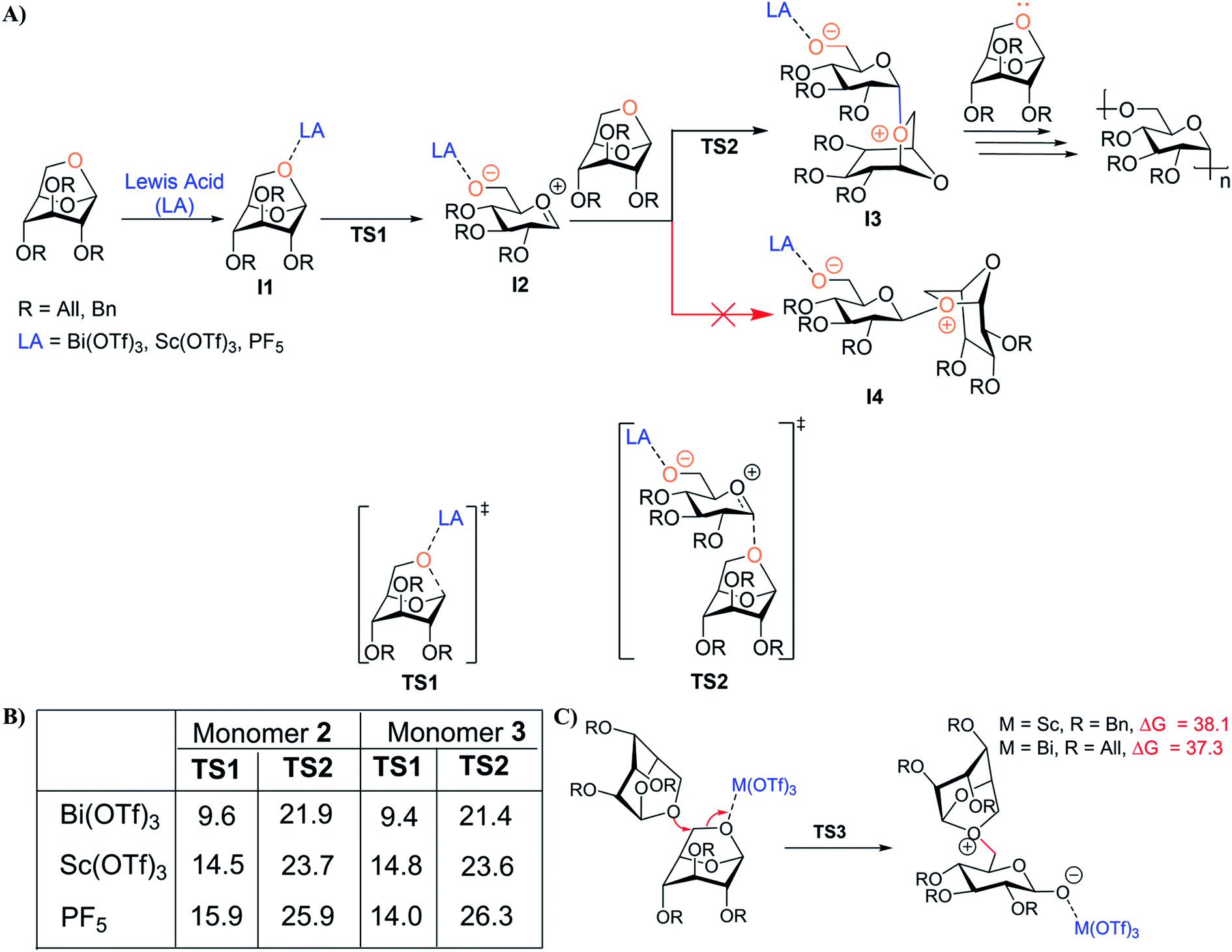 | ||
| Fig. 2 (A) Key mechanistic steps for cROP of 2 and 3 catalyzed by Lewis acid. Transition-state structures for the ring-opening of 1,6-anhydro linkages (TS1) and nucleophilic additions of monomers 2 and 3 (TS2) catalyzed by various Lewis acids. Computed energetics indicate that nucleophilic addition of monomers 2 or 3 to I2 to form intermediate I4 (pseudo-equatorial approach) is energetically disfavored compared to formation of intermediate I3 (pseudo-axial approach; Fig. S46†). (B) Table depicting computed energetics in kcal mol−1 for key mechanistic steps. (C) Alternative pathway for cROP of 2 catalyzed by Sc(OTf)3 and the cROP of 3 catalyzed by Bi(OTf)3. The corresponding free energies of activation (kcal mol−1) with respect to I1 at the SMD(DCM)/ωB97X-D/def2-TZVP, def2-TZVP|SDD(Bi)//SMD(DCM)/M06-L/6-31+G(d,p), LanL2DZ(Bi) level. | ||
Polymer stereoregularity
1H NMR analysis of poly(2) and poly(3) compared to the respective monomers indicates that the polymers possess 1,6-α glycosidic stereoregularity as the anomeric proton resonances (in the β-configuration) appeared at 4.94 ppm (Fig. 3B and D).41 Additionally 13C NMR of poly(2) and poly(3) also showed the α configuration as the anomeric carbon resonances appeared at 97.8 ppm and 97.4 ppm, respectively (Fig. S35 and S37†).41,49 The stereoregularity of the polymers was also confirmed via optical rotation measurements. The optical rotation values for poly(2) and poly(3) were measured to be +91.5 cm3 dm−1 g−1 and +84 cm3 dm−1 g−1 respectively, with the optical rotation values for 2 and 3 being −31.5 cm3 dm−1 g−1 and −45 cm3 dm−1 g−1, respectively. Overall, these results indicate that the cROP of 2 and 3 with metal triflates yields highly stereoregular levoglucosan polymers with 1,6-α glycosidic linkages. Notably, this work demonstrates that highly stereoregular levoglucosan polymers can be synthesized under mild conditions without the need for an energy intensive process involving high vacuum and low temperatures, as reported previously. 1H NMR analysis also indicates that the alkene functionality in poly(3) is intact during cROP with the vinyl proton resonances at 5.91 ppm, 5.27 ppm and 5.14 ppm. The successful synthesis of poly(3) while preserving multiple pendant allyl groups will further allow for click-chemistry modifications.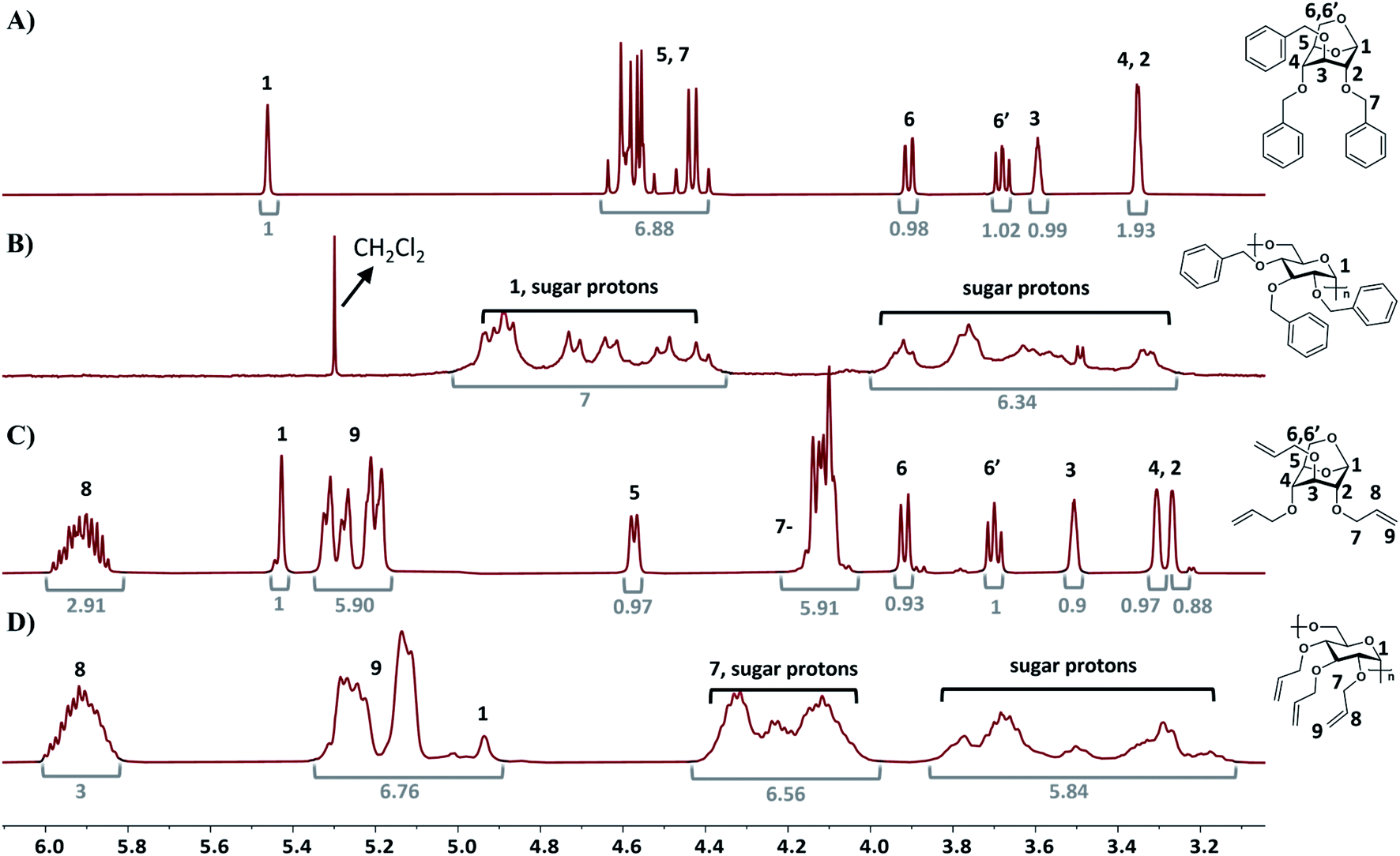 | ||
| Fig. 3 1H NMR spectra of (A) monomer 2, (B) poly(2), (C) monomer 3, and (D) poly(3). Corresponding integrations are depicted in grey numbers. | ||
Kinetics of polymerization
Apart from understanding the ring-opening mechanism, we were also interested in investigating the cROP kinetics for each monomer to determine optimum reaction time (ESI Section 5† for details). We observed a short induction period of ∼20 min during cROP of 2 (Table S7 and Fig. S5†), which reaches an equilibrium conversion of 64% in ≤24 h (Fig. 4A). Conversely, cROP of 3 has a much longer induction period of ∼4 h (Table S8†) and reaches an equilibrium conversion of 83% in ≤72 h (Fig. 4B). Remarkably, the induction period for cROP of 3, as evidenced by 1H NMR spectroscopy, was accompanied with a drastic color change in the solution throughout the 4 h period (Fig. S10 and S11†). We hypothesize that this long cROP induction period of 3 is due to the non-productive coordination of Bi(OTf)3 with the allylic ether oxygens and the glucopyranose ring oxygen. This was supported by DFT energetics calculations depicting that non-productive coordination of Bi with the allylic ether oxygens and the glucopyranose ring oxygen is favored at multiple locations (Fig. 4C). Furthermore, while comparing 2 and 3, we find that the relative free energies of binding the M(OTf)3 to each of these respective oxygen atoms vary slightly (Fig. 4C and S44†). However, the free energy trends and magnitudes are similar across both 2 and 3. Along with tracking conversion over time, the molar mass and dispersity of the growing polymer chains was also monitored throughout this kinetic study. For poly(2), the Mn increased up to 40% conversion (Fig. S18†), and for poly(3) the Mn increased up to 60% conversion (Fig. S19†). Lastly, narrow dispersities were maintained for poly(2) throughout the reaction duration (∼1.2 to 1.4, Fig. S20†), whereas for poly(3) dispersity was higher in the initial stages and gradually decreased to a stable value (∼1.5, Fig. S21†).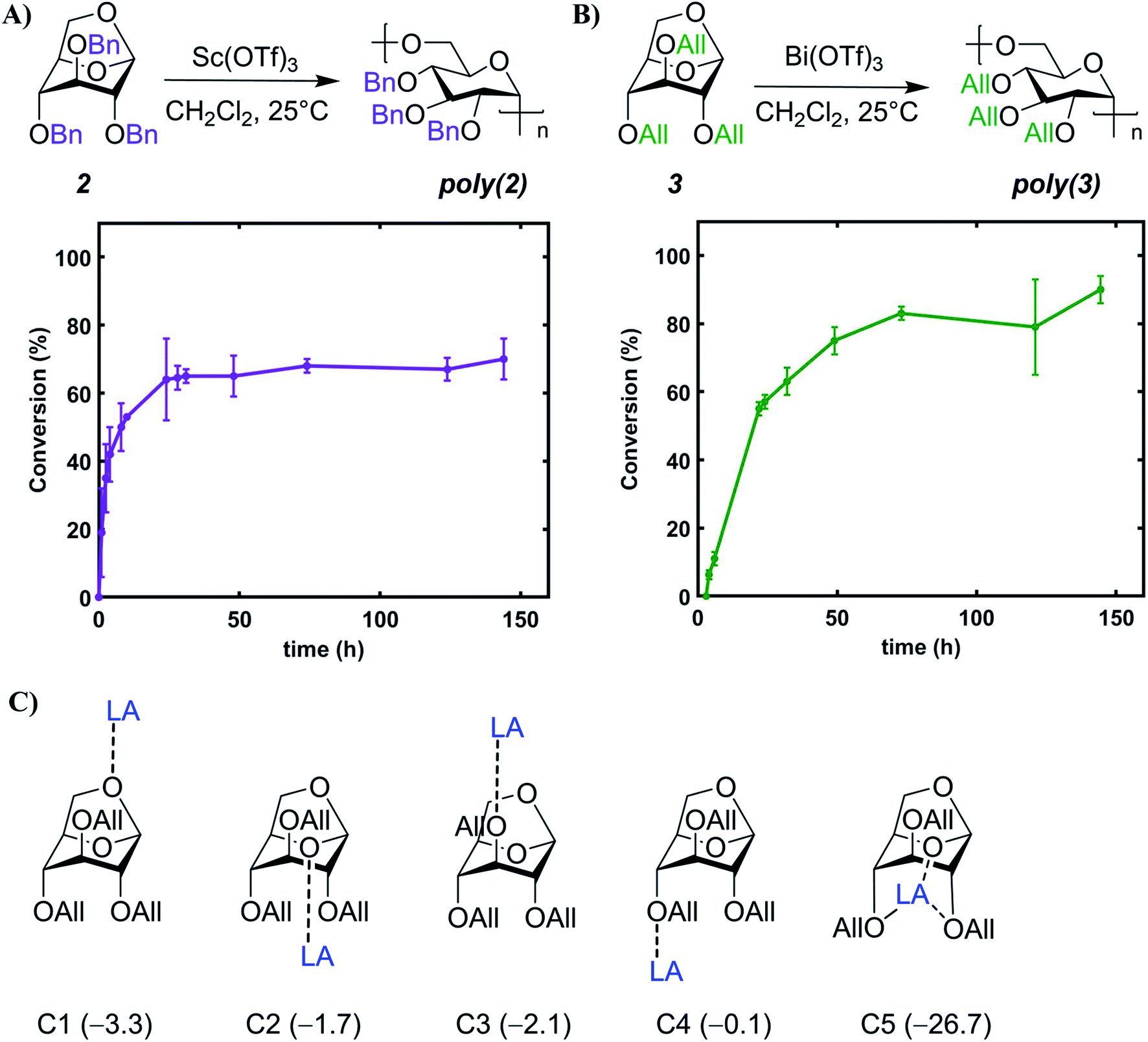 | ||
| Fig. 4 (A) Conversion-time plot for cROP of 2 with Sc(OTf)3. Polymerization conditions: [2]0 = 2 M, 2: Sc(OTf)3 = 200:1. (B) Conversion-time plot for cROP of 3 with Bi(OTf)3. Polymerization conditions: [3]0 = 7 M, 3:Bi(OTf)3 = 100:1. Monomer conversion determined by 1H NMR spectroscopy. Error bars based on true values. (C) Coordination modes for monomer 3 with LA (Bi(OTf)3) and the corresponding binding free energies (ΔG‡ in kcal mol−1) are provided in the parenthesis. | ||
Thiol–ene post polymerization modification
The allylic pendant groups enable facile post-polymerization modification of poly(3), which we envisioned could serve as a stereo- and regio-regular scaffold for rapid, UV-initiated thiol–ene click reactions for further tailoring polymer properties.61 Such a renewably-derived and stereoregular scaffold is attractive for many applications such as sustainable polymers and biologically active polymers. Thioglycerol and lauryl mercaptan were chosen as model thiols as they are structurally similar to renewable glycerol and lauryl alcohol.7,62,63 Additionally, the contrast in hydrophilicity/hydrophobicity of these thiols was expected to provide starkly different properties after modification (Fig. 5A). Real-time Fourier-transform infrared (RT-FTIR) spectroscopy was used to study the kinetics of thiol–ene reactions with poly(3). Fig. 5B depicts the kinetic data for the formation of poly(4) and poly(5) based on the conversion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C functional groups in poly(3). Both the reactions reach a plateau conversion of >99% in ∼1 min, suggesting near full consumption of the allylic pendant groups in poly(3). 1H NMR analysis of purified poly(4) and poly(5) indicates complete disappearance of vinylic proton signals as shown in Fig. 5C and D. Analysis of poly(4) and poly(5) by size-exclusion chromatography also showed an increase in Mn as compared to poly(3), but some of this increase may be due to fractionation as a result of purification steps (Table S9†). The solubility of synthesized polymers was tested in a range of solvents (Table S10†). As expected, poly(4) and poly(5) display starkly different solubility properties, with poly(4) being the only water-soluble polymer in the synthesized library.
C functional groups in poly(3). Both the reactions reach a plateau conversion of >99% in ∼1 min, suggesting near full consumption of the allylic pendant groups in poly(3). 1H NMR analysis of purified poly(4) and poly(5) indicates complete disappearance of vinylic proton signals as shown in Fig. 5C and D. Analysis of poly(4) and poly(5) by size-exclusion chromatography also showed an increase in Mn as compared to poly(3), but some of this increase may be due to fractionation as a result of purification steps (Table S9†). The solubility of synthesized polymers was tested in a range of solvents (Table S10†). As expected, poly(4) and poly(5) display starkly different solubility properties, with poly(4) being the only water-soluble polymer in the synthesized library.
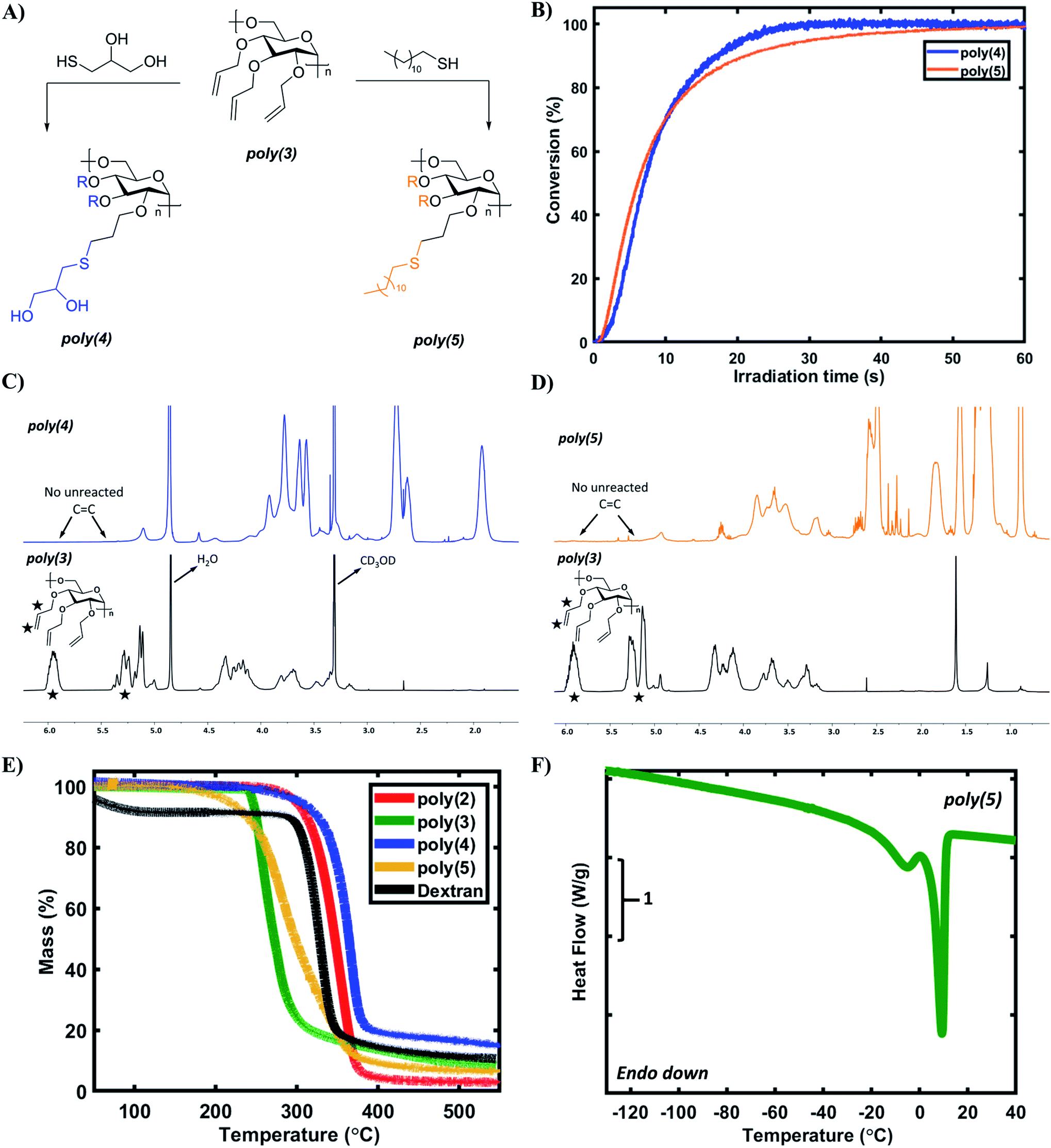 | ||
| Fig. 5 (A) Schematic depicting post-polymerization modification of poly(3) by UV-initiated thiol–ene reaction with 1-thioglycerol and lauryl mercaptan to create poly(4) and poly(5) respectively. (B) Post-polymerization modification kinetic data depicting conversion of the CC functional groups in poly(3) during UV irradiation. The ratio of thiol to ene groups in both the formulations is 1:1. (C) 1H NMR spectrum of poly(4) in CD3OD indicating complete disappearance of CC proton signals. (D) 1H NMR spectrum of poly(5) in CDCl3 indicating complete disappearance of CC proton signals. (E) TGA curves (under N2, 10 °C min−1) of the levoglucosan-based polymers and control dextran. (F) DSC thermogram (second heating, 10 °C min−1) of poly(5) depicting the double melting peak. | ||
Thermal properties
Lastly, the thermal properties of the synthesized polysaccharides were examined via thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) to understand thermal stability and thermal transitions respectively. Dextran, a natural and commercial polysaccharide, was used as a control because it also contains 1,6-α-glycosidic linkages and glucopyranose rings. Except for poly(3), each of the homopolymers demonstrated excellent thermal stability with a Td,10% > 300 °C (Fig. 5E), consistent with the thermal stability of dextran and trimethylated levoglucosan polymer (Td,10% = 347 °C).19 Comparatively, poly(3) had a slightly lower Td,10% (250 °C), which could be attributed to the fragmentation of pendant allylic ether groups at this temperature, consistent with literature.64 In general, levoglucosan-based polysaccharides possess excellent thermal stability even at moderate molecular weights, likely due to the rigid glucopyranose ring in the backbone. The glass transition temperature (Tg) of poly(2), poly(3), and poly(4) was observed to be 32 °C, 5 °C, and −14 °C, respectively (Fig. S24–S26†); no Tg was observed for poly(5) down to −150 °C, which is consistent with other lauryl-pendant polymers.62,65 The Tg values of poly(2), poly(3), and poly(5) are predictably lower than that of the control dextran (199 °C), potentially due to the strong hydrogen bonding between unsubstituted dextran chains. Surprisingly, poly(4) exhibited a sub-zero Tg despite the presence of pendant hydroxyl groups, potentially due to the added flexibility of the aliphatic methylene units. Additionally, the Tg for poly(2) is lower than that of trimethylated levoglucosan polymer (Tg ∼300 °C).19 It is hypothesized that this result can be explained based on the steric bulk of pendant groups. As compared to methyl groups, benzyl groups possess higher steric bulk, leading to higher free volume between adjacent polymer chains. A higher free volume will allow polymer chains to slide past each other more easily, thereby resulting in a lower Tg. Overall, DSC analysis demonstrates that the Tg of levoglucosan-based polysaccharides can be tailored based on the identity of the pendant groups, with a remarkable Tg window of >180 °C accessible with the few pendant groups evaluated in this study.DSC analysis also revealed an interesting double melting peak for poly(5) as shown in Fig. 5F (Tm1 = −5 °C, Tm2 = 9 °C). The crystallinity of poly(5) is most likely due to lauryl side chain crystallization, also observed in other polymeric systems with lauryl side chains.62,65 However, the double melting phenomenon could be caused by multiple factors. One explanation is the lamellar thickness model, which attributes the double melting behavior to the presence of lamellae with two different thicknesses.66–68 Other potential explanations include the crystallization of poly(5) chains with varying degrees of side chain functionalization and some extent of polysaccharide backbone crystallization. It is also worth highlighting that even at moderate molecular weights, poly(3) offers very high functionality for modification (for Mn = 6.7 kDa the number of allyl groups = 72), enabling the facile synthesis of highly functional materials. The high thermal stability, wide range of accessible Tg values, and the potential crystallization phenomena of these derivatives demonstrate that poly(3) is an excellent renewable scaffold for post-polymerization reactions to tailor properties to desired applications in a variety of applications. Furthermore, the levoglucosan platform provides easy access to fully functionalized dextran derivatives with the ability to install the desired pendant group both pre- and post-polymerization.
Conclusions
In conclusion, we have synthesized functional stereoregular 1,6-α linked polysaccharides with tunable thermal properties from cellulose-derived levoglucosan via cROP and post-polymerization thiol–ene click reactions. Through systematic screening experiments, we have identified green and recyclable metal triflate catalysts for cROP of levoglucosan derivatives under mild conditions. We have also provided kinetic, thermodynamic, and mechanistic insights into cROP of these derivatives. Computational studies reveal that initial ring-opening of levoglucosan derivatives is energetically favored at the 1,6 anhydro linkage and subsequently nucleophilic addition of monomer to the carbenium intermediate leads to 1,6-α glycosidic linkages in a regio- and stereo-specific manner. The allyl-functional polymer has been identified as an excellent scaffold for synthesis of highly functional materials via rapid, UV-meditated thiol–ene modifications. Levoglucosan-based polysaccharides generally demonstrate excellent thermal stability and a vast Tg window of >180 °C is accessible with the few pendant groups highlighted in this study. We believe that levoglucosan exhibits great potential as a renewable feedstock for the development of next generation sustainable and biocompatible polymers with tailored properties.Data availability
The datasets supporting this article are provided in the ESI.†Author contributions
M. K. P. wrote the manuscript and performed all experimental studies and data analysis. Y. R. performed all computational studies and contributed to the computation related text and figures in the manuscript. D. J. S. provided mentorship and edited the manuscript. C. J. C. provided mentorship for computational studies and edited the computation related parts of the manuscript. C. J. E. and T. M. R. provided mentorship, research direction, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported and funded by the NSF Center for Sustainable Polymers at the University of Minnesota; a National Science Foundation supported Center for Chemical Innovation (CHE-1901635). The authors acknowledge John Beumer for preparing the TOC graphic.References
- R. Geyer, in Plastic Waste and Recycling, ed. Trevor M. Letcher, 2020, pp. 13–32 Search PubMed.
- M. Hong and E. Y.-X. Chen, Future Directions for Sustainable Polymers, Trends Chem., 2019, 1, 148–152 CrossRef CAS.
- L. A. Lucia, Lignocellulosic biomass: A potential feedstock to replace petroleum, BioResources, 2008, 3, 981–982 Search PubMed.
- J. C. Serrano-Ruiz, R. Luque and A. Sepúlveda-Escribano, Transformations of biomass-derived platform molecules: From high added-value chemicals to fuels via aqueous-phase processing, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- O. Olatunji, S. Akinlabi and N. Madushele in Valorization of Biomass to Value-Added Commodities, Springer Nature Switzerland AG, ed. A. O. A. M. O. Daramola, 2020, pp. 3–20 Search PubMed.
- R. M. O’dea, J. A. Willie and T. H. Epps, 100th Anniversary of Macromolecular Science Viewpoint: Polymers from Lignocellulosic Biomass. Current Challenges and Future Opportunities, ACS Macro Lett., 2020, 9, 476–493 CrossRef.
- F. H. Isikgor and C. R. Becer, Lignocellulosic biomass: a sustainable platform for the production of bio-based chemicals and polymers, Polym. Chem., 2015, 6, 4497–4559 RSC.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- Z. Zhang, J. Song and B. Han, Catalytic Transformation of Lignocellulose into Chemicals and Fuel Products in Ionic Liquids, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- N. Brun, P. Hesemann and D. Esposito, Expanding the biomass derived chemical space, Chem. Sci., 2017, 8, 4724–4738 RSC.
- S. Maduskar, V. Maliekkal, M. Neurock and P. J. Dauenhauer, On the Yield of Levoglucosan from Cellulose Pyrolysis, ACS Sustain. Chem. Eng., 2018, 6, 7017–7025 CrossRef CAS.
- M. R. Rover, A. Aui, M. M. Wright, R. G. Smith and R. C. Brown, Production and purification of crystallized levoglucosan from pyrolysis of lignocellulosic biomass, Green Chem., 2019, 21, 5980–5989 RSC.
- I. Itabaiana Junior, M. Avelar Do Nascimento, R. O. M. A. De Souza, A. Dufour and R. Wojcieszak, Levoglucosan: A promising platform molecule?, Green Chem., 2020, 22, 5859–5880 RSC.
- I. G. Hakeem, P. Halder, M. H. Marzbali, S. Patel, S. Kundu, J. Paz-Ferreiro, A. Surapaneni and K. Shah, Research progress on levoglucosan production via pyrolysis of lignocellulosic biomass and its effective recovery from bio-oil, J. Environ. Chem. Eng., 2021, 9, 105614 CrossRef CAS.
- J. Wang, Z. Lu and A. Shah, Techno-economic analysis of levoglucosan production via fast pyrolysis of cotton straw in China, Biofuels, Bioprod. Biorefining, 2019, 13, 1085–1097 CrossRef CAS.
- A. Zheng, T. Chen, J. Sun, L. Jiang, J. Wu, Z. Zhao, Z. Huang, K. Zhao, G. Wei, F. He and H. Li, Toward Fast Pyrolysis-Based Biorefinery: Selective Production of Platform Chemicals from Biomass by Organosolv Fractionation Coupled with Fast Pyrolysis, ACS Sustain. Chem. Eng., 2017, 5, 6507–6516 CrossRef CAS.
- K. Wu, H. Wu, H. Zhang, B. Zhang, C. Wen, C. Hu, C. Liu and Q. Liu, Enhancing levoglucosan production from waste biomass pyrolysis by Fenton pretreatment, Waste Manag., 2020, 108, 70–77 CrossRef CAS PubMed.
- S. Penczek, P. Kubisa and K. Matyjaszewski, Bicyclic Monomers in Cationic Ring-Opening Polymerization, Adv. Polym. Sci., 1985, 139–176 CrossRef.
- D. Yoshida and T. Yoshida, Elucidation of High Ring-Opening Polymerizability of Methylated 1,6-Anhydro Glucose, J. Polym. Sci. Part A Polym. Chem., 2009, 47, 1013–1022 CrossRef CAS.
- T. Yoshida, Synthesis of polysaccharides having specific biological activities, Prog. Polym. Sci., 2001, 26, 379–441 CrossRef CAS.
- M. Grinstaff, R. Xiao and M. W. Grinstaff, Chemical Synthesis of Polysaccharides and Polysaccharide Mimetics Progress in Polymer Science Chemical synthesis of polysaccharides and polysaccharide mimetics, Prog. Polym. Sci., 2017, 74, 78–116 CrossRef.
- G. L. Gregory, E. M. Lopez-Vidal and A. Buchard, Polymers from sugars: cyclic monomer synthesis, ring-opening polymerisation, material properties and applications, Chem. Commun., 2017, 53, 2198–2217 RSC.
- A. S. Balijepalli and M. W. Grinstaff, Poly-Amido-Saccharides (PASs): Functional Synthetic Carbohydrate Polymers Inspired by Nature, Acc. Chem. Res., 2020, 53, 2167–2179 CrossRef CAS PubMed.
- M. Varghese, R. S. Rokosh, C. A. Haller, S. L. Chin, J. Chen, E. Dai, R. Xiao, E. L. Chaikof and M. W. Grinstaff, Sulfated poly-amido-saccharides (sulPASs) are anticoagulants: In vitro and in vivo, Chem. Sci., 2021, 12, 12719–12725 RSC.
- K. Kobayashi, H. Sumitomo and H. Ichikawa, Regioselectively Modified Stereoregular Polysaccharides. 8. Synthesis and Functions of Partially 3-O-Octadecylated (1→ 6) -α-D- Glucopyranans, Macromolecules, 1986, 19, 529–535 CrossRef CAS.
- M. D. Gallovic, S. Bandyopadhyay, H. Borteh, D. G. Montjoy, M. A. Collier, K. J. Peine, B. E. Wyslouzil, E. M. Bachelder and K. M. Ainslie, Microparticles formulated from a family of novel silylated polysaccharides demonstrate inherent immunostimulatory properties and tunable hydrolytic degradability, J. Mater. Chem. B, 2016, 4, 4302–4312 RSC.
- S. Iqbal, R. Marchetti, A. Aman, A. Silipo, S. A. U. Qader and A. Molinaro, Enzymatic and acidic degradation of high molecular weight dextran into low molecular weight and its characterizations using novel Diffusion-ordered NMR spectroscopy, Int. J. Biol. Macromol., 2017, 103, 744–750 CrossRef CAS PubMed.
- F. Diot-Néant, E. Rastoder, S. A. Miller and F. Allais, Chemo-Enzymatic Synthesis and Free Radical Polymerization of Renewable Acrylate Monomers from Cellulose-Based Lactones, ACS Sustain. Chem. Eng., 2018, 6, 17284–17293 CrossRef.
- T. Debsharma, Y. Yagci and H. Schlaad, Cellulose-Derived Functional Polyacetal by Cationic Ring-Opening Polymerization of Levoglucosenyl Methyl Ether, Angew. Chem., Int. Ed., 2019, 58, 18492–18495 CrossRef CAS PubMed.
- T. Debsharma, F. N. Behrendt, A. Laschewsky and H. Schlaad, Ring-Opening Metathesis Polymerization of Biomass-Derived Levoglucosenol, Angew. Chemie, 2019, 131, 6790–6793 CrossRef.
- P. Ray, T. Hughes, C. Smith, M. Hibbert, K. Saito and G. P. Simon, Development of bio-acrylic polymers from Cyrene™: Transforming a green solvent to a green polymer, Polym. Chem., 2019, 10, 3334–3341 RSC.
- K. Kaya, T. Debsharma, H. Schlaad and Y. Yagci, Cellulose-based polyacetals by direct and sensitized photocationic ring-opening polymerization of levoglucosenyl methyl ether, Polym. Chem., 2020, 11, 6884–6889 RSC.
- S. Fadlallah, A. A. M. Peru, A. L. Flourat and F. Allais, A straightforward access to functionalizable polymers through ring-opening metathesis polymerization of levoglucosenone-derived monomers, Eur. Polym. J., 2020, 138, 109980 CrossRef CAS.
- M. G. Banwell, X. Liu, L. A. Connal and M. G. Gardiner, Synthesis of Functionally and Stereochemically Diverse Polymers via Ring-Opening Metathesis Polymerization of Derivatives of the Biomass-Derived Platform Molecule Levoglucosenone Produced at Industrial Scale, Macromolecules, 2020, 53, 5308–5314 CrossRef CAS.
- S. Fadlallah, A. A. M. Peru, L. Longé and F. Allais, Chemo-enzymatic synthesis of a levoglucosenone-derived bi-functional monomer and its ring-opening metathesis polymerization in the green solvent Cyrene™, Polym. Chem., 2020, 11, 7471–7475 RSC.
- F. Diot-Néant, L. Mouterde, S. Fadlallah, S. A. Miller and F. Allais, Sustainable Synthesis and Polycondensation of Levoglucosenone-Cyrene-Based Bicyclic Diol Monomer: Access to Renewable Polyesters, ChemSusChem, 2020, 13, 2613–2620 CrossRef PubMed.
- T. Debsharma, B. Schmidt, A. Laschewsky and H. Schlaad, Ring-Opening Metathesis Polymerization of Unsaturated Carbohydrate Derivatives: Levoglucosenyl Alkyl Ethers, Macromolecules, 2021, 54, 2720–2728 CrossRef CAS.
- E. R. Ruckel and C. Schuerch, Preparation of High Polymers from l,6-Anhydro-2,3,4-tri-O-Substituted β-D-Glucopyranose, J. Org. Chem., 1966, 31, 2233–2239 CrossRef CAS.
- Y. Du Hongmei Liu, S. Cheng, J. Liu, Y. Du and Z. Bai, Synthesis of Pentasaccharide and Heptasaccharide Derivatives and Their Effects on Plant Growth, J. Agric. Food Chem., 2008, 56, 5634–5638 CrossRef PubMed.
- A. Fürstner, M. Albert, J. Mlynarski, M. Matheu and E. DeClercq, Structure Assignment, Total Synthesis, and Antiviral Evaluation of Cycloviracin B1, J. Am. Chem. Soc., 2003, 125, 13132–13142 CrossRef PubMed.
- T. Kakuchi, A. Kusuno, M. Miura and H. Kaga, Cationic ring-opening polymerization of 1,6-anhydro- 2,3,4-tri-O-allyl-b-D-glucopyranose as a convenient synthesis of dextran, Macromol. Rapid Commun., 2000, 21, 1003–1006 CrossRef CAS.
- T. Laird, Special feature section: Safety of chemical processes, Org. Process Res. Dev., 2002, 6, 876 CrossRef CAS.
- Q. Yang, M. Sheng, J. J. Henkelis, S. Tu, E. Wiensch, H. Zhang, Y. Zhang, C. Tucker and D. E. Ejeh, Explosion Hazards of Sodium Hydride in Dimethyl Sulfoxide, N, N-Dimethylformamide, and N, N-Dimethylacetamide, Org. Process Res. Dev., 2019, 23, 2210–2217 CrossRef CAS.
- T. Uryu, H. Tachikawa, K.-I. Ohaku, K. Terui and K. Matsuzaki, Synthesis of 2,3,4-tri-O-benzyl-[1→6]-α-D-glucopyranan and [1→6]-α-D-glucopyranan with high molecular weight by polymerization of 1,6-anhydro-2,3,4-tri-O-benzyl-β-D-glucopyranose, Makromol. Chem., 1977, 178, 1929–1940 CrossRef CAS.
- J. Zachoval and C. Schuerch, Steric Control in the Polymerization of 1,6-Anhydro-B-D-glucopyranose Derivatives, J. Am. Chem. Soc., 1969, 91, 1165–1169 CrossRef CAS.
- T. Kakuchi, A. Kusuno, M. Mori, T. Satoh, M. Miura, M. Sharfuddin and H. Kaga, Precision synthesis of (1→6)-α-D-glucopyranan by cationic ring-opening polymerization of 1,6-anhydro-2,3,4-tri-O-allyl-β-D-glucopyranose, Macromol. Symp., 2002, 181, 101–106 CrossRef CAS.
- A. Kusuno, M. Mori, T. Satoh, M. Miura, H. Kaga and T. Kakuchi, Enantioseparation properties of (1→6)-α-D-glucopyranan and (1→6)-α-D-mannopyranan tris(phenylcarbamate)s as chiral stationary phases in HPLC, Chirality, 2002, 14, 498–502 CrossRef CAS PubMed.
- L. You, T. E. Hogen-Esch, Y. Zhu, J. Ling and Z. Shen, Brønsted acid-free controlled polymerization of tetrahydrofuran catalyzed by recyclable rare earth triflates in the presence of epoxides, Polymer, 2012, 53, 4112–4118 CrossRef CAS.
- L. Fu, L. Li, J. Wang, K. Knickelbein, L. Zhang, I. Milligan, Y. Xu, K. O'Hara, L. Bitterman and W. Du, Synthesis of clickable amphiphilic polysaccharides as nanoscopic assemblies, Chem. Commun., 2014, 50, 12742–12745 RSC.
- N. Teramoto, T. Motoyama, R. Yosomiya and M. Shibata, Synthesis, thermal properties, and biodegradability of propyl-etherified starch, Eur. Polym. J., 2003, 39, 255–261 CrossRef CAS.
- C. Lorenzini, D. L. Versace, C. Gaillet, C. Lorthioir, S. Boileau, E. Renard and V. Langlois, Hybrid networks derived from isosorbide by means of thiol-ene photoaddition and sol-gel chemistry, Polymer, 2014, 55, 4432–4440 CrossRef CAS.
- D. J. Saxon, M. Nasiri, M. Mandal, S. Maduskar, P. J. Dauenhauer, C. J. Cramer, A. M. Lapointe and T. M. Reineke, Architectural Control of Isosorbide-Based Polyethers via Ring-Opening Polymerization, J. Am. Chem. Soc., 2019, 141, 5107–5111 CrossRef CAS PubMed.
- M. Lahcini, H. Qayouh, T. Yashiro, S. M. Weidner and H. R. Kricheldorf, Bismuth-triflate-catalyzed polymerizations of ε-caprolactone, Macromol. Chem. Phys., 2011, 212, 583–591 CrossRef CAS.
- N. Nomura, A. Taira, T. Tomioka and M. Okada, Catalytic approach for cationic living polymerization: Sc(OTf)3-catalyzed ring-opening polymerization of lactones, Macromolecules, 2000, 33, 1497–1499 CrossRef CAS.
- A. E. Neitzel, T. J. Haversang and M. A. Hillmyer, Organocatalytic Cationic Ring-Opening Polymerization of a Cyclic Hemiacetal Ester, Ind. Eng. Chem. Res., 2016, 55, 11747–11755 CrossRef CAS.
- M. Helou, J. M. Brusson, J. F. Carpentier and S. M. Guillaume, Functionalized polycarbonates from dihydroxyacetone: Insights into the immortal ring-opening polymerization of 2,2-dimethoxytrimethylene carbonate, Polym. Chem., 2011, 2, 2789–2795 RSC.
- P. Kubisa and J. P. Vairon, Ring-Opening Polymerization of Cyclic Acetals, Polymer Science: A Comprehensive Reference 10 Vol. Set, 2012, vol. 4, pp. 183–211 Search PubMed.
- B. T. Whiting and G. W. Coates, Synthesis and Polymerization of Bicyclic Ketals : A Practical Route to High-Molecular Weight Polyketals, J. Am. Chem. Soc., 2013, 135(30), 10974–10977 CrossRef CAS PubMed.
- M. Kawalec, M. Śmiga-Matuszowicz and P. Kurcok, Counterion and solvent effects on the anionic polymerization of β-butyrolactone initiated with acetic acid salts, Eur. Polym. J., 2008, 44, 3556–3563 CrossRef CAS.
- G. Odian, Principles of Polymerization, John Wiley & Sons, 2004 Search PubMed.
- C. E. Hoyle and C. N. Bowman, Thiol-ene click chemistry, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- H. Sajjad, W. B. Tolman and T. M. Reineke, Block Copolymer Pressure-Sensitive Adhesives Derived from Fatty Acids and Triacetic Acid Lactone, ACS Appl. Polym. Mater., 2020, 2, 2719–2728 CrossRef CAS.
- S. Wang, S. Vajjala Kesava, E. D. Gomez and M. L. Robertson, Sustainable thermoplastic elastomers derived from fatty acids, Macromolecules, 2013, 46, 7202–7212 CrossRef CAS.
- T. K. Vardareli, S. Keskin and A. Usanmaz, Thermal degradation of poly(allyl methacrylate) by mass spectroscopy and TGA, J. Macromol. Sci. Part A Pure Appl. Chem., 2006, 43, 1569–1581 CrossRef CAS.
- D. J. Saxon, E. A. Gormong, V. M. Shah and T. M. Reineke, Rapid Synthesis of Chemically Recyclable Polycarbonates from Renewable Feedstocks, ACS Macro Lett., 2021, 10, 98–103 CrossRef CAS.
- A. Alizadeh, L. Richardson, J. Xu, S. McCartney, H. Marand, Y. W. Cheung and S. Chum, Influence of structural and topological constraints on the crystallization and melting behavior of polymers. 1. Ethylene/1-octene copolymers, Macromolecules, 1999, 32, 6221–6235 CrossRef CAS.
- C. Schick, Differential scanning calorimetry (DSC) of semicrystalline polymers, Anal. Bioanal. Chem., 2009, 395, 1589–1611 CrossRef CAS PubMed.
- O. P. Bajpai, S. Panja, S. Chattopadhyay and D. K. Setua, Process-structure-property relationships in nanocomposites based on piezoelectric-polymer matrix and magnetic nanoparticles, Elsevier Ltd., 2015 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00146b |
| ‡ Co-second authors. |
| This journal is © The Royal Society of Chemistry 2022 |