
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn unexpected all-metal aromatic tetranuclear silver cluster in human copper chaperone Atox1†
Xiuxiu
Wang
ab,
Zong-Chang
Han
c,
Wei
Wei
 *abf,
Hanshi
Hu
c,
Pengfei
Li
d,
Peiqing
Sun
a,
Xiangzhi
Liu
b,
Zhijia
Lv
g,
Feng
Wang
g,
Yi
Cao
d,
Zijian
Guo
*ae,
Jun
Li
*ch and
Jing
Zhao
*abef
*abf,
Hanshi
Hu
c,
Pengfei
Li
d,
Peiqing
Sun
a,
Xiangzhi
Liu
b,
Zhijia
Lv
g,
Feng
Wang
g,
Yi
Cao
d,
Zijian
Guo
*ae,
Jun
Li
*ch and
Jing
Zhao
*abef
aState Key Laboratory of Coordination Chemistry, Chemistry and Biomedicine Innovation Center (ChemBIC), School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: jingzhao@nju.edu.cn
bSchool of Life Sciences, Nanjing University, Nanjing 210023, China
cDepartment of Chemistry and Key Laboratory of Organic Optoelectronics & Molecular Engineering of Ministry of Education, Tsinghua University, Beijing 100084, China
dNational Laboratory of Solid State Microstructure, Department of Physics, Nanjing University, Nanjing 210023, China
eNanchuang (Jiangsu) Institute of Chemistry and Health, Nanjing 210023, China
fShenzhen Research Institute, Nanjing University, Shenzhen 518000, China
gElias James Corey Institute of Biomedical Research, Wuxi Biortus Biosciences Co., Ltd, Jiangyin 214437, China
hDepartment of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China
First published on 30th May 2022
Abstract
Metal clusters, such as iron–sulfur clusters, play key roles in sustaining life and are intimately involved in the functions of metalloproteins. Herein we report the formation and crystal structure of a planar square tetranuclear silver cluster when silver ions were mixed with human copper chaperone Atox1. Quantum chemical studies reveal that two Ag 5s1 electrons in the tetranuclear silver cluster fully occupy the one bonding molecular orbital, with the assumption that this Ag4 cluster is Ag42+, leading to extensive electron delocalization over the planar square and significant stabilization. This bonding pattern of the tetranuclear silver cluster represents an aromatic all-metal structure that follows a 4n + 2 electron counting rule (n = 0). This is the first time an all-metal aromatic silver cluster was observed in a protein.
Introduction
Silver ions and silver nanoparticles have been widely used in medicine with a long history.1–4 They are best known as antibacterial agents with excellent biocompatibility in industrial, healthcare and domestic applications. Over the past few decades, the antibacterial mechanisms of silver and silver nanoparticles were intensively studied; the major mechanisms include the destruction of the [4Fe–4S] clusters of proteins,5,6 the displacement of the catalytic metals in metalloenzymes7 and the membrane disruption.8–11 The interaction between silver-containing substances and metal transporters directly affects the toxicity and metabolism processes of silver in biosystems. Studies on the precise reaction mode between silver and endogenous metalloproteins will not only provide an important basis for understanding the metabolism and biotransformation of silver in organisms, but also explore new coordination modes of silver in cells. Due to their tight coordination with sulfhydryl groups in proteins, it was suggested that silver ions interact with copper proteins in human cells.12–15 For example, recently, Batista and Zhuang et al. reported that the copper-regulated human odorant receptor OR2T11 showed a similar effect with ionic and nanoparticulate silver.16 Nevertheless, there is still scarce structural evidence on the interaction between copper chaperones and Ag.In human cells, copper regulation and trafficking is strictly controlled by complex systems including many cytosolic copper chaperones.17 As an Atx1-like copper chaperone, Atox1 is mainly in charge of delivering intracellular Cu(I) in eukaryotic cells from copper transporter Ctr1 at the plasma membrane and the metal-binding domain (MBD) of copper-transporting ATPase with a conserved CysXXCys metal-binding motif.18 An inspiring report by He et al. showed that small molecules significantly attenuate cancer cell proliferation by inhibiting the human copper trafficking of proteins Atox1 and CCS.19 Notably, an XAS investigation of Ag coordination in Atox1 demonstrated that Ag binds in digonal coordination to the Cu(I) binding loop in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry.20 Inspired by these pioneering studies, we set out to explore the structural basis of the interaction between silver ions and Atox1.
:1 stoichiometry.20 Inspired by these pioneering studies, we set out to explore the structural basis of the interaction between silver ions and Atox1.
Results and discussion
Mass and NMR spectroscopy measurements
We first determined the binding conditions between Atox1 and excessive silver ions (molar concentration: Atox1 = 0.135 mM, Ag = 1.35 mM) in the solution state by liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS). The data suggested that one equivalent of Atox1 could bind three to four equivalents of Ag under 0 to 0.1 mM dithiothreitol (DTT) treatment (Fig. S1A–C†). Since there is only one reported metal-binding site in Atox1 (Cys12 and Cys15), the silver ions might form a complex as a result of the protein–metal interaction. However, when the concentration of DTT was raised to 1 mM, the binding of Ag with Atox1 appeared to be minimal (Fig. S1D†). The binding sites of Ag in Atox1 were further analyzed by 1H and 15N-HSQC NMR spectroscopy on the 15N isotopic labeled Atox1 (Fig. S2†). The overlap of two spectra in Fig. S2† showed that the binding of Ag to apo-Atox1 resulted in a significant shift of several peaks (Cys12, Gly14, Cys15 and Ala16), while the majority of signals were barely perturbed by the Ag binding. This result suggested that Cys12 and Cys15 residues are the binding sites of Ag in solution, and the overall protein structure remains unchanged. The data are also in agreement with the NMR measurement of Cu(I) binding to Atox1.21,22Mechanical stability study of the Ag complex in Atox1
Then we set out to study the mechanical stability of the Ag complex in Atox1. Recently, the mechanical stability of metal complexes within proteins has been studied in detail using atomic force microscopy (AFM) based single molecule force spectroscopy.23–27 The mechanical stability of metal complexes was found to be directly related to the folding, stability and function of metalloproteins. We have revealed that the mechanical strength of the Au–S bond in gold-binding protein GolB is much weaker than that in the non-proteinaceous complexes, which may provide insights into the gold detoxification mechanism mediated by this protein.28 However, it remains technically challenging to study inter-protein metal complexes using single molecule AFM, because breaking intermolecular metal coordination bonds does not provide a clear mechanical signature in the force-extension curves.As most of the reported metal–Atox1 structures are dimers (Fig. S3†), we developed a novel single-molecule assay by engineering a loop bypass in between two Atox1 domains to reliably measure the mechanical stability of the Ag complex.29,30 The experimental design is depicted in Fig. 1A, which combines a loop bypass to recognize the breakage of a metal complex with clear mechanical fingerprinting domains to identify single molecule events, and specific molecular anchoring sites for efficiently picking up protein molecules. The engineered chimeric polyprotein included two Atox1 domains with a long unstructured loop in the center. Rupturing the metal cluster would release the length of the loop as well as the unfolded Atox1 sequences in between two metal binding sites in the force-extension curves. Besides the two Atox1 domains, four GB1 domains were flanked on both ends to serve as a mechanical fingerprint to identify single molecule events. The C-terminal SNAP domain was used to covalently link the polyprotein to the glass surface and the N-terminal Cohesin domain was used to form strong reversible binding with the XMod-Dockerin motif linked to the cantilever tip to reliably pickup single molecules. Then, the AFM experiment in buffer was conducted. In Tris buffer without adding Ag ions, the force-extension curves showed mainly the mechanical features of unfolding of GB1 domains (Fig. 1B) with contour length increments (ΔLc) of ∼18 nm (Fig. 1C) and unfolding forces of ∼150 pN (Fig. 1D) at a pulling speed of 400 nm s−1. The rupture of Cohesin with XMod-Dockerin could occur in either a single step or two steps (Fig. S4†). The unfolding of apo-Atox1 occurred at forces below the detection limit of our AFM (∼5 pN). However, when 1 μg mL−1 of Ag ions was added to the system, in ∼70% of the events showing a clear mechanical fingerprint, we observed an additional peak preceding the unfolding events of GB1 and the ΔLc value of this peak was ∼63 nm, consistent with the expected length change for the breakage of the Ag complex (Fig. 1C). In a few rare cases, we observed that the rupture of the Ag complex proceeded in two steps (Fig. S3†). Prior to the rupture of the metal complex, partial unfolding of the sequence outside the metal complex in an Atox1 domain could also be observed. The rupture forces of the Ag complex in these events were similar to that without partial unfolding of Atox1. Nonetheless, observation of the rupturing signature of the Ag complex suggests that it can remain stable in solution before force is applied. The low frequency of occurrence of this event (∼70%) can be attributed to its low stability in solution. The low stability is also evidenced by the low average rupture forces for the Ag complex of ∼64 pN (Fig. 1D), which is much weaker than the mechanical stability of GB1 and other metal chelation bonds reported in the literature.23–28,31 By comparing with other reported systems containing metal–thiol bonds in proteins, the very low mechanical stability of the Ag–Atox1 complex indicated that there might not be a simple Ag–S bond formed when Ag interacted with Atox1 protein under solution conditions (Table S1†).2,12,32–35
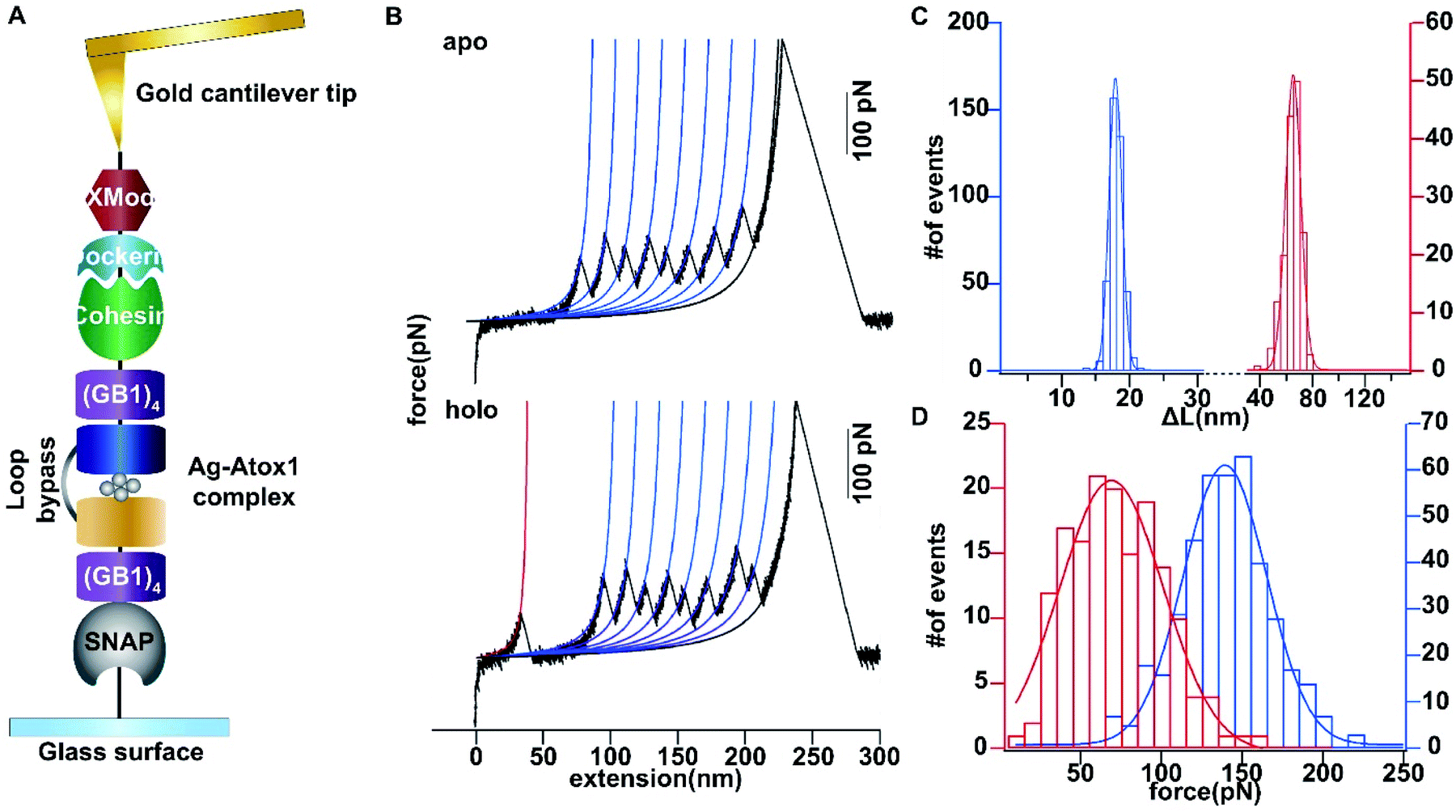 | ||
| Fig. 1 Single-molecule force spectroscopy analysis of the mechanical stability of the Ag4–(Atox1)2 complex. (A) Depiction of the experimental pulling configuration, with XMod-Doc attached to the gold cantilever tip and Coh-(GB1)4-Atox1-linker-Atox1-(GB1)4-SNAP-tag attached to the glass surface. (B) Single-molecule force spectroscopy experiments on the engineered chimeric polyprotein in the absence and presence of Ag. The representative force-extension traces for the stretching of the polyprotein in the apo and holo forms are shown. (C) The distributions of contour length increments for the stretching of the polyprotein in the apo and holo forms are shown respectively. Given that Atox1 and the linker comprise 68 and 54 amino acids, respectively, and the N–C distance of the structured Atox1–Ag–Atox1 complex is 5.6 nm, the contour length increment is expected to be 63.75 nm ((68 aa × 2 + 54 aa) × 0.365 nm/aa − 5.6 nm). (D) The unfolding force histogram for the stretching of the polyprotein in the apo and holo forms is shown respectively. | ||
Crystal structure of Ag-bound Atox1 protein
To reveal the detailed structure of the Ag complex in Atox1, next, we started to resolve the crystal structure of Ag-bound Atox1 protein. As an Atx1-like copper chaperone, Atox1 is mainly responsible for delivering intracellular Cu(I) in eukaryotic cells, with very little perturbation by the Ag binding. The data are in agreement with the NMR measurement of Cu(I) binding to Atox1 from the copper transporter Ctr1 at the plasma membrane and the metal binding domain of copper-transporting ATPase with a conserved CysXXCys metal binding motif.18 Pivotal progress was made by Rosenzweig et al. on several Atox1 crystal structures in the presence of Cu(II), Hg(II), Cd(II), and cisplatin, and they elucidated the molecular details of the metal transfer process between Atox1 and its target MBDs (Fig. S3†).29,30At first, a lower resolution structure of the dimeric silver Ag4–(Atox1)2 was determined with 2.70 Å resolution (Fig. S5†) (PDB accession code 5F0W), In this structure, the two cysteine residue pairs are coordinated to the Ag ion with average Ag–Cys(S) distances of 2.34 Å. And to solve its phase problems, the method of molecular replacement was performed. In this case, we considered the occupancy of all the non-hydrogen atoms to be 1, including the Ag atoms. Instead, we introduced the B factor as a signal to describe the variance of each atom. For the four Ag atoms, the B factors are 35.62, 34.41, 30.60, and 32.94, respectively. The average B factor of these Ag atoms is 33.39. That means the four Ag atoms have relative stationary and fixed locations, suggesting the existence and validity of the tetranuclear silver clusters. It is worth noting that the two Atox1 proteins in the asymmetric unit are linked by two other Ag ions with an average distance of 2.95 Å from the cysteine coordinating Ag ions, indicating an unexpected Ag–Ag bond in the Ag4–(Atox1)2 structure (Fig. S6†).36,37 However, the 2.70 Å resolution structure of 5F0W is not sufficient to fully determine the possible existence of the Ag–Ag bond. So, we continued to optimize the crystallization conditions to get better quality crystals. After several rounds of screening, a better structure of Ag4–(Atox1)2 was determined with 1.75 Å resolution (Fig. S7A†) (PDB accession code 7DC1), and the superimposed 2Fo − Fc electron density map is shown in Fig. S7B.†
The tetrasilver clusters in Atox1 dimer
The details of the tetrasilver cluster in Atox1 dimer (7DC1) is shown in Fig. 2. In addition, the S–Ag–S bond angles of 168° are also consistent with the average values in linear and two-coordinate Ag–S complexes that are found in the small-molecule Cambridge Structural Database.38 Notably, the four silver ions have four Ag–Ag linkages with 3.17 Å and 2.83 Å. The average dihedral angle of the four Ag–Ag linkages is 154°.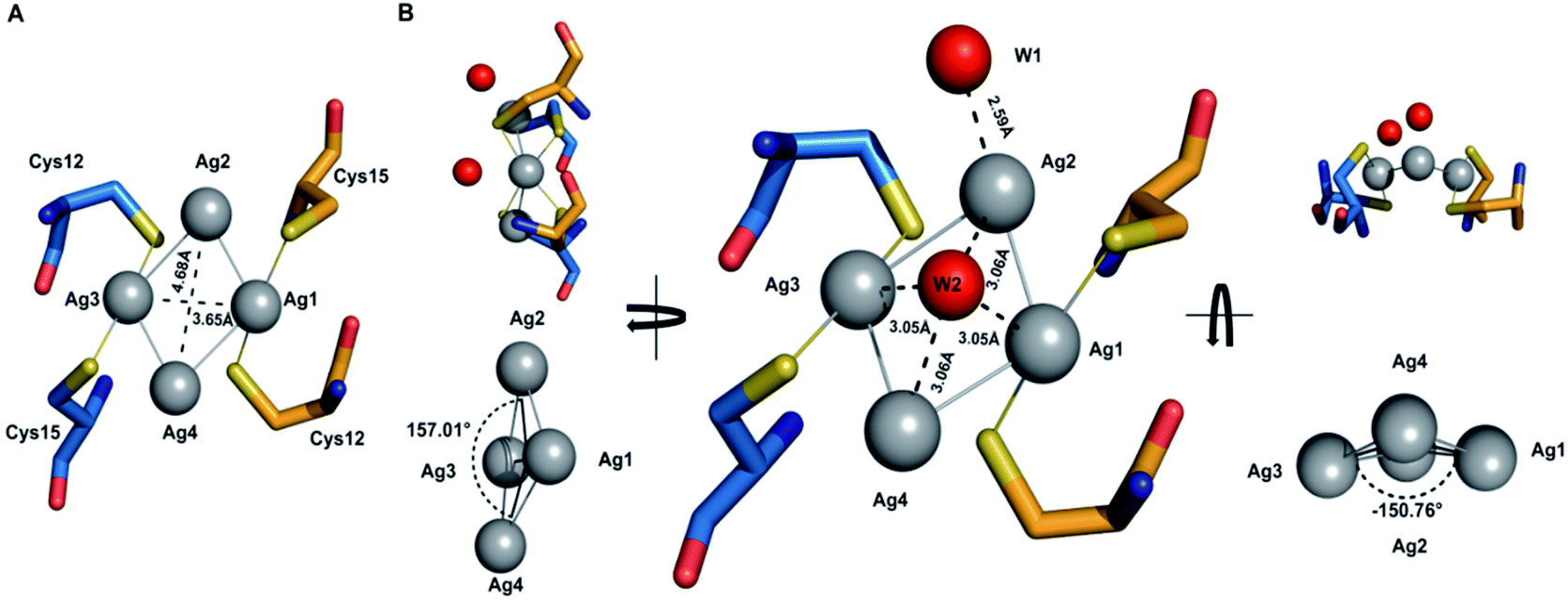 | ||
| Fig. 2 Close-up view showing details of the tetrasilver cluster in Atox1 dimer. The distances are: Ag1⋯Ag4 = 3.17 Å, Ag1⋯Ag3 = 3.65 Å, Ag1⋯Ag2 = 2.83 Å, and Ag2⋯Ag4 = 4.68 Å. The atomic angles are ∠Ag4–Ag1–Ag2 = 102.38° and ∠Ag1–Ag2–Ag3 = 74.69°. | ||
Interestingly, the only reported structure of Ag bound to MNK4 containing an MTCXXC metal binding domain is a monomer with one equivalent of Ag.15 And in all the reported structures of metal bridged Atox1 dimer, only one metal (Cu/C/Hg/cisPt) holds the two Atox1 monomers together with the extended hydrogen bonding network near the metal binding site (Fig. S3†). Our symmetrical Ag4–(Atox1)2 structure is more similar to the reported structural geometry of a tetranuclear copper cluster in the Bacillus subtilis Atx1-like copper chaperone protein CopZ, but the coordination of the [Cu4(S-Cys)4(N-His)2] cluster is different.39
On the other hand, Wang et al. reported a designed synthesis of tetranuclear silver clusters – by utilizing organic acetylide ligands. In their structures, the silver atoms form a square planar tetrasilver cluster through an Ag–Ag linkage (average length of 2.96 Å), which is held together by both σ- and π-bonding of the tert-butylacetylide anion in the μ4-η1,η1,η1,η2 mode.40 In our Ag4–(Atox1)2 structure (7DC1), there are only two oxygen atoms W1 and W2 from water around the four silver ions. W1 is near Ag2 with the distance of 2.59 Å, and the other oxygen atom W2 is onto the four silver ions with the average distance of 3.06 Å. These two oxygen atoms probably act as ligands to support and stabilize the tetranuclear silver clusters. Interestingly, although the mass spectrometry and AFM data implied that an uncertain polynucleated Ag cluster might form in Atox1 in the solution state, the crystal structure of Ag-bound Atox1 protein demonstrated the formation of a more stable tetranuclear silver cluster in the solid state. Some previous studies indicated that the growth of protein crystals promotes the packing of silver ions and direct silver cluster formation through specific interactions with Atox1.41
Quantum chemical investigation of the [Ag4]q+ cluster
To gain insight on the electronic structure and chemical bonding of the tetra-Ag cluster, quantum chemical calculations are performed by using density functional theory (DFT). Although it is difficult to determine the charge state of the [Ag4]q+ cluster directly through experimental approaches, EPR experiment shows no signal corresponding to the [Ag4]q+ cluster, as shown in Fig. S9,† implying that the system has a closed-shell electron configuration. We therefore carried out constrained DFT geometry optimizations on the [Ag4]q+ clusters in the cavity of 7DC1 by assuming Ag44+, Ag42+ and Ag40, respectively. As shown in Table S9,† the constrained DFT optimization result of Ag42+ is in reasonable agreement with the experimentally measured structure at 1.7 Å resolution. In contrast, the optimization of Ag44+ leads to much larger deviation of both Ag–Ag distances and dihedral angles and the optimization of Ag40 does not lead to a converged structure.The partially reduced Ag42+ cluster is not completely surprising given the usage of TCEP (tris(2-carboxyethyl)phosphine), which is a relatively strong reductive reagent with a redox potential of −0.33 V at pH 7. As a common redox reagent also known as Cleland's reagent, TCEP can help to reduce Ag+ ions to form Ag42+ in the preparation of Ag–Atox1, especially because of the extra stability of the aromatic Ag42+ cluster (see below):
| 4Ag+ + 2R–SH → R–S–S–R + Ag42+ + 2H+ |
The Ag42+ cluster is particularly stable because according to the Hückel molecular orbital (HMO) analysis of the three types of [Ag4]q+ cluster with D2h, D4h and Td symmetries, respectively (Tables S4–S6†), the four 5s AOs form one bonding MO (ag), one non-bonding MO (bu), and two antibonding ones (bu + ag) in D2h; one bonding MO (a1g), two non-bonding MOs (eu), and one antibonding MO (b1g) in D4h; and one bonding MO (a1) and three antibonding MOs (t2) in Td. All the three types of HMO analyses show that there is only one strong bonding MO and thus leads to a 2-electron counting rule, which shows that the two s-electron system of Ag42+ is the most stable one for the [Ag4]q+ cluster. The calculated frontier MOs based on the experimentally measured cavity and optimized D2h, D4h and Td geometries of Ag42+ are shown in Table S7,† which confirms the above conclusion. The MO contours of these four clusters show that the Ag42+ with C2 symmetry is qualitatively closer to a pseudo D2h structure. The slightly reduced C2 symmetry is a result of the ligand effects and intermolecular interactions in the complicated protein environment.
The correlation diagram for the orbital interaction along with the frontier canonical Kohn–Sham valence MOs of Ag42+ are shown in Fig. 3.42 The delocalization bonding contributed by the HOMO in the Ag42+ cation results in considerable aromatic stabilization, where it follows the 2-electron counting rule, also described by the Hückel 4n + 2 rule (n = 0). The normalized multicenter bond index of 0.46 between the four Ag atoms is comparable to 0.46 in square C4H42+. The ELF color-filled map (Fig. S8†) also shows non-negligible electron-pair density in the center of the cluster, supporting a type of delocalized, albeit weak, 4-center bonding interaction. The calculated nucleus-independent chemical shift (NICS) indices at the molecule-center, two triangle face-center, and 1 Å above the molecule-center display considerable negative values, which are comparable to those in benzene, also confirming the all-metal sigma-aromaticity of the Ag42+ cluster (Table S8†).43–46
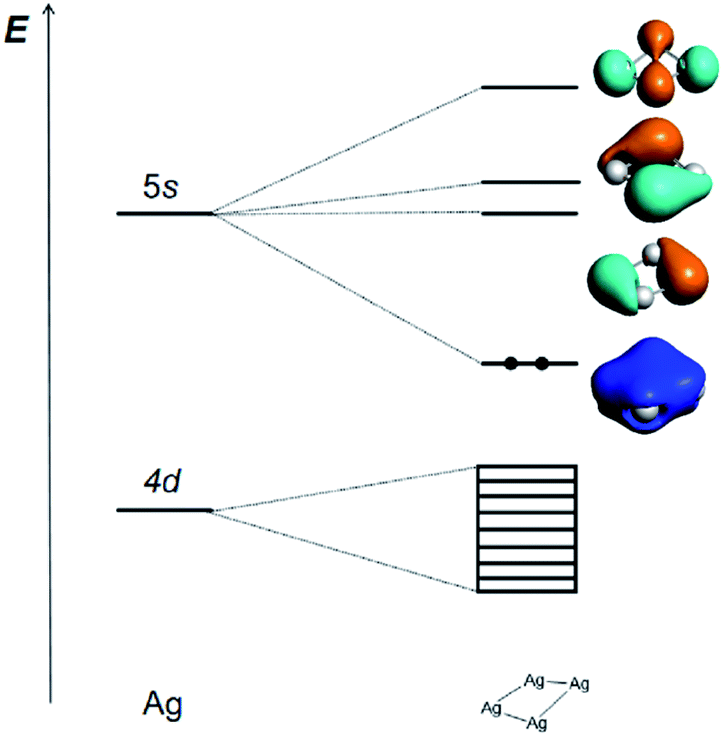 | ||
| Fig. 3 Schematic representation of the Kohn–Sham MO energy levels and the contours of four frontier Kohn–Sham canonical valence MOs (isosurface = 0.03 a.u.) at the PBE/TZP level of the Ag42+ cation, illustrating the bonding interactions between Ag atom and the Ag42+ dication. | ||
Conclusions
In summary, we have demonstrated the interactions between Ag ions and human copper chaperon Atox1 in crystal states. A tetranuclear silver cluster with 1.75 Å resolution is obtained, and the structural analysis of the Ag bound crystal structures and quantum chemical calculations have revealed surprising Ag–Ag bonding features and σ-aromaticity regarding the tetrasilver cluster geometry.Our results might have implication on the involvement of Atox1 in the silver transportation and detoxification. The Ag4 clusters were largely buried in two Atox1 domains and became less solvent accessible, which may also potentially minimize their impact on the redox equilibrium of the cell. As atox1 is a key protein involved in Cu(I) homeostasis in eukaryotic cells, these results suggest that silver ion toxicity relies on its direct binding to human copper chaperon and leads to the formation of an unusual complex. By studying the interaction between cisplatin and Atox1, Rosenzweig et al. suggested that there was a direct relationship between cisplatin resistance and copper homeostasis in vivo.30,47–49 Recently, Lombi et al. reported that the biotransformation of AgNPs was dominated by sulfidation in cells, which can be viewed as one of the cellular detoxification pathways for Ag.69 In view of the relatively weak stability of the Ag–Atox1 complex, the interaction between the Ag cluster and Atox1 is quite dynamic. Further work is desirable on the detailed crystal structure analysis of silver clusters and metalloproteins. Atox1 may be required to competitively bind toxic silver ions without affecting the function of copper transporters. This mechanically weak Ag–S binding enabled Atox1 to robustly maintain its biological function, suggesting a potential principle of metal ion transport in vivo. Our work might contribute to the understanding of the silver trafficking and detoxification mechanism of cellular internalized Ag from a precise structural point of view. This work may also stimulate future research towards interactions between silver and Atox1 in vivo.
On the other hand, the size-controlled synthesis of nanosilver clusters has been a challenge. Nanosilver materials, composed of several to a few hundred silver atoms, are not only best known as antibacterial agents with excellent biocompatibility in industrial and healthcare applications, but are also useful for their unique optical, catalytic, electronic and magnetic properties.50–53 The intrinsic properties of nanosilvers including clusters and particles are mainly determined by their size, shape, and structure.54,55 Hence, simple and size-controlled synthesis of nanosilver products has received great attention.54,56,57 In addition to the commonly reported synthesis of silver nanoparticles, some significant studies on silver nanoclusters were reported for their unique optical properties,53,58 such as Ag6, Ag8, and Ag12 silver nanoclusters with predictable sizes and emission energies,59 protein-based silver (Ag) nanoclusters with fluorescence excitation and excitation anisotropy spectra for revealing spectral and structural patterns,60 and highly luminescent Ag9 and Ag16 nanoclusters with tunable emissions.61 Thus, our work might suggest a new approach to synthesize nanosilver clusters by using protein template with applications probably in antibacterial studies, catalysis, or imaging.
More importantly, not only for aromatic compounds but also for all-metal clusters, aromaticity is a significant property to account for their stability. Since the groundbreaking work on the aromaticity observation in a series of bimetallic clusters was first reported by Boldyrev and Wang in 2001, the aromaticity concept was expanded into the arena of all-metal species.62 A large number of aromatic all-metal clusters including coinage metal clusters have been discovered in the gas phase.63–66 However, traditional chemical synthesis of the aromatic all-metal clusters often requires high temperatures, and complex procedures. Since the last two decades, proteins have been used as templates for the synthesis of metal nanomaterials in solution, and these approaches have been used to successfully encapsulate and stabilize a number of different enzymes with applications in biological catalysis.41,67,68 The aromatic tetrasilver clusters formed by Atox1 dimer herein may point out a new approach to the biocompatible protein-templated synthesis of aromatic metallic clusters. The chemical and physical properties of the reported aromatic tetrasilver cluster are under investigation. The metalloaromatic clusters are likely to attract much interest for their unique structures.
Data availability
All experimental data and detailed procedures are available in the ESI.†Author contributions
X. W., W. W. and J. Z. designed the research; X. W., Z.-C. H., W. W., H. H., P. L., P. S., X. L., Z. L., F. W. and Y. C. performed the research; X. W. and W. W. analyzed the data; Z.-C. H., H. H. and J. L. performed the theoretical study; and X. W., W. W., Z. G. and J. Z. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (21671099, 22025701), and the Shenzhen Basic Research Program (JCYJ20170413150538897, JCYJ20180508182240106), supported by the Fundamental Research Funds for the Central Universities (020514380139). The theoretical work was supported by the National Natural Science Foundation of China (22033005, J. L.) and by Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002, J. L.). The calculations were done using supercomputers at the Center for Computational Science and Engineering at SUSTech, the CHEM high-performance supercomputer cluster (CHEM-HPC) located at the Department of Chemistry of SUSTech, and the Computational Chemistry Laboratory of the Department of Chemistry under the Tsinghua Xuetang Talents Program.Notes and references
- S. Eckhardt, P. S. Brunetto, J. Gagnon, M. Priebe, B. Giese and K. M. Fromm, Chem. Rev., 2013, 113, 4708–4754 CrossRef CAS PubMed.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef CAS PubMed.
- S. Silver, L. T. Phung and G. Silver, J. Ind. Microbiol. Biotechnol., 2006, 33, 627–634 CrossRef CAS PubMed.
- J. R. Morones, J. L. Elechiguerra, A. Camacho, K. Holt, J. B. Kouri, J. T. Ramirez and M. J. Yacaman, Nanotechnology, 2005, 16, 2346–2353 CrossRef CAS PubMed.
- H. J. Park, J. Y. Kim, J. Kim, J. H. Lee, J. S. Hahn, M. B. Gu and J. Yoon, Water Res., 2009, 43, 1027–1032 CrossRef CAS PubMed.
- H. Y. Xu, F. Qu, H. Xu, W. H. Lai, Y. A. Wang, Z. P. Aguilar and H. Wei, BioMetals, 2012, 25, 45–53 CrossRef CAS PubMed.
- M. R. Ciriolo, P. Civitareale, M. T. Carri, A. Demartino, F. Galiazzo and G. Rotilio, J. Biol. Chem., 1994, 269, 25783–25787 CrossRef CAS PubMed.
- C. N. Lok, C. M. Ho, R. Chen, Q. Y. He, W. Y. Yu, H. Z. Sun, P. K. H. Tam, J. F. Chiu and C. M. Che, J. Proteome Res., 2006, 5, 916–924 CrossRef CAS PubMed.
- M. Yamanaka, K. Hara and J. Kudo, Appl. Environ. Microbiol., 2005, 71, 7589–7593 CrossRef CAS PubMed.
- W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed.
- Q. L. Feng, J. Wu, G. Q. Chen, F. Z. Cui, T. N. Kim and J. O. Kim, J. Biomed. Mater. Res., 2000, 52, 662–668 CrossRef CAS PubMed.
- C. F. Shaw, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS PubMed.
- M. R. Ciriolo, P. Civitareale, M. T. Carri, A. Demartino, F. Galiazzo and G. Rotilio, J. Biol. Chem., 1994, 269, 25783–25787 CrossRef CAS PubMed.
- J. Lee, M. M. O. Peña, Y. Nose and D. J. Thiele, J. Biol. Chem., 2002, 277, 4380–4387 CrossRef CAS PubMed.
- J. Gitschier, B. Moffat, D. Reilly, W. I. Wood and W. J. Fairbrother, Nat. Struct. Biol., 1998, 5, 47–54 CrossRef CAS PubMed.
- S. Li, L. Ahmed, R. Zhang, Y. Pan, H. Matsunami, J. L. Burger, E. Block, V. S. Batista and H. Zhuang, J. Am. Chem. Soc., 2016, 138, 13281–13288 CrossRef CAS PubMed.
- B.-E. Kim, T. Nevitt and D. J. Thiele, Nat. Chem. Biol., 2008, 4, 176–185 CrossRef CAS PubMed.
- S. Lutsenko, A. Gupta, J. L. Burkhead and V. Zuzel, Arch. Biochem. Biophys., 2008, 476, 22–32 CrossRef CAS PubMed.
- J. Wang, C. Luo, C. Shan, Q. You, J. Lu, S. Elf, Y. Zhou, Y. Wen, J. L. Vinkenborg, J. Fan, H. Kang, R. Lin, D. Han, Y. Xie, J. Karpus, S. Chen, S. Ouyang, C. Luan, N. Zhang, H. Ding, M. Merkx, H. Liu, J. Chen, H. Jiang and C. He, Nat. Chem., 2015, 7, 968–979 CrossRef CAS PubMed.
- G. Veronesi, T. Gallon, A. Deniaud, B. Boff, C. Gateau, C. Lebrun, C. Vidaud, F. Rollin-Genetet, M. Carrière, I. Kieffer, E. Mintz, P. Delangle and I. Michaud-Soret, Inorg. Chem., 2015, 54, 11688–11696 CrossRef CAS PubMed.
- A. K. Wernimont, D. L. Huffman, A. L. Lamb, T. V. O'Halloran and A. C. Rosenzweig, Nat. Struct. Mol. Biol., 2000, 7, 766 CrossRef CAS PubMed.
- J. Gitschier, B. Moffat, D. Reilly, W. I. Wood and W. J. Fairbrother, Nat. Struct. Mol. Biol., 1998, 5, 47–54 CrossRef CAS PubMed.
- J. Perales-Calvo, A. Lezamiz and S. Garcia-Manyes, J. Phys. Chem. Lett., 2015, 6, 3335–3340 CrossRef CAS PubMed.
- A. E. M. Beedle, A. Lezamiz, G. Stirnemann and S. Garcia-Manyes, Nat. Commun., 2015, 6, 7894 CrossRef CAS PubMed.
- P. Zheng, G. M. Arantes, M. J. Field and H. Li, Nat. Commun., 2015, 6, 7569 CrossRef CAS PubMed.
- P. Zheng and H. Li, J. Am. Chem. Soc., 2011, 133, 6791–6798 CrossRef CAS PubMed.
- P. Zheng and H. Li, Biophys. J., 2011, 101, 1467–1473 CrossRef CAS PubMed.
- W. Wei, Y. Sun, M. Zhu, X. Liu, P. Sun, F. Wang, Q. Gui, W. Meng, Y. Cao and J. Zhao, J. Am. Chem. Soc., 2015, 137, 15358–15361 CrossRef CAS PubMed.
- A. K. Wernimont, D. L. Huffman, A. L. Lamb, T. V. O'Halloran and A. C. Rosenzweig, Nat. Struct. Biol., 2000, 7, 766–771 CrossRef CAS PubMed.
- A. K. Boal and A. C. Rosenzweig, J. Am. Chem. Soc., 2009, 131, 14196–14197 CrossRef CAS PubMed.
- Y. Cao and H. Li, Nat. Mater., 2007, 6, 109–114 CrossRef CAS PubMed.
- G. D. Yuan, H. X. Liu, Q. Ma, X. Li, J. Y. Nie, J. L. Zuo and P. Zheng, J. Phys. Chem. Lett., 2019, 10, 5428–5433 CrossRef CAS PubMed.
- G. B. Song, F. Tian, H. X. Liu, G. Q. Li and P. Zheng, J. Phys. Chem. Lett., 2021, 12, 3860–3867 CrossRef CAS PubMed.
- W. Wei, Y. Sun, M. L. Zhu, X. Z. Liu, P. Q. Sun, F. Wang, Q. Gui, W. Y. Meng, Y. Cao and J. Zhao, J. Am. Chem. Soc., 2015, 137, 15358–15361 CrossRef CAS PubMed.
- R. K. Ainavarapu, J. Brujic, H. H. Huang, A. P. Wiita, H. Lu, L. W. Li, K. A. Walther, M. Carrion-Vazquez, H. B. Li and J. M. Fernandez, Biophys. J., 2007, 92, 225–233 CrossRef PubMed.
- M. Hong, W. Su, R. Cao, W. Zhang and J. Lu, Inorg. Chem., 1999, 38, 600–602 CrossRef CAS PubMed.
- E. Bosch and C. L. Barnes, Inorg. Chem., 2002, 41, 2543–2547 CrossRef CAS PubMed.
- A. Changela, K. Chen, Y. Xue, J. Holschen, C. E. Outten, T. V. O'Halloran and A. Mondragón, Science, 2003, 301, 1383–1387 CrossRef CAS PubMed.
- S. Hearnshaw, C. West, C. Singleton, L. Zhou, M. A. Kihlken, R. W. Strange, N. E. Le Brun and A. M. Hemmings, Biochemistry, 2009, 48, 9324–9326 CrossRef CAS PubMed.
- C.-Y. Gao, L. Zhao and M.-X. Wang, J. Am. Chem. Soc., 2011, 133, 8448–8451 CrossRef CAS PubMed.
- H. Wei, Z. D. Wang, J. Zhang, S. House, Y. G. Gao, L. M. Yang, H. Robinson, L. H. Tan, H. Xing, C. J. Hou, I. M. Robertson, J. M. Zuo and Y. Lu, Nat. Nanotechnol., 2011, 6, 93–97 CrossRef CAS PubMed.
- ADF2019, SCM Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2018, https://www.scm.com/.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 16 Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- Z. F. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. B. Howell, R. Safaei, C. A. Larson and M. J. Sailor, Mol. Pharmacol., 2010, 77, 887–894 CrossRef CAS PubMed.
- K. Katano, A. Kondo, R. Safaei, A. Holzer, G. Samimi, M. Mishima, Y.-M. Kuo, M. Rochdi and S. B. Howell, Cancer Res., 2002, 62, 6559–6565 CAS.
- Z. Xi, W. Guo, C. Tian, F. Wang and Y. Liu, Chem. Commun., 2013, 49, 11197–11199 RSC.
- X. Chen and H. Schluesener, Toxicol. Lett., 2008, 176, 1–12 CrossRef CAS PubMed.
- K. Chaloupka, Y. Malam and A. M. Seifalian, Trends Biotechnol., 2010, 28, 580–588 CrossRef CAS PubMed.
- Y. Tao, M. Li, J. Ren and X. Qu, Chem. Soc. Rev., 2015, 44, 8636–8663 RSC.
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- Y. Wu, D. Wang and Y. Li, Chem. Soc. Rev., 2014, 43, 2112–2124 RSC.
- T. Klaus, R. Joerger, E. Olsson and C. G. Granqvist, Proc. Natl. Acad. Sci., 1999, 96, 13611–13614 CrossRef CAS PubMed.
- R. R. Naik, S. J. Stringer, G. Agarwal, S. E. Jones and M. O. Stone, Nat. Mater., 2002, 1, 169–172 CrossRef CAS PubMed.
- C.-Y. Chiu, L. Ruan and Y. Huang, Chem. Soc. Rev., 2013, 42, 2512–2527 RSC.
- V. A. Morozov and M. Y. Ogawa, Inorg. Chem., 2013, 52, 9166–9168 CrossRef CAS PubMed.
- T. S. Sych, Z. V. Reveguk, V. A. Pomogaev, A. A. Buglak, A. A. Reveguk, R. R. Ramazanov, N. M. Romanov, E. V. Chikhirzhina, A. M. Polyanichko and A. I. Kononov, J. Phys. Chem. C, 2018, 122, 29549–29558 CrossRef CAS.
- X. Yuan, M. I. Setyawati, A. S. Tan, C. N. Ong, D. T. Leong and J. P. Xie, NPG Asia Mater., 2013, 5, e39 CrossRef CAS.
- X. Li, A. E. Kuznetsov, H.-F. Zhang, A. I. Boldyrev and L.-S. Wang, Science, 2001, 291, 859–861 CrossRef CAS PubMed.
- J. M. Mercero, A. I. Boldyrev, G. Merino and J. M. Ugalde, Chem. Soc. Rev., 2015, 44, 6519–6534 RSC.
- C. S. Wannere, C. Corminboeuf, Z.-X. Wang, M. D. Wodrich, R. B. King and P. v. R. Schleyer, J. Am. Chem. Soc., 2005, 127, 5701–5705 CrossRef CAS PubMed.
- F.-X. Pan, L.-J. Li, Y.-J. Wang, J.-C. Guo, H.-J. Zhai, L. Xu and Z.-M. Sun, J. Am. Chem. Soc., 2015, 137, 10954–10957 CrossRef CAS PubMed.
- A. I. Boldyrev and L.-S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed.
- B. R. Heywood, Nature, 1991, 349, 684–687 CrossRef.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935–4978 CrossRef CAS PubMed.
- A. Malysheva, A. Ivask, C. L. Doolette, N. H. Voelcker and E. Lombi, Nat. Nanotechnol., 2021, 16, 926–932 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1sc07122j |
| This journal is © The Royal Society of Chemistry 2022 |