
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting biomolecule adsorption on MoS2 nanosheets with high structural fidelity†
Le Nhan
Pham
 * and
Tiffany R.
Walsh
*
* and
Tiffany R.
Walsh
*
Institute for Frontier Materials, Deakin University, Geelong, Victoria 3216, Australia. E-mail: tiffany.walsh@deakin.edu.au
First published on 16th March 2022
Abstract
A new force field, MoSu-CHARMM, for the description of bio-interfacial structures at the aqueous MoS2 interface is developed, based on quantum chemical data. The force field describes non-covalent interactions between the MoS2 surface and a wide range of chemistries including hydrocarbon, alcohol, aldehyde, ketone, carboxylic acid, amine, thiol, and amino acid groups. Density functional theory (DFT), using the vdW-DF2 functional, is employed to create training and validation datasets, comprising 330 DFT binding energies for 21 organic compounds. Development of MoSu-CHARMM is guided by two criteria: (i) minimisation of energetic differences compared to target DFT data and (ii) preservation of the DFT energetic rankings of the different binding configurations. Force-field performance is validated against existing high-quality structural experimental data regarding adsorption of four 26-residue peptides at the aqueous MoS2 interface. Adsorption free energies for all twenty amino acids in liquid water are calculated to provide guidance for future peptide design, and interpret the properties of existing experimentally-identified MoS2-binding peptides. This force field will enable large-scale simulations of biological interactions with MoS2 surfaces in aqueous media where an emphasis on structural fidelity is prioritised.
Introduction
Molybdenum disulphide (MoS2) as a 2D material has emerged as a promising candidate for a wide array of applications such as biosensing,1–5 gas sensing,6–8 water purification,9–11 and drug delivery.12 These potential applications involve non-covalent interfacial interactions between the basal surface of MoS2 layers and adsorbates or analytes. Insights into these non-covalent interactions can provide guidance to adapt, functionalise, and activate the surface of MoS2, thus broadening its potential applications.Accordingly, both experimental and theoretical studies to investigate the interaction between MoS2 and several types of adsorbates have been reported. Experimental data revealed that 2D MoS2 layers have affinity with several small inorganic compounds.7,13–15 Other experimental studies have focussed on MoS2 interaction with a variety of larger organic molecules.15–19 For larger biological molecules such as proteins and peptides, MoS2 manifests a variable interaction.20–22 In addition, several attempts have sought to theoretically study the interactions between the MoS2 surface and a wide range of compounds to shed light on these interfaces and explain experimentally observed properties. Density functional theory (DFT) has been used to study small-size adsorbates23–29 in the gas phase environment because its capabilities to (usually) provide a solid understanding of the energetic behaviours and geometrical structures of adsorbates/analytes on surfaces. For more complicated and large-scale systems for which DFT approaches are not practical, molecular dynamics (MD) simulations, based on empirical inter-atomic potentials (force-fields), can be employed as a compromise between the rigour of the description and the relevance of the structural model. Peptides, proteins, and DNA20,22,30–33 are such typical systems in this respect. However, the quality of the force-field used to describe this non-covalent bio-interface is critical to the success of the MD simulations. Specifically, the non-bonded interactions (Enonbond) across the molecule–surface interface must be adequately captured and balanced. In the CHARMM22* force-field, the Enonbond term consists of Coulombic electrostatics and van der Waals terms. The van der Waals contribution is approximated by the Lennard-Jones (LJ) 12-6 potential. Identifying (i.e. fitting) the parameters that inform the interfacial non-bonded interactions, and verifying the resultant force-field performance, are the chief tasks required to create a bio-interfacial force-field.
In general, two types of fitting philosophy/strategy can be seen in literature for obtaining these parameters. For the MoS2 2D surface, the most direct strategy to obtain the LJ contribution is to fit the four separate homo-atomic parameters (εMo–Mo, εS–S,σMo–Mo and σS–S), perhaps also including heteroatomic LJ parameters34 (εMo–S, σMo–S), and subsequently rely on the relevant (force-field dependent) mixing rules to describe all remaining LJ interactions between the MoS2 layers and its adsorbates. The key advantage of this strategy is simplicity; the maximum number of required homo-atomic van der Waals parameters required for the surface is small (2n, where n is the number of chemical elements in the surface, for example n = 2 for MoS2). Several MD simulation studies22,30,32,35 on MoS2 interfaces have implemented this philosophy. With this type of parameter set, the force-field may or may not adequately recover features of interest for these interfaces, for example microscopic structural traits of adsorbates on the MoS2 surface, as recently demonstrated for water.36
An alternative but more elaborate strategy is to incorporate a greater number of hetero-atomic van der Waals parameters into the parameter set that can be focussed and tailored to address contributions that the mixing rules may not be sufficiently robust to recover. This bespoke type of force-field parametrisation strategy has been realised for several surface materials including boron nitride,37 gold,38–40 and graphene.41 A general feature of this type of force-field is that the number of van der Waals parameters is much greater than those obtained from the mixing-rule strategy, but in turn might be able to better capture key structural interfacial properties of adsorbates on surfaces.36 In pragmatic terms, the two strategies mentioned above may ultimately reflect the unavoidable nature of compromise, which is an ever-present aspect associated with using an empirical inter-atomic potential in large-scale MD simulations. In other words, it requires a choice based on the differing priorities and purpose(s) of the intended MD simulations, with an emphasis on thermodynamic properties (e.g. surface energies, etc.) for the mixing-rule-based force-field, and an emphasis on adsorbate (including liquid) structural ensembles for the bespoke-type force-field.
As discussed above, the mixing-rule parametrisation strategy may or may not well recover the interaction structures of adsorbates on the MoS2 surface. A bespoke (i.e. non-mixing rule) force-field that can more specifically describe at least the leading interactions between the MoS2 surface and adsorbates is highly desirable. Such a force-field, which is currently not available, can enable large-scale MD-simulation-based investigations into MoS2-based bio-interfacial phenomena with the aim of maximising structural reliability of the results. Therefore, in this work a bespoke set of hetero-atomic van der Waals parameters aimed at describing the configuration-specific interactions between the MoS2 surface and several families of adsorbates (hydrocarbon, alcohol, aldehyde, ketone, carboxylic acid, amine, thiol, amino acid, and arbitrary combinations of these functional groups) have been developed. The development process is based on a high-quality quantum chemical dataset that was validated against gas-phase experimental data. The resultant force-field was also validated, in this instance against existing high quality structural data obtained from sum frequency generation (SFG) spectroscopy of peptides adsorbed at the aqueous MoS2 interface.20 This paper is organised into three sections. The first summarises generation of the DFT database required for the fitting process. The second covers the parameter fitting process, and in the third, validation and the first applications of the newly developed force-field are accomplished to probe the interaction with MoS2 of amino acids and peptides in an aqueous environment.
Methodology
Specifically, benchmarked DFT calculations were used to generate the database of configuration-specific surface adsorption energies. Following this, a fitting process was implemented to determine the best possible van der Waals parameters. In the last stage, MD simulations using the newly developed force-field were conducted to validate performance against experimental structural data. In addition, the free energies of adsorption between MoS2 and the 20 naturally-occurring amino acids in water were obtained using umbrella sampling simulations. Full technical details of DFT calculations, parameter fitting, and MD simulations are provided in the ESI.†Quantum chemical calculations
Due to the paramount importance of role of the database, the quantum chemical calculations must be meticulously conducted at the most reliable level of theory. In doing so, two steps were done to ensure the selection of an appropriate functional and of calculation setting parameters.The functional selection and calculation settings were determined following benchmarking against experimental gas-phase adsorption data and probing the convergence of energy. Such gas-phase experimental data are sparse, with data points only available for thiophene15,16 and butene.15 Therefore, gas phase MoS2 adsorption energies of thiophene and butene were calculated with eight van der Waals-corrected functionals (vdW-DF-c09,42–44 vdW-DF-cx,45 vdW-DF-ob86,46 vdW-DF-obk8,47 vdW-DF2-b86r, vdW-DF2-c09,42,48 vdW-DF,43,44 and vdW-DF2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48) and benchmarked against these experimental adsorption data. These data (ESI, Table S1†) indicate that the vdW-DF2 functional can recover the experimental data well, within an error of less than 3 kJ mol−1, and therefore this functional was chosen to calculate configuration-specific interaction energies of all 21 adsorbates (the full list of molecules is provided in the Results and discussion) used for the parametrisation. Energy convergence testing suggested a kinetic cutoff of 50 Ry and charge density cutoff of 300 Ry in combination with a uniform k-point mesh of 5 × 5 × 1. To improve the accuracy, these parameters were increased to 75 Ry and 500 Ry respectively, and 7 × 7 × 1 to calculate single point energies of optimised configurations. All further details of the calculation settings are provided in the ESI† and were based on previous work.36 The adsorption energies were calculated using the PBE projector augmented-wave (PAW) pseudopotential using Quantum Espresso 6.6.49,50
48) and benchmarked against these experimental adsorption data. These data (ESI, Table S1†) indicate that the vdW-DF2 functional can recover the experimental data well, within an error of less than 3 kJ mol−1, and therefore this functional was chosen to calculate configuration-specific interaction energies of all 21 adsorbates (the full list of molecules is provided in the Results and discussion) used for the parametrisation. Energy convergence testing suggested a kinetic cutoff of 50 Ry and charge density cutoff of 300 Ry in combination with a uniform k-point mesh of 5 × 5 × 1. To improve the accuracy, these parameters were increased to 75 Ry and 500 Ry respectively, and 7 × 7 × 1 to calculate single point energies of optimised configurations. All further details of the calculation settings are provided in the ESI† and were based on previous work.36 The adsorption energies were calculated using the PBE projector augmented-wave (PAW) pseudopotential using Quantum Espresso 6.6.49,50
Parametrisation process
The vdW-DF2 adsorption data were used to fit and validate various hetero-atomic LJ parameters for families of adsorbates. The DFT dataset was divided into two subsets, namely fitting and validation sets; the former was used to fit the data while the latter was used to validate the optimal parameters obtained from the fitting process. Each subset featured compounds from the same adsorbate-chemistry families.The optimised geometries of the adsorbates on the basal plane of MoS2 and the adsorption energy data were used for the fitting process. The vdW-DF2 adsorbate–MoS2 geometrical configurations were kept unchanged during the fitting process, except for reduction of the vertical distances from the adsorbates to MoS2 surface. As has been done previously, because the vdW-DF functional was found to overestimate the distance from adsorbates to surfaces,37,41,51 the molecule–surface distance was systematically reduced by 0.2 Å. The MD simulation supercell sizes and dimensions of the periodic cells are provided in Fig. S3 of the ESI.† To make the fitting process feasible, all bespoke hetero-atomic parameters (σij and εij) were fitted within a space of values ranging from 0 to 8, as determined from preliminary fitting evaluations. The fitting space was then narrowed gradually to locate as many wells as possible where the optimal values of parameters can be obtained; each well corresponds to a region where the difference in energies between the DFT fitting dataset and force-field outputs is smaller. Further fitting processes were conducted for each well to refine and locate the best possible parameters. Final parameters were selected based on two criteria: (i) that the difference in adsorption energies produced at the vdW-DF2 and current force-field was as small as possible, and (ii) the energetic rankings of the MoS2-adsorbate configurations determined at the vdW-DF2 level were conserved as much as possible with the current force-field. This protocol of force field development has been previously demonstrated to be essential and robust in recovering the ensemble configurations for interfacial systems.36 The fitted parameters were then validated by calculating adsorption energies for the adsorbates in the relevant subset of the validation set.
Following a process previously used for obtaining the gold-peptide force field GolP-CHARMM,38 the fitting process was implemented in a sequential boot-strapping scheme following the increasing structural complexity of functional groups to take into account all LJ interactions that will contribute to the interfacial interactions. The homo-atomic parameters (εMo–Mo, εS–S,σMo–Mo and σS–S) were fitted first to reproduce the adsorption energies of alkanes, based on mixing rules. Hetero-atomic van der Waals parameters were then derived for chemically-specific interactions. Specifically, hetero-atomic parameters for aromatic carbon were next fitted after alkane carbon, since this type of carbon will serve to fit other bio-based functional groups (phenol and indole) later on as alkane did. After this point, bespoke parameters for the hydroxyl, thiol, and amine groups were then fitted. Once the parameters for description of the hydroxyl group were available, the carboxylic and amide groups were treated next. Separate parameters were also required to be fitted specifically for indole/imidazole.
Molecular dynamics simulations
MD fitting and simulations were conducted using the GROMACS 2020.4 package.52 All MD simulations were conducted in the NVT ensemble at 300 K, and the system temperature was maintained with the Nosé–Hoover thermostat.53,54 The Particle-Mesh Ewald (PME)55,56 algorithm was used at every integration time step of 1 fs for the electrostatics calculation, within a real-space cutoff of 11.0 Å, smoothly tapering from 10.0 Å.The newly obtained force field was then validated extensively against experimental SFG spectroscopic data. In this experimental study,20 SFG observations were used to infer the possible interaction configurations between each of four different α-helical peptides and a monolayer MoS2 sheet in liquid water. These peptides were 26 amino acids long and based on the antimicrobial peptide, denoted here as X (KWKLFKKIGIGAVLKVLTTGLPALIS), and its three mutants denoted A, B, C (peptide sequences provided in the ESI†). To implement the MD validation simulations, a periodic cell of dimensions 6.82 nm × 7.86 nm × 11.00 nm containing a monolayer MoS2 sheet (with all substrate atoms fixed in space) and a single peptide chain solvated with liquid water was subjected to multiple 10 ns simulations in the NVT ensemble. For the peptide X, mutant A, and mutant B, eight simulations with different initial configurations of MoS2-peptide were performed to probe the tilting behaviours of peptides; for mutant C, 16 NVT simulations were conducted to investigate the adsorbed orientation.
The adsorption free energies between each of the 20 amino acids and the monolayer MoS2 sheet in liquid water were predicted using the newly developed force-field. The umbrella sampling technique57 was used and the weighted histogram analysis method (WHAM)58 was employed to analyse these data. The reaction coordinate for umbrella sampling was defined as the vertical centre-of-mass (COM) distance from amino acid to the MoS2 nanosheet plane (all substrate atoms were fixed in space). A total of 45 windows was used, with a spacing of 0.05 nm along the reaction coordinate, and a restraint potential of 3000 kJ mol−1 nm−2 applied to the adsorbate reference site. Simulations in the NVT ensemble were run for 100 ns per window, amounting to a total simulation duration of 4500 ns per adsorbate. Full details of the umbrella sampling simulations, and the methods used to convert the potential of mean force into a binding free energy, are provided in the ESI.†
Results and discussion
Quantum chemical adsorption energies
The strongest gas-phase adsorption energies of 21 compounds on the basal plane of MoS2 generated by the quantum chemical calculations are summarised in Table 1. The data for all 330 DFT calculations (geometries and energies for all configurations of the 21 compounds) are available as additional ESI Materials as indicated in the ESI† (comprising CIF files for the structures and a spreadsheet for the corresponding energies). Two general trends can be observed from adsorption energies: (i) the interaction is generally proportional to the sizes of adsorbates, and (ii) larger interfacial interaction areas facilitate markedly stronger interaction between adsorbates and the MoS2 surface. For a given adsorbate–MoS2 interaction, the strongest adsorbate–surface interaction was typically the result of maximising interfacial contact; Fig. 1 illustrates the case of benzene. Variation of interaction energies was also dependent on the van der Waals radii of constituent atoms (ethanol 28.9 kJ mol−1vs. ethanethiol 34.1 kJ mol−1). The interfacial size-dependence of interaction energy implies that gas-phase behaviours of adsorbates on the basal plane of MoS2 are chiefly governed by the van der Waals interactions.24| Training set | Validation set | ||
|---|---|---|---|
| Adsorbate | E ad | Adsorbate | E ad |
| Methane | 13.9 | Butane | 35.9 |
| Ethane | 21.3 | Benzene | 47.1 |
| Hexane | 50.9 | Ethanol | 28.9 |
| Toluene | 55.7 | Acetone | 35.6 |
| Methanol | 23.8 | Methanamide | 27.9 |
| Phenol | 53.5 | Methylamine | 28.7 |
| Methanoic acid | 24.2 | Indole | 65.9 |
| Ethanamide | 27.9 | Diethyl sulfide | 53.2 |
| Ethylamine | 29.3 | Methanethiol | 28.6 |
| Imidazole | 41.1 | ||
| Dimethyl sulfide | 39.4 | ||
| Ethanethiol | 34.1 | ||
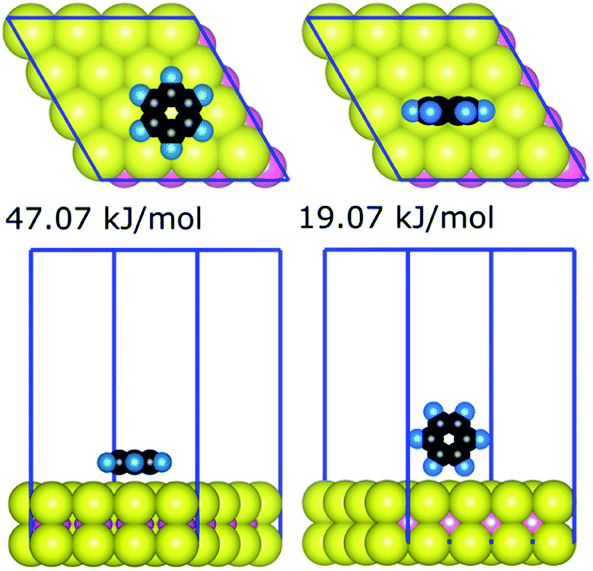 | ||
| Fig. 1 Top and side views of two benzene–MoS2 configurations, illustrating the dependence of contact area on binding strength determined from the DFT calculations. Blue lines indicate the periodic cell boundaries. | ||
The adsorption energies of all 21 compounds on MoS2 followed a consistent trend as noted for other surfaces such as boron nitride37 and gold.38 Adsorbates that featured aromatic ring structures manifested stronger interaction, for example benzene (47.1 kJ mol−1), phenol (53.5 kJ mol−1), toluene (55.7 kJ mol−1), and indole (65.9 kJ mol−1) whereas smaller structures such as ethane, methane, and methanol supported weaker interaction. Also, the long hydrocarbon chain yielded a stronger interaction (hexane, 50.9 kJ mol−1). This suggests that any larger adsorbate constructed from the molecular building blocks with stronger interaction energies might have stronger gas-phase interaction in comparison to those formed with weaker blocks. In an approximate sense, the suggested energy trending based on building blocks can be seen in the series of amino acids studied at the DFT level.59,60
Notably, the adsorption energies provided in Table 1 have been systematically reported at reliable level of theory (vdW-DF2, as indicated by the benchmarking data, ESI†). Adsorption energies of six of the organic compounds considered here were previously reported for different levels of theory (benzene,24 toluene,23 acetone,61,62 ethane,63,64 methanol,61 and ethanol62). As mentioned above, choice of a reliable level of theory is important in determination of the adsorption energy, and amongst the above previously reported data, only the adsorption energy of benzene24 (45.4 kJ mol−1), which is in the same range of the reported value in the current work 47.1 kJ mol−1, was determined with one of the benchmarked functionals (vDW-DF), and reported an adsorption structure comparable with that reported here. The adsorption energies of the other previously reported adsorbates were determined from geometries not determined using van der Waals functionals (either vdW-DF functionals or empirically corrected functionals) and/or a sufficient number of initial configurations, and differed significantly with the vdW-DF2 values reported here. With a wide range of adsorbate families and benchmarked level of theory, the adsorption energy set reported in the current is anticipated to provide a comprehensive and reliable dataset for future reference.
Force field parametrisation
The entire adsorption energy dataset was divided into two subsets (training and validation sets) to fit the bespoke hetero-atomic LJ parameters. The final parameters were required to meet two strict criteria, simultaneously satisfying DFT energetic ranking of the configurations and minimal energy differences between the force-field and DFT energies.To initiate the fitting process, the atomic partial charges (qi and qj) of the two surface atom types Mo and S must be determined. Based on similar efforts to determine the water–MoS2 interaction via the same strategy,36 the pair of charges +0.50 and −0.25e for Mo and S, respectively, were adopted. These charges were obtained from random phase approximation65 calculations and were independently used to successfully recover both macro- and microscopic traits of water droplets on the MoS2 surface.36,66 In combination with the non-bonded parameters developed36 previously for description of water–MoS2 interaction, a full force-field, denoted here as MoSu-CHARMM, was defined.
As mentioned in the Methods, a step-wise bootstrapping process was followed for the fitting. First, for hydrocarbons, homo-atomic ε and σ for Mo and S were fitted to ensure that regular mixing rules were capable of describing four types of interactions Mo–alkane carbon, Mo–alkane hydrogen, S–alkane carbon, and S–alkane hydrogen; therefore only four parameters were obtained (see Table 2). For the remaining adsorbates, bespoke hetero-atomic LJ parameters were fitted and validated for specific families of compounds separately. The optimal non-bonded parameters for specific families of adsorbates are listed in Table 2. It was noted that parameters fitted for ketones and alcohols worked well for carboxylic acid adsorbates. However, parameters fitted for the amine and ketone groups could not be similarly applied to the amides; therefore, bespoke parameters were specifically fitted for the whole amide group.
| Group | Interaction | Parameter | ||
|---|---|---|---|---|
| σ (nm) | ε (kJ mol−1) | |||
| Hydrocarbon | R | Mo | 0.395 | 0.103 |
| S | 0.338 | 3.404 | ||
| Aromatic ring | R | Mo–C | 0.130 | 0.103 |
| S–C | 0.314 | 1.175 | ||
| Alcohol | R-OH | Mo–O | 0.303 | 2.766 |
| S–O | 0.301 | 2.105 | ||
| Mo–H | 0.070 | 0.035 | ||
| S–H | 0.290 | 0.075 | ||
| Phenol | R-OH | Mo–O | 0.303 | 2.600 |
| S–O | 0.301 | 0.600 | ||
| Mo–H | 0.070 | 0.035 | ||
| S–H | 0.290 | 0.075 | ||
| Ketone | R-CO-R | Mo–O | 0.310 | 0.260 |
| S–O | 0.295 | 1.480 | ||
| Mo–C | 0.621 | 0.093 | ||
| S–C | 0.293 | 3.320 | ||
| Amine | R-C–NH2 | Mo–N | 0.100 | 0.103 |
| S–N | 0.300 | 2.680 | ||
| Mo–C | 0.100 | 0.100 | ||
| S–C | 0.301 | 1.200 | ||
| Amide | R-Cα–CO–NH2 | Mo–N | 0.100 | 0.103 |
| S–N | 0.300 | 3.360 | ||
| Mo–C | 0.490 | 0.240 | ||
| S–C | 0.288 | 0.700 | ||
| Mo–O | 0.306 | 0.070 | ||
| S–O | 0.292 | 1.350 | ||
| Mo–Cα | 0.401 | 1.460 | ||
| S–C | 0.251 | 0.110 | ||
| Diazole | S–N | 0.290 | 2.700 | |
| Mo–N | 0.400 | 0.103 | ||
| S–C | 0.279 | 0.020 | ||
| Mo–C | 0.413 | 0.660 | ||
| Thiol and thioether | R-C–S–H | Mo–S | 0.389 | 2.200 |
| R-C–S–C-R | S–S | 0.339 | 1.175 | |
| Mo–C | 0.370 | 0.050 | ||
| S–C | 0.342 | 1.000 | ||
In total, 72 non-bonded parameters were developed. A general feature of this parameter set is that the well depth εij and some of the sigma values of most interactions with the involvement of the MoS2 sulfur atoms were significantly larger than those of molybdenum (Table 2). This suggests that the sulfur atomic layers of the MoS2 surface dominate the van der Waals contributions to the total MoS2–adsorbate interaction. This is understandable given the sulfur atomic layers cover the Mo layer and are exposed more directly to the adsorbates.
All parameters provided in Table 2 were obtained using the fitting set, for which the molecule–surface interaction energies recovered by the newly developed force-field were in high correlation with those determined at the vdW-DF2 level of theory (RMSD = 1.76 kJ mol−1) evaluated from ∼190 data points (∼190 geometrical configurations). On this basis, the new force-field covers most of the important configurations in the geometrical space of the fitting set, and is expected to perform well for different, but related, compounds. Indeed, the interaction energies of about ∼140 geometrical configurations of the validation set were energetically predicted with a promising RMSD value of 2.57 kJ mol−1. Both fitting and validation RMSD values are below chemical accuracy of 4 kJ mol−1. With such small values of fitting and validation RMSD, MoSu-CHARMM is believed to meet the first criterion of minimising energetic differences with the DFT values.
The correlations between the force-field energies and vdW-DF2 energies are summarised in Fig. 2. It is noted that for some very weak interaction configurations observed for e.g. some cases of methane (fitting set) and some cases of e.g. benzene (validation set), the actual vdW-DF2 interaction of such weak interactions should be significantly weaker than has been calculated here, due to the high proportional contribution of the zero-point energy to the final interaction energy, as has been demonstrated in previous work.36 In other words, the true adsorption energies of these weak configurations are overshot, and as a result such overestimation of weak interaction energy leads to the apparent discrepancy between the vdW-DF2 and FF energies for the very weak-binding regions on the potential hypersurface of adsorbate–MoS2 interfacial interaction. The fitted parameters accommodate this, such that the force field confers a stronger interaction for these weak cases.
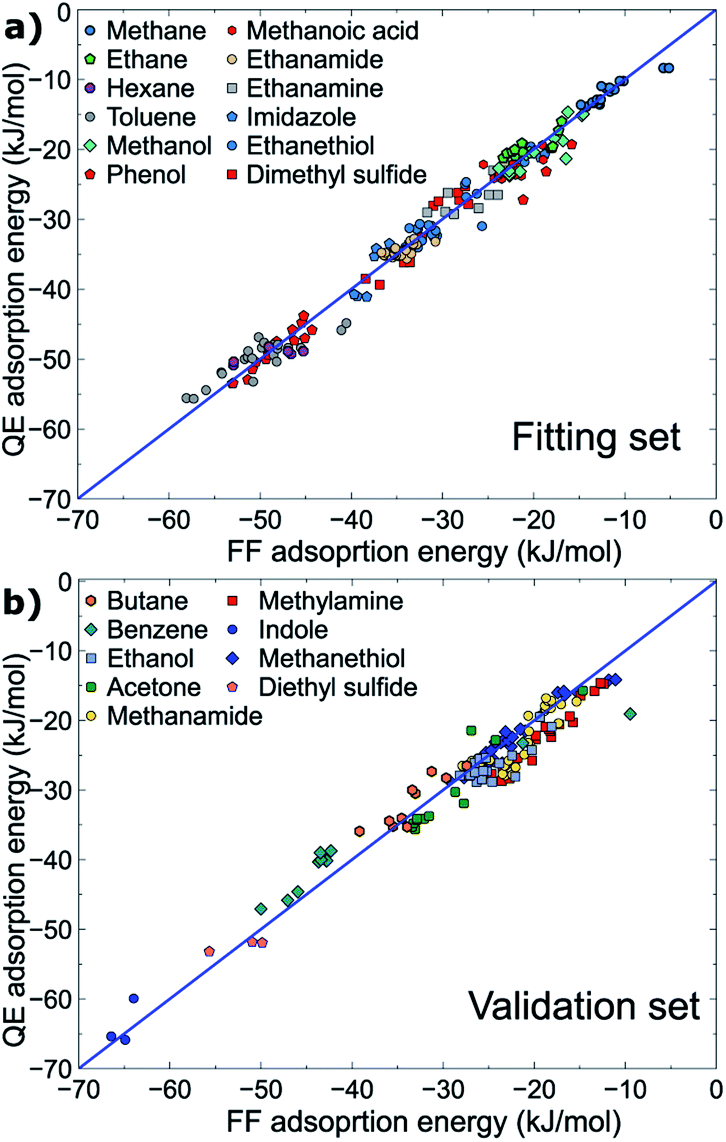 | ||
| Fig. 2 Correlation between energies produced at the vdW-DF2 level and the MoSu-CHARMM force-field. Data for the (a) fitting and (b) validation sets are plotted separately. | ||
MoSu-CHARMM was designed to capture as much as possible the microscopic structural ensemble traits of the adsorbates at the MoS2 surface. This feature is incorporated into the new force-field via reproduction of the first-principles energetic ranking of any set of MoS2-adsorbate configurations. To elaborate, in principle, the energetic orderings determined at the vdW-DF2 level should be precisely recovered at the MoSu-CHARMM level. In practise, such orderings were broadly recovered, meaning that the new force field can distinguish between the groups of strongest and weakest interactions in a set of configurations, and any configurations in between these two extremities of ranking (referred to as “mid-range”). Such energetic orderings were strictly applied for the fitting set, and their fulfilment demanded as a performance metric in the validation set. Fig. 3 exemplifies the energetic ranking of MoS2–phenol interaction configurations. Clearly, MoSu-CHARMM can resolve and capture the energetic trends of phenol adsorbed on MoS2, including the pattern of the three weakest interactions. Application of the same vdW-DF DFT-fitting strategy and the same two criteria in the development of a MoS2/water force-field resulted in recovery of microscopic ensemble data as obtained from RPA calculations, which gives a vote of confidence in the same approach as applied here.36
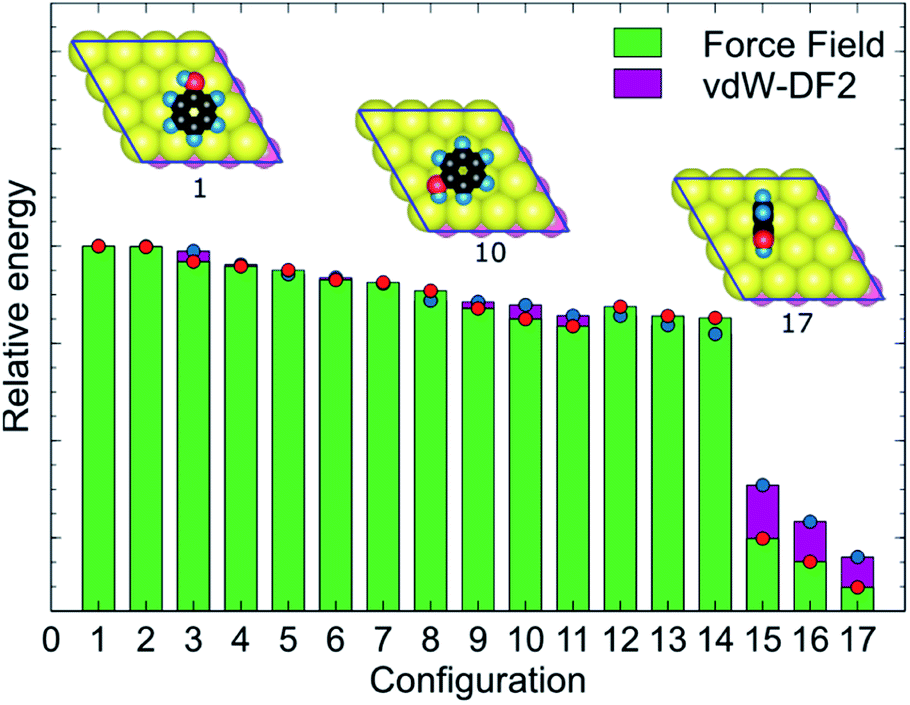 | ||
| Fig. 3 Example of the fidelity of the DFT/force-field energetic ranking of the 17 MoS2–phenol interaction configurations along with three top-view geometries representing a typical strongest, mid-range, and weakest interaction between MoS2 and phenol. The red and blue circles highlight the difference between FF and DFT energies. In the molecular images, S, C, O, and H atoms are represented with yellow, black, red, and blue colours. | ||
Peptide adsorption structures at the aqueous MoS2 interface
A key step in the development of the MoSu-CHARMM force-field is to ensure recovery of experimental structural data obtained from aqueous interfaces. The critical role of interfacial solvent structuring at the aqueous/solid interface and its impact on peptide adsorption are widely accepted.67–73 In the case of the aqueous MoS2 interface, the parameters for the MoS2/water interaction were fitted (following the same procedure as used here) and tested, again for recovery of structural fidelity, in addition to reproducing the known water droplet contact angle.36However, peptide/surface adsorption data for aqueous interfaces are typically rare; however, the adsorption structures of four α-helical peptides at the aqueous MoS2 interface experimentally probed using Sum Frequency Generation (SFG) spectroscopy20 formed the basis of this validation process. In this instance, the SFG observations were related to the degree of tilt in the surface-adsorbed configuration, which could be related back to ratios of the signal intensity of the amide I spectral peak collected using different combinations of s and p polarisation of the incident and detected radiation. It is noted that SFG is not a single-molecule technique; the SFG observations produce data that are ensemble-averaged.
As strictly interpreted from these SFG spectroscopy data,20 the interfacial interaction configuration of the four α-helical peptides (parent peptide X, and three mutants A, B, and C) can be divided into two unambiguous categories; those that exclusively adsorbed with their helix oriented parallel with the surface (defined as a tilt angle of 180°, corresponding to an absence of SFG signal regardless of polarisation), such that all adsorbed molecules in the ensemble must adopt this conformation, or, a presence of SFG signal that indicates at least some fraction of the ensemble adopting an upright/tilted conformation with respect to the surface plane. The SFG data reported by Xiao et al.20 suggest that mutant C followed the former scenario, whereas parent X and mutants A and B followed the latter. In particular, the ability of the force-field to recover the exclusive behaviour of mutant C would provide a strong test of structural fidelity.
The MD simulations using MoSu-CHARMM indicate that peptide X and mutants A and B could adopt a tilted adsorption configuration, whereas mutant C consistently adopted a parallel configuration on the MoS2 surface. The simulations in the current work indicate that peptide X and its two mutants A and B interacted with the MoS2 surface via surface attachment at one terminus, leaving the remaining terminus projected into the solution (Fig. 4). More precisely, the peptide X and mutant B interacted via the N-terminus, with the C-terminus protruding into the solvent, whereas mutant A's behaviour was reversed, with surface attachment via the C-terminus. It is noted that the SFG spectroscopic data cannot distinguish which terminus end makes surface contact – only the ensemble-averaged orientation of the helix may be deduced from these experimental data. The interaction schemes of the four peptides differ slightly with those inferred from coarse-grained MD simulation data20 that claimed that C-terminal contact with the MoS2 surface was responsible for all tilted configurations. However, this coarse-grained potential was not fitted to specific MoS2 interactions. In contrast, mutant C was found to consistently adopt a parallel configuration on the surface of MoS2, further confirmed by additional simulations, the number of which exceeded those for the other three peptides.
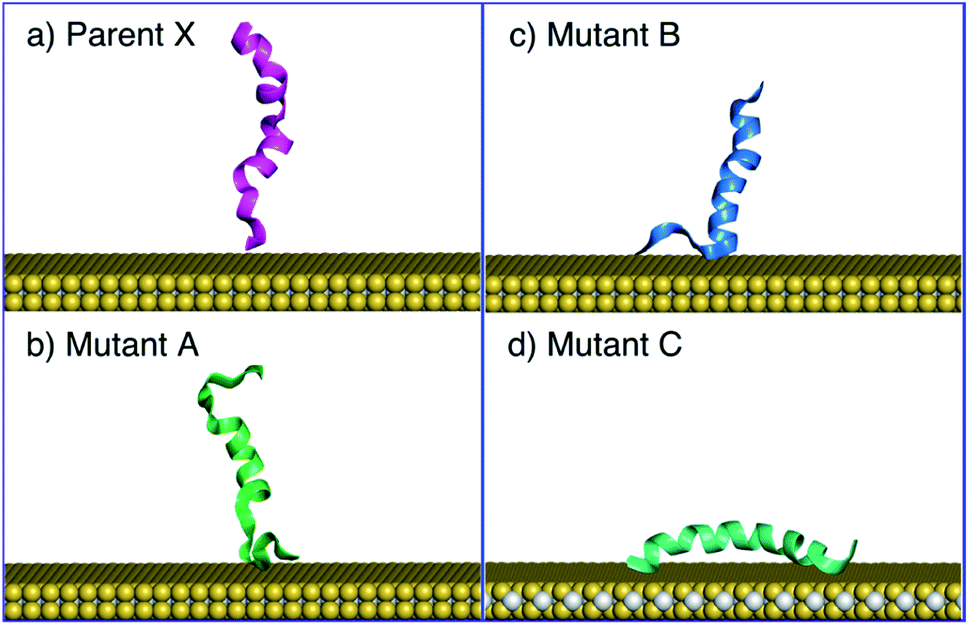 | ||
| Fig. 4 Side views of predicted adsorption structures illustrating tilting behaviours in panels (a)–(c) of the parent peptide (X), mutant A, and mutant B, whereas mutant C adopted a parallel configuration on MoS2 and the helicity of its secondary structure was conserved during the simulations, panel (d). Water not shown for clarity. | ||
In addition to the tilting behaviours of peptides adsorbed on the basal plane of MoS2, the α-helical structure of mutant C was also monitored in the MD simulations of the current work (Fig. 4d). In the experimental data reported by Xiao et al.20 the helicity of mutant C in the surface-adsorbed state was experimentally inferred from circular dichroism (CD) spectroscopic observations. The MD simulations in the current work found that some helicity of mutant C was retained in all cases (noting that all simulations of mutant C produced a parallel configuration). Therefore, MD simulations with the MoSu-CHARMM force-field can recover the relevant structural features of peptides adsorbed on the MoS2 surface, sufficient for description of biological phenomena at the aqueous interface with MoS2.
Amino acid adsorption free energies
The MoSu-CHARMM force-field was also used to predict the interaction free energy between each of the 20 naturally-occurring amino acids and the aqueous MoS2 interface. These simulations were done to evaluate and rank the strength of the amino acid interactions with the MoS2 surface in an aqueous environment, with data summarised in Fig. 5 (numerical data provided in Table S2, ESI†). Three selected configurations of adsorbed amino acids are provided in Fig. 6 and corresponding images for all amino acids are shown in Fig. S7, ESI.†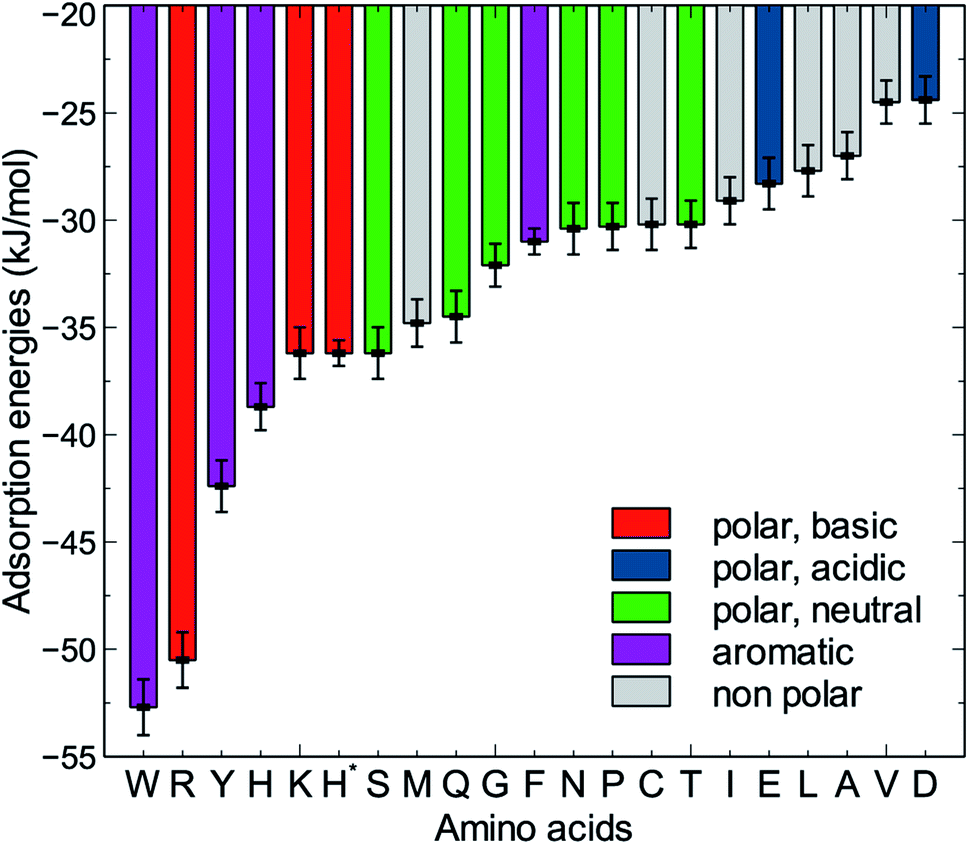 | ||
| Fig. 5 Adsorption free energies of the twenty amino acids at the aqueous interface of the MoS2 basal plane. H* is the protonated state of histidine. | ||
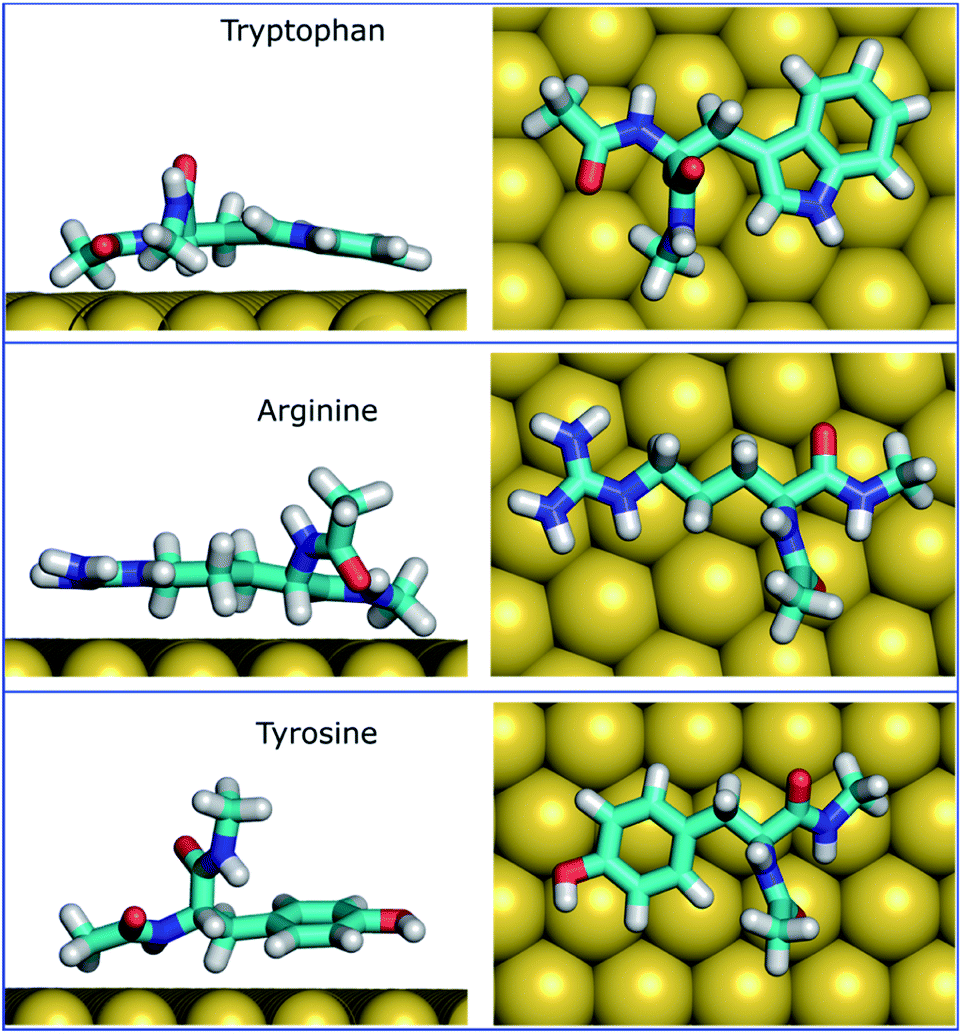 | ||
| Fig. 6 The most populated configurations of three strongest binding amino acids in the surface adsorbed state, predicted from the umbrella sampling simulations (side and top views). Water not shown for clarity. | ||
To date, there has been little clarity regarding which residues contribute to strong peptide binding at the aqueous MoS2 interface; these data are anticipated to provide the initial clues to address this question. These data in Fig. 5 indicate that the amino acids featured comparable or stronger adsorption to the MoS2 surface than those reported for adsorption at the Au,74 Ag,74 graphene,75 and boron nitride37 interfaces. However, the general trend of binding strength to MoS2 is similar to some of these other materials, in which tryptophan, arginine and tyrosine are among the amino acids with the strongest binding, whereas alanine and aspartate support some of the weakest binding. However, there are notable contrasts with the energetic ranking of amino acid adsorption compared with h-BN,37 in particular that Ile and Leu were ranked considerably stronger in relative binding strength on h-BN vs. MoS2, whereas Lys was of the weakest amino acids on h-BN, yet relatively strong on MoS2. These data provide a baseline not only for designing new MoS2-binding peptides, but also gives insights into how to achieve 2D-materials-binding selectivity via sequence design. Moreover, the use of such amino acid data was a critical factor in recent work exploiting small data machine learning approaches based on Bayesian optimization to search for new materials-selective peptide sequences for Ag and Au surfaces.76
Experimental biocombinatorial screening studies have identified several peptide sequences with affinity for the MoS2 surface.22,77,78 Specifically, four peptide sequences (MoSBP1,77 HLL,78 MoS2-P15,22 and MoS2-P2822 listed in Table 3 were determined to have strong affinity to MoS2, and two others (MoSBP2077 and MoS2-P322) were identified as weak binders. The four strong binding sequences share a common trait in that they feature one or more residues corresponding with the predicted strong affinity amino acids (Trp, Tyr, and Arg), which might anchor the peptide–MoS2 contact, analogous to Arg and Trp which are thought to be anchors on the gold surface.68 In contrast, the weak binding peptides (MoSBP20 and MoS2-P3) do not contain any of the predicted strong affinity residues in their sequences. Bearing in mind that peptide binding strength is not merely an additive function of the individual residue binding tendencies, nonetheless, appearance of strong binding residues in the strong-binding sequences, and the absence of these residues in the weak-binding sequences, can be considered as early indication of the predictive performance of MoSu-CHARMM, and a vote of confidence in the current approach.
![[Y with combining low line]](https://www.rsc.org/images/entities/b_char_0059_0332.gif)
![[R with combining low line]](https://www.rsc.org/images/entities/b_char_0052_0332.gif)
![[W with combining low line]](https://www.rsc.org/images/entities/b_char_0057_0332.gif)
It is clear that strongly interacting amino acids feature a longer alkane side-chain (Arg and Lys), or an aromatic ring (Trp or Tyr, and to a lesser extent, Phe). The most populated configurations (Fig. 6) obtained from the umbrella sampling simulations indicate that maximisation of contact area on the surface is a key trait. However, contact area per se does not appear to be the distinguishing feature, since bulkier amino acids that also have potentially large surface contact areas such as Val, Leu and Ile, are amongst the weakest binders. This suggests possible entropic effects, e.g. bulkier non-polar residues may cause greater disruption of the solvent structuring in the near-surface region, compared with the more planar aromatic groups. The dependence of binding strength in the gas-phase compared with the in-solvent cases also suggests solvation entropy effects; the aromatic groups in the gas phase are also of the strongest in the DFT dataset, however, the bulkier alkanes are not correspondingly weak adsorbates in the gas phase.
Conclusions
This work introduces an all-atom force-field, MoSu-CHARMM, developed for describing biointerface structures with MoS2 surfaces under aqueous conditions. Although peptides and proteins are the key targets of this new force-field, a wide range of adsorbate families can be described. MoSu-CHARMM has been designed with a particular emphasis on recovering and predicting interfacial structural data. This was accomplished via imposition of two rigorous criteria, satisfying a minimum difference compared with the DFT dataset (330 points) obtained using the vdW-DF2 functional (fitting and validation RMSDs of 1.76 and 2.57 kJ mol−1, respectively) and preserving a maximum fidelity of configuration-specific energetic rankings. The new force-field successfully recovered experimental structural data of peptides adsorbed at the aqueous MoS2 surface and reproduced characteristics regarding the helical secondary structure in the surface-adsorbed state. The adsorption free energies of all twenty naturally-occurring amino acids at the aqueous MoS2 interface were determined using umbrella sampling simulations, yielding amino acid binding traits that are consistent with experimentally-identified MoS2-binding sequences. These data give insights into the composition of the currently limited set of known MoS2-binding peptides and provides a pathway to systematically designing and testing new binding sequences. Overall, this force-field will enable advances in fundamental understanding of structure–property relationships for biointerfaces with MoS2 nanosheets.Data availability
All crystallographic information files (CIFs) and energies (associated Excel spreadsheet) of the structures used for the fitting and validation processes are provided in the ESI† bundle. These data can be accessed through the github repository https://github.com/lenhanpham/MoS2-FF-paper.Author contributions
Conceptualisation: TRW. Design: both authors. Investigation: LNP. Formal analysis: LNP. Writing – original draft: both authors. Writing – reviewing and editing: both authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Air Force Office of Scientific Research Grant FA9550-18-1-0329. Computational resources were provided by the National Computational Infrastructure (NCI), Canberra, and the Pawsey Supercomputing Centre, Perth, under the NCMAS scheme.References
- K. Liu, J. Feng, A. Kis and A. Radenovic, ACS Nano, 2014, 8, 2504–2511 CrossRef CAS PubMed.
- A. B. Farimani, K. Min and N. R. Aluru, ACS Nano, 2014, 8, 7914–7922 CrossRef CAS PubMed.
- C. H. Naylor, N. J. Kybert, C. Schneier, J. Xi, G. Romero, J. G. Saven, R. Liu and A. T. C. Johnson, ACS Nano, 2016, 10, 6173–6179 CrossRef CAS PubMed.
- S. Barua, H. S. Dutta, S. Gogoi, R. Devi and R. Khan, ACS Appl. Nano Mater., 2018, 1, 2–25 CrossRef CAS.
- K. Kalantar-zadeh and J. Z. Ou, ACS Sens., 2016, 1, 5–16 CrossRef CAS.
- Q. He, Z. Zeng, Z. Yin, H. Li, S. Wu, X. Huang and H. Zhang, Small, 2012, 8, 2994–2999 CrossRef CAS PubMed.
- H. Li, Z. Yin, Q. He, H. Li, X. Huang, G. Lu, D. W. H. Fam, A. I. Y. Tok, Q. Zhang and H. Zhang, Small, 2012, 8, 63–67 CrossRef CAS PubMed.
- B. Liu, L. Chen, G. Liu, A. N. Abbas, M. Fathi and C. Zhou, ACS Nano, 2014, 8, 5304–5314 CrossRef CAS PubMed.
- Y. Liu, Y. Zhao, X. Zhang, X. Huang, W. Liao and Y. Zhao, Chem. Eng. J., 2021, 422, 130082 CrossRef CAS.
- S. Roobakhsh, Z. Rostami and S. Azizian, Sep. Purif. Technol., 2018, 200, 23–28 CrossRef CAS.
- K. Ai, C. Ruan, M. Shen and L. Lu, Adv. Funct. Mater., 2016, 26, 5542–5549 CrossRef CAS.
- B. L. Li, M. I. Setyawati, L. Chen, J. Xie, K. Ariga, C.-T. Lim, S. Garaj and D. T. Leong, ACS Appl. Mater. Interfaces, 2017, 9, 15286–15296 CrossRef CAS PubMed.
- D. J. Late, Y.-K. Huang, B. Liu, J. Acharya, S. N. Shirodkar, J. Luo, A. Yan, D. Charles, U. V. Waghmare, V. P. Dravid and C. N. R. Rao, ACS Nano, 2013, 7, 4879–4891 CrossRef CAS PubMed.
- Y. Liu, L. Hao, W. Gao, Z. Wu, Y. Lin, G. Li, W. Guo, L. Yu, H. Zeng, J. Zhu and W. Zhang, Sens. Actuators, B, 2015, 211, 537–543 CrossRef CAS.
- M. Salmeron, G. A. Somorjai, A. Wold, R. Chianelli and K. S. Liang, Chem. Phys. Lett., 1982, 90, 105–107 CrossRef CAS.
- S. L. Peterson and K. H. Schulz, Langmuir, 1996, 12, 941–945 CrossRef CAS.
- F. K. Perkins, A. L. Friedman, E. Cobas, P. M. Campbell, G. G. Jernigan and B. T. Jonker, Nano Lett., 2013, 13, 668–673 CrossRef CAS PubMed.
- L. Ye, H. Xu, D. Zhang and S. Chen, Mater. Res. Bull., 2014, 55, 221–228 CrossRef CAS.
- Z. Wu, Q. Duan, X. Li and J. Li, Environ. Sci. Pollut. Res., 2019, 26, 31344–31353 CrossRef CAS PubMed.
- M. Xiao, S. Wei, Y. Li, J. Jasensky, J. Chen, C. L. Brooks and Z. Chen, Chem. Sci., 2018, 9, 1769–1773 RSC.
- X. Sun, J. Fan, C. Fu, L. Yao, S. Zhao, J. Wang and J. Xiao, Sci. Rep., 2017, 7, 10290 CrossRef PubMed.
- S. Cetinel, W.-Z. Shen, M. Aminpour, P. Bhomkar, F. Wang, E. R. Borujeny, K. Sharma, N. Nayebi and C. Montemagno, Sci. Rep., 2018, 8, 3374 CrossRef PubMed.
- U. Patil and N. M. Caffrey, J. Chem. Phys., 2018, 149, 094702 CrossRef PubMed.
- P. G. Moses, J. J. Mortensen, B. I. Lundqvist and J. K. Nørskov, J. Chem. Phys., 2009, 130, 104709 CrossRef PubMed.
- C. González, B. Biel and Y. J. Dappe, Phys. Chem. Chem. Phys., 2017, 19, 9485–9499 RSC.
- V. M. Bermudez, J. Phys. Chem. C, 2020, 124, 15275–15284 CrossRef CAS.
- M. Sahoo, J. Wang, Y. Zhang, T. Shimada and T. Kitamura, J. Phys. Chem. C, 2016, 120, 14113–14121 CrossRef CAS.
- M. Schleicher and M. Fyta, ACS Appl. Electron. Mater., 2020, 2, 74–83 CrossRef CAS.
- L. Kou, A. Du, C. Chen and T. Frauenheim, Nanoscale, 2014, 6, 5156–5161 RSC.
- J. Liu, J. Zeng, C. Zhu, J. Miao, Y. Huang and H. Heinz, Chem. Sci., 2020, 11, 8708–8722 RSC.
- B. Luan and R. Zhou, ACS Nano, 2018, 12, 3886–3891 CrossRef CAS PubMed.
- B. Luan and R. Zhou, J. Phys. Chem. Lett., 2018, 9, 3409–3415 CrossRef CAS PubMed.
- T. R. Walsh and M. R. Knecht, Bioconjugate Chem., 2019, 30, 2727–2750 CrossRef CAS PubMed.
- V. Varshney, S. S. Patnaik, C. Muratore, A. K. Roy, A. A. Voevodin and B. L. Farmer, Comput. Mater. Sci., 2010, 48, 101–108 CrossRef CAS.
- Z. Gu, W. Li, L. Hong and R. Zhou, J. Chem. Phys., 2016, 144, 175103 CrossRef PubMed.
- L. N. Pham and T. R. Walsh, Chem. Commun., 2021, 57, 3355–3358 RSC.
- A. Budi and T. R. Walsh, Langmuir, 2019, 35, 16234–16243 CrossRef CAS PubMed.
- L. B. Wright, P. M. Rodger, S. Corni and T. R. Walsh, J. Chem. Theory Comput., 2013, 9, 1616–1630 CrossRef CAS PubMed.
- L. B. Wright, P. M. Rodger, T. R. Walsh and S. Corni, J. Phys. Chem. C, 2013, 117, 24292–24306 CrossRef CAS.
- F. Iori, R. Di Felice, E. Molinari and S. Corni, J. Comput. Chem., 2009, 30, 1465–1476 CrossRef CAS PubMed.
- Z. E. Hughes, S. M. Tomásio and T. R. Walsh, Nanoscale, 2014, 6, 5438–5448 RSC.
- V. R. Cooper, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 161104 CrossRef.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- T. Thonhauser, S. Zuluaga, C. A. Arter, K. Berland, E. Schröder and P. Hyldgaard, Phys. Rev. Lett., 2015, 115, 136402 CrossRef CAS PubMed.
- K. Berland and P. Hyldgaard, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 035412 CrossRef.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- K. Lee, É. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101 CrossRef.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- P. Giannozzi, O. Baseggio, P. Bonfà, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. de Gironcoli, P. Delugas, F. Ferrari Ruffino, A. Ferretti, N. Marzari, I. Timrov, A. Urru and S. Baroni, J. Chem. Phys., 2020, 152, 154105 CrossRef CAS PubMed.
- Z. E. Hughes, L. B. Wright and T. R. Walsh, Langmuir, 2013, 29, 13217–13229 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- J. Song, M. Li, H. Liang and H. Lou, Comput. Theor. Chem., 2017, 1118, 115–122 CrossRef CAS.
- P. Zhang, Z. Wang, L. Liu, L. H. Klausen, Y. Wang, J. Mi and M. Dong, Appl. Mater. Today, 2019, 14, 151–158 CrossRef.
- F. Mehmood and R. Pachter, J. Appl. Phys., 2014, 115, 164302 CrossRef.
- X.-Q. Tian, L. Liu, X.-R. Wang, Y.-D. Wei, J. Gu, Y. Du and B. I. Yakobson, J. Mater. Chem. C, 2017, 5, 1463–1470 RSC.
- N. Yu, L. Wang, M. Li, X. Sun, T. Hou and Y. Li, Phys. Chem. Chem. Phys., 2015, 17, 11700–11704 RSC.
- J. Ren, H. Liu, Y. Xue and L. Wang, Nanoscale Res. Lett., 2019, 14, 293 CrossRef PubMed.
- M. Heiranian, Y. Wu and N. R. Aluru, J. Chem. Phys., 2017, 147, 104706 CrossRef PubMed.
- V. Sresht, A. Govind Rajan, E. Bordes, M. S. Strano, A. A. H. Pádua and D. Blankschtein, J. Phys. Chem. C, 2017, 121, 9022–9031 CrossRef CAS.
- D. A. Cannon, N. Ashkenasy and T. Tuttle, J. Phys. Chem. Lett., 2015, 6, 3944–3949 CrossRef CAS PubMed.
- L. B. Wright, J. P. Palafox-Hernandez, P. M. Rodger, S. Corni and T. R. Walsh, Chem. Sci., 2015, 6, 5204–5214 RSC.
- J. Schneider and L. Colombi Ciacchi, J. Am. Chem. Soc., 2012, 134, 2407–2413 CrossRef CAS PubMed.
- A. A. Skelton, T. Liang and T. R. Walsh, ACS Appl. Mater. Interfaces, 2009, 1, 1482–1491 CrossRef CAS.
- J.-W. Shen, T. Wu, Q. Wang, Y. Kang and X. Chen, ChemPhysChem, 2009, 10, 1260–1269 CrossRef CAS PubMed.
- R. Notman and T. R. Walsh, Langmuir, 2009, 25, 1638–1644 CrossRef CAS PubMed.
- A. Mungikar and D. Forciniti, Biomacromolecules, 2006, 7, 239–251 CrossRef CAS PubMed.
- J. P. Palafox-Hernandez, Z. Tang, Z. E. Hughes, Y. Li, M. T. Swihart, P. N. Prasad, T. R. Walsh and M. R. Knecht, Chem. Mater., 2014, 26, 4960–4969 CrossRef CAS.
- Z. E. Hughes and T. R. Walsh, J. Mater. Chem. B, 2015, 3, 3211–3221 RSC.
- Z. E. Hughes, M. A. Nguyen, J. Wang, Y. Liu, M. T. Swihart, M. Poloczek, P. I. Frazier, M. R. Knecht and T. R. Walsh, ACS Nano, 2021, 15, 18260–18269 CrossRef CAS PubMed.
- J. Chen, E. Zhu, J. Liu, S. Zhang, Z. Lin, X. Duan, H. Heinz, Y. Huang and J. J. De Yoreo, Science, 2018, 362, 1135–1139 CrossRef CAS PubMed.
- C. Muratore, A. T. Juhl, A. J. Stroud, D. Wenbi Lai, A. M. Jawaid, K. M. Burzynski, J. M. Dagher, G. M. Leuty, C. Harsch, S. S. Kim, Y. H. Ngo, N. R. Glavin, R. J. Berry, M. F. Durstock, P. A. Derosa, A. K. Roy, E. M. Heckman and R. R. Naik, Appl. Phys. Lett., 2018, 112, 233704 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Method details and data for DFT calculations, including benchmarking against experiment; force field fitting details; method details for umbrella sampling simulations and amino acid binding free energy data. See DOI: 10.1039/d1sc06814h |
| This journal is © The Royal Society of Chemistry 2022 |