
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiazido macrocyclic sulfates as a platform for the synthesis of sequence-defined polymers for antibody drug conjugates†
Neil L.
Forsythe
,
Mikayla F.
Tan
and
Heather D.
Maynard
 *
*
Department of Chemistry and Biochemistry, California NanoSystems Institute, University of California, Los Angeles, California 90095-1569, USA. E-mail: maynard@chem.ucla.edu
First published on 7th March 2022
Abstract
To improve the efficacy of antibody drug conjugates (ADCs), there has been significant focus on increasing the drug-to-antibody ratio (DAR) in order to deliver more payload. However, due to the hydrophobicity of many cytotoxics, highly-loaded conjugates often have lower physicochemical stability and poorer pharmacokinetic outcomes, requiring the development of new hydrophilic linkers. Herein, we report a platform for the preparation of functional, sequence-defined polymers for conjugation to antibodies. We demonstrate the successful synthesis of novel diazido macrocyclic sulfate monomers of varied size ranging from 4 to 7 ethylene glycol repeat units. These monomers were then successively ring-opened to produce sequence-defined polymers that contained either 4 or 6 azides for post-synthesis functionalization. Given the hydrophilic ethylene glycol backbone and chemically defined nature of the polymers, we envisioned this as a useful strategy in the preparation of highly-loaded ADCs. To demonstrate this, we prepared a model polymer-fluorophore scaffold composed of 4 coumarin molecules and conjugated it to Herceptin. We fully characterized the conjugate via mass spectrometry, which yielded a polymer-to-antibody ratio of 6.6, translating to a total of 26 fluorophores conjugated to the antibody at the inter-chain disulfides. We believe this technology to not only be a meaningful contribution to the field of sequence-defined polymers and conjugates, but also as a general and tunable platform for drug delivery.
Introduction
Antibody drug conjugates (ADCs) are an increasingly important therapeutic modality for the treatment of cancer. The attachment of a cytotoxic agent to an antibody against antigen-presenting cancer cells is a proven formula that has yielded the approval of 10 drugs with many more promising candidates in clinical development.1,2 This strategy of targeted delivery of potent, apoptotic agents such a tubulin binders, DNA-damaging molecules, spliceosome inhibitors, and RNA polymerase inhibitors typically offers larger therapeutic windows over traditional chemotherapy due to reduced toxicity towards healthy cells.3 To further expand on the success of this platform, a variety of strategies have been employed to improve the therapeutic index of ADCs. For one, improvements in antigen selection and discovery are important in order to successfully target cancer cells over healthy tissue. Considerations such as antigen overexpression, cell surface availability, and internalization pathways are considered when engineering antibodies for more effective ADCs.2,4In addition to these antigen discovery and antibody engineering efforts, new conjugation and linker chemistries are explored for next-generation ADCs to further improve the therapeutic indices of the modality. One method that has been pursued to reduce the minimum effective dose of these drugs is to increase the drug to antibody ratio (DAR) of the final conjugate. In theory, higher drug loadings would allow for increased delivery of cytotoxics, being especially effective in cancer cells with lower antigen presentation or when using lower potency payloads.5 However, it has been demonstrated that while higher DAR ADCs do often show increased potency in vitro, they often show lower tolerability and have poorer pharmacokinetic outcomes.6,7 This observed discrepancy is thought to be the result of rapid, immune-mediated clearance of the more hydrophobic, highly-loaded conjugates. In addition to poor in vivo performance, highly loaded ADCs can suffer from decreased stability as the hydrophobic payloads can cause aggregation and precipitation.8 While previous attempts to mitigate these challenges have focused on targeting DARs between 2–4, new polymeric linkers have the potential to both provide increased DARs while masking the hydrophobicity of payloads.9
Hydrophilic polymers can confer significant advantages over traditional ADC linkers given their multivalency for drug attachment and solubilizing characteristics. Indeed, polymeric linkers based on poly(ethylene glycol),9,10 dextran,11,12 and acrylamides13,14 demonstrate the ability to achieve a higher DAR while maintaining hydrophilicity. However, when using these scaffolds, the inherent dispersity of the polymer can lead to challenges in obtaining precise drug loading and are more difficult to characterize. Sequence-defined polymers offer the unique advantage of being chemically defined, allowing for more facile characterization and consistent drug loading. A number of innovative strategies have been employed in the synthesis of chemically-defined polymers including: Passerini reactions,15 esterification/amidation reactions,16–18 click reactions,19–21 photochemical transformations,22 radical reactions,23,24 and many more.25 Furthermore, a portion of these chemistries have been employed directly as linkers for ADCs with step-growth polysarcosines18 and oligothioetheramide backbones20,26 demonstrating the added benefit of sequence-defined polymers in the ADC field.
In this work, we offer a new strategy for the support-free synthesis of chemically defined polymers based on the iterative, stepwise polymerization of azide-functionalized, macrocyclic sulfate monomers. Inspired by the work of Jiang and coworkers,27 we envisioned that incorporation of an azide into an ethylene glycol (EG) macrocycle (Fig. 1) would allow for the efficient synthesis of sequence-defined oligoethylene glycols that could be functionalized with a desired payload. We believe this strategy is generally a valuable contribution to the field of chemically defined polymers as it allows for the rapid synthesis of linear polyethers with azide-bearing side chains. Furthermore, this chemistry is protecting-group free, and each ring opening step can be purified rapidly via flash chromatography. This feature of the synthesis could therefore accommodate a larger scale, which could be more economical in both time and cost when compared to methods that rely on preparatory HPLC purification for each chain-extending step. Furthermore, this method results in polymers with poly(ethylene glycol) backbones, which is a proven scaffold for the solubilization of hydrophobic, organic molecules. It should be noted that similar structures can be generated using techniques that employ molecular sieving for purifying intermediates.28 However, in contrast to previous reports, the synthesis reported herein allows for the direct incorporation of varied end-group functionalities into the polyether, which we find particularly advantageous especially when considering bioconjugation applications.
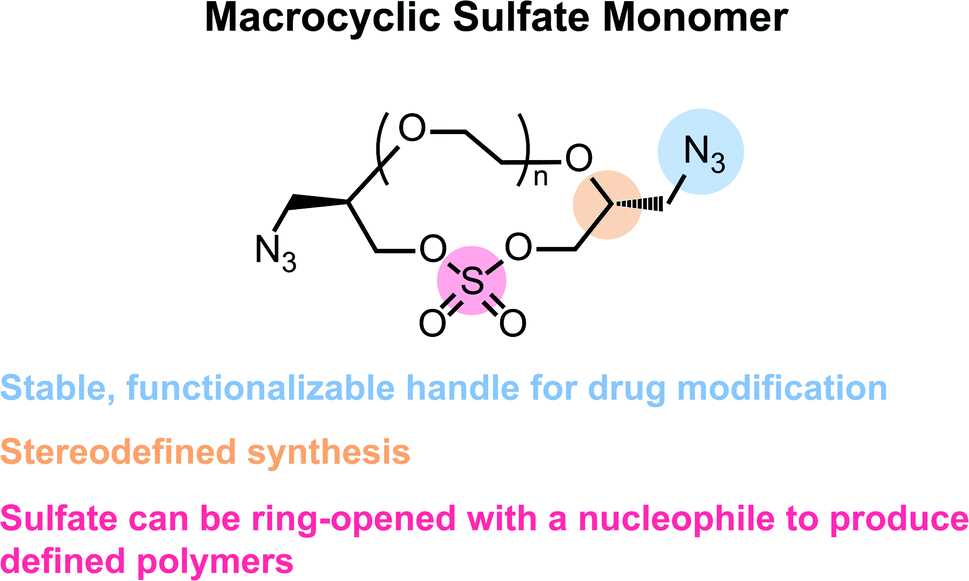 | ||
| Fig. 1 Advantages of the macrocyclic sulfate monomer in this research. | ||
Results and discussion
When considering the synthesis of these azido macrocyclic sulfates (AzMCS) monomers, we determined two criteria to be essential. First, the incorporation of the azide functionality needed be accomplished in a stereodefined manner to avoid diastereomeric mixtures during the polymerization process. Second, the synthesis needed to be amenable to the production of a diverse set of macrocycles such that the platform is tunable in terms of the size of the macrocycles, spacing of azides in the polymer, and eventual spacing of drugs in the sequence-defined polymers. To address the first requirement, we started with (R)-epichlorohydrin, which could be purchased cheaply at $0.50 USD per gram (Scheme 1). Installation of benzyl alcohol and ring opening with sodium azide then proceeded in high yields to produce a secondary alcohol that could be diversified with ditosylated EGs. The resulting di-benzylated products were deprotected non-reductively using NaBrO3 and sodium dithionite to yield the diazido ethylene glycols.29,30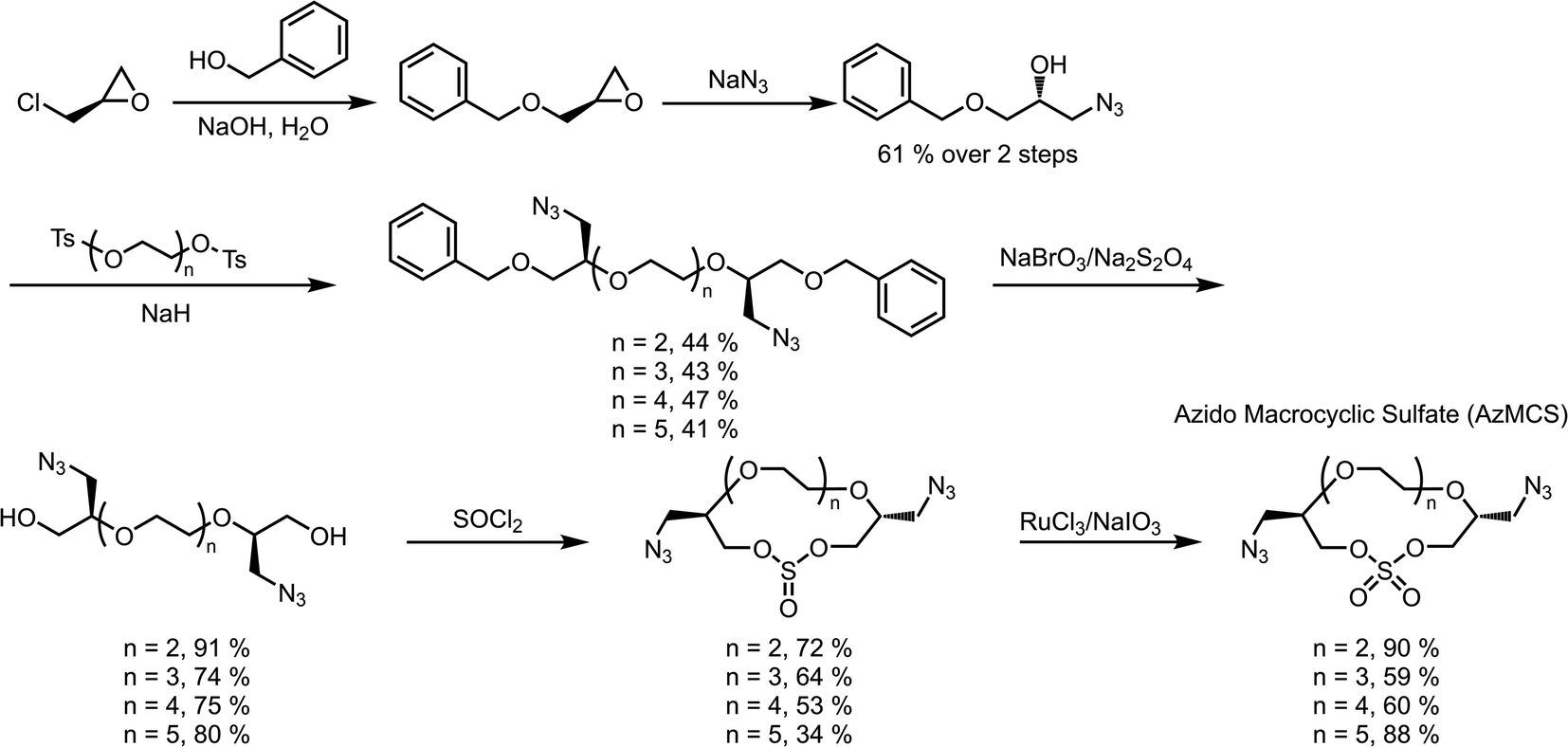 | ||
| Scheme 1 Synthesis of di-azido macrocyclic sulfate monomers of varied size. | ||
While good yields were obtained in the preparation of the diazido ethylene glycols, we observed a pattern of decreasing yields for larger EGs during the macrocyclization reaction. While it may be expected that this reaction would be less efficient for larger EGs, we further noticed that the larger macrocycles, particularly, the heptaethylene glycol (n = 5 in Scheme 1), are less stable and can degrade during flash chromatography. While a one-pot macrocyclization and oxidation could help avoid exposure of the less stable macrocyclic sulfite to silica, we found that such methods resulted in unacceptable amounts of impurities that proved difficult to remove. Despite the lower yield of the di-azido heptaethylene glycol, we were pleased that macrocyclization for the smaller (n = 2 through n = 4 in Scheme 1) diazido EGs could be achieved in good yields (53–72%). Final oxidation of the sulfite intermediates to the more stable sulfate additionally proceeded in high yields to provide the AzMCS monomers.
For the polymerization reactions, we decided to demonstrate the methodology by proceeding with the highest yielding tetraethylene glycol (n = 2 in Scheme 1) AzMCS. We chose to use 1,4 propargyloxy benzyl alcohol in the first ring opening step as it would provide a future handle for modification through a 1,3 dipolar cycloaddition and is UV active, allowing for more facile purification during flash chromatography. While this benzyl alcohol suites our applications well, it should be noted that a wide range of nucleophiles are compatible with this ring opening procedure.27 Flexibility in type of nucleophile is an additional advantageous feature of this synthesis as it could allow for the installation of wide range of functionalities at the beginning of the polymerization process. The ring opening of the AzMCS monomer using 1,4 propargyloxy benzyl alcohol proceeded with a high yield of 79%, and the pure product was rapidly obtained via flash chromatography (Fig. 2). Two additional cycles of ring opening and chain extension were performed with similarly high yields and purity to obtain the third generation hexa-azide dodeca-ethylene glycol polymer.
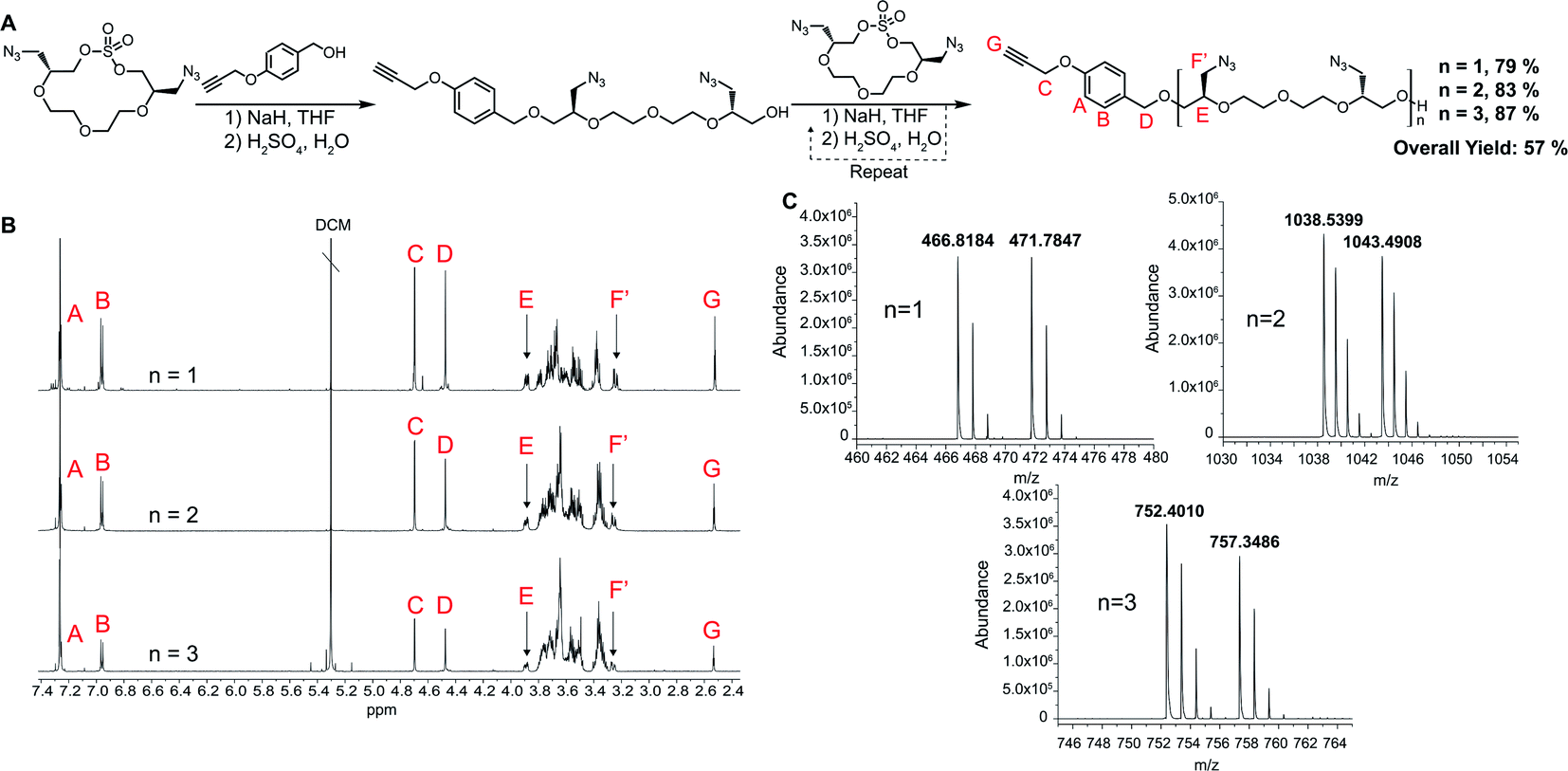 | ||
| Fig. 2 Synthesis of AzMCS polymers. (A) Synthesis of propargyl hexaazide polymer, (B) purified NMR of each ring-opening step, and (C) mass spectrum of each chain extension (NH4+ and Na+ adducts). | ||
One advantage of iterative, step-growth polymerization is the ability to tailor the sequence and composition of the polymer exactly. To demonstrate this in our system, we performed the same procedure of successive ring-opening polymerization reactions but alternated AzMCS monomers with unfunctionalized macrocyclic sulfates to make a tetraazide hexadeca-ethylene glycol polymer (Fig. 3A). Alternating monomers such as this allows for greater spacing between the azide functionalities and illustrates how these molecules can be tailored in terms of azide composition, spacing, and overall size.
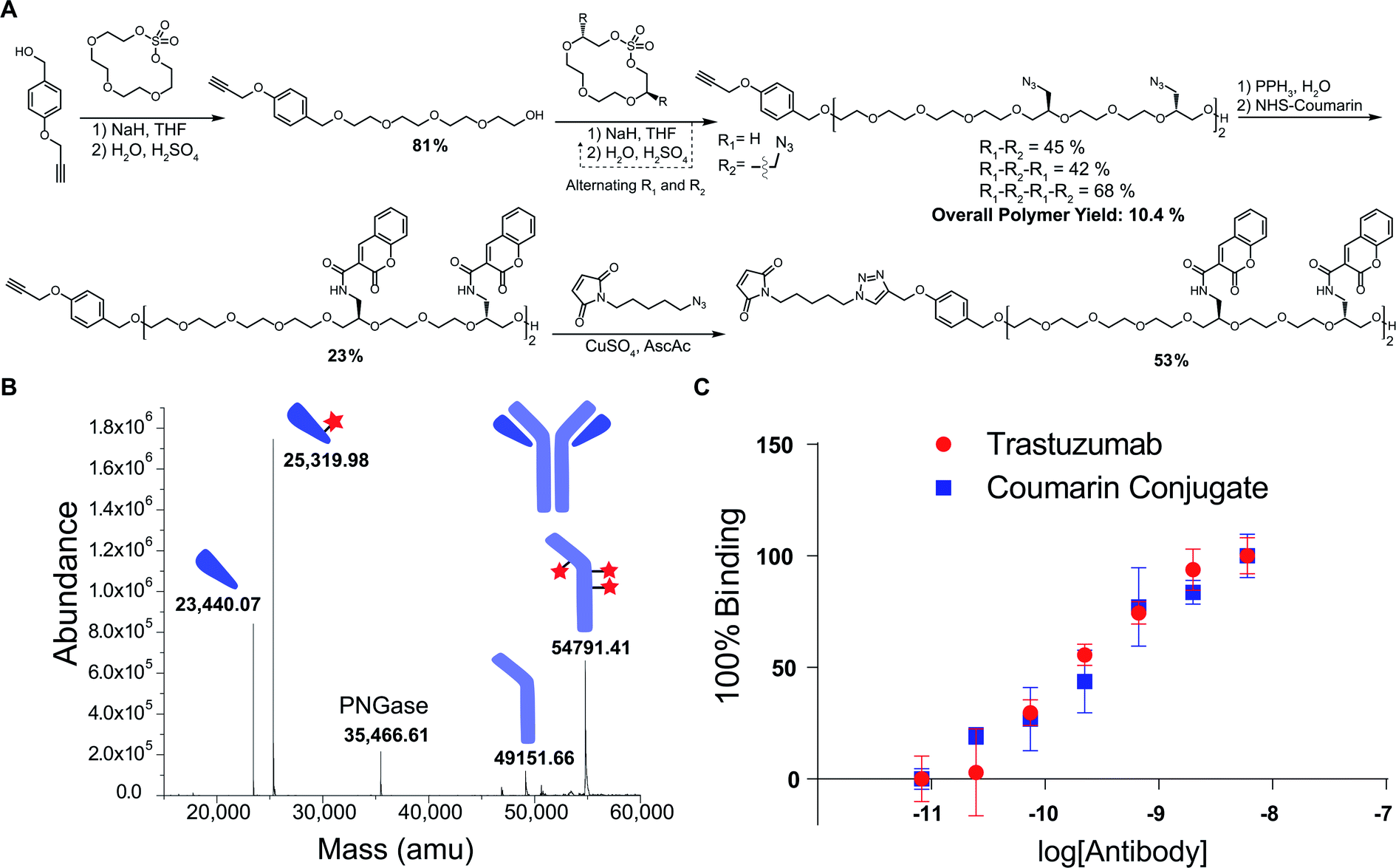 | ||
| Fig. 3 (A) Scheme for the synthesis of tetraazide poly(ethylene glycol) along with coumarin functionalization and maleimide installation. Yields noted in the polymerization process are reported as the individual, elongation reactions, not the overall yield. (B) Mass spectrometry of digested and reduced antibody–coumarin conjugate. Red stars represent the number of polymers modifying each antibody subunit. (C) Activity of trastuzumab and coumarin conjugate towards HER2 antigen as measured by ELISA. | ||
We next explored a proof-of-concept application for these polymers. Given the previously noted challenges with obtaining high-DAR ADCs, we envisioned this as a useful scaffold in achieving highly-conjugated antibodies. Using a coumarin derivative as a model, we first reduced the tetrazide hexadeca-ethylene glycol polymer using a Staudinger reduction to form the tetra-amine product (Fig. 3A). In one pot, we then alkylated the amines with N-hydroxysuccinimide (NHS)-activated coumarin to form the tetra-coumarin product. Finally, we installed a maleimide for thiol conjugation through a copper catalyzed 1,3 dipolar cycloaddition to provide the final product for antibody conjugation in 53% yield.
The polymer was then conjugated to trastuzumab, which is a commonly employed antibody in ADCs for the treatment of HER2 positive breast cancer. By first reducing the interchain disulfide bonds of the antibody using TCEP, we were able to successfully conjugate the maleimide-containing tetracoumarin polymer to the antibody. During this conjugation, the use of 20% DMF cosolvent was required to solubilize the polymer, suggesting that future use of cytotoxics or more hydrophobic ligands will necessitate increased PEG content in the polymer to improve solubility. After purification, the conjugate was characterized via liquid chromatography mass-spectrometry (LCMS), and the polymer-to-antibody ratio was determined to be 6.6, which translates to approximately 26 coumarin molecules per antibody (Fig. 3B). Additionally, in order to ensure that the conjugate maintained activity towards the HER2 antigen, we analyzed affinity via indirect ELISA. In this experiment, we observed no significant difference in binding between our conjugate and unmodified trastuzumab (Fig. 3C), which reinforces that these polymers do not have a significant impact on the affinity of the antibody. In addition to demonstrating the successful conjugation, we wanted to access the stability of the coumarin conjugate. Using size exclusion chromatography (SEC), we observed no appreciable aggregation of the conjugate, even after storing the material for 5 days at 22 °C (Fig. S1†). However, during this stress test, we did observe the evolution of a low molecular weight shoulder that we believe to be evidence of half-antibody fragmentation. Further evidence of this was provided by light scattering analysis from the SEC that reported the molecular weight of the conjugate 39% smaller than that of unmodified trastuzumab, consistent with the presence of half–antibody conjugate. We hypothesize that our conjugates could favor this dissociation due to the large size of the conjugated polymers, which in turn cause alterations of the antibody structure. We do believe that this could be significantly mitigated through use site-specific cysteine engineered antibodies, which would not require the reduction and alkylation of interchain disulfides.31 Furthermore, this would allow for the formation of a completely homogenous product, which is advantageous from both a characterization and manufacturing perspective.
The success of this model system indicates the applicability of these chemically-defined polymers as scaffolds for ADCs. The advantage of this approach is the facile characterization of the conjugate via LCMS and the large total number of coumarins that could be conjugated through the multivalent, hydrophilic linker without a reduction in binding activity. Furthermore, the lack of aggregation observed via SEC is encouraging for the continued development of the platform. Future work includes investigation of these scaffolds for ADCs by site-specifically incorporating cytotoxic payloads, which are more hydrophobic than coumarin, and comparing the performance of these sequence-defined scaffolds to current linker technology.
Conclusions
In summary, we present a new method for the preparation of sequence-defined polymers based on diazido macrocyclic sulfate building blocks. This strategy offers several benefits over other methodologies, primarily that each successive chain-extending step can be purified rapidly via flash chromatography, and it allows for the direct incorporation of functional end groups. Additionally, due to the makeup of the macrocycles, the final polymer contains a backbone solely composed of hydrophilic, non-ionic ethylene glycol units. We hypothesize that these unique characteristics are ideally suited for ADC linkers and look forward to continued investigation of this application. Additionally, due to the tunable and modular nature of this platform, we envision it to be useful in a variety of polymeric drug-delivery settings.Data availability
Experimental details and additional data can be found in the attached ESI.†Author contributions
N. F. and M. T. conducted the synthesis, conjugation, and experimentation. H. M. oversaw the project. All authors contributed to the writing and editing of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Research reported in this publication was supported by the National Institutes of Health under award number R01EB020676 (HDM) and T32GM008496 (NLF). We also acknowledge the Dr Myung Ki Hong Professor in Polymer Science Chair (HDM) for additional funding.Notes and references
- N. Joubert, A. Beck, C. Dumontet and C. Denevault-Sabourin, Pharmaceuticals, 2020, 13, 245 CrossRef CAS.
- P. Khongorzul, C. J. Ling, F. U. Khan, A. U. Ihsan and J. Zhang, Mol. Cancer Res., 2020, 18, 3–19 CrossRef CAS PubMed.
- L. N. Tumey, in Innovations for Next-Generation Antibody-Drug Conjugates, ed. M. Damelin, Springer International Publishing, Cham, 2018, pp. 187–214 Search PubMed.
- W. R. Strohl, Protein Cell, 2018, 9, 86–120 CrossRef CAS.
- N. Bodyak and A. V. Yurkovetskiy, in Innovations for Next-Generation Antibody-Drug Conjugates, ed. M. Damelin, Springer International Publishing, Cham, 2018, pp. 215–240 Search PubMed.
- X. Sun, J. F. Ponte, N. C. Yoder, R. Laleau, J. Coccia, L. Lanieri, Q. Qiu, R. Wu, E. Hong, M. Bogalhas, L. Wang, L. Dong, Y. Setiady, E. K. Maloney, O. Ab, X. Zhang, J. Pinkas, T. A. Keating, R. Chari, H. K. Erickson and J. M. Lambert, Bioconjugate Chem., 2017, 28, 1371–1381 CrossRef CAS PubMed.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS PubMed.
- J. W. Buecheler, M. Winzer, J. Tonillo, C. Weber and H. Gieseler, Mol. Pharmaceutics, 2018, 15, 2656–2664 CrossRef CAS.
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-LaFord, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Nat. Biotechnol., 2015, 33, 733–735 CrossRef CAS PubMed.
- P. J. Burke, J. Z. Hamilton, S. C. Jeffrey, J. H. Hunter, S. O. Doronina, N. M. Okeley, J. B. Miyamoto, M. E. Anderson, I. J. Stone, M. L. Ulrich, J. K. Simmons, E. E. McKinney, P. D. Senter and R. P. Lyon, Mol. Cancer Ther., 2017, 16, 116–123 CrossRef CAS PubMed.
- H. Schneider, L. Deweid, T. Pirzer, D. Yanakieva, S. Englert, B. Becker, O. Avrutina and H. Kolmar, ChemistryOpen, 2019, 8, 354–357 CrossRef CAS PubMed.
- A. V. Yurkovetskiy, M. Yin, N. Bodyak, C. A. Stevenson, J. D. Thomas, C. E. Hammond, L. Qin, B. Zhu, D. R. Gumerov, E. Ter-Ovanesyan, A. Uttard and T. B. Lowinger, Cancer Res., 2015, 75, 3365–3372 CrossRef CAS PubMed.
- T. Etrych, J. Strohalm, L. Kovár, M. Kabesová, B. Ríhová and K. Ulbrich, J. Controlled Release, 2009, 140, 18–26 CrossRef CAS.
- L. Zhang, Y. Fang, J. Kopeček and J. Yang, Eur. J. Pharm. Sci., 2017, 103, 36–46 CrossRef CAS PubMed.
- S. C. Solleder, D. Zengel, K. S. Wetzel and M. A. R. Meier, Angew. Chem., Int. Ed., 2016, 55, 1204–1207 CrossRef CAS PubMed.
- K. Takizawa, C. Tang and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 1718–1726 CrossRef CAS.
- K. Takizawa, H. Nulwala, J. Hu, K. Yoshinaga and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5977–5990 CrossRef CAS.
- W. Viricel, G. Fournet, S. Beaumel, E. Perrial, S. Papot, C. Dumontet and B. Joseph, Chem. Sci., 2019, 10, 4048–4053 RSC.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS PubMed.
- M. Porel and C. A. Alabi, J. Am. Chem. Soc., 2014, 136, 13162–13165 CrossRef CAS.
- S. Pfeifer, Z. Zarafshani, N. Badi and J.-F. Lutz, J. Am. Chem. Soc., 2009, 131, 9195–9197 CrossRef CAS.
- W. Konrad, C. Fengler, S. Putwa and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 7133–7137 CrossRef CAS PubMed.
- D. Oh, M. Ouchi, T. Nakanishi, H. Ono and M. Sawamoto, ACS Macro Lett., 2016, 5, 745–749 CrossRef CAS.
- Y. Hibi, M. Ouchi and M. Sawamoto, Nat. Commun., 2016, 7, 11064 CrossRef CAS PubMed.
- P. Nanjan and M. Porel, Polym. Chem., 2019, 10, 5406–5424 RSC.
- J. A. Walker, M. R. Sorkin, F. Ledesma, S. R. Kabaria, R. M. Barfield, D. Rabuka and C. A. Alabi, Bioconjugate Chem., 2019, 30, 2982–2988 CrossRef CAS PubMed.
- H. Zhang, X. Li, Q. Shi, Y. Li, G. Xia, L. Chen, Z. Yang and Z.-X. Jiang, Angew. Chem., 2015, 127, 3834–3838 CrossRef.
- R. Dong, R. Liu, P. R. J. Gaffney, M. Schaepertoens, P. Marchetti, C. M. Williams, R. Chen and A. G. Livingston, Nat. Chem., 2019, 11, 136–145 CrossRef CAS PubMed.
- M. Adinolfi, G. Barone, L. Guariniello and A. Iadonisi, Tetrahedron Lett., 1999, 40, 8439–8441 CrossRef CAS.
- M. Niemietz, L. Perkams, J. Hoffman, S. Eller and C. Unverzagt, Chem. Commun., 2011, 47, 10485–10487 RSC.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, methods, HPLC, MS, and 1H NMR spectra. See DOI: 10.1039/d1sc06242e |
| This journal is © The Royal Society of Chemistry 2022 |