
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of cooling rate in crystallization-driven block copolymer self-assembly†
Shaofei
Song
 a,
Jingjie
Jiang
a,
Ehsan
Nikbin
b,
Jane Y.
Howe
abd,
Ian
Manners
c and
Mitchell A.
Winnik
*ad
a,
Jingjie
Jiang
a,
Ehsan
Nikbin
b,
Jane Y.
Howe
abd,
Ian
Manners
c and
Mitchell A.
Winnik
*ad
aDepartment of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada. E-mail: m.winnik@utoronto.ca; Tel: +1-416-978-6495
bDepartment of Materials Science and Engineering, University of Toronto, 184 College Street, Toronto, Ontario M5S 3E4, Canada
cDepartment of Chemistry, University of Victoria, Victoria, British Columbia V8P 5C2, Canada
dDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, Toronto, Ontario M5S 3E2, Canada
First published on 2nd December 2021
Abstract
Self-assembly of crystalline-coil block copolymers (BCPs) in selective solvents is often carried out by heating the mixture until the sample appears to dissolve and then allowing the solution to cool back to room temperature. In self-seeding experiments, some crystallites persist during sample annealing and nucleate the growth of core-crystalline micelles upon cooling. There is evidence in the literature that the nature of the self-assembled structures formed is independent of the annealing time at a particular temperature. There are, however, no systematic studies of how the rate of cooling affects self-assembly. We examine three systems based upon poly(ferrocenyldimethylsilane) BCPs that generated uniform micelles under typical conditions where cooling took pace on the 1–2 h time scale. For example, several of the systems generated elongated 1D micelles of uniform length under these slow cooling conditions. When subjected to rapid cooling (on the time scale of a few minutes or faster), branched structures were obtained. Variation of the cooling rate led to a variation in the size and degree of branching of some of the structures examined. These changes can be explained in terms of the high degree of supersaturation that occurs when unimer solutions at high temperature are suddenly cooled. Enhanced nucleation, seed aggregation, and selective growth of the species of lowest solubility contribute to branching. Cooling rate becomes another tool for manipulating crystallization-driven self-assembly and controlling micelle morphologies.
Introduction
Block copolymers (BCPs) are amphiphilic molecules and undergo self-assembly in bulk or in selective solvents and provide an effective approach to access regular nano- and microstructures.1–8 Especially in solution, self-assembly has proved to be a low-cost processing protocol and afforded structures with morphological diversity and complexity, ranging from spheres, vesicles, and cylinders to hierarchical assemblies.9,10 These amphiphilic polymeric materials have been widely used for applications including drug delivery, catalysis, epoxy resin reinforcement, and water purification.11–14 In selective solvents, the solvophobic block undergoes microphase separation to form the core. When the core polymer is amorphous, the morphology is determined largely by the ratio of the soluble-to-insoluble block, and the solvophilic block provides colloidal stability to the micelles. When the core-forming block can crystallize, crystallization can serve as the main driving force for self-assembly, and lead to a new set of morphologies characterized by low curvature of the core-corona interface. This process is often referred to as crystallization-driven self-assembly (CDSA).15–29 For core-crystalline micelles, the morphology is largely determined by the block ratio, in which BCPs with long corona chains tend to form one-dimensional (1D) fibers and those with shorter corona chains tend to form 2D platelets.30 One of the most notable features of CDSA is that when the nucleation and growth steps can be separated, one can often obtain uniform morphologies.There are two general approaches to induce CDSA of crystalline-coil BCPs in solution. In the “solvent switch” approach, one mixes a solution of the BCP in a good solvent for both blocks with a solvent selective for the corona forming block. This promotes micelle formation via crystallization of the core-forming block. Alternatively, one can heat a suspension of the BCP in a selective solvent to promote BCP solubility. In this thermal processing approach, crystallization of the core-forming block occurs upon cooling. Because CDSA operates under kinetic rather than thermodynamic control, a number of other factors can influence the morphologies obtained. These include choice of solvent-non-solvent mixtures, heating or final cooling temperature,31 BCP concentration,32 the presence of salts or other small molecules in the solvent,33–36 and the presence of homopolymer corresponding to the crystalline block.37–41
For example, Schmelz et al.20 compared the self-assembly of polystyrene-block-polyethylene-block-poly(methyl methacrylate) (PS-b-PE-b-PMMA) triblock terpolymer under thermal processing conditions in two solvents of different quality for PE. The polymer dissolved upon heating in both solvents, but in dioxane, the PE phase-separated above its melting temperature, leading to spherical micelles that confined the crystallization of PE upon further cooling. In toluene, PE remained soluble until it crystalized, leading to cylindrical micelles. In this study, the authors also examined the effect of quenching to different crystallization temperatures as well as some aspects of the crystallization kinetics. Xu and coworkers31 examined the influence of crystallization temperature on the morphology of poly(ε-caprolactone)-block-poly(ethylene oxide) (PCL-b-PEO) micelles in aqueous solution. They used water-THF mixtures to prepare the micelles in water, and then heated the solution above the melting temperature of the PCL block. The samples with a shorter PEO corona block formed lamellar micelles when cooled to 20 °C, but cylindrical micelles when cooled to 0 °C. In contrast, the samples with a longer PEO block showed converse behavior, and lamellar and cylindrical micelles were formed at the lower crystallization temperature. Yang et al.34 described the effect of small molecule additives on the micelle morphologies. In a PCL-b-PEO micelle solution, addition of small amounts of phenol fragmented the 1D cylindrical micelles into short cylinders or spherical micelles, whereas L-threonine induced the transformation of spherical micelles into cylinders. These additives enable a reversible micelle morphological transformation. van de Ven and Eisenberg37 showed that addition of a short PCL homopolymer to a self-assembling PCL-b-PEO BCP led to morphological transformations of micelles from spheres to rods and finally to lamellae. In a remarkable series of experiments, Qiu et al.38 used rod-like micelles of PFS-b-PDMS (PDMS = polydimethylsiloxane) as seeds for the addition of a mixture of short PFS homopolymer and PFS-b-P2VP (2VP = 2-vinylpyridine). Living CDSA led to the formation of uniform rectangular platelets of controlled size.
There are other examples where the combination of solvent and temperature have been examined for their influence on micelle morphology,42,43 but there are few studies that have paid attention to the influence of the cooling protocol. Schmalz, Ballauff and coworkers44–46 examined the crystallization and aggregation behavior of a PB-b-PEO (PB = poly(1,2-butadiene)) BCP in heptane. Heptane is a poor solvent for PEO. The BCP self-assembled into spherical micelles composed of a liquid PEO core and a soluble PB corona at 70 °C. They compared two cooling pathways. Slow cooling to 30 °C led to meander structures, whereas rapid cooling by immersing a sample into liquid nitrogen led to spherical micelles. Zhu and coworkers47 compared the self-assembly of PE-b-PEO in trichlorobenzene (TCB) with that in dimethylformamide (DMF). When a solution of the BCP in TCB was heated at 140 °C and then slowly cooled to 25 °C, the BCP formed diamond-shaped micelles with a mono-layer crystallized PE lamella core. The BCP was less soluble in DMF. At 140 °C, it formed spherical micelles composed of a molten PE core and a soluble PEO corona. As the temperature was decreased quickly to 25 °C, the micelle morphology changed from spherical to platelet-like micelles with a double-layer crystallized PE lamella core sandwiched by the solvent-swollen PEO corona, suggesting that the cooling rate accompanying with solvents significantly influenced resultant micelle morphologies.
The experiments reported here were prompted by an apparent contradiction in two sets of experiments previously reported from this laboratory. In 2015,48 we reported a series of co-self-assembly experiments carried out by heating and then cooling mixtures of PFS-b-PI in decane with different amounts of PFS homopolymer. The sample vials were heated in an oil bath to dissolve the polymer and then cooled in air to room temperature (RT 23 °C) by removing the samples from the oil bath. While the PFS-b-PI samples employed formed long cylindrical micelles under these conditions, the mixtures with PFS homopolymer formed insect-like branched structures with narrow platelet centers and long spider-leg-like protrusions from each end. Our focus in this first set of experiments was on mixtures containing substantial (>5 w/w %) homopolymer. More recently, we returned to the topic of homopolymer-BCP blends and showed that adding only traces of crystalline PFS homopolymer to a PFS BCP could lead to fiber-like micelles uniform in length by a single heating−cooling step in a selective solvent.40,41 In these experiments, we heated the sample vials in an oil bath and cooled them by turning off the heater and allowed the hot oil bath and vials to cool to RT in air. We noticed, however, that some samples in the first set of experiments (relatively rapid cooling) (e.g., 5.76 w/w% PFS) formed insect-like branched structures at compositions (even up to 10 w/w% PFS) that led to rod-like micelles of uniform length in the second set of experiments (relatively slow cooling).40 This led us to hypothesize that the cooling protocol played a more important role in determining the micelle morphology than the blend composition.
In the work reported here, we examine the influence of cooling rate on micelle morphologies for a series of PFS BCPs including their mixtures with PFS homopolymer. We show that cooling rate can greatly influence the size and shape of the colloidal structures formed. Rapid cooling promotes branching, particularly in BCP mixtures with PFS homopolymer. This can lead to new and uniform nano- and microstructures, and thus the cooling rate becomes another aspect of CDSA that one can use to manipulate micelle morphologies.
Results and discussion
We begin by describing more precisely the cooling protocols similar to those described above in our previous experiments. Samples were in 13 mm diameter vials containing 2 mL solution initially heated in an oil bath at 90 °C as shown in Fig. 1a. In the quick cooling protocol, the vial was removed from the oil bath and allowed to cool in air. The cooling profile is shown by the blue curve in Fig. 1b. We can describe a characteristic cooling time t1/2, eqn (1), as the time needed for the temperature of the sample in the vial to decrease from the initial temperature Ti (here 90 °C) to Ti − ΔT/2, where Tf is the final temperature upon cooling (here 23 °C).| t1/2 = t(Ti−ΔT/2); ΔT = Ti − Tf | (1) |
 | ||
| Fig. 1 (a) A representative simple equipment for block copolymer self-assembly. For cooling in the air, the vial was heated in the oil bath first and then taken out for cooling. (b) Temperature changes vs. time during cooling procedure. | ||
The quick cooling process is characterized by t1/2 = 1.8 min. In the slow cooling profile, the hot plate is turned off, and the vial in the oil bath cools at the same slow rate as the oil bath. This process is characterized by t1/2 = 61 min. The red and green curves in Fig. 1b show that the temperature inside the vial tracks closely that of the oil bath itself. Later in the paper, we show that these cooling profiles can reasonably be fitted to an exponential curve.
In Scheme 1, we show the structure of the homopolymer and BCP samples examined in this report. We have chosen one PFS homopolymer with DP = 45 (DP = degree of polymerization) and six different PFS BCPs with four different corona-forming blocks. The synthesis and characterization of each of these samples have been described in previous publications. See the ESI† for details.
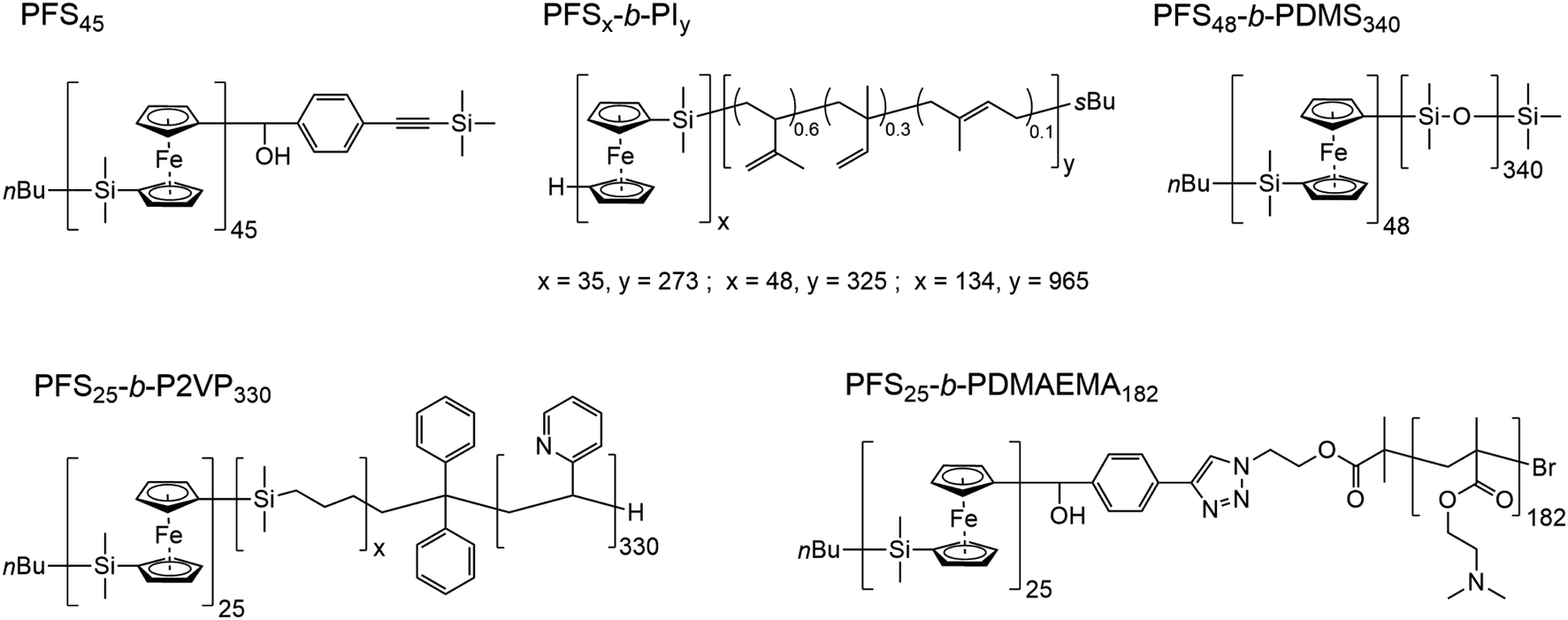 | ||
| Scheme 1 Structures of the homopolymer and block copolymers employed in this work. PFS, poly(ferrocenyldimethylsilane); PI, polyisoprene; PDMS, poly(dimethylsiloxane); P2VP, poly(2-vinylpyridine); PDMAEMA, poly(2-(N,N-dimethylamino)ethyl methacrylate). The subscripts refer to the number average degrees of polymerization. | ||
Homopolymer crystallization and self-assembly of BCPs with a narrow molecular weight distribution
We begin by probing the effects of cooling rates on the structures formed by PFS homopolymer at low concentration in decane when subjected to thermal annealing followed by a cooling cycle similar to that described in Fig. 1. For homopolymers in the melt state, the cooling rate is known to affect the crystallization temperature and, in turn, the subsequent melting temperature of a sample.49 For example, rapid cooling would reduce the crystallinity, since the polymer chains do not have enough time to pack into the ordered structures. Thus many defects may appear. In contrast, slow cooling would lead to relatively high crystallinity. However, there are few examples describing how cooling rate affects the crystallization behaviors of homopolymers or crystalline-coil BCPs in solution.44–47We also use these samples to define a sample preparation protocol, with the intent of providing each sample with a common thermal history. To define the thermal history, a sample in THF solution (50 μL, 1 mg mL−1) was introduced into a 13 mm diameter vial. The THF was evaporated under a stream of dry air. Solvent (1.0 mL) was introduced to afford a polymer concentration of 0.05 mg mL−1. Then the mixture was heated for 30 min to a sample-dependent annealing temperature followed by a cooling protocol.
Two samples of PFS45 suspended in decane at 0.05 mg mL−1 were heated for 30 min at 140 °C. By eye, the samples appeared to fully dissolve. The sample subjected to quick cooling (here t1/2 = 2.5 min) led to flower-like structures, 10–15 μm in diameter, seen in the transmission electron microscopy (TEM) images in Fig. 2a and ESI Fig. S1.† At higher magnification, one of the petals appears to be multi-layered packing platelets truncated at the ends. The sample subjected to slower cooling (here t1/2 = 50 min) also led to flower-like structures, but here the flowers are larger in size (>30 μm), and the petals are longer and thinner. While these differences are not dramatic, they are consistent with more rapid nucleation upon rapid cooling, leading to a larger number of smaller structures. They also allow us to set a baseline for observations reported below.
 | ||
| Fig. 2 TEM images of structures formed by homopolymer PFS45 through (a) quick cooling and (b) slow cooling process after heating in decane (0.05 mg mL−1, 1 mL) at 140 °C for 30 min and then cooling to RT. TEM images of micelles formed by PFS25-b-P2VP330 through (c) quick cooling and (d) slow cooling process after heating in 2-propanol (0.5 mg mL−1, 1 mL) at 80 °C for 30 min and cooling to RT. TEM images of micelles formed by PFS35-b-PI273 through (e) quick cooling and (f) slow cooling process after heating in decane (0.5 mg mL−1, 1 mL) at 80 °C for 30 min and cooling to RT. | ||
A few additional comments are in order about some ambiguities in experiments with PFS homopolymer samples. Samples subjected to heating and cooling lead to a significant fraction of solid precipitate accompanied by smaller objects suspended in solution. In these and in earlier experiments with PFS homopolymer,29,40,41,48,50 we examined only aliquots from the supernatant. There is no guarantee that these structures are representative of the solid at the bottom of the vial.
We next turn to examples consisting of three BCPs with long corona-forming blocks and narrow molecular weight distributions, PFS25-b-P2VP330, PFS35-b-PI273 and PFS48-b-PI325. These BCPs normally form elongated one-dimensional (1D) fiber-like micelles with a broad length distribution upon direct self-assembly by a heating–cooling protocol. Pairs of vials containing samples in 1.0 mL solvent were prepared at 0.5 mg mL−1 as described above (2-propanol for PFS25-b-P2VP330; decane for PFS35-b-PI273 and PFS48-b-PI325). These vials were sealed and placed into an oil bath at 80 °C and annealed for 30 min. Then one of the two vials for each BCP was taken out of the oil bath and allowed to cool in air (quick cooling, here t1/2 = 2.4 min). The other vial was left in the oil bath to cool to RT (slow cooling, t1/2 = 37 min). After aging for 24 h at RT, these samples were subjected to TEM measurements.
Results for PFS25-b-P2VP330 in 2-propanol are presented in Fig. 2c and d. Fiber-like micelles, uniform in width but polydisperse in length can be seen in both images. Quick cooling (Fig. 2c) led to much shorter micelles (<3 μm) than slow cooling (Fig. 2d, > 10 μm). Similar results are seen for PFS35-b-PI273 in decane (Fig. 2e and f) and for PFS48-b-PI325 in decane (ESI, Fig. S2†): elongated 1D micelles polydisperse in length, with much shorter micelles formed upon rapid cooling. The polydisperse nature (Lw/Ln > 1.25) of the 1D micelles indicates, first that nucleation and growth occurred simultaneously, and second, that the BCP samples dissolved completely upon annealing. Surviving crystallites would lead to micelles of uniform length as one observes in self-seeding experiments. These results are also consistent with rapid cooling leading to faster nucleation. Since the amount of BCP sample is the same in both vials, formation of more nuclei will lead to the formation of a larger number of shorter micelles. We keep these ideas in mind as a design principle as we examine other systems that generate more complex morphologies.
Co-self-assembly of homopolymer/BCP mixtures
As mentioned in the Introduction, we recently reported that addition of a trace of PFS homopolymer to the types of PFS BCPs described in the previous section could lead to fiber-like micelles of uniform length in a single heating-cooling step.40,41 Our idea at the time was that some homopolymer crystallites might survive the heating process and nucleate growth of the fiber-like micelles upon cooling. In an earlier paper that looked at self-assembly of PFS homopolymer/block copolymer mixtures,48 we reported that insect-like branched structures were formed. Once we realized that there was an overlap in the composition of these different experiments, we hypothesized that the differences in the structures obtained by self-assembly were due to differences in cooling rate. Here we examine a new set of samples to test this idea.We begin by looking at a sample in decane of PFS48-b-PI325 in the presence of 1 w/w % PFS45 homopolymer. In our notation, we refer to this mixture as PFS45/PFS48-b-PI325 (1 w/w%). The samples were prepared as described above and then heated to 90 °C for 30 min. In Fig. 3 and ESI Fig. S3,† we compare structures obtained upon quick cooling in air (t1/2 = 1.8 min) and slow cooling in the oil bath (t1/2 = 61 min). Quick cooling led to branched structures consisting of an elongated central platelet from which one or two long fibers protrude from the ends. When we measure the longest dimensions of these branched structures, we find that they are rather uniform. Analysis of more than 200 micelles in several images with the software ImageJ showed that the mean length Ln = 5370 ± 920 nm (see Fig. S3a and c†). Slow cooling led to long single fibers in which one can often identify a darker platelet-like feature in the center. These micelles were also uniform in length, with Ln = 6012 ± 636 nm (Fig. S3b and d†). These results confirm that the cooling rate played a vital role in determining the micelle morphologies. Furthermore, election diffraction patterns (Fig. S4†) demonstrate the single crystal nature of these PFS homopolymer/block copolymer assemblies. In both the center core and the fiber branches, they show with essentially identical patterns of three pairs of diffraction spots separated by ca. 60°, indicating that the PFS core of the structures exhibit a single crystal order with monoclinic symmetry.
 | ||
| Fig. 3 TEM images of micelles formed via co-self-assembly of PFS45/PFS48-b-PI325 (1 w/w%) in decane (1 mg mL−1) under different cooling protocols. Both samples were heated at 90 °C for 30 min then cooled to 23 °C: (a) quick cooling; (b) slow cooling. Scale bars: 1 μm. | ||
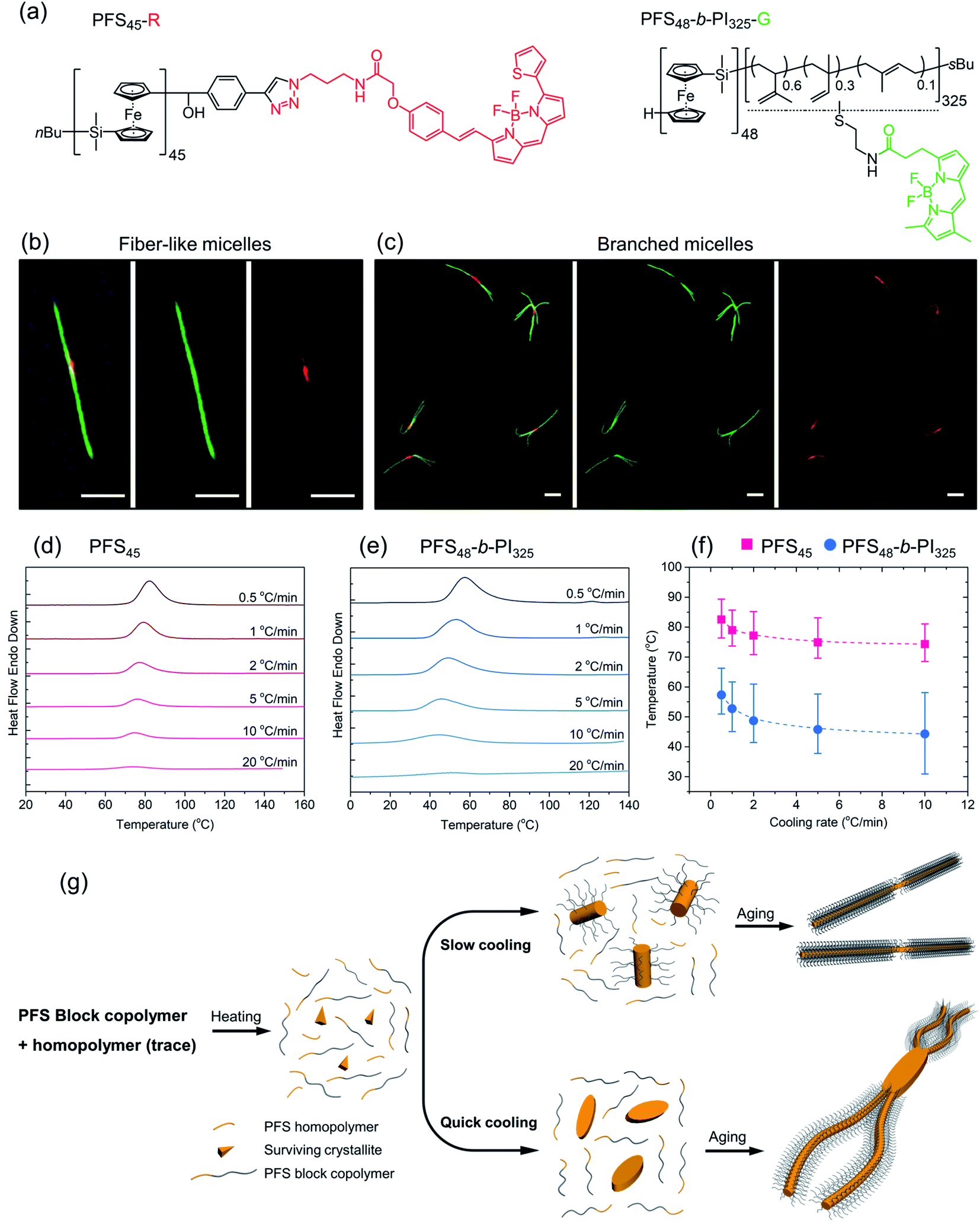 | ||
| Fig. 4 (a) Chemical structures of PFS homopolymer (PFS45-R) and block copolymer (PFS48-b-PI325-G) labeled by fluorescent dyes BDP 630/650 and BDP FL, respectively. (b and c) LCFM images of micelles by co-self-assembly of PFS45-R/PFS48-b-PI325-G (1.5 w/w%) (1 mg mL−1, 90 °C), (b) fiber-like micelles via slow cooling; (c) branched micelles by quick cooling. Scale bars: 2 μm. DSC traces of (d) homopolymer PFS45 and (e) block copolymer PFS48-b-PI325 in decane at different cooling rates after heating to 160 °C. (f) Plots showing that the crystallization temperatures Tc decreased with increasing cooling rates. The error bars describe the full width at half of the maximum (FWHM) of the crystallization peak. (g) Proposed co-self-assembly process. Most PFS homopolymer will dissolve as free unimers upon heating to 90 °C, although some tiny crystallites may survive to nucleate micelle growth. For the slow cooling process, initial seed micelles were formed primarily by homopolymer as well as some block copolymers. In the quick cooling process, the initial seeds were formed almost entirely by PFS homopolymers. | ||
Subsequently, a mixture PFS45-R/PFS48-b-PI325-G (1.5 w/w%) in decane at 1 mg mL−1 was heated to 90 °C for 30 min and then allowed to cool slowly to RT. The middle LCFM images in Fig. 4b for the green channel show that the green color associated with the BCP appears along the entire structure, whereas the right-hand image for the red channel shows that the homopolymer can be detected exclusively in the center. The merged image at the left shows the yellow color associated with superposition of the red and green fluorescence. Fig. 4c shows the corresponding results for the homopolymer/BCP mixture subjected to quick cooling. The middle image from the green channel shows the green BCP fluorescence in the branched structures, but close inspection of the structures shows apparent dark regions near the branch sites. These are the sites where the red fluorescence can be seen in the right-hand panel indicating the presence of homopolymer. In the merged image at the left, the red color is much more prominent than in the merged image of the linear micelles obtained upon slow cooling. Additional images are presented in the ESI Fig. S6.† The main message here is that upon rapid cooling, homopolymer crystallization dominates and that these homopolymer crystallites serve as branch sites for the growth of BCP fibers.
To obtain additional insights into the crystallization behavior of PFS45 homopolymer and PFS48-b-PI325 BCP we examined cooling curves from hot solutions by micro-differential scanning calorimetry (μDSC). We could not obtain a measureable signal at low concentrations. Hence samples of the homopolymer at 3.5 mg mL−1 and of the BCP at 10 mg mL−1 in decane in sealed pans were examined by μDSC. Each sample was first heated to 160 °C (above its melting point, 125–130 °C,53,54 to remove the thermal history), then cooled to 0 for the BCP or 20 °C for the homopolymer. The cooling runs at different cooling rates (i.e., 20 °C min−1, 10 °C min−1, 5 °C min−1, 2 °C min−1, 1 °C min−1, 0.5 °C min−1) are presented in Fig. 4d, e and ESI Fig. S7.† We found that the crystallization peaks (Tcs) of both homopolymer and BCP shifted to lower temperature with increasing cooling rates (Fig. 4f). The amplitudes also decrease. While the full width at half of the maximum (FWHM) of the crystallization peaks (see ESI Fig. S7†) broadened for faster cooling, the area under the curve, a measure of the crystallization enthalpy, strongly decreased. Tc values for the BCP were about 15 °C lower than those for the homopolymer, and the peaks were broader for the BCP.
These experiments suggest that when a hot solution containing homopolymer and BCP is cooled, the homopolymer will be the first to crystallize. If the cooling rate is slow, these crystallites will nucleate growth of BCP fibers along with homopolymer remaining in solution. Both components will appear in the center region of the 1D micelles that are formed. In contrast, rapid cooling will promote rapid crystallization of less perfect homopolymer crystals. These may be polycrystalline or crystallite aggregates that can nucleate branched growth of BCP fibers.50
We summarize the self-assembly mechanism in Fig. 4g. When the homopolymer/BCP/decane mixtures were heated to 90 °C, the BCP dissolved and most if not all of the homopolymer dissolved. We allow for the possibility that some crystallites of the homopolymer might survive the annealing process. Upon cooling, in the slow cooling process, homopolymer nucleated co-crystallization of BCP unimer and the remaining homopolymer in solution. After aging, 1D fiber-like micelles were obtained. In the quick cooling process, the initial seeds mainly came from homopolymer nucleation or deposition on surviving crystallites. These seeds often provided more than one nucleation site at each end. Upon aging, branched structures formed.
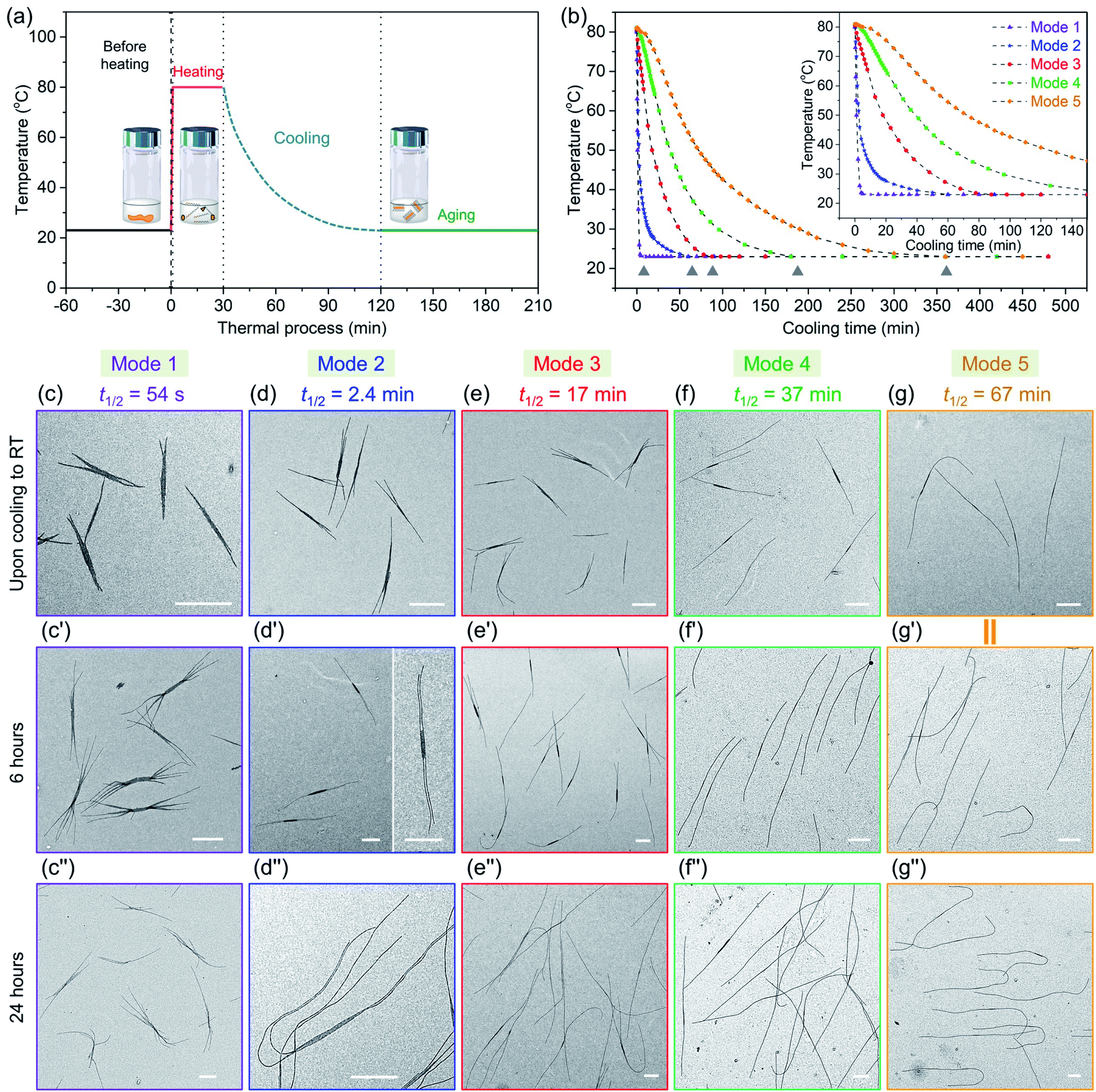 | ||
| Fig. 5 (a) Scheme for all thermal processes of the samples, PFS45/PFS48-b-PI325 (0.25 w/w%) in decane (0.5 mg mL−1) through preheating at 80 °C for 30 min and then cooling to 23 °C (RT). (b) Temperature changes vs. time in different cooling procedures in this work. We employed five different cooling protocols (modes). The grey triangle symbols label the points upon cooling to RT. TEM images of micelles formed in different cooling protocols for co-self-assembly, (c–g) upon cooling to RT; (c′–g′) after cooling to RT for 6 h; (c′′–g′′) after cooling to RT for 24 h. (g and g′) are from the same sample. Scale bars: 1 μm. | ||
| Cooling modes | t 1/2 | Time (from 80 °C to 28 °C) |
|---|---|---|
| Mode 1 | 54 s | 2.8 min |
| Mode 2 | 2.4 min | 19 min |
| Mode 3 | 17 min | 53 min |
| Mode 4 | 37 min | 105 min |
| Mode 5 | 67 min | 205 min |
We then took aliquots of each of the sample solutions, placed them on a TEM grid, and imaged the samples, as a function of aging time. For the most rapidly cooled sample (Mode 1), the first sample was taken after approximately 3 min after removal from the oil bath and immersion in water at 23 °C. To minimize any changes caused by slow evaporation of decane, we injected a 100 μL aliquot into 900 μL hexanes before placing a 10 μL droplet on a carbon-coated copper TEM grid. Fig. 5c, c′′ and S9† show the structures formed at different growth stages for this sample. The initial structures formed after 3 min are branched assemblies with a platelet center and fiber-like protrusions at the two termini. Even though they had not reached their final morphologies, their size was narrowly distributed. We measured the longest distance from the ends of the fibers at both ends as shown in ESI Fig. S9 and S10.† By analysis of multiple micelles with ImageJ, determined by measuring more than 200 samples in several images, we found that these branched micelles were very uniform in length with Ln = 1571 nm, Lw/Ln = 1.02 (ESI Table S1†). The branches grew longer with aging time, reaching Ln = 4290 nm (Lw/Ln = 1.02) after 6 h and Ln = 5323 nm (Lw/Ln = 1.03) after 24 h. The branch number at each end varied from 2 to 5.
In the Mode 2 cooling process, we still found branched structures at all aging times (Fig. 5d, d′′ and S10†). In the first sample taken after cooling for ca. 20 min, elongated platelets can be seen with fiber-like protrusions. These micelles were characterized by Ln = 2453 nm, Lw/Ln = 1.03. The micelles continue to grow, characterized by Ln = 4474 nm, Lw/Ln = 1.03. Scarf-like micelles with long parallel tassels can be seen in Fig. 5d′ after aging 6 h. There are commonly only two fibers from each chain end, different from that seen in micelles formed by cooling protocol Mode 1. After cooling for 24 h, these micelles reached a length of Ln = 8830 nm, Lw/Ln = 1.03 (see ESI Fig. S10 and Table S1†). They are much longer than the branched assemblies formed in Mode 1, even though the total amount of homopolymer and BCP unimer in each vial was the same. Two factors can affect the overall length. Micelles formed via Mode 1 cooling have more branches and the average length of the branches are shorter. Also, more nuclei are likely to have formed upon more rapid cooling, leading to a larger number of shorter micelles.
A similar situation can be seen for Mode 3. Many branched micelles can be seen, accompanied by a few 1D structures (Fig. 5e). Over time, the micelles grew much longer. The initial sample, cooled to ca. 28 °C after 55 min consisted of elongated lenticular platelets with short protruding fibers characterized by Ln = 2768 nm, Lw/Ln = 1.03. After 6 h and 24 h aging, mixtures containing 1D fiber-like micelles and branched micelles were obtained (Fig. 5e′ and 5e′′), with mean lengths of 4629 nm and 9258 nm, respectively and narrow dispersities (ESI Fig. S11 and Table S1†). Meanwhile, their overall lengths still showed low dispersity. These micelles are somewhat longer than those obtained in Mode 2, possibly due to the reduced branching number. For the self-assembly through Mode 4 and 5, we did not observe branched structures, including samples examined immediately upon cooling to RT and after aging for 24 h. For Mode 4, the initial sample taken after 105 min cooling was characterized by Ln = 3216 nm, Lw/Ln = 1.03 and grew to mean lengths of 5083 nm after 6 h and 9566 nm after 24 h, all with narrow length dispersities. For the slowest cooling protocol, the initial sample, with Ln = 5050 nm, Lw/Ln = 1.02, was taken after 6 h. These micelles grew to Ln = 9602 nm, Lw/Ln = 1.02 after 24 h. Note that the final micelle length after 24 h aging was essentially the same for cooling protocols Mode 4 (t1/2 = 37 min) and Mode 5 (t1/2 = 67 min) (Fig. 5f, f′′, g, g′′, ESI Fig. S12, S13, Table S1 and S2†).
We can draw a number of interesting conclusions from this systematic study of the effect of cooling rates on the co-self-assembly of PFS45/PFS48-b-PI325 (0.25 w/w%) in decane. First, rapid cooling led to branched structures. Slower cooling led to fewer branches, and the slowest cooling rate resulted in 1D fiber-like micelles. Overall, the more highly branched structures were characterized by shorter branches. Second, all of these structures for this mixture were uniform in length. Thus in all cases, nucleation occurred on a faster time scale than growth. Finally, we note that BCP unimer deposition on the growing micelles was relatively slow and took place largely at room temperature. A substantial fraction of growth occurred between aging times of 6 and 24 h. We note that cooling rate becomes another tool that one can employ to control micelle morphology.
Co-self-assembly of homopolymer + two BCPs
At the next level of complexity, we examine the co-self-assembly of mixtures of two BCPs in the presence of small amounts of homopolymer. Two factors influence the self-seeding behavior of a mixture of two BCPs that share a common crystallizable block.55 The first factor is the characteristic dissolution temperature of the micelles, and the second factor is the rate at which unimer adds to the growing micelles upon cooling. Only the second factor is important if the annealing temperature is high enough that one component undergoes nearly complete dissolution. The length of the PFS block plays a dominant role in both processes, and a kinetic study by Boott et al.56 showed that the micelle growth rate was particularly sensitive to the length of the PFS block. If the two BCPs have similar PFS block lengths and the corona chains lead to similar growth rates in the cooling step, one anticipates a near random distribution of corona chains in the comicelles. If one of the blocks can be selectively stained in TEM images of the micelles, one anticipates finding a patchy structure to the comicelles.In the first set of experiments, we chose the homopolymer PFS45 and two BCPs PFS48-b-PI325 and PFS48-b-PDMS340 with the same core-forming block but different corona-forming blocks. Experimentally, aliquots of the homopolymer (0.5 mg mL−1 in THF) and each BCP (10 mg mL−1 in THF) were added successively to a vial and then the THF was evaporated in a stream of dry air, leading to a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mass ratio of the two BCPs PFS48-b-PI325 and PFS48-b-PDMS340 containing 0.5 w/w% homopolymer (PFS45/(PFS48-b-PI325 + PFS48-b-PDMS340)). The samples were subjected to similar co-self-assembly conditions (c = 1 mg mL−1 in decane; heating to 90 °C for 30 min) but different cooling modes followed by aging at RT for 24. For rapid cooling (t1/2 = 1.2 min, similar to Mode 1 in Fig. 5), the vial was taken out from the oil bath and immersed in water at RT. For slow cooling (t1/2 = 61 min, similar to the one in Fig. 1 and Mode 4 in Fig. 5), the vials were allowed to cool unperturbed in the oil bath. These different cooling protocols led to distinct micelle morphologies. To enhance the contrast in the TEM images shown in Fig. 6, we treated the comicelle dispersions with Karstedt's catalyst, which selectively adds platinum nanoparticles (PtNPs) to the PI corona. In Fig. 6b and ESI Fig. S14,† we see that the rapid cooling mode led to uniform branched micelles with patchy details along the branches (inset in Fig. 6b). They have low dispersity in length (Ln = 9245 nm, Lw/Ln = 1.02) as determined with ImageJ by measuring 200 positions in different samples and different TEM images (see ESI Fig. S14†). Around 3–5 branches appeared at each side. In contrast, only 1D patchy comicelles were obtained in the sample subjected to the slow cooling mode (Fig. 6c). The uniform patchy fibers have a somewhat shorter length (Ln = 8724 nm, Lw/Ln = 1.02) (see ESI Fig. S15†) compared with the patchy branched micelles.
:3 mass ratio of the two BCPs PFS48-b-PI325 and PFS48-b-PDMS340 containing 0.5 w/w% homopolymer (PFS45/(PFS48-b-PI325 + PFS48-b-PDMS340)). The samples were subjected to similar co-self-assembly conditions (c = 1 mg mL−1 in decane; heating to 90 °C for 30 min) but different cooling modes followed by aging at RT for 24. For rapid cooling (t1/2 = 1.2 min, similar to Mode 1 in Fig. 5), the vial was taken out from the oil bath and immersed in water at RT. For slow cooling (t1/2 = 61 min, similar to the one in Fig. 1 and Mode 4 in Fig. 5), the vials were allowed to cool unperturbed in the oil bath. These different cooling protocols led to distinct micelle morphologies. To enhance the contrast in the TEM images shown in Fig. 6, we treated the comicelle dispersions with Karstedt's catalyst, which selectively adds platinum nanoparticles (PtNPs) to the PI corona. In Fig. 6b and ESI Fig. S14,† we see that the rapid cooling mode led to uniform branched micelles with patchy details along the branches (inset in Fig. 6b). They have low dispersity in length (Ln = 9245 nm, Lw/Ln = 1.02) as determined with ImageJ by measuring 200 positions in different samples and different TEM images (see ESI Fig. S14†). Around 3–5 branches appeared at each side. In contrast, only 1D patchy comicelles were obtained in the sample subjected to the slow cooling mode (Fig. 6c). The uniform patchy fibers have a somewhat shorter length (Ln = 8724 nm, Lw/Ln = 1.02) (see ESI Fig. S15†) compared with the patchy branched micelles.
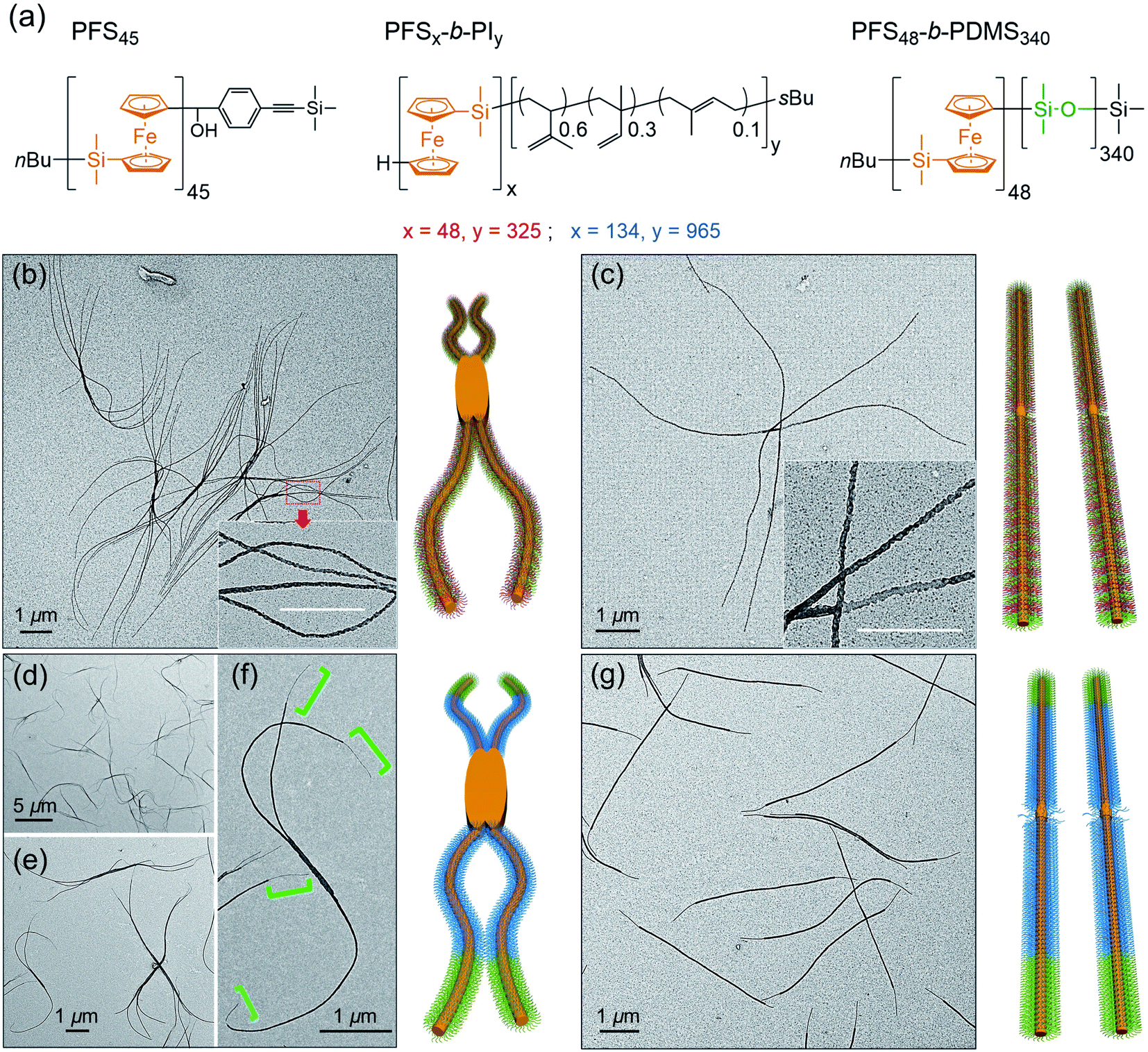 | ||
| Fig. 6 (a) Chemical structures of the PFS homopolymer and the three block polymers employed. In the drawings to the right of each set of TEM images we use orange to indicate the location of PFS, red/blue to indicate the location of the PI corona chains, and green to indicate the location of the PDMS corona chains. (b and c): TEM images of the micelles formed by PFS45/(PFS48-b-PI325 + PFS48-b-PDMS340) (0.5 w/w%, BCP mass ratio 2:3). The mixture was heated at 90 °C for 30 min. (b) Branched structures formed upon rapid cooling; (c) linear structures formed upon slow cooling. (d–f): TEM images of the micelles formed by PFS45/(PFS134-b-PI965 + PFS48-b-PDMS340) (0.5 w/w%, BCP mass ratio 5:1). The mixture was heated at 100 °C for 30 min. Branched block comicelle structures formed upon rapid cooling. (g) Same sample as in (d–f) but linear block comicelle structures formed upon slow cooling. The PI bloc was stained with Karstedt's catalyst to show the patchy structure of the corona in (b and c), and the block comicelle structure in (d–g). | ||
In the second set of experiments, we examine a blend of two BCPs with very different PFS block lengths. Here we consider a 5:1 w/w mixture of PFS134-b-PI965 and PFS48-b-PDMS340 in the presence of 0.5 w/w % PFS45 homopolymer in decane. In these experiments, we employed a common annealing temperature (100 °C), annealing time (30 min) and polymer concentration (1 mg mL−1), conditions for which essentially all of the PFS48-b-PDMS340 and most of the PFS134-b-PI965 would dissolve. Upon cooling, we anticipate that the more rapid growth of the BCP with the longer PFS block will promote the formation of block comicelles. The new aspect of the experiments reported here is the variation in cooling rate.
As shown in Fig. 6d–f rapid cooling (immersion in water at RT, t1/2 = 1 min) led to the formation of branched assemblies with 1–5 branches at each end. By measuring more than 200 objects by ImageJ, we found these branched structures were rather uniform in their longest dimension (Ln = 7879 nm, Lw/Ln = 1.02, see ESI Fig. S16†). By staining the PI blocks of each sample with Karstedt's catalyst, we found that these structures consisted of separate blocks, and the terminal block was PDMS (Fig. 6f). In contrast, the slow cooling protocol (t1/2 = 48 min) led only to 1D triblock fiber-like comicelles (Fig. 6g). The total length and the length of each block were also uniform (Ln = 5390 nm, Lw/Ln = 1.03; center block, Ln = 4133 nm, Lw/Ln = 1.03; terminal block, Ln = 617 nm, Lw/Ln = 1.03) (see ESI Fig. S17†).
While the results in the previous paragraph establish unambiguously that rapid cooling promotes branching for these BCPs, there is another feature of the results that merits further comment. It is curious that the branched structures formed upon rapid cooling have a longer mean length (Ln = 7879 nm) than the 1D comicelle counterparts (Ln = 5390 nm). This is different from the situation seen in Fig. 5 where micelle length increased as the cooling rate decreased. To explain how rapid cooling can lead to branched micelles longer than the linear micelles obtained by slow cooling, we have to assume that rapid cooling led to a smaller overall number of nuclei. While the nature of these nuclei are not yet known, they likely involve participation of PFS134-b-PI965 with the long PFS block.
Self-assembly of BCPs with polydisperse corona-forming blocks
In the final example, we examine the influence of cooling rate on the self-seeding behavior of a PFS BCP with a short PFS block and a polydisperse corona chain. BCPs with a short PFS block and a much longer corona-forming block under direct self-assembly conditions in a selective solvent normally form 1D fiber-like micelles of uniform width but with a broad distribution of lengths. In a recent publication,57 we reported that the BCP PFS25-b-PDMAEMA182 (PDMAEMA = poly(2-(N,N-dimethylamino)ethyl methacrylate)) with a polydisperse corona chain self-assembled to form uniform 1D structures in a single heating and cooling step. Under these conditions, several similar BCPs with more narrowly disperse corona chains formed micelles with a broad length distribution. DSC cooling traces indicate that BCPs with a polydisperse corona chain exhibit a broader crystallization exotherm compared to BCPs with more narrowly disperse corona chains.57 We showed that the change in self-assembly behavior was a consequence of BCPs in the sample with the shortest corona chains. These either nucleated first upon cooling or survived as crystallites during the heating step, so that nucleation occurred much faster than micelle growth. Here we revisit these experiments to see how sensitive they are to a change in cooling rate.The structure of PFS25-b-PDMAEMA182 is shown in Fig. 7a and its GPC analysis is shown in Fig. 7b. TEM images of micelles formed by BCPs with the same PFS block but more monodisperse corona chains are presented in the ESI Fig. S18.†
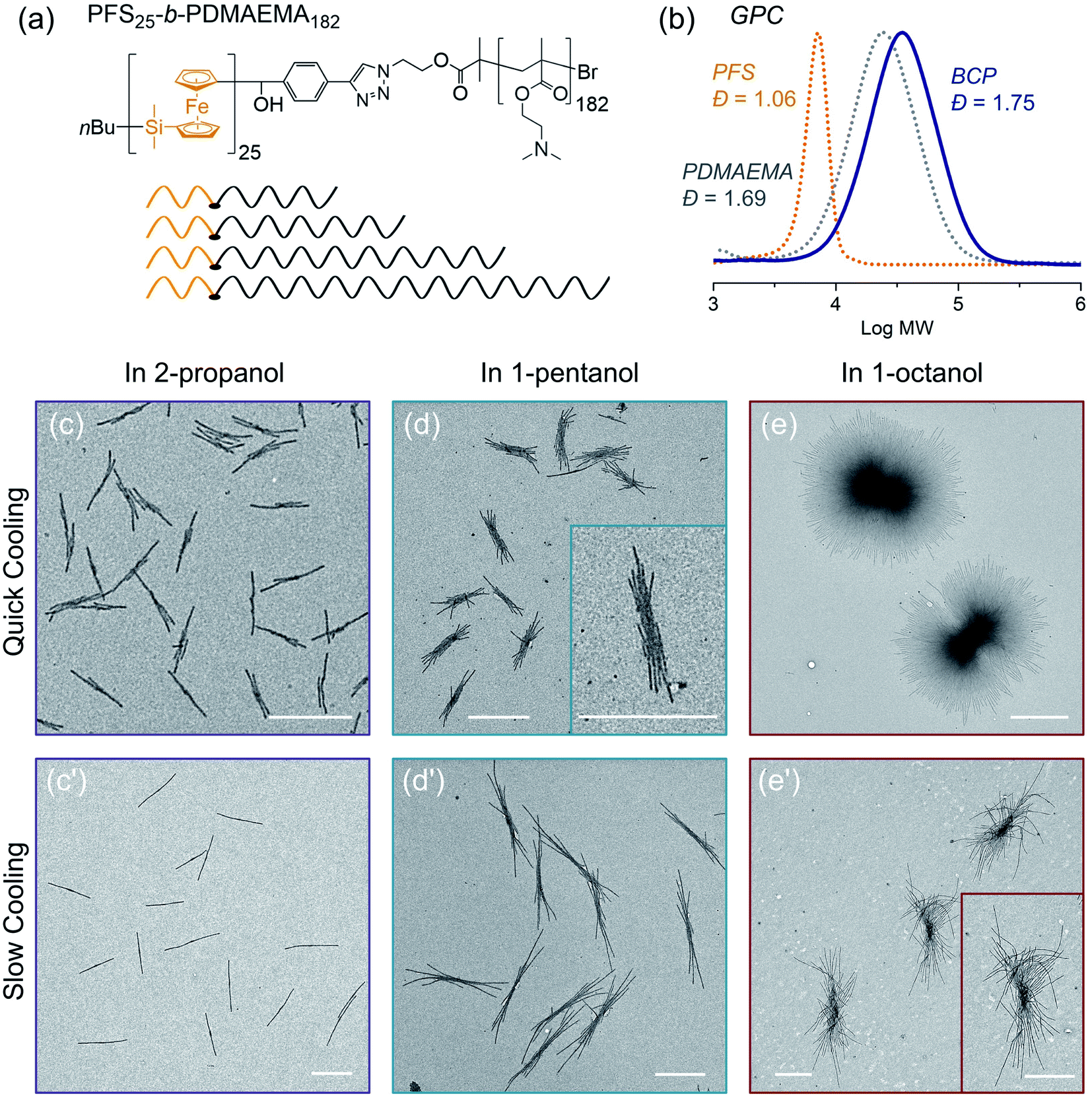 | ||
| Fig. 7 (a) Structure of PFS25-b-PDMAEMA182 and the molecular chain scheme showing uniform PFS blocks (orange) and polydisperse PDMAEMA blocks (grey). (b) GPC traces of PFS, PDMAEMA and the BCP PFS25-b-PDMAEMA182. TEM images of structure formed by PFS25-b-PDMAEMA182 in different solvents (1 mg mL−1, 1 mL) through (c, d, e) rapid cooling and (c′, d′, e′) slow cooling, respectively, after heating at 80 °C for 30 min and cooling to RT and aging for 24 h. For rapid cooling, t1/2 = 2.4 min; for slow cooling, t1/2 = 37 min. Scale bars: 1 μm. The insets in (d and e′) show structures at higher magnification. | ||
To prepare samples of PFS25-b-PDMAEMA182 for the self-assembly experiments reported here, a solution of the polymer was injected into ethanol. The precipitate was collected and dried at RT. We used a microbalance to weigh the dry samples and placed them into 4 mL vials. One of three selective solvents, 2-propanol, 1-pentanol or 1-octanol, was added to afford polymer concentrations of 1 mg mL−1 in 1 mL solvent. Subsequently, the sealed vials were placed into the oil bath (preheated to 80 °C) for 30 min. Then the samples were cooled to RT and aged for another 24 hours. We employed the two cooling processes shown in Fig. 1. For each selective solvent, one vial was taken out of the oil bath and cooled in air; the other one remained in the oil bath, which was turned off and allowed to cool to RT. TEM images of the micelles obtained are presented in Fig. 7.
In 2-propanol (Fig. 7c and c′), we found that slow cooling led to 1D rod-like micelles, as we reported previously.57 In contrast, rapid cooling led to branched structures with one or two fibers growing out of a central point. Both sets of structures are uniform in size. By measuring more than 200 micelles using ImageJ, we found the 1D rods formed by slow cooling in 2-propanol to have a mean length, Ln = 1201 nm, Lw/Ln = 1.01. The branched counterparts obtained by rapid cooling are characterized by Ln = 743 nm, Lw/Ln = 1.04 (see ESI Fig. S19†).
The choice of solvent has a major effect on the self-assembly of this BCP.58–62 For example, in 1-pentanol, branched structures are formed even with slow cooling (Fig. 7d, d′ and ESI Fig. S20†). There was a greater extent of branching in the insect-like structures formed by slow cooling in 1-pentanol than by rapid cooling in 2-propanol. However, the extent of branching increased for the micelles formed by rapid cooling. Both sets of structures were uniform in size. As in the case of 1-pentanol, rapid cooling process led to relatively shorter micelles (Ln = 1131 nm, Lw/Ln = 1.03) than those (Ln = 2333 nm, Lw/Ln = 1.01) obtained with the slow cooling rate. Results for self-assembly experiments in 1-octanol are shown in Fig. 7e, e′′ and ESI Fig. S21, S22.† The structures formed in 1-octanol are more highly branched than those formed in 1-pentanol, again with big differences associated with the cooling rate. The assemblies formed under slow cooling showed more than 10 branches on each side. The length along the longest direction was uniform (Ln = 3173 nm, Lm/Ln = 1.01) (see ESI Fig. S22†). In contrast, rapid cooling led to star-like or pompom-like micelles with short fiber-like cilia protruding from the entire surface of these objects. Their length also is uniform (Ln = 2698 nm, Lm/Ln = 1.01) (Fig. S21†), but they are much wider.
In summary, two factors affect the extent of branching in the self-assembly of PFS25-b-PDMAEMA182 with polydisperse corona chains. The structures formed upon heating-then-cooling in 1-pentanol and in 1-octanol are more branched than those formed in 2-propanol, and rapid cooling increases the extent of branching. Our explanation for the polydispersity effect involves the fraction of the sample with the shortest corona block. These molecules will have the lowest solubility upon heating, and crystallites enriched in this component may survive the annealing step. They can serve as nuclei during the cooling step. In our view, slow cooling allowed more time for BCP molecules with relatively longer corona chains to trap the initial seed micelles. This led to elongated (1D) seeds or seeds with fewer sites for unimer deposition, and in this way, led to 1D rod-like micelles (in 2-propanol) or to multiple rods growing from a central core (1-pentanol, 1-octanol). In contrast, rapid cooling promoted additional nucleation and aggregation of seed crystallites. These seeded the growth of BCPs with long corona chains, leading to more highly branched structures consisting of multiple fiber-like micelles attached to a common core.
Summary and conclusions
We described several examples of thermally-induced crystallization-driven self-assembly of PFS BCPs in which the cooling rate plays an important role in determining micelle morphology. We chose examples in which samples annealed to the point where they appear to fully dissolve generated uniform structures, often rod-like 1D micelles, upon slow cooling (over ca. 2 h). Rapid cooling (over 5–10 min) led to branched structures, and in one example where modestly branched structures formed upon slow cooling (e.g., PFS25-b-PDMAEMA182 with polydisperse corona chains in 1-pentanol), much more highly branched structures formed upon more rapid cooling. Even with the increase in branching, the structures formed by CDSA remained uniform in size.Polymer crystallization is driven by supersaturation. In the case of CDSA of block copolymers, heating promotes unimer dissolution, and cooling leads to supersaturated solutions. Since the solubility of the dissolved unimer decreases as the solution cools, rapid cooling leads to a higher degree of supersaturation early in the self-assembly process. This is analogous to large undercoolings in melt crystallization of semicrystalline polymers. High supersaturation can promote nucleation, particularly of the least soluble components of a mixture, and crystallite aggregation can lead to seed particles with multiple faces for initiating the growth of BCP micelles.
For simple PFS diblock copolymers with long corona chains of uniform length, direct self-assembly with slow cooling of samples that dissolve completely upon heating generates 1D micelles that are polydisperse in length. Here self-nucleation is a rare event and nucleation and growth compete, leading to a distribution of micelle lengths. In the presence of small amounts of PFS homopolymer, homopolymer nucleation is promoted upon cooling, enhancing the formation of micelles of uniform length as seen in Fig. 3 and 4. As seen in Fig. 4, rapid nucleation and growth of homopolymer crystals upon rapid cooling leads to platelets that nucleate the subsequent growth of fiber-like micelles.
Fig. 5 shows two important features of the self-assembly process. First, a faster rate of cooling led to more highly branched structures. Perhaps more important, it shows that a substantial portion of the self-assembly took place after the sample had cooled to room temperature. This tells us that the extent of branching is determined very early in the self-assembly process while the sample is in the initial stages of cooling.
Fig. 6 extends these experiments to mixtures of PFS BCPs with different corona chains that include a trace of PFS homopolymer. Slow cooling of heated solutions led to linear micelles with a patchy or blocky corona structure, depending upon the choice of BCP. Rapid cooling led to branched structures with one or two long fibers growing out of a central platelet, but with the same patchy or blocky corona structure as in the case of slow cooling. These branched structures are often novel morphologies that are not accessible in any other way.
The main lesson from this study is that the cooling rate becomes another tool that one can use to manipulate crystallization-driven self-assembly and control micelle morphologies. It will be interesting to see how this concept can be applied to BCPs with a crystallizable block with important optical or electronic properties.
Data availability
Data for this paper, including TEM images, confocal images and NMR spectra are available at https://doi.org/10.5683/SP3/XLJEB7.Author contributions
S. S. and M. A. W. conceived the project and designed the experiments. S. S. carried out the experiments of synthesis, self-assembly, and other characterizations. J. J. provided helpful discussion on cooling modes. E. N. and J. Y. H. conducted the election diffraction measurements. M. A. W. and S. S. analyzed the data and wrote the manuscript with input from I. M. M. A. W. supervised the project. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The Toronto authors thank NSERC Canada (Grant Number: RGPIN-2017-03741) for their support of this research. Election diffraction measurements were carried out in the CFI-funded Open Centre for the Characterization of Advanced Materials.References
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- D. J. Pochan, Z. Y. Chen, H. G. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94–97 CrossRef CAS PubMed.
- J. X. Zheng, H. Xiong, W. Y. Chen, K. Lee, R. M. Horn, R. P. Quirk, B. Lotz, E. L. Thomas, A. C. Shi and S. Z. D. Cheng, Macromolecules, 2006, 39, 641–650 CrossRef CAS.
- J. Dupont, G. Liu, K. Nihara, R. Kimoto and H. Jinnai, Angew. Chem., Int. Ed., 2009, 48, 6144–6147 CrossRef CAS PubMed.
- W. N. He and J. T. Xu, Prog. Polym. Sci., 2012, 37, 1350–1400 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. H. Gröschel and A. H. E. Müller, Nanoscale, 2015, 7, 11841–11876 RSC.
- S. T. Russell, R. Raghunathan, A. M. Jimenez, K. Zhang, S. D. Brucks, C. Iacob, A. C. West, O. Gang, L. M. Campos and S. K. Kumar, Macromolecules, 2020, 53, 548–557 CrossRef CAS.
- Z. Deng and S. Y. Liu, Polymer, 2020, 207, 122914 CrossRef CAS.
- M. Karayianni and S. Pispas, J. Polym. Sci., 2021, 59, 1874–1898 CrossRef CAS.
- L. MacFarlane, C. Zhao, J. Cai, H. Qiu and I. Manners, Chem. Sci., 2021, 12, 4661–4682 RSC.
- Y. Geng, P. Dalhaimer, S. S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- J. Schöbel, M. Burgard, C. Hils, R. Dersch, M. Dulle, K. Volk, M. Karg, A. Greiner and H. Schmalz, Angew. Chem., Int. Ed., 2017, 56, 405–408 CrossRef PubMed.
- J. Cai, C. Li, N. Kong, Y. Lu, G. Lin, X. Wang, Y. Yao, I. Manners and H. B. Qiu, Science, 2019, 366, 1095–1098 CrossRef CAS PubMed.
- B. Lotz and A. J. Kovacs, Kolloid Z. Z. Polym., 1966, 209, 97–114 CrossRef CAS.
- T. Vilgis and A. Halperin, Macromolecules, 1991, 24, 2090–2095 CrossRef CAS.
- A. P. Gast, P. K. Vinson and K. A. Cogan-Farinas, Macromolecules, 1993, 26, 1774–1776 CrossRef CAS.
- X. S. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- Z. X. Du, J. T. Xu and Z. Q. Fan, Macromolecules, 2007, 40, 7633–7637 CrossRef CAS.
- J. Schmelz, M. Karg, T. Hellweg and H. Schmalz, ACS Nano, 2011, 5, 9523–9534 CrossRef CAS PubMed.
- T. Gädt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144–150 CrossRef PubMed.
- N. Petzetakis, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2011, 2, 955–960 RSC.
- X. Y. Li, Y. Gao, X. Xing and G. Liu, Macromolecules, 2013, 46, 7436–7442 CrossRef CAS.
- S. Ganda, M. Dulle, M. Drechsler, B. Förster, S. Förster and M. H. Stenzel, Macromolecules, 2017, 50, 8544–8553 CrossRef CAS.
- D. Tao, C. Feng, Y. Cui, X. Yang, I. Manners, M. A. Winnik and X. Huang, J. Am. Chem. Soc., 2017, 139, 7136–7139 CrossRef CAS PubMed.
- Y. Sha, M. A. Rahman, T. Zhu, Y. Cha, C. W. McAlister and C. B. Tang, Chem. Sci., 2019, 10, 9782–9787 RSC.
- S. Yang and T. L. Choi, Chem. Sci., 2020, 11, 8416–8424 RSC.
- Z. Z. Tong, Y. Su, Y. Jiang, Y. Xie, X. Chen and R. K. O'Reilly, Macromolecules, 2021, 54, 2844–2851 CrossRef CAS.
- S. F. Song, H. Zhou, S. Y. Ye, J. Tam, J. Y. Howe, I. Manners and M. A. Winnik, Angew. Chem., Int. Ed., 2021, 60, 10950–10956 CrossRef CAS PubMed.
- L. Cao, I. Manners and M. A. Winnik, Macromolecules, 2002, 35, 8258–8260 CrossRef CAS.
- Z. X. Du, J. T. Xu and Z. Q. Fan, Macromol. Rapid Commun., 2008, 29, 467–471 CrossRef CAS.
- G. Guerin, G. Molev, D. Pichugin, P. A. Rupar, F. Qi, M. Cruz, I. Manners and M. A. Winnik, Macromolecules, 2019, 52, 208–216 CrossRef CAS.
- W. N. He, J. T. Xu, B. Y. Du, Z. Q. Fan and X. S. Wang, Macromol. Chem. Phys., 2010, 211, 1909–1916 CrossRef CAS.
- J. X. Yang, B. Fan, J. H. Li, J. T. Xu, B. Y. Du and Z. Q. Fan, Macromolecules, 2016, 49, 367–372 CrossRef CAS.
- Z. Z. Tong, R. Zhang, P. Ma, H. Xu, H. Chen, Y. Li, W. Yu, W. Zhuo and G. Jiang, Langmuir, 2017, 33, 176–183 CrossRef CAS PubMed.
- X. Y. Wang, R. Y. Wang, B. Fan, J. T. Xu, B. Y. Du and Z. Q. Fan, Macromolecules, 2018, 51, 2138–2144 CrossRef CAS.
- G. Rizis, T. G. M. van de Ven and A. Eisenberg, Angew. Chem., Int. Ed., 2014, 53, 9000–9003 CrossRef CAS PubMed.
- H. B. Qiu, Y. Gao, C. E. Boott, O. E. Gould, R. L. Harniman, M. J. Miles, S. E. Webb, M. A. Winnik and I. Manners, Science, 2016, 352, 697–701 CrossRef CAS PubMed.
- B. Fan, R. Y. Wang, X. Y. Wang, J. T. Xu, B. Y. Du and Z. Q. Fan, Macromolecules, 2017, 50, 2006–2015 CrossRef CAS.
- S. F. Song, H. Zhou, G. Hicks, C. K. Rastogi, Q. Yu, I. Manners and M. A. Winnik, Chem. Commun., 2020, 56, 4595–4598 RSC.
- S. F. Song, X. M. Liu, E. Nikbin, J. Y. Howe, Q. Yu, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2021, 143, 6266–6280 CrossRef CAS PubMed.
- J. S. Qian, G. Guerin, Y. J. Lu, G. Cambridge, I. Manners and M. A. Winnik, Angew. Chem., Int. Ed., 2011, 50, 1622–1625 CrossRef CAS PubMed.
- J. S. Qian, X. Li, D. J. Lunn, J. Gwyther, Z. M. Hudson, E. Kynaston, P. A. Rupar, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2014, 136, 4121–4124 CrossRef CAS PubMed.
- A. M. Mihut, A. Chiche, M. Drechsler, H. Schmalz, E. Di Cola, G. Krausch and M. Ballauff, Soft Matter, 2009, 5, 208–213 RSC.
- A. M. Mihut, J. J. Crassous, H. Schmalz and M. Ballauff, Colloid Polym. Sci., 2010, 288, 573–578 CrossRef CAS.
- A. M. Mihut, J. J. Crassous, H. Schmalz, M. Drechsler and M. Ballauff, Soft Matter, 2012, 8, 3163–3173 RSC.
- Z. Li, R. Liu, B. Mai, W. Wang, Q. Wu, G. Liang, H. Gao and F. Zhu, Polymer, 2013, 54, 1663–1670 CrossRef CAS.
- G. Cambridge, M. J. Gonzalez-Alvarez, G. Guerin, I. Manners and M. A. Winnik, Macromolecules, 2015, 48, 707–716 CrossRef CAS.
- J. E. K. Schawe, J. Appl. Polym. Sci., 2016, 133, 42977 CrossRef.
- H. B. Qiu, G. Cambridge, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2013, 135, 12180–12183 CrossRef CAS PubMed.
- D. J. Lunn, C. E. Boott, K. E. Bass, T. A. Shuttleworth, N. G. McCreanor, S. Papadouli and I. Manners, Macromol. Chem. Phys., 2013, 214, 2813–2820 CrossRef CAS.
- C. E. Boott, D. J. Lunn and I. Manners, J. Polym. Sci., Part A: Polym. Chem., 2015, 54, 245–252 CrossRef.
- R. G. H. Lammertink, M. A. Hempenius, I. Manners and G. J. Vancso, Macromolecules, 1998, 31, 795–800 CrossRef CAS.
- J. Duvigneau, E. Kutnyanszky, I. Y. Phang, H.-J. Chung, H. Wu, L. Dos Ramos, T. Gädt, S. F. M. Yusoff, M. A. Hempenius, I. Manners and G. J. Vancso, Polymer, 2014, 55, 2716–2724 CrossRef CAS.
- J. P. Xu, H. Zhou, Q. Yu, G. Guerin, I. Manners and M. A. Winnik, Chem. Sci., 2019, 10, 2280–2284 RSC.
- C. E. Boott, E. M. Leitao, D. W. Hayward, R. F. Laine, P. Mahou, G. Guerin, M. A. Winnik, R. M. Richardson, C. F. Kaminski, G. R. Whittell and I. Manners, ACS Nano, 2018, 12, 8920–8933 CrossRef CAS PubMed.
- S. F. Song, H. Zhou, I. Manners and M. A. Winnik, Chem, 2021, 7, 2800–2821 CAS.
- S. Pearce, X. He, M. S. Hsiao, R. L. Harniman, L. R. MacFarlane and I. Manners, Macromolecules, 2019, 52, 6068–6079 CrossRef CAS.
- S. Yang, S. Y. Kang and T. L. Choi, J. Am. Chem. Soc., 2019, 141, 19138–19143 CrossRef CAS PubMed.
- S. F. Song, Q. Yu, H. Zhou, G. Hicks, H. Zhu, C. K. Rastogi, I. Manners and M. A. Winnik, Chem. Sci., 2020, 11, 4631–4643 RSC.
- S. F. Song, M. Puzhitsky, S. Y. Ye, M. Abtahi, C. K. Rastogi, E. Lu, G. Hicks, I. Manners and M. A. Winnik, Macromolecules, 2020, 53, 6576–6588 CrossRef CAS.
- S. F. Song, H. Zhou, M. Puzhitsky, Y. F. Zhang, G. Hicks, Y. Lu, I. Manners and M. A. Winnik, Macromolecules, 2021, 54, 930–940 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05937h |
| This journal is © The Royal Society of Chemistry 2022 |