
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA homogeneous high-DAR antibody–drug conjugate platform combining THIOMAB antibodies and XTEN polypeptides†
Neelie
Zacharias‡
a,
Vladimir N.
Podust‡
b,
Kimberly K.
Kajihara
a,
Douglas
Leipold
 a,
Geoffrey
Del Rosario
a,
Desiree
Thayer§
b,
Emily
Dong
a,
Maciej
Paluch
a,
David
Fischer
a,
Kai
Zheng
a,
Corinna
Lei
a,
Jintang
He
a,
Carl
Ng
a,
Dian
Su
a,
Luna
Liu
a,
Shabkhaiz
Masih
a,
William
Sawyer
a,
Jeff
Tinianow
a,
Jan
Marik
a,
Victor
Yip
a,
Guangmin
Li
a,
Josefa
Chuh
a,
J. Hiroshi
Morisaki
a,
Summer
Park¶
a,
Bing
Zheng
a,
Hilda
Hernandez-Barry
a,
Kelly M.
Loyet
a,
Min
Xu
a,
Katherine R.
Kozak
a,
Gail Lewis
Phillips
a,
Ben-Quan
Shen
a,
Cong
Wu
a,
Keyang
Xu
a,
Shang-Fan
Yu
a,
Amrita
Kamath
a,
Rebecca K.
Rowntree
a,
Dorothea
Reilly
a,
Thomas
Pillow
a,
Andrew
Polson
a,
Volker
Schellenberger
*b,
Wouter L. W.
Hazenbos||
*a and
Jack
Sadowsky**
*a
a,
Geoffrey
Del Rosario
a,
Desiree
Thayer§
b,
Emily
Dong
a,
Maciej
Paluch
a,
David
Fischer
a,
Kai
Zheng
a,
Corinna
Lei
a,
Jintang
He
a,
Carl
Ng
a,
Dian
Su
a,
Luna
Liu
a,
Shabkhaiz
Masih
a,
William
Sawyer
a,
Jeff
Tinianow
a,
Jan
Marik
a,
Victor
Yip
a,
Guangmin
Li
a,
Josefa
Chuh
a,
J. Hiroshi
Morisaki
a,
Summer
Park¶
a,
Bing
Zheng
a,
Hilda
Hernandez-Barry
a,
Kelly M.
Loyet
a,
Min
Xu
a,
Katherine R.
Kozak
a,
Gail Lewis
Phillips
a,
Ben-Quan
Shen
a,
Cong
Wu
a,
Keyang
Xu
a,
Shang-Fan
Yu
a,
Amrita
Kamath
a,
Rebecca K.
Rowntree
a,
Dorothea
Reilly
a,
Thomas
Pillow
a,
Andrew
Polson
a,
Volker
Schellenberger
*b,
Wouter L. W.
Hazenbos||
*a and
Jack
Sadowsky**
*a
aGenentech, Inc., 1 DNA Way, South San Francisco, CA 94080, USA. E-mail: whazenbos@vir.bio; jack.sadowsky@gmail.com
bAmunix Pharmaceuticals, Inc., 2 Tower Place, South San Francisco, CA 94080, USA. E-mail: vschellenberger@amunix.com
First published on 28th January 2022
Abstract
The antibody–drug conjugate (ADC) is a well-validated modality for the cell-specific delivery of small molecules with impact expanding rapidly beyond their originally-intended purpose of treating cancer. However, antibody-mediated delivery (AMD) remains inefficient, limiting its applicability to targeting highly potent payloads to cells with high antigen expression. Maximizing the number of payloads delivered per antibody is one key way in which delivery efficiency can be improved, although this has been challenging to carry out; with few exceptions, increasing the drug-to-antibody ratio (DAR) above ∼4 typically destroys the biophysical properties and in vivo efficacy for ADCs. Herein, we describe the development of a novel bioconjugation platform combining cysteine-engineered (THIOMAB) antibodies and recombinant XTEN polypeptides for the unprecedented generation of homogeneous, stable “TXCs” with DAR of up to 18. Across three different bioactive payloads, we demonstrated improved AMD to tumors and Staphylococcus aureus bacteria for high-DAR TXCs relative to conventional low-DAR ADCs.
Introduction
Antibody–drug conjugates (ADCs) have become mainstays as cell-targeted delivery vehicles for small molecules and are poised to have broad impact as therapeutics for a range of human diseases.1,2 The first generation of ADCs directed potent, broad-spectrum cytotoxic agents to tumor cells; after decades of research several such ADCs have gained approval for clinical use against various cancers.1,3 The scope of payloads benefitting from antibody-mediated delivery (AMD) to human cells has in recent years been expanding and now includes steroids,4,5 TLR agonists,6 oligonucleotides,7 bifunctional degraders,8 epigenetic modulators9 and other molecules.2 We recently described antibody–antibiotic conjugates (AACs) for the treatment of Staphylococcus aureus infections, exemplifying successful AMD to bacteria.10 Despite these advances, there continue to exist biological limits to AMD effectiveness, including receptor expression level, internalization rate, tissue penetration and circulation half-life.11 These barriers have historically prevented all but the most potent of payloads and most highly expressed of antigens from being addressed with conventional ADCs. Innovations in antibody engineering, linker design and payload chemistry will likely be essential to expand the scope of payloads and antigens for which AMD will be successful.Maximizing the drug-to-antibody ratio (DAR) represents a key approach to increasing ADC payload delivery efficiency that, in principle, overcomes intrinsic biological limits.12 A typical antibody has >70 lysines and 8 cysteines that can be modified by a suitably-reactive payload.13,14 However, payload attachment increases conjugate hydrophobicity and can at high DAR lead to aggregation, fast clearance and poor efficacy. Since early ADC studies demonstrated such deleterious effects at DAR = 8,14 a longstanding practical limit has existed: the vast majority of reported ADCs, including most clinically-approved agents, have DAR < 4 (Scheme 1A).2,3 Motivation to deliver novel payloads via AMD has inspired efforts to increase payload loading although few have led to ADCs with DAR > 4 that are also effective in vivo. The most precedented approach involves incorporation of PEG or another hydrophilic moiety between antibody and payload, which has led to effective ADCs wherein all 8 interchain Cys residues are conjugated (i.e., DAR = 8; Scheme 1B).15–19 Other researchers have described efficacious ADCs with DAR up to approximately 20, wherein 3–5 antibody lysines or cysteines are conjugated stochastically to a payload-loaded hydrophilic polyacetal polymer dubbed “Fleximer” (Scheme 1B).20,21 While these strategies represent significant advances, conjugation of payloads or polymers to endogenous antibody cysteines is not ideal as these residues form stabilizing inter and intramolecular disulfides.22 Incomplete payload conjugation to antibody residues is also not ideal as the resulting ADC is heterogeneous, which can complicate characterization and result in toxicity.23
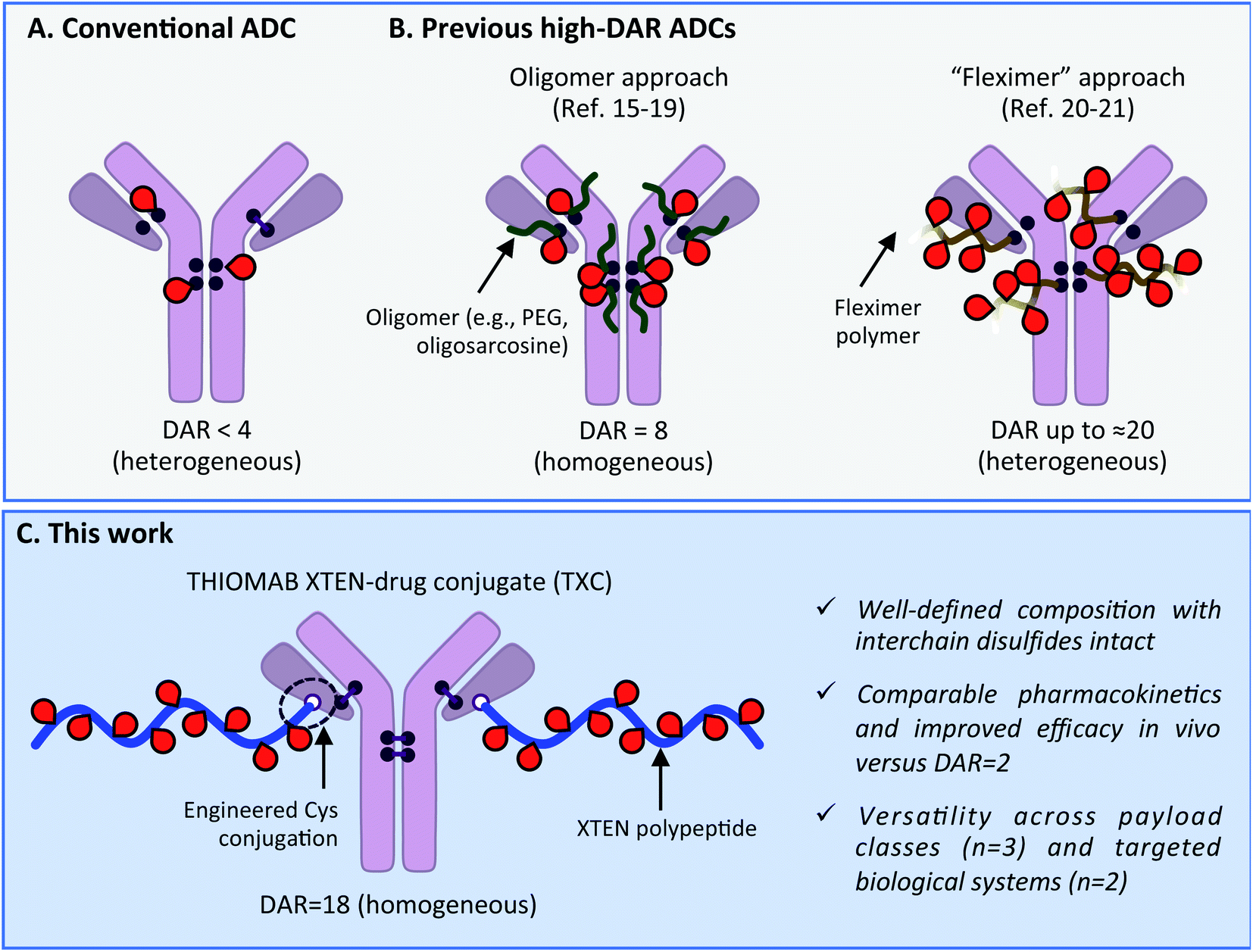 | ||
| Scheme 1 (A) Conventional ADC with DAR < 4. (B) Previous approaches to increase drug-to-antibody ratio (DAR) for ADCs. (C) Present high-DAR TXC strategy. | ||
We describe herein successful merging of the XTEN polypeptide scaffold with cysteine-engineered THIOMAB antibodies to generate homogeneous and highly efficacious THIOMAB antibody drug conjugates (TXCs) with DAR of up to 18 (Scheme 1C). The XTEN polypeptide is composed of a pseudo-repeating pattern of hydrophilic and small neutral or negatively-charged amino acids (Ala, Gly, Pro, Ser, Thr, Glu) and was developed originally as an alternative to PEG for half-life extension of protein and peptide therapeutics.24 In this work, we hypothesized that an XTEN linker could offset hydrophobicity of attached payloads in an ADC. Because it is expressed recombinantly, XTEN is well-defined in sequence and length, an advantage relative to heterogeneous polymeric scaffolds.24 Similarly, cysteine-engineered THIOMAB antibodies enable site-specific, homogeneous conjugation of payloads to antibodies.23,25 Thus, combining XTEN and THIOMABs in TXCs was expected to provide fine control over all aspects of conjugate composition and avoid payload insertion at interchain disulfide linkages, capabilities lacking in previous high-DAR ADC approaches. We applied our TXC approach across three different payloads – a microtubule destabilizing agent, a DNA monoalkylator and an antibiotic. When compared to their conventional low-DAR ADC counterparts in cells and mice, the high-DAR TXC molecules in each case showed an at least proportional increase in in vivo efficacy while, importantly, maintaining favorable stability and pharmacokinetic profiles. Based on our results, we believe the TXC platform we describe has the potential to enhance AMD of both traditional and novel payloads to tumor and non-tumor cells.
Results & discussion
Our high-DAR TXC conjugation strategy involved three main steps from XTEN, a linker-payload and a Cys-engineered THIOMAB antibody (Fig. 1A). First, XTEN modified recombinantly to incorporate multiple Cys residues (ESI Fig. 1†) was reacted with an iodoacetamide payload. Second, a maleimide handle was installed on XTEN via reaction with an amine-reactive linker (e.g., SMCC); since XTEN contains no Lys residues, we expected this reaction to be selective for its N-terminus.26 Last, the mal-XTEN-payload intermediate was reacted with the two engineered cysteines of a THIOMAB antibody to give the final TXC. As a proof-of-concept, this scheme was carried out with a 432-amino acid XTEN with 9 Cys introduced evenly along its length, an iodoacetamide derivative of the antimitotic ADC payload maytansine (iodo-PEG-May), and a THIOMAB derived from the anti-Her2 antibody trastuzumab with an engineered Cys introduced in place of Val at position 205 in each light chain (Fig. 1B). The theoretical maximum DAR for this TXC is 18, although initial conditions for conjugation of iodo-PEG-May to XTEN resulted in a heterogeneous product as determined by LCMS and SEC, giving a final TXC with an average DAR of ∼16 (αHer2/XTEN-May16; Fig. 1C and D). Optimization of conjugation and purification conditions led ultimately to homogeneous XTEN-payload intermediates (Fig. 1C) and TXCs with DAR = 18 (described in Materials and Methods and below), but for expediency we decided in parallel with these efforts to further characterize αHer2-/XTEN-May16.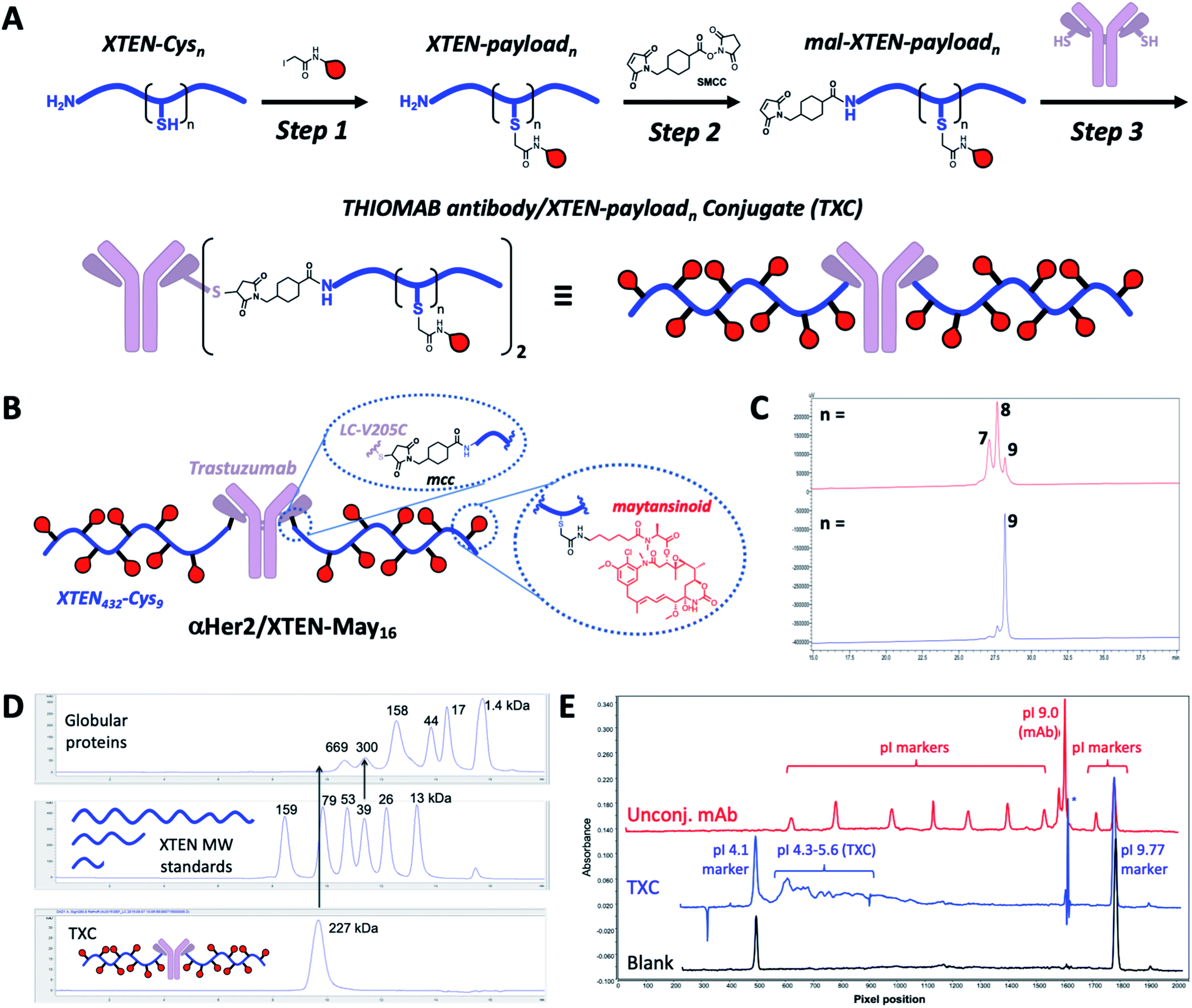 | ||
| Fig. 1 TXC conjugation strategy and biophysical characterization. (A) Iodoacetamide-modified payloads are conjugated to engineered cysteines on XTEN. The XTEN-payload intermediate is reacted with SMCC to install a maleimide at the N-terminus. Finally, mal-XTEN-payloadn is conjugated to the two engineered Cys of a THIOMAB antibody to give the final THIOMAB antibody/XTEN-payloadn conjugate (TXC). (B) Structure of αHer2/XTEN-May16 TXC (average DAR = 16); (C) HPLC chromatogram of purified mal-XTEN-Mayn intermediate generated via initial conjugation methods, giving a heterogeneous product with an average n = 8 (red trace) and optimized conjugation methods giving a more homogeneous product with n = 9 (blue trace); (D) analytical size-exclusion chromatogram (SEC) of globular protein standards, XTEN standards of different nominal molecular weights indicated in kDa, and αHer2/XTEN-May16; (E) IC-IEF fingerprint for αHer2/XTEN-May16 (blue) from which an approximate pI value of 4.1–5.6 was derived. Unconjugated antibody spiked with pI markers (red) and blank (black) are shown for reference. | ||
Biophysical analysis of the αHer2/XTEN-May16 TXC suggested a dominant influence of the XTEN component on TXC charge and size, consistent with studies of XTEN fusion proteins.26 We observed, for example, by SEC that the elution time of the TXC is inconsistent with its molecular weight as compared to globular protein standards, likely due to an extended conformation of the attached XTEN moieties (Fig. 1D)27 Extrapolating from a plot of the MW versus retention time for the globular protein standards, the TXC behaves as a ∼4.9 MDa globular protein. Capillary electrophoresis isoelectric focusing (cIEF) showed that the TXC had an isoelectric point (pI) of approximately 4.1–5.6, considerably lower than the pI of the unconjugated antibody (trastuzumab, pI = 9.0) and consistent with the net negative charge of XTEN due to prevalence of glutamic acid residues (Fig. 1E and ESI Fig. 1†).26
TXC pharmacokinetics
Given that fast clearance is a typical reason why high-DAR (e.g., DAR = 8) ADCs are not efficacious in vivo,12,14 we first evaluated pharmacokinetics of the αHer2/XTEN-May16 TXC in non-tumor-bearing SCID mice. We included several controls in the study: unconjugated trastuzumab (αHer2), a traditional THIOMAB antibody–drug conjugate (TDC) bearing a similar maytansinoid payload with DAR = 2 (αHer2/May2), and a TXC generated from the unmodified 432-amino acid XTEN lacking payloads (αHer2/XTEN).The TXC αHer2/XTEN-May16 displayed exposure (AUC) and half-life within ∼1.5-fold that of αHer2, αHer2/May2 and αHer2/XTEN controls (Fig. 2 and ESI Table 1†). Interestingly, the distribution (alpha) phase for αHer2/XTEN-May16 appeared to be longer and shallower relative to the TDC (αHer2/May2) or unconjugated αHer2 antibody, suggesting some differential distribution of the TXC. Since the atypical clearance profile was not observed for the TXC lacking maytansine payloads, αHer2/XTEN, this behavior exhibited by αHer2/XTEN-May16 is likely attributable to the attached payloads. Overall, these results were encouraging given previous studies showing rapid clearance of conventional ADCs with DAR ≥ 8.28
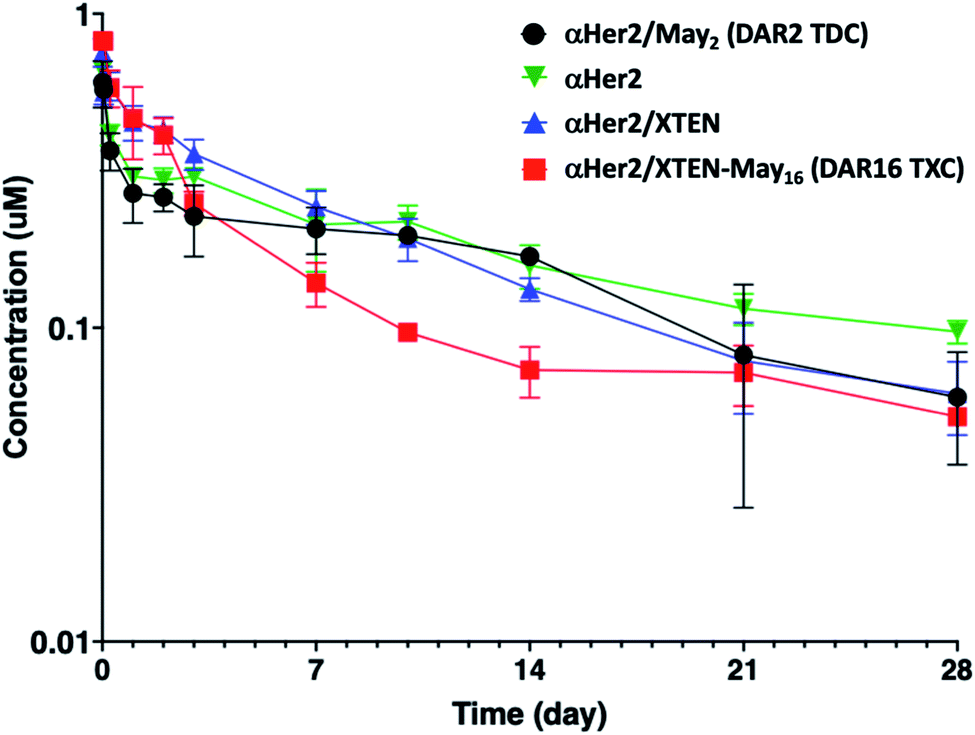 | ||
| Fig. 2 Pharmacokinetic analysis of high-DAR maytansinoid TXC (αHer2/XTEN-May16) versus XTENylated antibody lacking payloads (αHer2/XTEN), low-DAR TDC (αHer2/May2), and THIOMAB antibody control (αHer2). Concentrations measured are normalized to the molar doses of each conjugate. | ||
TXC stability
We next sought to assess stability in biological milieu, hypothesizing that the TXC could be susceptible to deconjugation of the payload from the XTEN, the XTEN from the antibody or proteolytic degradation of XTEN itself in serum, any of which could explain the promising PK data for αHer2/XTEN-May16 in Fig. 2. Assessment of stability by mass spectrometry of an intact TXC proved to be challenging given differential assay sensitivities for the various species that could result from the possible disconnections above (data not shown). Thus, to obtain an overall picture of TXC stability, we performed a battery of in vitro and in vivo experiments, either with XTEN-payload fragments amenable to mass spectrometry-based measurements or with full TXCs employing methods not based on mass spectrometry.Stability of the chemical bond between the payload and XTEN as well as XTEN proteolytic integrity were assessed in mouse serum, using two N-terminally-biotinylated 432-amino acid XTENs, each conjugated to three MMAE payloads via a valine–citrulline (Val–Cit) dipeptide linker (Fig. 3A). One conjugate (biotin/XTEN-mc-vc-MMAE) was generated by reacting the XTEN cysteines with a maleimide version of Val–Cit-MMAE while the other was generated by reaction with an iodoacetamide derivative (biotin/XTEN-iodo-vc-MMAE). The latter is more akin to how iodo-PEG-May payloads were attached to XTEN in αHer2/XTEN-May16 (Fig. 1B). Given that the Val–Cit linker and maleimide/Cys-derived thiosuccinimide connections can be highly labile in biological milieu,29,30 we saw the above studies as stringent tests for XTEN/payload linker stability.
 | ||
| Fig. 3 In vitro stability of XTEN-payload conjugate. (A) Structures of biotinylated XTEN-payload conjugates generated from an iodoacetamide or a maleimide Val–Cit-PAB-MMAE payload. Mass spectrometry data of (B) the iodoacetamide-derived conjugate or (C) the maleimide-derived conjugate before and after 48 or 96 hours of incubation at 37 °C in mouse serum. Peak marked with “*” corresponds to XTEN missing the C-terminal Arg residue. Peak marked with “#” is due to mass spectrometry-induced fragmentation after the Val–Cit dipeptide linker. | ||
For biotin/XTEN-iodo-vc-MMAE we observed minimal deconjugation and no cleavage of the Val–Cit dipeptide in mouse serum for 96 hours (Fig. 3B). We could not assess whether cleavage processes had occurred for biotin/XTEN-mc-vc-MMAE, given that we observed loss of signal beginning at 48 hours of incubation (Fig. 3C). It is possible that a heterogeneous mixture of deconjugated products formed from this conjugate. For both XTEN conjugates, we observed minor species consistent with a loss of the C-terminal Arg residue on the XTEN, possibly cleaved off by a plasma/serum carboxypeptidase.31 The high plasma stability of the thioether bond in biotin/XTEN-iodo-vc-MMAE is consistent with previous studies showing high stability of the same thioether connection in other contexts.32 The stability of the Val–Cit dipeptide in this conjugate was on the other hand surprising given that the Val–Cit linker in conventional ADCs is readily cleaved enzymatically in mouse plasma.29,31 A recent study showed that introduction of a glutamic acid N-terminal to the Val–Cit dipeptide in a conventional ADC protects the linker from plasmatic cleavage.33 It is possible that the negative charge of the Glu residues in XTEN surrounding the payload conjugation sites similarly protects the Val–Cit linker.
Stability of TXCs in vivo was next assessed via two orthogonal methods, one employing a radiolabeled XTEN antibody conjugate bearing no payloads, 89Zr-αCD22/XTEN, and another employing a TXC bearing six Val–Cit-PAB-MMAE payloads, αHer2/XTEN-MMAE6 (Fig. 4A). First, we observed no discernible change in the size of the radiolabeled XTEN conjugate (89Zr-αCD22/XTEN), as isolated from mice over the course of 5 days, by SEC, suggesting that major XTEN cleavage or deconjugation events do not occur to significant extents (Fig. 4B). Second, we measured the cytotoxic potency in SkBR3 cells unresponsive to αHer2 alone of the αHer2/XTEN-MMAE6 TXC isolated from a mouse one day after dosing and quantitated (Fig. 4C). No discernible shift in IC50 or maximum cell-killing of the dosed TXC relative to the input TXC was observed, indicating that no cleavage events had occurred to the conjugate in vivo that reduce the number of MMAE payloads attached, namely antibody-XTEN or XTEN-payload bond cleavage. Taken altogether, our stability data suggest that a TXC using iodoacetamide/Cys chemistry for the payload/XTEN connection and maleimide/Cys chemistry for the XTEN/antibody connection (employing the LC V205C site on the antibody) is constitutionally stable in vitro and in vivo.
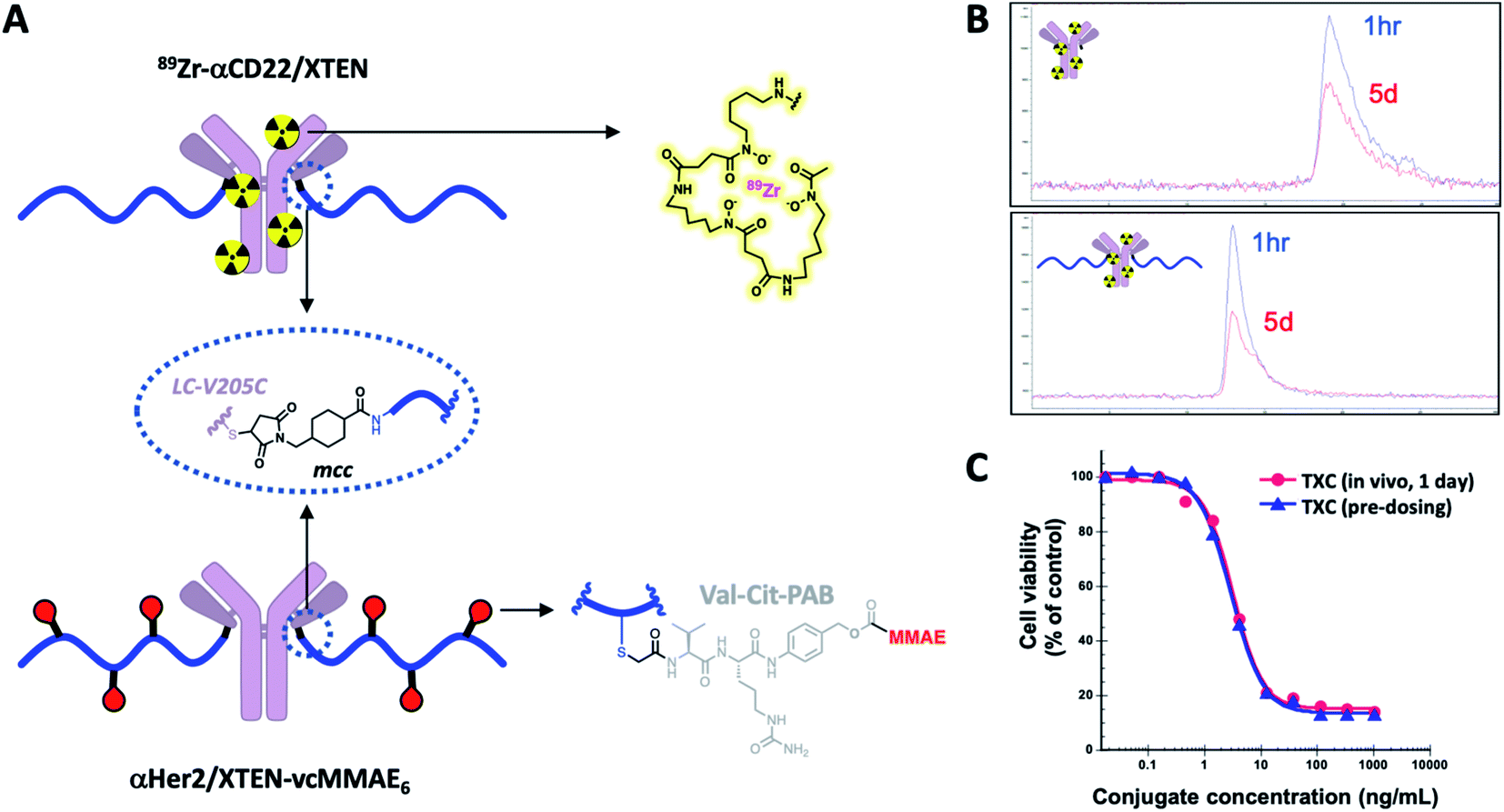 | ||
| Fig. 4 In vivo stability of intact TXCs. (A) Structure of 89Zr-αCD22/XTEN (radiolabeled) and αHer2/XTEN-vcMMAE6 conjugates used to evaluate in vivo stability. (B) SEC-based tracking of 89Zr-radiolabeled antibody or TXC isolated from mice 1 hour or 5 days after dosing. (C) Ex vivo potency-based assessment in Her2-positive SkBR3 cells of αHer2/XTEN-vcMMAE6 TXC isolated from mice 1 day after dosing in comparison to the input (pre-dosed) TXC. | ||
Anti-tumor high-DAR TXCs
We sought next to evaluate the TXC platform for payload delivery to tumor cells. As the tumor antigen, we chose CD22, a receptor on malignant and normal B cells and an ideal model system for AMD as it has been targeted successfully by conventional ADCs with a variety of payloads and linkers.34,35 For one TXC, we chose the same antimitotic maytansinoid and linker as was used for the generation of αHer2/XTEN-May16, constructing αCD22/XTEN-May16 in the same manner (Fig. 1B). For the other TXC, we chose a DNA alkylator payload, a pyrrolobenzodiazepine (PBD) monoamide (PBDma), connected to the XTEN via a peptide-based Val–Ala linker. However, we employed an alternative conjugation route to obtain the TXC (ESI Fig. 2†). Rather than conjugating the PBDma payload to XTEN cysteines first we conjugated an iodoacetamide-cyclooctyne derivative. Subsequent attachment of the N-terminal maleimide and reaction with a THIOMAB antibody thus gave a TXC precursor with cyclooctynes that could be reacted in the final step with an azide-modified payload via strain-promoted azide–alkyne cycloaddition (SPAAC). To offset hydrophobicity introduced by the cyclooctynes, short PEG spacers were incorporated in both cyclooctyne and payload components. The SPAAC method enabled from the same 432-amino acid, 9-cysteine XTEN and anti-CD22 THIOMAB antibody described above generation of the αCD22/XTEN-PBDma18 TXC (Fig. 5A). Characterization by LCMS (Fig. 5B) and SEC (Fig. 5C) confirmed a homogeneous DAR of 18. As comparators to the TXCs above, we generated low-DAR THIOMAB-drug conjugates (TDCs) αCD22/May1.7 and αCD22/PBDma2 (Fig. 5A and ESI Fig. 3†) with DAR = 1.7 and 2.0, respectively.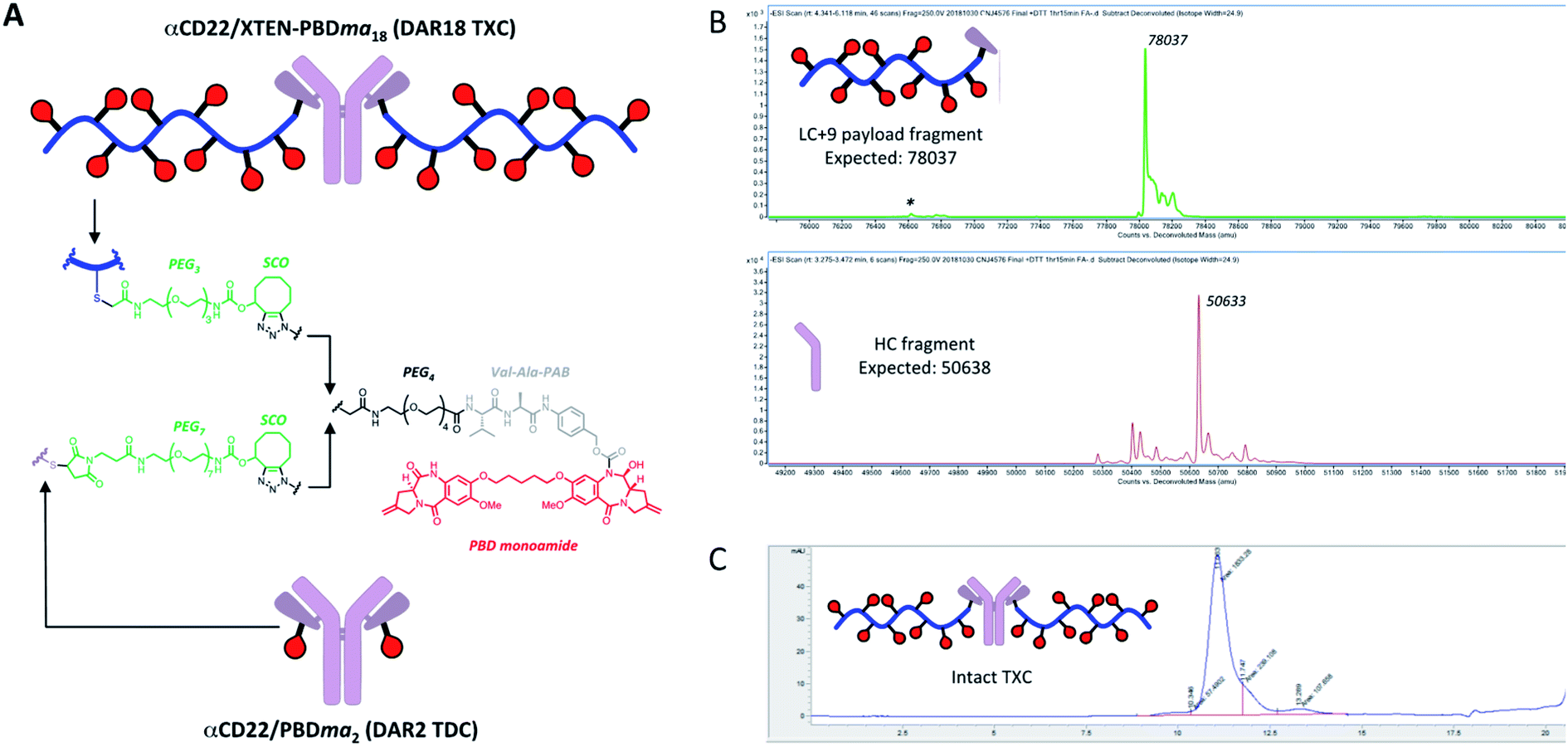 | ||
| Fig. 5 (A) Structures of αCD22/XTEN-PBDma18 TXC and TDC control αCD22/PBDma2. Analysis of (B) dissociated light and heavy chains of DTT-reduced TXC by LCMS and (C) intact TXC by size-exclusion chromatagraphy indicate a final DAR of 18. | ||
The anti-CD22 TXCs, TDCs and Her2 TXC control were evaluated for anti-tumor efficacy in a CD22-expressing BJAB cell-derived mouse xenograft model (Fig. 6). At a single payload-matched dose of 44 nmol payload per kg, both αCD22/May1.7 and αCD22/XTEN-May16 were barely capable of inhibiting tumor growth (<50% TGI; Fig. 6A). Strikingly, however, at a higher payload dose (109 nmol kg−1) αCD22/XTEN-May16 was highly effective, causing tumor regression (115% TGI), while αCD22/May1.7 remained only weakly efficacious (56% TGI). The anti-Her2 TXC, αHer2/XTEN-May16, was not efficacious at the higher 109 nmol kg−1 payload dose, as expected given BJAB cells do not express Her2.
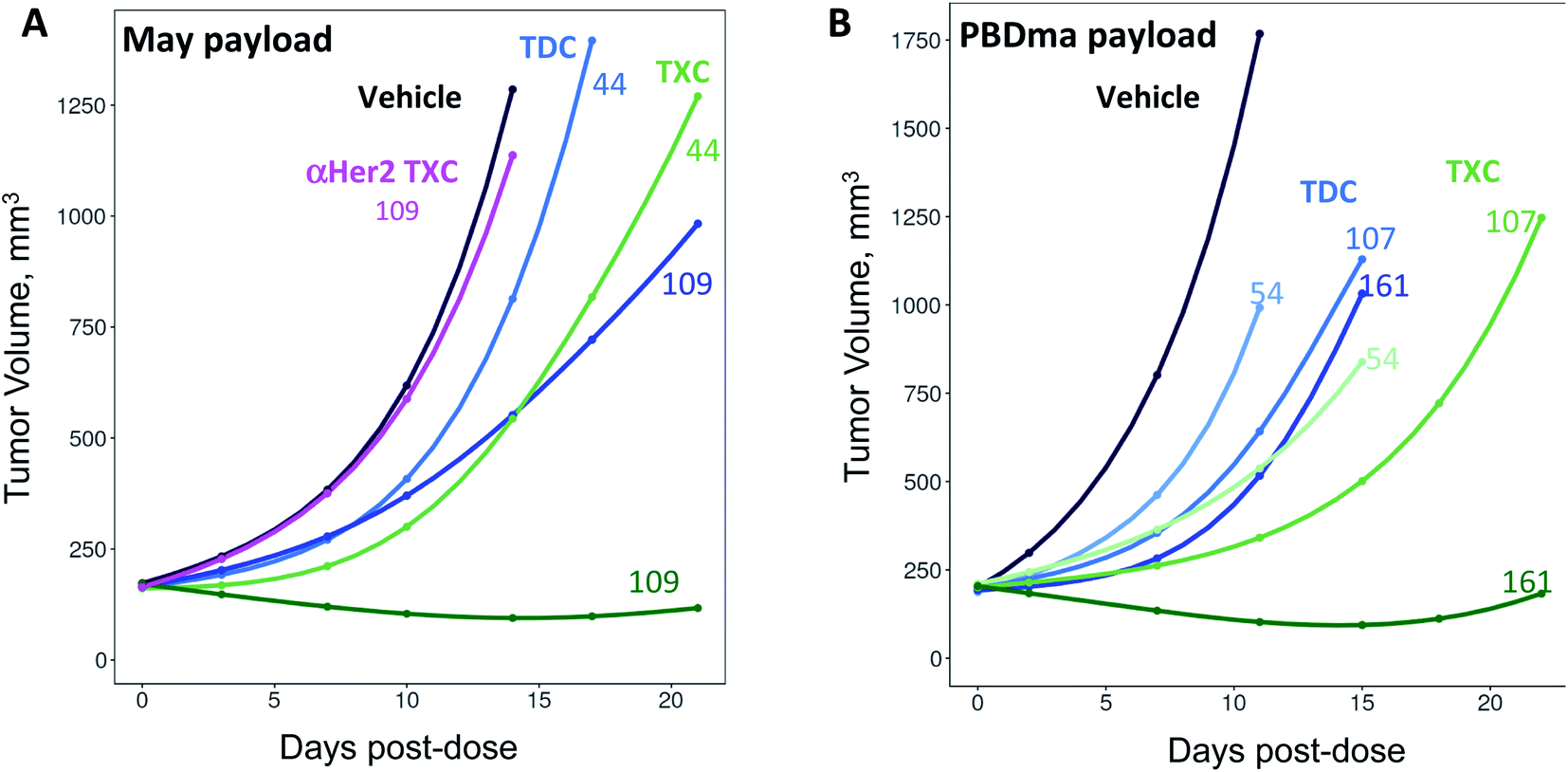 | ||
| Fig. 6 In vivo anti-tumor efficacy of high-DAR TXCs (A) αCD22/XTEN-May16 and (B) αCD22/XTEN-PBDma18versus corresponding low-DAR TDC controls αCD22/May1.7 and αCD22/PBDma2, respectively. Cubic spline fitted tumor volumes are plotted for each treatment group (n = 5/group) over the duration of study with curves for anti-CD22 TXCs and TDCs in green and blue, respectively, and curves for vehicle and anti-Her2 TXC controls in black and magenta, respectively. Single intravenous doses in nmol kg−1 payload are shown above each curve. | ||
As with the maytansine-loaded TXC we also observed increased potency per payload for αCD22/XTEN-PBDma18 as compared to the TDC control in the BJAB xenograft model (Fig. 6B). This effect was most significant at a payload-matched dose of 161 nmol kg−1, at which the αCD22/PBDma2 was modestly effective (∼50% TGI) and αCD22/XTEN-PBDma18 caused tumor regression (110% TGI). Sparse sampling of TXCs (and TDCs) over the course of the xenograft studies indicated that the high-DAR TXCs did not have qualitatively altered pharmacokinetics versus TDC controls (ESI Fig. 4†). No body weight loss was observed for maytansine- or PBDma-loaded conjugates at any doses indicating all were well-tolerated (ESI Fig. 5†). Overall, our results confirm supralinear improvements in anti-tumor efficacy at matched payload doses of the high-DAR TXC versus low-DAR TDC for both payloads tested. We observed a supralinear effect on potency in cultured cells as well (ESI Fig. 6†).
The higher potency of anti-tumor TXCs versus TDCs at the same payload dose suggests the former can be more efficient in delivering payload. At a matched payload dose, the molar antibody dose of a DAR = 18 TXC is ∼9-fold lower than that of a DAR = 2 TDC. Thus, it is possible that at matched payload doses (lower antibody doses) where high-DAR TXCs are more efficacious than TDCs that binding to the receptors on the tumor is at or below saturation by the TXC and above saturation by the TDC. In such a scenario, a significant proportion of the administered TDC would be expected to not be able to bind and deliver payload to the tumor whereas a greater proportion of the TXC does so, contributing to the disconnect in payload-normalized efficacies. We considered the alternate possibility that the greater per-payload efficacy of TXCs versus TDCs was due, for some reason, to direct enhancement by XTEN and not differences in DAR per se. To test this, we tested the impact of XTEN attachment on activity of a conventional ADC with maytansinoid payloads attached to antibody lysines at DAR = 4.5 (ESI Fig. 7A†). In the BJAB xenograft model, the XTENylated ADC (XADC) showed lower efficacy relative to the non-XTENylated conventional ADC at matched payload doses, suggesting a modest negative impact of XTEN conjugation (ESI Fig. 7B†). Thus, XTEN itself appears not to be the driver for improved per-payload potency of high-DAR TXCs versus low-DAR controls.
Anti-bacterial high-DAR TXCs
Finally, we established whether TXCs could enhance delivery of an antibiotic to methicillin-resistant Staphylococcus aureus bacteria. Previously, we reported anti-S. aureus activity of a TDC molecule with DAR = 2, in which an antibody specific for the βGlcNAc epitope of the bacterial surface antigen wall teichoic acid (αWTA) was connected to the rifamycin analog dimethyl DNA31 (dmDNA31) antibiotic through a Val–Cit linker.10 The mechanism of action for the anti-WTA TDC is more complicated than that of antitumor ADCs and includes binding to the bacterial surface (opsonization), ingestion of the TDC-bound bacteria by phagocytic cells, cleavage of the linker in the phago-lysosome, and intracellular killing of the bacteria by the released antibiotic.10 Employing fluorophore-labeled anti-WTA antibodies conjugated and unconjugated to XTEN, we observed that the 432-amino acid XTEN used for antitumor TXCs impaired both binding of the conjugate to whole S. aureus USA300 bacteria (ESI Fig. 8A†), as well as uptake of pre-opsonized S. aureus into macrophages (ESI Fig. 8B†). Increasing XTEN size from 432 to 864 amino acids further reduced binding to bacteria and association with macrophages while reducing the XTEN length to 144 amino acids enabled both processes to occur unimpeded. Thus, we generated an anti-S. aureus WTA TXC with a 144-residue XTEN, a dmDNA31 antibiotic payload linked via a Val–Ala linker at a homogeneous DAR of 18 using the SPAAC conjugation strategy (αWTA/XTEN144-dmDNA3118) and a corresponding DAR = 2 TDC control employing a similar linker, (αWTA/dmDNA312, Fig. 7A and ESI Fig. 9†). | ||
| Fig. 7 Enhanced in vitro antibacterial potency of high-DAR TXC. (A) Structures of αWTA/XTEN144-dmDNA3118 TXC (DAR = 18) and αWTA/dmDNA312 TDC (DAR = 2). (B and C) S. aureus USA300 bacteria were incubated with conjugates (TXC in blue or TDC in black) or free mAb (purple, used at 100 nM) and ingested by macrophages. After further incubation to enable intracellular killing, bacterial viability was assessed by enumerating colony-forming units (CFU). Anti-S. aureus activity was expressed as a function of molar concentration of (B) conjugate or (C) payload. LOD, limit of detection. Data represent average ±SD of 3 experiments. | ||
In an in vitro assay measuring killing of opsonized S. aureus bacteria in the presence of macrophages, the αWTA/XTEN144-dmDNA3118 TXC was ∼9-fold more potent per conjugate or equipotent per payload versusαWTA/dmDNA312 (Fig. 7B and C). These potency trends are consistent qualitatively with mass spectrometry-based measurements of free intracellular dmDNA31 antibiotic released from the two conjugates (ESI Fig. 10†). The DAR-proportional increase in anti-S. aureus potency and released antibiotic from the TXC versus TDC indicates that the TXC is at least as efficient at delivering the antibiotic intracellularly as the TDC.
In mice infected systemically with methicillin-resistant S. aureus, a dose of 23 nmol conjugate per kg of the DAR = 18 TXC was equipotent with 210 nmol conjugate per kg of the DAR = 2 TDC control at reducing the number of bacterial colony-forming units (CFUs) in the kidneys, although only ∼50% of the animals were cured of infection (Fig. 8A). Increasing the TXC dose to 210 nmol conjugate per kg reduced kidney CFUs to undetectable levels in all animals, while the DAR = 2 control showed only partial efficacy. When dosed per payload, at either 140 nmol payload per kg or 420 nmol payload per kg, the TXC and TDC gave equivalent efficacy (Fig. 8B). The concentration of conjugate detected in the serum at 24 h and 48 h after IV injection of αWTA/XTEN144-dmDNA3118 was within 2-fold that of αWTA/dmDNA312 and of free αWTA antibody (ESI Fig. 11†). In summary, these data demonstrated enhanced antibody-normalized efficacy of the DAR = 18 TXC molecule in a therapeutic S. aureus infection model compared to a DAR = 2 TDC, and that the increase in efficacy was approximately proportional to the increase in DAR (i.e., ∼9 fold). These results differ from those obtained with anti-tumor TXCs in that efficacy was correlated linearly with payload dose for both high and low-DAR conjugates. To establish the cause for such differences, further studies into XTEN effects on antigen binding, opsonization, internalization and payload release for TXCs are required.
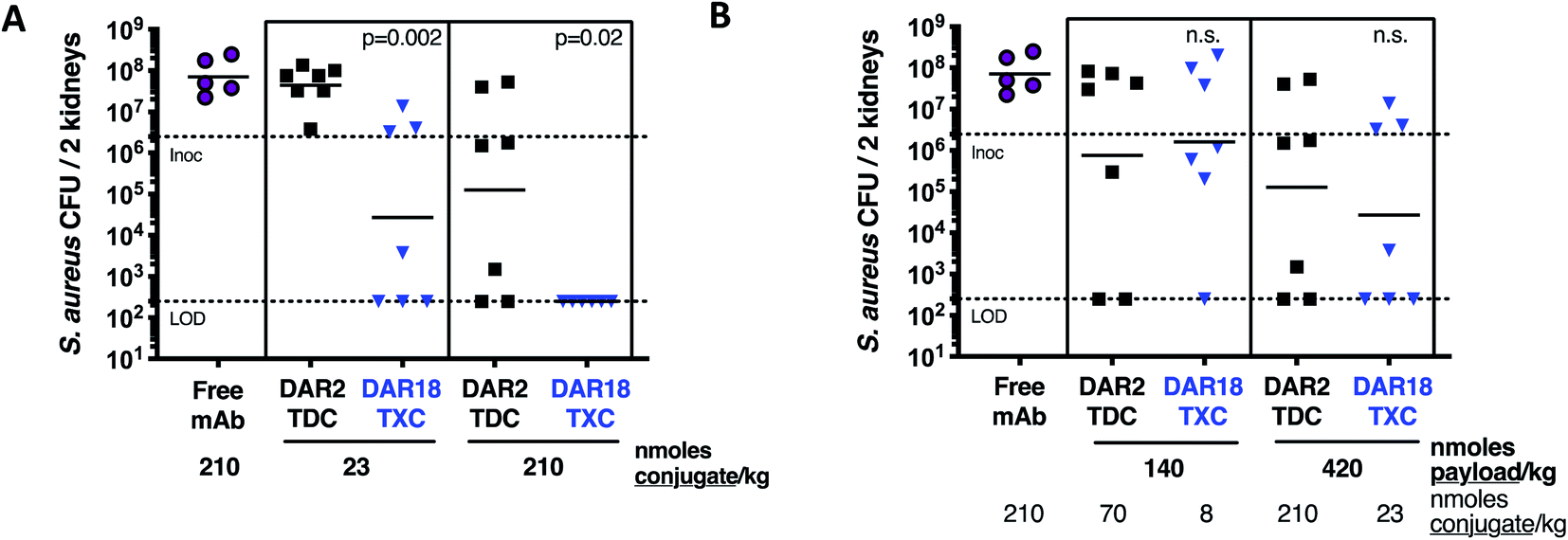 | ||
| Fig. 8 Enhanced in vivo antibacterial efficacy of DAR18 TXC. One day after i.v. infection with S. aureus USA300, mice were treated with a single i.v. dose of αWTA/XTEN144-dmDNA3118 (DAR18 TXC, blue), αWTA/dmDNA312 (DAR2 TDC, black), or free mAb (purple). Four days after infection, bacterial burden in kidneys was determined by CFU determination. (A) When treated with an equimolar dose of either 23 nmoles mAb per kg (i.e., nmoles conjugate per kg) or 210 nmoles of mAb per kg, DAR18 TXC showed significantly higher kidney CFU reduction compared to DAR2 TDC. (B) When doses were compared by matching payload molarity side-by-side, efficacy of DAR18 TXC and DAR2 TDC was similar, indicating that the enhancement in efficacy in (A) was approximately proportional to the 9-fold increase in DAR. Bars, geometric means; lower dashed line, lower limit of detection; upper dashed line, i.v. infection inoculum. P values indicate difference between DAR18 TXC and DAR2 TDC, as determined by Mann–Whitney test; n.s., not significant. | ||
Conclusion
In summary, we have described a conjugation platform combining XTEN and site-specific antibody conjugation (THIOMABs) to give well-defined TXCs with simultaneously high DAR and enhanced payload delivery capacity in vivo. Our results represent significant advances relative to previous ADC optimization efforts in terms of the DAR achieved (up to 18), homogeneity of the conjugates and versatility demonstrated across different payload classes (antimitotic, DNA alkylator, antibiotic), linkers (cleavable versus uncleavable) and biological systems (tumor versus bacterial cells). Because payloads are attached at the last step of the optimized SPAAC method via a simple click reaction, we expect it will be possible to prepare libraries of high-DAR TXCs with a variety of payloads and linkers, facilitating rapid testing prior to in vivo studies.Across two different cytotoxic payloads, a microtubule and DNA damaging agent, we observed anti-tumor efficacy for high DAR TXCs that was increased versus low DAR controls, even when the total payload dose for each was equivalent. We hypothesize that this supralinear effect is driven by differences in levels of receptor saturation on the tumor by the TXC and TDC. We presume that toxicity of ADCs is not supralinearly dependent on total payload dose given that it appears to be driven by non-receptor-mediated (pinocytotic) uptake in normal cells.36 Thus, an improvement in therapeutic index might be achievable with high-DAR TXCs with proper selection of antigen, payload and dose.
For AMD of antibiotics to treat infectious diseases, the improvement realized by high-DAR TXCs versus low-DAR conjugates is impactful. The DAR = 2 AAC we described previously required a dose to achieve efficacy in mice ∼10 times that required for a typical cytotoxic ADC.10 We were able in the studies herein to employ a dose of an antibacterial TXC approximately 9-fold lower than that of a low-DAR ADC while maintaining comparable efficacy, closing the potency gap between AMD of an antibiotic versus cytotoxic payload. The high-DAR antibacterial TXC may facilitate the use of a lower conjugate dose in the clinic, potentially simplifying formulation and lowering cost of manufacturing. Alternatively, the TXC at a high dose may increase exposure of bacteria to more payload and therefore drive more durable bactericidal activity.
Methods
General
XTEN polypeptides were expressed and purified as described elsewhere.37 Anti-CD22, anti-Her2 and anti-WTA THIOMAB antibodies for conjugate generation are Cys mutants of those discovered previously and were deblocked prior to conjugation.10,25,38,39 All other reagents were obtained from commercial vendors unless otherwise indicated. All animal studies were carried out in compliance with National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee at Genentech, Inc.Linker-payload synthesis
Synthesis of maytansinoid, PBDma and dmDNA31 linker-payloads was carried out as described in the ESI.† SMCC-DM1 (Levana Biopharma, SET0101), iodoacetamido-Val–Cit-PAB-MMAE (custom order synthesis, Levena, P1803L009131), iodo-maytansinoid (Levena, P1501047027), mal-PEG7-SCO (SiChem, SC-8305), mal-PEG4-DBCO (Aldrich, 760676), and sulfo-SMCC (Sigma-Aldrich, M6035) were obtained from commercial sources.Conjugation and analytical characterization
To perform conjugation of payloads to XTEN bearing engineered Cys, XTEN-Cysn polypeptides were reduced with TCEP at 1.3-fold molar excess over XTEN polypeptides and incubated at 80 °C for 30 min in HEPES pH 8.0. The pH of the solution was adjusted to 7.2 with 1 M HEPES buffer and an iodoacetamide-payload, freshly prepared as a stock solution in DMF, was added at 3-fold molar excess versus XTEN Cys concentration. The conjugation reaction was conducted with a final composition of 50% DMF at room temperature overnight. The XTEN-payloadn product of the conjugation reactions was purified by reverse phase HPLC (BioBasic 4 column, ThermoFisher) with a mobile phase of water, 0.1% formic acid (A) and acetonitrile, 0.1% formic acid (B). Purified material was lyophilized and dissolved in HEPES pH 7.2. To functionalize the N-terminal α-amino group with a maleimide, sulfo-SMCC was added at a 10-fold molar excess over XTEN polypeptide. The final mal-XTEN-payloadn was formulated by dialysis (Slide-A-Lyzer cassette, 10 kDa MWCO, Thermo Fisher Scientific) into 10 mM succinate, 150 mM NaCl, 2 mM EDTA. The extent of conjugation was determined by reverse-phase LC-MS in negative ionization mode (TOF ESI, Agilent).Conjugation of mal-XTEN-payloadn (and other maleimide-functionalized XTENs) to THIOMAB antibodies was conducted in HEPES, pH 7.2 using 2.5 molar excess of mal-XTEN-payloadn per unpaired reduced Cys for >3 hours at room temperature. The TXC was purified by anion exchange chromatography (HiTrap CaptoQ ImpRes column, Cytiva) using mobile phases of (A) 50 mM Tris, pH 8 and (B) 50 mM Tris, pH 8, 1 M NaCl.
The modified SPAAC procedure to generate TXCs bearing the PBDma and dmDNA31 payloads (outlined in ESI Fig. 2†) involved first conjugation of an iodoacetamide-functionalized cyclooctyne (e.g., iodo-PEG3-SCO) to XTEN-Cys9, followed by modification of N-terminal α-amino group of XTEN with sulfo-SMCC and attachment of mal-XTEN-SCO9 to a THIOMAB antibody as described above. This intermediate antibody-XTEN-cyclooctyne conjugate was then reacted with 2.2-fold molar excess per cyclooctyne of the azide-functionalized payload in HEPES pH 7.2 and 20% DMF at room temperature until completion. The TXC was purified by size exclusion chromatography (Superdex 26/600 200 PG column, GE) in 200 mM KP04, 250 mM KCl, 15% isopropanol, pH 6.2. Purified conjugate was concentrated and formulated into 20 mM Tris, 100 mM NaCl, 0.02% Tween 20, pH 7.5 or PBS using a centrifugal spin concentrator (Amicon 50 kDa MWCO, Millipore).
XTENylated ADCs (XADCs, ESI Fig. 7a†) were generated by first conjugating an amine-reactive payload (NHS ester) to lysine residues of a THIOMAB antibody to achieve the desired DAR using standard methods. Attachment of maleimide-functionalized XTEN to the antibody engineered Cys proceeded as described above.
After purification all conjugates were analyzed by reverse-phase LC-MS (TOF ESI, Agilent) and analytical size-exclusion chromatography (SEC) (Yarra SEC4000 column, Phenomenex) to assess DAR and XTEN-to-antibody ratio, respectively. For a representative anti-Her2 (trastuzumab-derived) TXC and antibody, pI was determined by imaged capillary isoelectric focusing (ICIEF) using an iCE3 analyzer (ProteinSimple) with a fluorocarbon (Fc) coated capillary cartridge. An ampholyte mixture containing nine pI markers was used to establish a linear gradient for the separation. The pI for the TXC was determined by linear regression analysis of the measured pixel position and vendor-assigned pI valued for each marker.
In vivo pharmacokinetics
For full multi-timepoint PK analysis, a total of 99 naïve SCID.bg mice were randomly assigned to four groups of 33 animals in each group. Animals in group 1 received a single IV bolus dose of 5 mg kg−1αHer2/XTEN-May16 (TXC), group 2 received a single IV bolus dose of 5 mg kg−1αHer2/XTEN and animals in group 3 received a single IV bolus dose of 5 mg kg−1αHer2 (Trastuzumab). Group 4 received a single IV bolus dose of 5 mg kg−1αHer2/May2 (DAR2 TDC). Formulation buffer was used as the medium for injections. At selected times throughout the study, plasma from 3 animals in each group was obtained via cardiac puncture and assayed for total antibody (TAB) concentrations using an LC-MS/MS assay (PPD, Inc.). The total antibody plasma concentration vs. time data was normalized using molar mass of each conjugate for exposure comparison across molecules. The total antibody plasma concentration vs. time data was analyzed using a non-compartmental approach (Phoenix™ WinNonlin® v.6.3; Pharsight Corporation; Mountain View, CA) to provide estimation of PK parameters. For mice, the total antibody plasma concentration vs. time data were naïve-pooled together (sparse sampling approach) to provide PK parameter estimations. Parameters calculated include the maximum concentration (Cmax); area under the plasma concentration–time curve from time 0 to 28 days (AUC0-28); area under the plasma concentration–time curve from time 0 to infinity (AUC0-inf); and the total clearance (CL).Aliquots (10 μL) of serum from mice infected with S. aureus and treated with TXC, TDC or free mAb were transferred to a 96-well protein LoBind plate containing 300 μL of 4% BSA-PBST (1x PBST/BSA; 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4;/w), followed by addition of 20 μL of 1 μg mL−1 SILuMab (Sigma-Aldrich) internal standard solution. The samples were incubated with biotinylated sheep anti-human IgG (Abcam) at 8 °C overnight with shaking. Next, 25 μL of streptavidin magnetic beads (Dynabeads MyOne, Streptavidin T1, ThermoFisher) was added and mixtures were further incubated for 1 h at RT with 950 rpm shaking. After washing the magnetic beads twice with 300 μL Buffer B (0.05% CHAPS in 1x PBS) and once with a mix of 100 μL of Buffer B and 200 μL of 1x PBS, the captured TXC, TDC or free mAb along with the SILuMab internal standard were eluted in 150 μL of 25 mM hydrochloric acid (HCl). After elution, the samples were neutralized with 20 μL of 1 M ammonium bicarbonate buffer (pH 8.0) resulting in a pH between 7 and 8. The neutralized samples were subjected to reduction by adding 10 μL of 100 mM DTT in 1 M Tris–HCl (pH 8.0) and incubating the samples at 55 °C for 45 min with shaking. The reduced samples were then alkylated by adding 10 μL of 240 mM iodoacetamide in deionized water at room temperature in the dark for 30 min with shaking. 25 μL of 0.1 μg μL−1 trypsin in ammonium bicarbonate was added for digestion at 37 °C for 3 hours. The trypsin digestion was quenched by adding 30 μL 10% formic acid followed by storage at 4 °C prior to LC-MS/MS analysis.
:4;/w), followed by addition of 20 μL of 1 μg mL−1 SILuMab (Sigma-Aldrich) internal standard solution. The samples were incubated with biotinylated sheep anti-human IgG (Abcam) at 8 °C overnight with shaking. Next, 25 μL of streptavidin magnetic beads (Dynabeads MyOne, Streptavidin T1, ThermoFisher) was added and mixtures were further incubated for 1 h at RT with 950 rpm shaking. After washing the magnetic beads twice with 300 μL Buffer B (0.05% CHAPS in 1x PBS) and once with a mix of 100 μL of Buffer B and 200 μL of 1x PBS, the captured TXC, TDC or free mAb along with the SILuMab internal standard were eluted in 150 μL of 25 mM hydrochloric acid (HCl). After elution, the samples were neutralized with 20 μL of 1 M ammonium bicarbonate buffer (pH 8.0) resulting in a pH between 7 and 8. The neutralized samples were subjected to reduction by adding 10 μL of 100 mM DTT in 1 M Tris–HCl (pH 8.0) and incubating the samples at 55 °C for 45 min with shaking. The reduced samples were then alkylated by adding 10 μL of 240 mM iodoacetamide in deionized water at room temperature in the dark for 30 min with shaking. 25 μL of 0.1 μg μL−1 trypsin in ammonium bicarbonate was added for digestion at 37 °C for 3 hours. The trypsin digestion was quenched by adding 30 μL 10% formic acid followed by storage at 4 °C prior to LC-MS/MS analysis.
Digested samples were analyzed by a Waters Acquity UPLC coupled online to a Sciex API 5000 mass spectrometer. Chromatographic separation was performed on Waters BEH C4 column (1.0 mm × 50 mm, 1.7 μm) using a gradient of mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile) at a flow rate of 200 μL min−1. A 10 min gradient from 10% B (0 min) to 15% B (1.0 min) to 22% B (4.0 min) to 30% B (4.5 min) to 45% (6.0 min) to 95% (6.1 min) to 95% (6.5 min) was used followed by wash and re-equilibration. The MS instrument was operated in positive ionization mode and the key parameters were set as follows: temperature: 500C°; duration; 9 min; cycles: 540; cycle: 1.0 second; collision gas (CAD): 8; curtain gas (CUR): 15; ion source gas (GS1): 50; ion source gas 2 (GS2): 50; MRM detection window: 90 seconds. Target scan time: 1 second; ion spray voltage (IS): 5000; entrance potential (EP): 10.0; collision cell exit potential (CXP): 18.0. The Analyst 1.5.1 software (SCIEX) was used for data analysis.
In vitro and in vivo stability
In vitro DAR stability was assessed qualitatively in mouse serum and plasma using affinity capture followed by LC-MS. Biotinylated XTEN conjugate was diluted to 1 mg mL−1 in a blocking buffer of 1X PBS, 0.5% BSA, 15 ppm Proclin before spiking into whole blood at a final concentration of 100 μg mL−1. CB17 SCID mouse serum and plasma were prepared within 36 hours prior to use (sourced from BioIVT, lithium heparin used as anti-coagulant). A negative control of biotinylated XTEN conjugate spiked into the aforementioned blocking buffer was also included. After spiking into mouse serum and plasma, a 0 hour time-point was collected (n = 3) and diluted 16-fold in HBS-EP buffer before storing at −80 °C. Remaining mouse serum and plasma samples were incubated for 48 and 96 hours (n = 3) at 37 °C with agitation before 16-fold dilution in HBS-EP buffer and storage at −80 °C. Prior to use, mouse serum and plasma samples were thawed at room temperature for 15 minutes. Affinity capture was performed on a Thermo KingFisher™ 96 using streptavidin magnetic beads (Invitrogen Dynabeads™ M-280 Streptavidin). Beads were diluted to 1.25 mg mL−1 in HBS-EP buffer and 400 μL per well were aliquoted into a 96 deep-well block. Beads were washed twice in HBS-EP buffer before being mixed into the mouse serum and plasma samples. The samples and bead mixture were incubated at room temperature for 2 hours with agitation. After incubation, beads were washed twice in HBS-EP buffer and twice in LC-MS grade water before being deposited into a 96 well elution plate containing 100 μL of LC-MS grade water. The elution plate was heat-sealed with a non-permeable foil seal and incubated at 70 °C for 3 minutes to release the biotinylated XTEN conjugates. After elution, a magnetic plate was used to remove the beads. The eluate was analyzed by reversed-phase LC-MS on a Waters microLC (Agilent PLRP-S column, 1000 Å pores, 5 μm particles, 50 × 1 mm) coupled to a Waters Synapt G2 Q-TOF mass spectrometer.In vivo stability of TXCs was assessed qualitatively by a radioactivity-based method. Briefly, antibody (αCD22) or XTEN conjugate (αCD22/XTEN) were conjugated on lysine residues to desferrioxamine (TFP-Nsuc-DF-Fe, WuXi Apptec) and purified by gel filtration using a NAP25 gel filtration column (GE Healthcare), eluting with 10 mM sodium succinate, 240 mM sucrose 0.02% PS20, pH 5.5. Zirconium-89 (89Zr) chelation proceeded upon incubation of the desferrioxamine-labeled conjugates with 89Zr(IV) oxalate (2–4 mCi, 3D imaging) at room temperature for 3 min after which 0.5 M HEPES buffer (0.15 mL) was added and the radiolabeled protein was purified from free 89Zr using a NAP10 desalting column, eluting with 20 mM histidine acetate, 120 mM sucrose, 0.02% PS20 pH 5.5 (1.5 mL). The 89Zr-labeled αCD22 conjugate was obtained with a radiochemical yield of 93%, radiochemical purity of 97% and specific activity of 1.5 mCi mg−1. The 89Zr-labeled αCD22/XTEN conjugate was obtained with a radiochemical yield of 91%, a radiochemical purity of 100%, and a specific activity of 1.5 mCi mg−1 as determined by SEC, monitoring UV absorbance and radioactivity (BioSep-SEC-S 3000 column, Phenomenex).
An alternative method to measure in vivo stability involved injecting SCID mice with an anti-Her2 MMAE-linked TXC, collecting blood one day following injection, affinity capture and quantification of the conjugate with an LC-MS/MS assay (as employed to monitor pharmacokinetics) and assessment for cytotoxicity in Her2-expressing SkBR3 cells in a standard 5 day assay (Cell Titer Glo, Promega). The same TXC, as a pure conjugate pre-dosing, was tested in parallel in SkBR3 cells as a control for the in vivo-derived sample. Formulation buffer was used as the medium for animal injections.
In vitro cytotoxicity assays
In vitro cytotoxicity assays with tumor cell lines were performed in a manner analogous to that described previously.35,38In vitro anti-bacterial assays
Wild-type (WT) and protein A deficient (Δspa) methicillin-resistant Staphylococcus aureus USA300 bacteria were obtained and grown as described previously.40,41 Binding of Alexa488-labeled XTENylated mAbs targeting wall teichoic acid (WTA) to S. aureus Δspa USA300 bacteria was assessed as described with minor modifications.63 To assess association of XTENylated mAb-bound S. aureus bacteria with macrophages, the murine macrophage cell line RAW 264.7 (ATCC TIB71; ATCC, Manassas, VA) was cultured in DMEM with 10 mM HEPES and 10% fetal calf serum (GIBCO, Waltham, MA) in 24-well tissue culture plates (Corning, Corning, NY) at 37 °C in a humidified incubator with 5% CO2. Macrophages (3 × 105 cells per well) were incubated with 2.5 × 106 CFU per well of log-phase WT S. aureus bacteria, which had been pre-incubated with XTENylated or free mAb, for 2 h at 37 °C to induce phagocytosis, and washed. Fluorescence, analyzed by FACS Fortessa (Becton Dickinson, Franklin Lakes, NJ), was used as measure for bacteria–macrophage association. To assess intracellular bacterial killing, macrophages were incubated with XADC-preincubated S. aureus bacteria for 2 h to enable phagocytosis, as described above. Intracellular killing was assessed after a 2 day incubation post phagocytosis as described previously.10To assess antibiotic concentrations released by TXC or TDC upon phagocytosis, macrophages were incubated with bacteria that were pre-incubated with 30 nM of conjugate as described above. After 2 h of phagocytosis, macrophages were washed, and cell lysates were prepared and precipitated by incubation with acetonitrile for 60 minutes at RT. Extracts were lyophilized by evaporation under N2 (TurboVap; Biotage, Charlotte, NC) and reconstituted in 100 μL of 50% ACN and 0.1% formic acid (FA), filtered using a 0.45 μm glass fiber filter plate (Phenomenex, Torrance, CA) and analyzed by LC/MS/MS (Triple Quad 6500, Ab Sciex). Quantitation of released dmDNA31 antibiotic payload in culture and cell extracts was performed using a standard curve generated from known standard samples of the free dmDNA31 payload spiked into extracts from cells not treated with conjugates. Concentrations of dmDNA31 were calculated with MultiQuant software (Ab Sciex), and converted to estimated intracellular molar concentration of free dmDNA31 antibiotic in macrophages (CM) based on a cell number of 3 × 105 per well, and an average cell radius of approximately 6 μm as determined by microscopy. The latter corresponds to an average spheric/cylindric volume of 0.55 × 10−12 L per cell, and a total macrophage volume of [3 × 105] × [0.55 × 10−12] = 1.65 × 10−7 L per well, resulting in a conversion from pmoles per well (CW) to μM (CM) as follows: CM = 106 × [CW/(1.65 × 10−7)].
Mouse efficacy experiments
The BJAB human non-Hodgkin Lymphoma cell line was used to establish subcutaneous xenograft model for evaluating anti-tumor activity of anti-CD22 conjugates. This cell line was obtained from the Genentech cell line repository and authenticated by short tandem repeat (STR) profiling using the Promega PowerPlex 16 System and compared with external STR profiles of cell lines to determine cell line ancestry.Tumor cells (20 million cells suspended in 0.2 mL of Hank's Balanced Salt Solution) were inoculated in the flank area of female C.B-17 Fox Chase SCID mice (Charles River Lab, Hollister, CA). When tumors reached the desired volume (∼200 mm3), animals were divided into groups of n = 5–8 with a similar distribution of tumor volumes, and received an intravenous dose of vehicle (20 mM histidine acetate, 240 mM sucrose, 0.02% polysorbate-20, pH 5.5) or ADC through the tail vein (referred to as day 0). The treatment information was not blinded during measurement. Tumors were measured in two dimensions (length and width) using calipers and tumor volume was calculated using the formula: tumor size (mm3) = 0.5 × (length × width × width). Changes in body weights were reported as a percentage relative to the starting weight. Tumor sizes and mouse body weights were recorded twice weekly over the course of the study. Mice whose tumor volume exceeded 2000 mm3 or whose body weight loss was 20% of their starting weight were promptly euthanized per IACUC guidelines. Data were analyzed using the R statistical software system (R Foundation for Statistical Computing; Vienna, Austria), and mixed modeling was fit within R using the nlme package. Cubic regression splines were used to fit a non-linear profile to the time courses of body weight change or log2 tumor volume at each dose level. These non-linear profiles were then related to dose within the mixed model. This approach addresses both repeated measurements and modest dropouts due to any non-treatment-related removal of animals before study end. Results were plotted in natural scale as fitted body weight change or tumor volume of each group over time. Tumor growth inhibition (TGI) was calculated as percent area under the tumor volume–time curve (AUC) per day of each treatment group in relation to the vehicle, using the following formula: %TGI = 100 × [1 − (AUCtreatment/day ÷ AUCvehicle/day)].
The S. aureus in vivo infection model was performed as described previously.10 Briefly, seven week-old female CB17 scid mice (Charles River Laboratories, Hollister, CA), were infected with approximately 107 CFU of S. aureus USA300 in PBS by intravenous injection into the tail vein. At 24 h post infection, a single intravenous dose of antibody or antibody conjugate in PBS was given. At 4 d post infection, kidneys were homogenized using a GentleMACS dissociator (Miltenyi Biotec, San Diego, CA), and serial dilutions of the homogenates in PBS with 0.05% Tween were plated on TSA plates to determine the numbers of viable CFU.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
NZ designed, developed protocols for producing and generated antibody conjugates. VP designed, developed protocols for producing and generated XTEN and XTEN-payload conjugates. KK, HM and WH designed and conducted in vitro anti-bacterial assays. DL, PG, SM, and AK designed and performed PK studies. GDR, SY, and AP designed and performed mouse xenograft studies. ED, MP, PH and DR expressed and purified XTEN polypeptides. KZ conducted IC-IEF measurements. CL, JH, CN, DS, LL, CW and KX developed assays to measure TXC pharmacokinetics. WS, JM, VY, GL, BS and CW contributed to assessments of TXC stability. JC, BZ, KK, GP and AP designed and conducted cell-based cytotoxicity experiments. HHB and KL carried out measurements of intracellular antibiotics. SP, MX and WH designed and carried out in vivo antibacterial experiments. RR managed project. VP and TP designed and synthesized small molecule payloads. VS supervised project at Amunix. JS supervised project at Genentech and designed molecules. WH and JS designed studies and wrote the manuscript.Conflicts of interest
NZ, KK, DL, GDL, ED, MP, KZ, CL, JH, CN, DS, LL, PG, SM, WS, JT, JM, VY, GL, JC, JHM, SP, BZ, HHB, KL, MX, KRK, GLP, BQS, CW, KX, PH, SFY, AK, RR, DR, TP, AP, WH, and JS are or were employees of Genentech, Inc. at the time when this work was conducted. VNP and VS are employees of Amunix Pharmaceuticals, Inc..Acknowledgements
We thank Solomon Simunovic, Chi Sullivan, Jessica Richardson and Keri Zook at Genentech for assisting in establishing the collaboration with Amunix. The authors wish to thank Nancy Chiang and Gerald Nakamura for genetic engineering of antibodies. We acknowledge Rachana Ohri (Genentech) and WuXi Biologics for preparation of control ADCs and intermediate ADC reagents. We thank Pete Dragovich at Genentech for helpful discussions. Finally, we thank Shelly Xi and Zach Lange at Amunix for contributions to the work described.References
- J. Z. Drago, S. Modi and S. Chandarlapaty, Nat. Rev. Clin. Oncol., 2021, 18, 327–344 CrossRef PubMed.
- C. Theocharopoulos, P.-P. Lialios, M. Samarkos, H. Gogas and D. C. Ziogas, Vaccines, 2021, 9, 1111 CrossRef CAS PubMed.
- J. T. W. Tong, P. W. R. Harris, M. A. Brimble and I. Kavianinia, Molecules, 2021, 26, 5847 CrossRef CAS PubMed.
- M. Everts, R. J. Kok, S. A. Asgeirsdottir, B. N. Melgert, T. J. M. Moolenaar, G. A. Koning, M. J. A van Luyn, D. K. F. Meijer and G. Molema, J. Immunol., 2002, 168, 883–889 CrossRef CAS PubMed.
- J. C. Kern, M. Cancilla, D. Dooney, K. Kwasnjuk, R. Zhang, M. Beaumont, I. Figueroa, S. Hsieh, L. Liang, D. Tomazela, J. Zhang, P. E. Brandish, A. Palmieri, P. Stivers, M. Cheng, G. Feng, P. Geda, S. Shah, A. Beck, D. Bresson, J. Firdos, D. Gately, N. Knudsen, A. Manibusan, P. G. Schultz, Y. Sun and R. M. Garbaccio, J. Am. Chem. Soc., 2016, 138, 1430–1445 CrossRef CAS PubMed.
- A. J. R. Gadd, F. Greco, A. J. A. Cobb and A. D. Edwards, Bioconjugate Chem., 2015, 26, 1743–1752 CrossRef CAS PubMed.
- J. Dugal-Tessier, S. Thirumalairajan and N. Jain, J. Clin. Med., 2021, 10, 838 CrossRef CAS PubMed.
- T. H. Pillow, P. Adhikari, R. A. Blake, J. Chen, G. Del Rosario, G. Deshmukh, I. Figueroa, K. E. Gascoigne, A. V. Kamath, S. Kaufman, T. Kleinheinz, K. R. Kozak, B. Latifi, D. D. Leipold, C. S. Li, R. Li, M. M. Mulvihill, A. O'Donohue, R. K. Rowntree, J. D. Sadowsky, J. Wai, X. Wang, C. Wu, Z. Xu, H. Yao, S.-F. Yu, D. Zhang, R. Zang, H. Zhang, H. Zhou, X. Zhu and P. S. Dragovich, ChemMedChem, 2020, 15, 17–25 CrossRef CAS PubMed.
- E. Cini, V. Faltoni, E. Petricci, M. Taddei, L. Salvini, G. Giannini, L. Vesci, F. M. Milazzo, A. M. Anastasi, G. Battistuzzid and R. De Santis, Chem. Sci., 2018, 9, 6490–6496 RSC.
- S. M. Lehar, T. Pillow, M. Xu, L. Staben, K. K. Kajihara, R. Vandlen, L. DePalatis, H. Raab, W. L. Hazenbos, J. H. Morisaki, J. Kim, S. Park, M. Darwish, B.-C. Lee, H. Hernandez, K. M. Loyet, P. Lupardus, R. Fong, D. Yan, C. Chalouni, E. Luis, Y. Khalfin, E. Plise, J. Cheong, J. P. Lyssikatos, M. Strandh, K. Koefoed, P. S. Andersen, J. A. Flygare, M. W. Tan, E. J. Brown and S. Mariathasan, Nature, 2015, 527, 323–328 CrossRef CAS PubMed.
- C. M. Mckertish and V. Kayser, Biomedicines, 2021, 9, 872 CrossRef CAS PubMed.
- N. Bodyak and A. V. Yurkovetskiy, Innovations for Next-Generation Antibody-Drug Conjugates: Delivering More Payload (High DAR ADCs), ed. M. Damelin, Humana Press, 2018th edn, 2018 Search PubMed.
- M. T. Kim, Y. Chen, J. Marhoul and F. Jacobson, Bioconjugate Chem., 2014, 25, 1223–1232 CrossRef CAS PubMed.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS PubMed.
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-LaFord, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Nat. Biotechnol., 2015, 33, 733–735 CrossRef CAS PubMed.
- P. Strop, K. Delaria, D. Foletti, J. M. Witt, A. Hasa-Moreno, K. Poulsen, M. G. Casas, M. Dorywalska, S. Farias, A. Pios, V. Lui, R. Dushin, D. Zhou, T. Navaratnam, T.-T. Tran, J. Sutton, K. C. Lindquist, B. Han, S.-H. Liu, D. L. Shelton, J. Pons and A. Rajpal, Nat. Biotechnol., 2015, 33, 694–696 CrossRef CAS PubMed.
- D. M. Goldenberg and R. M. Sharkey, Expert Opin. Biol. Ther., 2020, 20, 871–885 CrossRef CAS PubMed.
- S. J. Keam, Drugs, 2020, 80, 501–508 CrossRef CAS PubMed.
- W. Viricel, G. Fournet, S. Beaumel, E. Perrial, S. Papot, C. Dumontetc and B. Joseph, Chem. Sci., 2019, 10, 4048–4053 RSC.
- A. V. Yurkovetskiy, M. Yin, N. Bodyak, C. A. Stevenson, J. D. Thomas, C. E. Hammond, L. L. Qin, B. Zhu, D. R. Gumerov, E. Ter-Ovanesyan, A. Uttar and T. B. Lowinger, Cancer Res., 2015, 75, 3365–3372 CrossRef CAS PubMed.
- A. V. Yurkovetskiy, N. D. Bodyak, M. Yin, J. D. Thomas, S. M. Clardy, P. R. Conlon, C. A. Stevenson, A. Uttard, L. L. Qin, D. R. Gumerov, E. Ter-Ovanesyan, C. Bu, A. J. Johnson, V. R. Gurijala, D. McGillicuddy, M. J. DeVit, L. L. Poling, M. Protopopova, L. Xu, Q. Zhang, P. U. Park, D. A. Bergstrom and T. B. Lowinger, Mol. Cancer Ther., 2021, 20, 885–895 CrossRef CAS PubMed.
- H. Liu and K. May, mAbs, 2012, 4, 17–23 CrossRef PubMed.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. L. Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- V. Schellenberger, C.-W. Wang, N. C. Geething, B. J. Spink, A. Campbell, W. To, M. D. Scholle, Y. Yin, Y. Yao, O. Bogin, J. L. Cleland, J. Silverman and W. P. C. Stemmer, Nat. Biotechnol., 2009, 27, 1186–1190 CrossRef CAS PubMed.
- P. Adhikari, N. Zacharias, R. Ohri and J. Sadowsky, Antibody-Drug Conjugates: Site-Specific Conjugation to Cys-Engineered THIOMABTM Antibodies, ed. L. N. Tumey, Humana Press, 2020, p. 2078 Search PubMed.
- V. N. Podust, B.-C. Sim, D. Kothari, L. Henthorn, C. Gu, C.-W. Wang, B. McLaughlin and V. Schellenberger, Protein Eng., Des. Sel., 2013, 26, 743–753 CrossRef CAS PubMed.
- V. N. Podust, S. Balan, B.-C. Sim, M. P. Coyle, U. Ernst, R. T. Peters and V. Schellenberger, J. Controlled Release, 2016, 240, 52–66 CrossRef CAS PubMed.
- X. Sun, J. F. Ponte, N. C. Yoder, R. Laleau, J. Coccia, L. Lanieri, Q. Qiu, R. Wu, E. Hong, M. Bogalhas, L. Wang, L. Dong, Y. Setiady, E. K. Maloney, O. Ab, X. Zhang, J. Pinkas, T. A. Keating, R. Chari, H. K. Erickson and J. M. Lambert, Bioconjugate Chem., 2017, 28, 1371–1381 CrossRef CAS PubMed.
- M. Dorywalska, P. Strop, J. A. Melton-Witt, A. Hasa-Moreno, S. E. Farias, M. G. Casas, K. Delaria, V. Lui, K. Poulsen, C. Loo, S. Krimm, G. Bolton, L. Moine, R. Dushin, T.-T. Tran, S.-H. Liu, M. Rickert, D. Foletti, D. L. Shelton, J. Pons and A. Rajpal, Bioconjugate Chem., 2015, 26, 650–659 CrossRef CAS PubMed.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- M. Dorywalska, R. Dushin, L. Moine, S. E. Farias, D. Zhou, T. Navaratnam, V. Lui, A. Hasa-Moreno, M. G. Casas, T.-T. Tran, K. Delaria, S.-H. Liu, D. Foletti, C. J. O'Donnell, J. Pons, D. L. Shelton, A. Rajpal and P. Strop, Mol. Cancer Ther., 2016, 15, 958–970 CrossRef CAS PubMed.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- Y. Anami, C. M. Yamazaki, W. Xiong, X. Gui, N. Zhang, Z. An and K. Tsuchikama, Nat. Commun., 2018, 9, 2512 CrossRef PubMed.
- A. F. Herrera and A. Molina, Clin. Lymphoma, Myeloma Leuk., 2018, 18, 452–468 CrossRef PubMed.
- A. G. Polson, J. Calemine-Fenaux, P. Chan, W. Chang, E. Christensen, S. Clark, F. J. de Sauvage, D. Eaton, K. Elkins, J. M. Elliott, G. Frantz, R. N. Fjkkjuji, A. Gray, K. Harden, G. S. Ingle, N. M. Kljavin, H. Koeppen, C. Nelson, S. Prabhu, H. Raab, S. Ross, J.-P. Stephan, S. J. Scales, S. D. Spencer, R. Vandlen, B. Wranik, S.-F. Yu, B. Zheng and A. Ebens, Cancer Res., 2009, 69, 2358–2364 CrossRef CAS PubMed.
- J. C. Masters, D. J. Nickens, D. Xuan, R. L. Shazer and M. Amantea, Invest. New Drugs, 2018, 36, 121–135 CrossRef CAS PubMed.
- S. Ding, M. Song, B.-C. Sim, C. Gu, V. N. Podust, C.-W. Wang, B. McLaughlin, T. P. Shah, R. Lax, R. Gast, R. Sharan, A. Vasek, M. A. Hartman, C. Deniston, P. Srinivas and V. Schellenberger, Bioconjugate Chem., 2014, 25, 1351–1359 CrossRef CAS PubMed.
- A. G. Polson, M. Williams, A. M. Gray, R. N. Fuji, K. A. Poon, J. McBride, H. Raab, T. Januario, M. Go, J. Lau, S.-F. Yu, C. Du, F. Fuh, C. Tan, Y. Wu, W.-C. Liang, S. Prabhu, J.-P. Stephan, J.-A. Hongo, R. C. Dere, R. Deng, M. Cullen, R. de Tute, F. Bennett, A. Rawstron, A. Jack and A. Ebens, Leukemia, 2010, 24, 1566–1573 CrossRef CAS PubMed.
- R. Fong, K. Kajihara, M. Chen, I. Hotzel, S. Mariathasan, W. L. W. Hazenbos and P. J. Lupardus, mAbs, 2018, 10, 979–991, DOI:10.1080/19420862.2018.1501252.
- I. R. Monk, I. M. Shah, M. Xu, M.-W. Tan and T. J. Foster, mBio, 2012, 20, e00277-11 Search PubMed.
- W. L. W. Hazenbos, K. K. Kajihara, R. Vandlen, J. H. Morisaki, S. M. Lehar, M. J. Kwakkenbos, T. Beaumont, A. Q. Bakker, Q. Phung, L. R. Swem, S. Ramakrishnan, J. Kim, M. Xu, I. M. Shah, B. A. Diep, T. Sai, A. Sebrell, Y. Khalfin, A. Oh, C. Koth, S. J. Lin, B.-C. Lee, M. Strandh, K. Koefoed, P. S. Andersen, H. Spits, E. J. Brown, M.-W. Tan and S. Mariathasan, PLoS Pathog., 2013, 9, e1003653 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05243h |
| ‡ These authors contributed equally. |
| § Present address: Atomwise, 717 Market Street, San Francisco, CA 94103. Email: desiree.thayer@gmail.com. |
| ¶ Present address: Ambys Medicines, 131 Oyster Point Blvd, South San Francisco, CA 94080. |
| || Current address: Vir Biotechnology, 499 Illinois St, San Francisco, CA 94158. |
| ** Present address: Carmot Therapeutics, 740 Heinz Avenue, Berkeley, CA 94710. |
| This journal is © The Royal Society of Chemistry 2022 |