
Enantioselective modification of sulfonamides and sulfonamide-containing drugs via carbene organic catalysis†
Runjiang
Song‡
a,
Yingguo
Liu‡
a,
Pankaj Kumar
Majhi‡
a,
Pei Rou
Ng
a,
Lin
Hao
 a,
Jun
Xu
ab,
Weiyi
Tian
*b,
Long
Zhang
c,
Hongmei
Liu
c,
Xinglong
Zhang
*d and
Yonggui Robin
Chi
*ae
a,
Jun
Xu
ab,
Weiyi
Tian
*b,
Long
Zhang
c,
Hongmei
Liu
c,
Xinglong
Zhang
*d and
Yonggui Robin
Chi
*ae
aDivision of Chemistry & Biological Chemistry, School of Physical & Mathematical Science, Nanyang Technological University, Singapore 637371, Singapore. E-mail: robinchi@ntu.edu.sg
bSchool of Pharmacy, Guizhou University of Traditional Chinese Medicine, Guiyang 550025, China. E-mail: tianweiyi@gzy.edu.cn
cInstitute of Nervous System Diseases, Xuzhou Medical University, Xuzhou 221002, China
dInstitute of High Performance Computing, A*STAR (Agency for Science, Technology and Research), 1 Fusionopolis Way, #16-16 Connexis, Singapore 138632, Singapore. E-mail: zhang_xinglong@ihpc.a-star.edu.sg
eKey Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Guizhou University, Huaxi District, Guiyang 550025, China
First published on 15th March 2021
Abstract
A carbene-catalyzed method for highly enantioselective modification of sulfonamides is disclosed. The reaction proceeds under mild conditions with broad substrate scope, wide functional group tolerance, and good to excellent yields. When multiple sulfonamides or amines are present in the same molecule, the reaction occurs in a highly chemo-selective manner. Application of our method allows for selective modification of sulfonamide-containing drug molecules to form the corresponding phthalidyl derivatives as potential prodrugs. Experimental observations and DFT calculations suggest that the reaction proceeds via a stepwise addition pathway, assisted by Li+ ions or protons. Non-covalent interactions, such as cation–π interactions, play important roles in enhancing the reactivity and controlling the enantioselectivity of the reaction.
1. Introduction
Sulfonamides are common functional moieties in organic molecules with applications in medicine and many other areas.1 A sizeable number of top-selling pharmaceuticals and molecules in the WHO's list of essential drugs contain sulfonamide moieties. Shown in Fig. 1a are several examples of sulfonamide-containing drugs, including hydrochlorothiazide,2 macitentan,3 sulfamethoxazole,4 pazopanib,5 and gliclazide.6 For instance, hydrochlorothiazide is a diuretic medication for the treatments of high blood pressure, congestive heart failure, diabetes insipidus, and renal tubular acidosis.2 These sulfonamide moieties often behave as critical functional groups in bioactive molecules.7 It is also established that further synthetic transformations of sulfonamides prepared from ammonia and primary amines can offer new structures and properties.8 For medicinal molecules, synthetic modifications of the sulfonamide moieties can provide potential prodrugs with enhanced performance and/or altered pharmacological properties. One such method is to react sulfonamides with phthalaldehydic acid derivatives (such as 3-bromophthalides) to form the corresponding phthalidyl sulfonamides (Fig. 1b).9b Unfortunately, with these present approaches it is difficult to control the enantioselectivity of the newly formed chiral center in the phthalidyl derivatives.9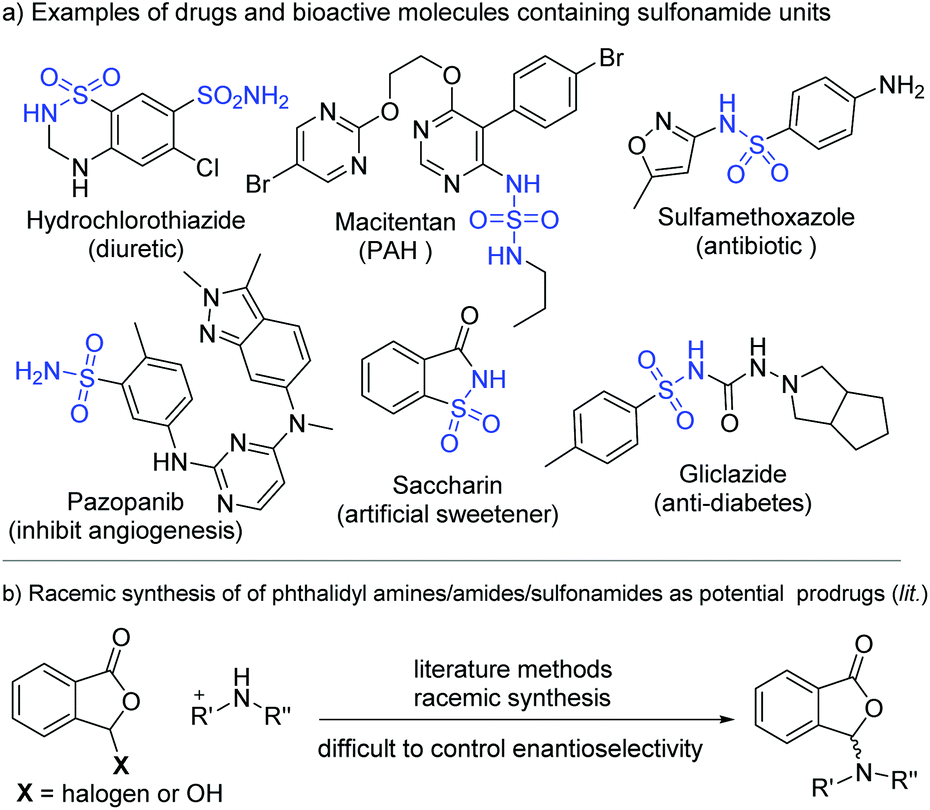 | ||
| Fig. 1 Sulfonamide drugs and the common racemic modification. | ||
Here we disclose an enantioselective modification of sulfonamides through a reaction with dialdehydes to afford optically enriched phthalidyl sulfonamides under N-heterocyclic carbene (NHC) organocatalysis (Fig. 2). DFT calculation suggests that this transformation proceeds via a stepwise process. Non-covalent interactions of the Li+ ion with catalyst-activated substrates are believed to play important roles in facilitating this reaction and controlling its enantio-selectivity. The operationally stable while physiologically labile phthalidyl units in our products have found proven applications in prodrugs.10 Our study constitutes the first enantioselective preparation of chiral phthalidyl amines/sulfonamides. It allows for facile modification of a diverse set of sulfonamides, including several commercial drug molecules.11 Preliminary in vitro bioactivity evaluations show that phthalidyl sulfamethoxazole offers enhanced antibacterial activities, when compared to the unmodified sulfamethoxazole.
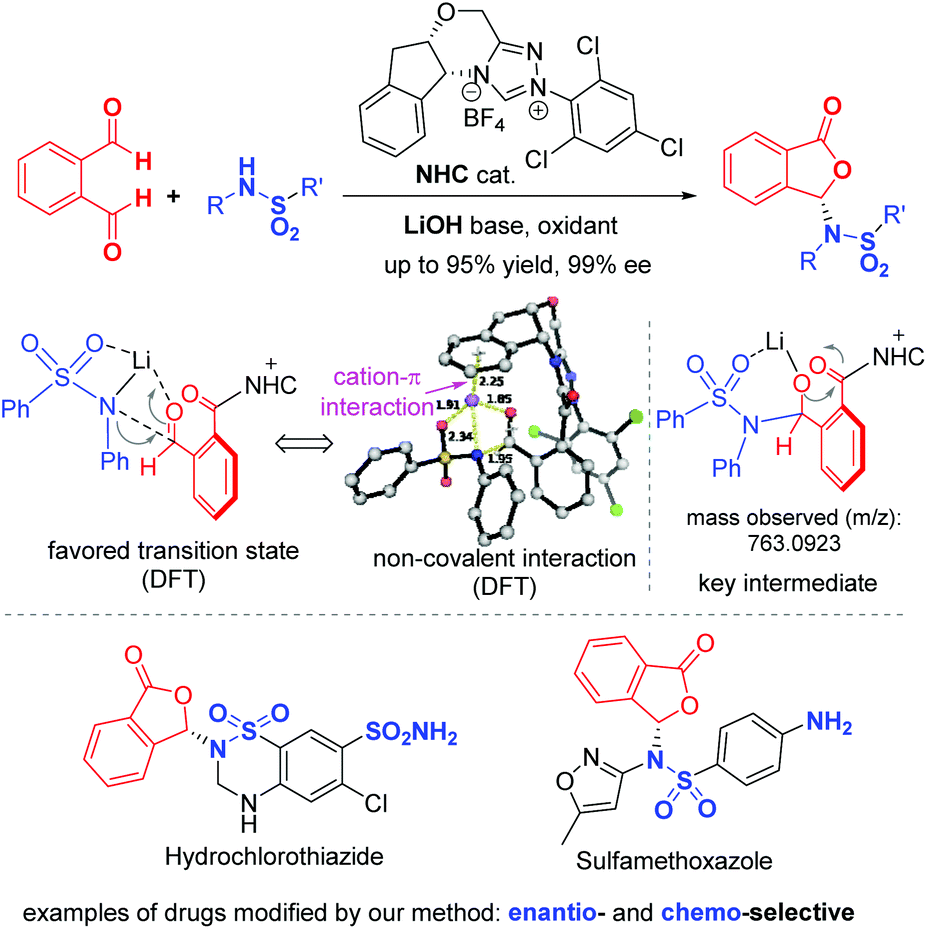 | ||
| Fig. 2 NHC-catalyzed enantioselective synthesis of phthalidyl sulfonamides (this work). | ||
2. Results and discussion
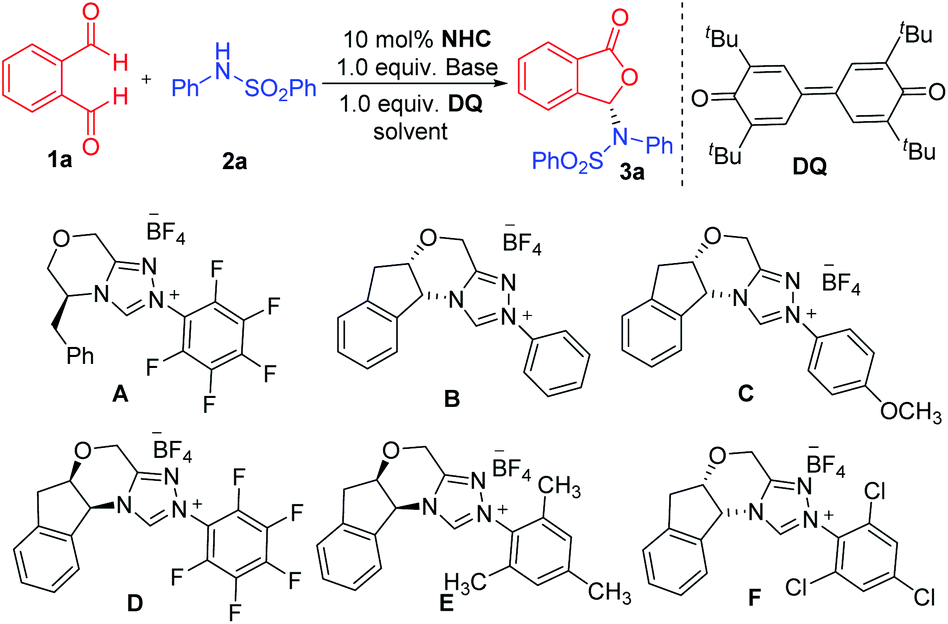
We initiated our studies using phthalaldehyde 1a and N-phenylbenzene sulfonamide 2a as the model substrates in the presence of DQ oxidant12 to search for suitable conditions (Table 1). We first used dichloromethane as the solvent and DBU as the base (entries 1–6). We examined amino acid-derived triazolium A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 as the NHC pre-catalyst and found the formation of the proposed product 3a in 77% yield and 46:54 er (entry 1). We then examined aminoindanol-derived NHC pre-catalysts (B to F). The aminoindanol-derived pre-catalyst with an N-phenyl substituent (B)14 led to 3a with an excellent yield, albeit with a very low 53:47 er value (entry 2). Introducing a methyoxyl unit on the para-position of the N-phenyl unit of B (to get catalyst C)15 gave the product in 88% yield and a slightly improved er (60:40 er, entry 3). A large increase in er value (16:84 er) was obtained when the NHC pre-catalyst with a N-pentafluorophenyl substituent (D)16 was used (entry 4). A slightly improved result (14:86 er) was obtained when a pre-catalyst (E)17 with an N-mesityl substituent was used (entry 5). Both pre-catalysts D and E led to lower yields. When NHC pre-catalyst F18 with a N-trichlorophenyl group was used, the reaction gave 3a in 92% yield and 86:14 er (entry 6). We next studied the effect of bases (entries 7 and 8) and found that the use of LiOH·H2O as the base could significantly improve the er value of 3a to 99:1 with 92% yield (entry 8). Other solvents such as PhCF3 and THF were also examined; a relatively small loss in yields or er values was observed (entries 9 and 10). The critical role of the Li+ ion (LiOH as the base) in enhancing the reaction enantioselectivity is unravelled using DFT calculations (vide infra).
13 as the NHC pre-catalyst and found the formation of the proposed product 3a in 77% yield and 46:54 er (entry 1). We then examined aminoindanol-derived NHC pre-catalysts (B to F). The aminoindanol-derived pre-catalyst with an N-phenyl substituent (B)14 led to 3a with an excellent yield, albeit with a very low 53:47 er value (entry 2). Introducing a methyoxyl unit on the para-position of the N-phenyl unit of B (to get catalyst C)15 gave the product in 88% yield and a slightly improved er (60:40 er, entry 3). A large increase in er value (16:84 er) was obtained when the NHC pre-catalyst with a N-pentafluorophenyl substituent (D)16 was used (entry 4). A slightly improved result (14:86 er) was obtained when a pre-catalyst (E)17 with an N-mesityl substituent was used (entry 5). Both pre-catalysts D and E led to lower yields. When NHC pre-catalyst F18 with a N-trichlorophenyl group was used, the reaction gave 3a in 92% yield and 86:14 er (entry 6). We next studied the effect of bases (entries 7 and 8) and found that the use of LiOH·H2O as the base could significantly improve the er value of 3a to 99:1 with 92% yield (entry 8). Other solvents such as PhCF3 and THF were also examined; a relatively small loss in yields or er values was observed (entries 9 and 10). The critical role of the Li+ ion (LiOH as the base) in enhancing the reaction enantioselectivity is unravelled using DFT calculations (vide infra).
|
|
|||||
|---|---|---|---|---|---|
| Entry | NHC | Base | Solvent | Yield (%) | er |
| a Reaction conditions: 1a (1.0 equiv.), 2a (1.5 equiv.), NHC (0.1 equiv.), base (1.0 equiv.), DQ (1.0 equiv.) = 3,3′,5,5′-tetra-tert-butyldiphenoquinone, solvent (2 mL). DBU = 1,8-diazabicyclo [5.4.0] undec-7-ene. Yields were isolated yields after SiO2 column chromatography. The er was determined via chiral-phase HPLC analysis. | |||||
| 1 | A | DBU | CH2Cl2 | 77 | 46:54 |
| 2 | B | DBU | CH2Cl2 | 84 | 53:47 |
| 3 | C | DBU | CH2Cl2 | 88 | 60:40 |
| 4 | D | DBU | CH2Cl2 | 66 | 16:84 |
| 5 | E | DBU | CH2Cl2 | 68 | 14:86 |
| 6 | F | DBU | CH2Cl2 | 92 | 86:14 |
| 7 | F | Cs2CO3 | CH2Cl2 | 79 | 96:4 |
| 8 | F | LiOH·H2O | CH2Cl2 | 92 | 99:1 |
| 9 | F | LiOH·H2O | PhCF3 | 80 | 97:3 |
| 10 | F | LiOH·H2O | THF | 74 | 92:8 |
With the optimized conditions in hand, we move to explore the substrate scope with respect to different aryl amine-derived sulfonamides (Scheme 1). The absolute configuration of 3a was confirmed via single crystal X-ray analysis. Sulfonamide containing different functional groups at the para-carbon of the phenyl ring were all well tolerated to furnish phthalidyl products (3b–i) with excellent enantiomeric ratios and good yields. These functional groups include halogen, cyano (–CN), nitro (–NO2), alkoxy (–OR), alkyl, alkenyl and alkynyl units (3b–i). Installing substituents at both the ortho and meta positions (–SCH3, –yne) of the phenyl ring led to a drop in the reaction yield but without diminishing the er value (3j, 3l). 2-Naphthylamine-derived sulfonamide was applied with 64% yield and 97:3 er (3m). It is also worth noting that pyridine amine-derived sulfonamides bearing various substituents and substitution patterns can fit in well with the present strategy (3n–3r).
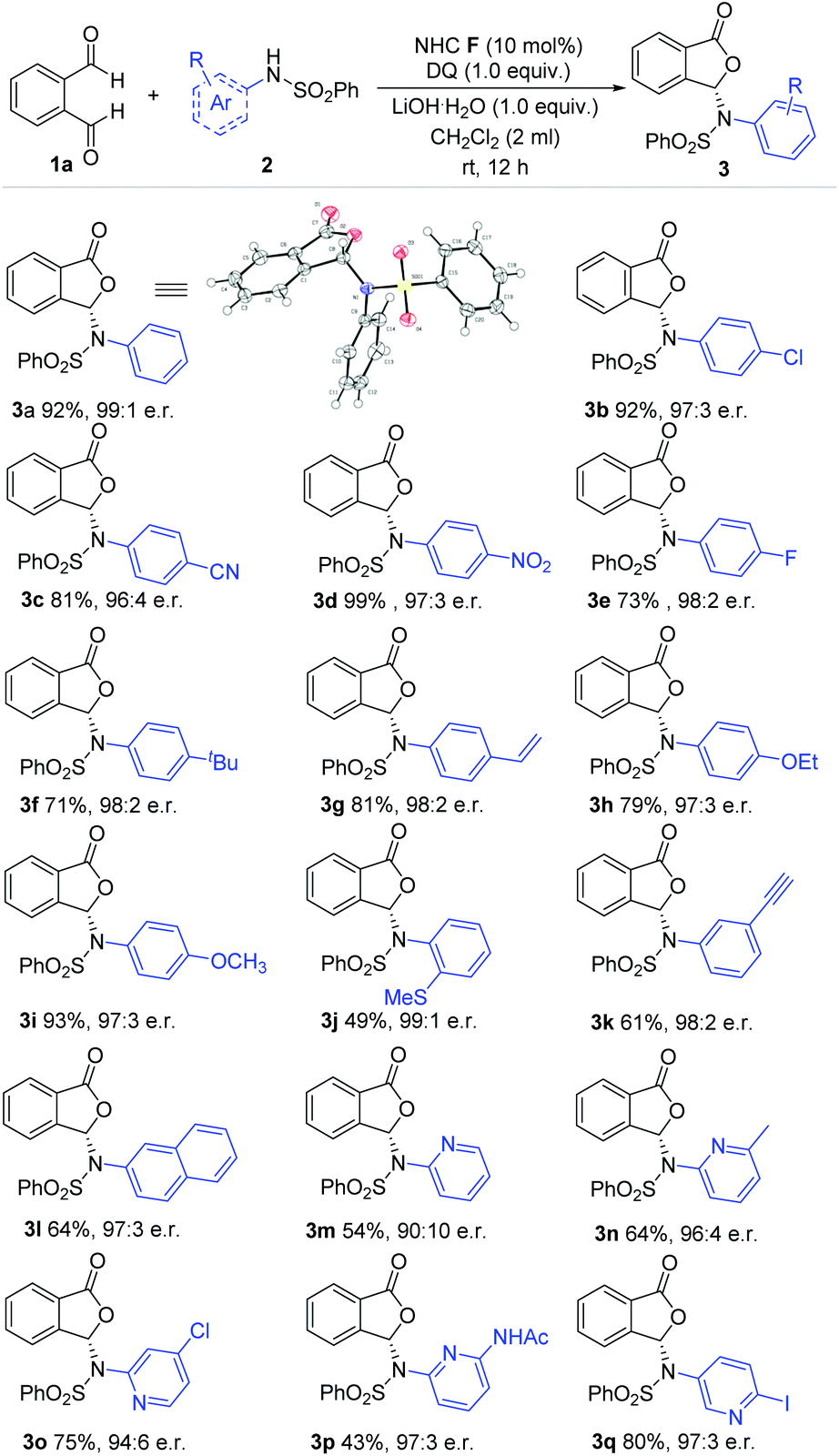 | ||
| Scheme 1 Examples of aryl amine-derived sulfonamides. Reaction conditions: 1a (1.0 equiv.), sulfonamides (1.5 equiv.), NHC F (10 mol%), DQ (1.0 equiv.), CH2Cl2 (2 mL), 12 h. DQ = 3,3′,5,5′-tetra-tert-butyldiphenoquinone. e.r. = enantiomeric ratio. Yields were isolated yields after SiO2 column chromatography. The e.r. was determined via chiral-phase HPLC analysis. 3a: CCDC No. 2045331.† | ||
We then moved to examine aliphatic amine-derived sulfonamides as substrates (Scheme 2). Aliphatic amine-based sulfonamides, especially N-benzyl substituted sulfonamides, afforded the desired product with high enantiomeric ratios and good yields (5a–5d). Halogen atoms on the phenyl ring were tolerated under our reaction conditions (5b–5c). The sulfonamide derivative from 2-(aminomethyl) pyridine also gave the desired phthalide derivative (5d) with high er value and good yield. When sulfonamides derived from methylamine and cyclohexylamine were used, the corresponding products (5e and 5f) were obtained with suppressed yields and er values (51% yield, 91:9 er for 5e; 57% yield, 92:8 er for 5f). Phthalaldehyde bearing substituents (5g–5h) did not significantly affect the reaction outcome.
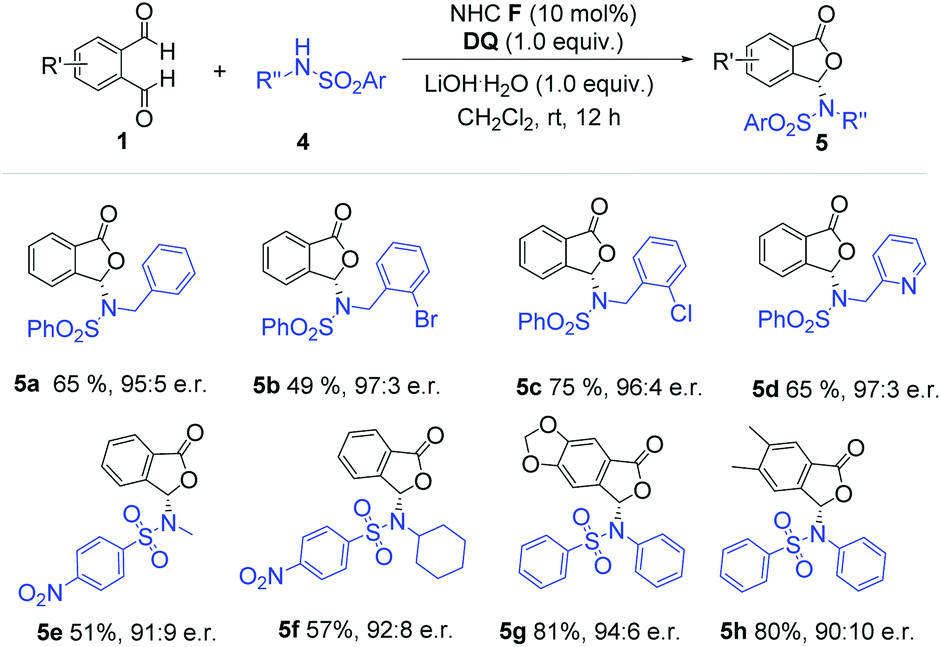 | ||
| Scheme 2 Examples of alkyl amine-derived sulfonamides. Reaction conditions: 1 (1.0 equiv.), sulfonamides (1.5 equiv.), NHC F (10 mol%), DQ (1 equiv.), CH2Cl2 (2 mL), 12 h. DQ = 3,3′,5,5′-tetra-tert-butyldiphenoquinone, er = enantiomeric ratio. Yields were isolated yields after SiO2 column chromatography. The er was determined via chiral-phase HPLC analysis. | ||
We next explored the use of our method for enantioselective modification of sulfonamide-containing food additives and drug molecules (Scheme 3). Saccharin is an artificial sweetener and could be modified with our method to give the corresponding phthalidyl sulfonamide 6a in 95% yield and 85:15 er. Hydrochlorothiazide is a diuretic medicine for the treatment of high blood pressure and swelling due to fluid buildup.2 This drug molecule could be converted to the phthalidyl derivative 6b with 80% yield and 91:9 er. Similarly, the antibiotic sulfamethoxazole could be modified to give 6c with 62% yield and 93:7 er. Remarkably, pre-protection of free amino groups in these molecules (hydrochlorothiazide and sulfamethoxazole) is not required. Our reaction selectively proceeded on one sulfonamide moiety without affecting the amine group (6b and 6c). In addition, when two sulfonamide moieties were present in the same molecule (hydrochlorothiazide), a product with selective reaction on the secondary amine-derived sulfonamide was observed (6b). This unusual chemo-selectivity, resulting from a complicated process involving multiple reversible reactions of these amine/sulfonamide groups, further highlights the robust features of our strategy.
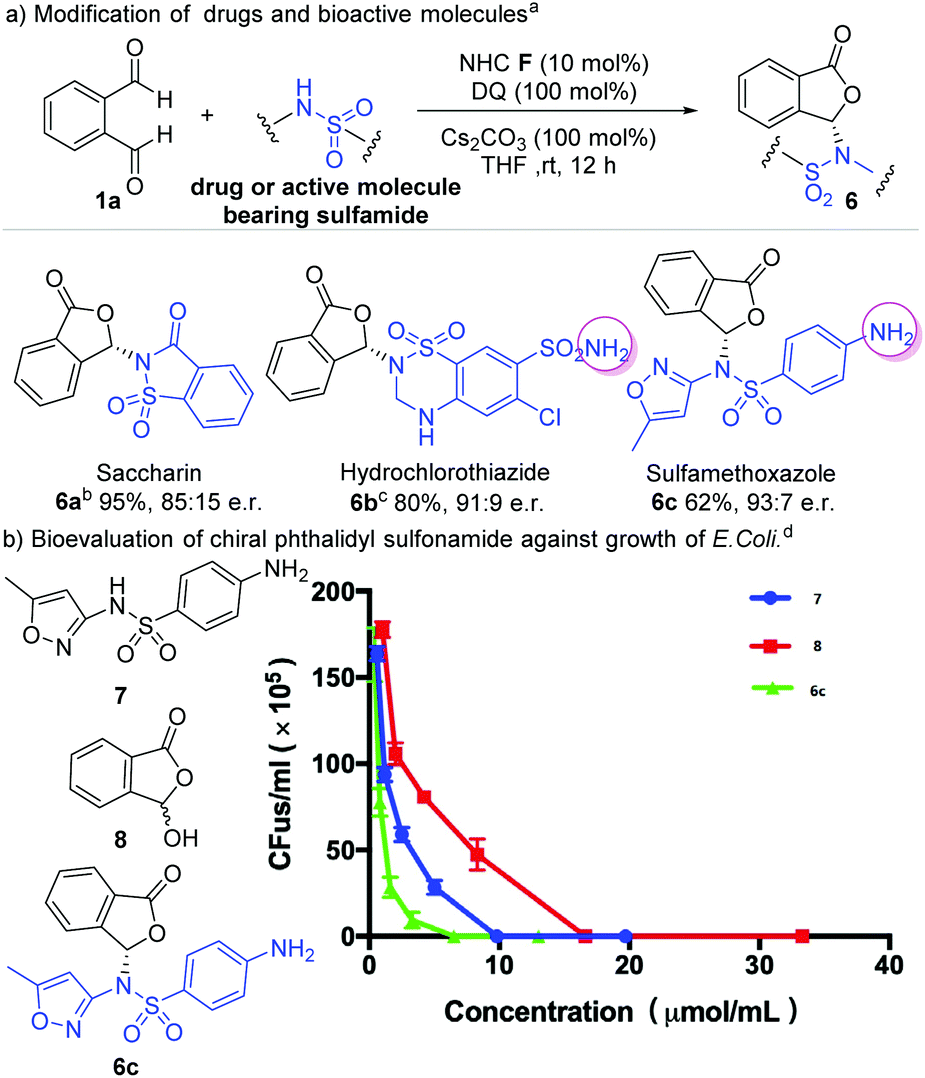 | ||
| Scheme 3 Modification of bioactive sulfonamides and preliminary bioactivity evaluation. Reaction conditions: 1a (1.0 equiv.), sulfonamides (1.5 equiv.), NHC F (10 mol%), DQ (1.0 equiv.), Cs2CO3 (1.0 equiv.), THF (2 mL), 12 h. DQ = 3,3′,5,5′-tetra-tert-butyldiphenoquinone, er = enantiomeric ratio. Yields were isolated yields after SiO2 column chromatography. The er was determined via chiral-phase HPLC analysis. aLiCl (1.0 equiv.) was added as the additive. bReplace Cs2CO3 with Et3N, 0 °C, 24 h. cBio-evaluation of chiral phthalidyl sulfonamides against the growth of E. coli (n = 3 biological replicates, mean ± SD). MIC values for 6c, 7 and 8 were 6.5 μmol mL−1, 9.9 μmol mL−1 and 16.7 μmol mL−1, respectively. | ||
We performed a preliminary antibiotic evaluation of our modified phthalidyl sulfamethoxazole 6c against the growth of E. coli.19 The hydroxyphthalide 7 and unmodified sulfamethoxazole 8 were tested as controls. The MIC (minimum inhibitory concentration) value (6.5 μmol mL−1) of our product 6c was found to be much lower than those of compounds 7 and 8 (MIC 9.9 μmol mL−1 and 16.7 μmol mL−1 respectively).
To elucidate the reaction pathway (Scheme 4) and unravel the origin of stereoselectivity of the present transformation, density functional theory (DFT) calculations were performed at the SMD(CH2Cl2)-M06-2X/def2-TZVP//SMD(CH2Cl2)-M06-2X/def2-SVP level of theory. The basis for the formation of Breslow intermediate (I) from the NHC catalyst F and the aldehyde substrate (1) has been previously established.20 This intermediate (I) is then oxidized by DQ to generate acyl azolium intermediate II that can be detected experimentally via high-resolution mass spectrometry. Intermediate II can undergo either concerted addition (not supported) or stepwise addition (supported by DFT studies) to yield the cyclized product (vide infra).
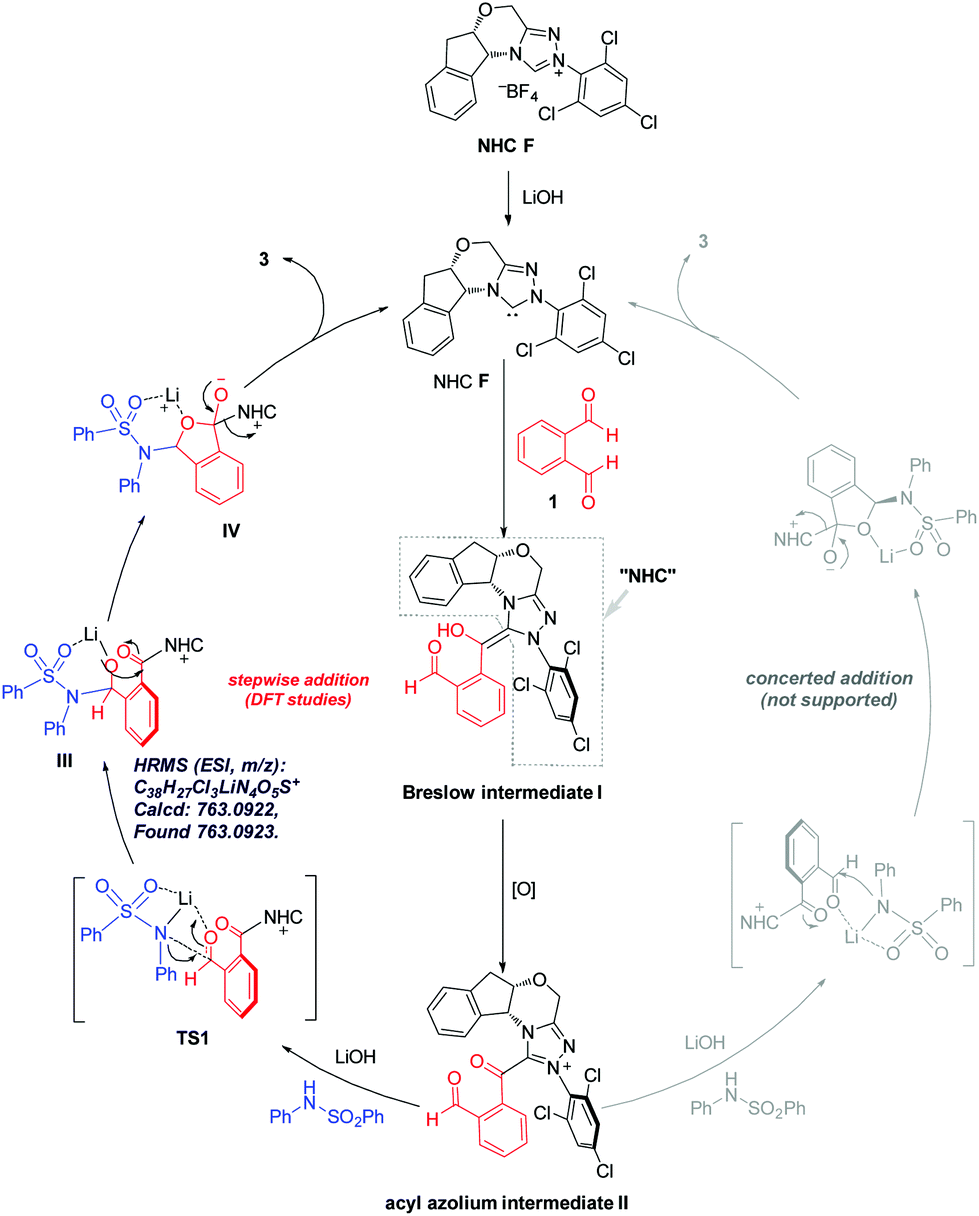 | ||
| Scheme 4 Postulated reaction mechanism. | ||
Computational results pinpoint the importance of the Li+ ion in affecting the observed enantioselectivity through the formation of cation–π interactions in key intermediates and transition states (Fig. 3). Two key conformers of acyl azolium intermediate II were found ((Si)-II and (Re)-II, Fig. 3; see ESI section V.1† for detailed conformational sampling). We note that for each conformer, only one face of the carbonyl group is open to attack by the incoming nucleophile as the other face is shielded from attack by forming π–π interactions between the aromatic ring of II and the 2,4,6-trichlorophenyl moiety of the NHC ligand. In the absence of the Li+ ion (see ESI section V.2† for a detailed mechanism), the negatively charged N-atom of the deprotonated sulfonamide attacks the carbonyl group of acyl azolium intermediate II without any appreciable barrier and with concomitant cyclization as the resulting carboanion attacks the adjacent carbonyl group without any barrier (Fig. S4†). This implies that in the absence of the Li+ ion, the cylization product forms immediately, whose product distribution will be determined by the thermodynamic stabilities of the reacting conformers of intermediate II ((Si)-II more stable). This mechanism predicts an er in favor of the (R)-phthalidyl sulfonamide product as 99.5:0.5 (Fig. S5†); thus, such a mechanism is unlikely in the present transformation. In the presence of the Li+ ion, the addition proceeds in a stepwise manner.
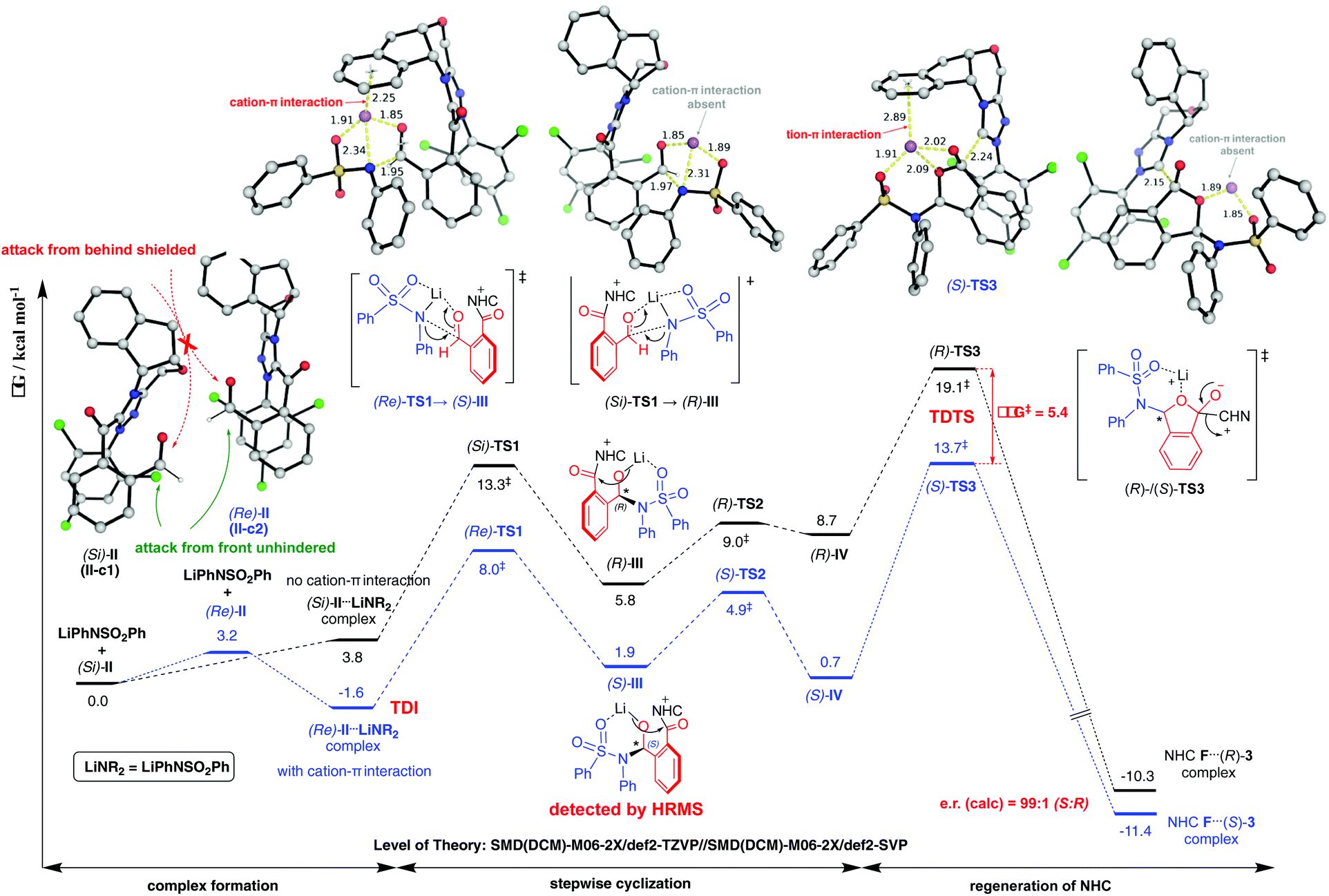 | ||
| Fig. 3 Gibbs energy profile computed at the SMD(CH2Cl2)-M06-2X/def2-TZVP//SMD(CH2Cl2)-M06-2X/def2-SVP level of theory for the reaction between NHC-bound intermediate II and lithium sulfonamide. Key DFT-optimized structures are given with key bond distances in Å. TDI = turnover frequency-determining intermediate; TDTS = turnover frequency-determining transition state. | ||
According to the “energetic span model”,21 the Li+ ion complexes with intermediate II to give the turnover frequency (TOF)-determining intermediate (TDI) (Re)-II⋯LiNR2 that is stabilized by the favorable Li+ cation–π interaction (absent in (Si)-II⋯LiNR2; Fig. S7†), thus reversing the thermodynamic favorability for (Si)-II in the absence of cation coordination to (Re)-II in the presence of cation coordination. The addition to the Re-face is now favored by 5.3 kcal mol−1 due to more favorable NCIs (non-covalent interactions) arising from the coordination of the Li+ ion to the aromatic ring in (Re)-TS1. This cation–π interaction exists in all transition states and intermediates for the favored pathway ((Re)-TS1, (S)-III/IV, (S)-TS2/TS3). The TDTSs are the regeneration of the NHC catalyst (S/R-TS3) with a barrier difference of 5.4 kcal mol−1 in favor of the (S)-phthalidyl sulfonamide product formation (calculated er of 99.9:0.1; Fig. 3), in excellent quantitative agreement with the experimental observation (entry 8, Table 1). We highlight the important role of the Li+ ion in facilitating a stepwise addition, allowing highly enantioselective cyclization to occur that could not be possible in the absence of cation participation (concerted, barrierless direct cyclization of the most thermodynamically stable (Si)-II conformer). We further note that the overall energetic span of 15.3 kcal mol−1 is consistent with the high yield observed at room temperature. The important role of the Li+ ion is further evidenced by the fact that its replacement by (protonated) DBU reduces the enantioselectivity (entry 6, Table 1) due to DBU's less coordinating nature than the Li+ ion (see ESI section V.5†).
3. Conclusions
In summary, we have developed a new approach for highly enantio- and chemo-selective modification of sulfonamides. The reaction starts with the addition of a carbene catalyst to one aldehyde moiety of phthalaldehyde as the initial step. The subsequent oxidation and a number of stepwise additions with sulfonamides furnish the corresponding phthalidyl sulfonamides with excellent yields and enantioselectivities. The possible reaction pathways were investigated via both experimental and DFT calculation methods, and non-covalent interactions (such as cation-π interactions) are believed to play crucial roles in promoting high enantioselectivities. Our new method uses mild conditions and tolerates various functional groups. Sulfonamide-containing drug molecules can be selectively modified to form the corresponding phthalidyl derivatives as potential prodrugs. Further explorations of our mechanistic observations for new catalysis development and applications of our methods in bioactive molecule modifications are in progress in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Yongxin Li (NTU) for assistance with X-ray structure analysis. We acknowledge the financial support from the Singapore National Research Foundation under its NRF Investigatorship (NRF-NRFI2016-06) and the Competitive Research Program (NRF-CRP22-2019-0002); the Ministry of Education, Singapore, under its MOE AcRF Tier 1 Award (RG108/16, RG5/19, RG1/18), MOE AcRF Tier 2 (MOE2019-T2-2-117), and MOE AcRF Tier 3 Award (MOE2018-T3-1-003); the Agency for Science, Technology and Research (A*STAR) under its A*STAR AME IRG Award (A1783c0008, A1783c0010); the GSK-EDB Trust Fund; Nanyang Research Award Grant, Nanyang Technological University; the National Natural Science Foundation of China (21772029, 21801051, 21807019, 21961006, 22071036, 22061007, 82360589, 81360589), the Frontiers Science Center for Asymmetric Synthesis and Medicinal Molecules, Department of Education, Guizhou Province [Qianjiaohe KY number (2020)004]; the 10 Talent Plan (Shicengci) of Guizhou Province ([2016]5649); the Science and Technology Department of Guizhou Province ([2018]2802, [2019]1020); the Program of Introducing Talents of Discipline to Universities of China (111 Program, D20023) at Guizhou University; the Guizhou Province First-Class Disciplines Project [(Yiliu Xueke Jianshe Xiangmu)-GNYL(2017)008], Guizhou University of Traditional Chinese Medicine (China); Guizhou University. X. Z thanks the Institute of High Performance Computing (IHPC), A*STAR for financial support and acknowledges the partial use of supercomputers in the A*STAR Computational Resource Centre (ACRC) and the National Supercomputing Centre (NSCC), Singapore (https://www.nscc.sg) for computations performed in this work.Notes and references
- (a) J. E. Lesch, The First Miracle Drugs: How the Sulfa Drugs Transformed Medicine, Oxford University Press, 2006 Search PubMed; (b) J. J. Li and E. J. Corey, Drug Discovery., Practices, Processes, and Perspectives, Wiley, 2013 CrossRef; (c) B. R. Smith, C. M. Eastman and J. T. Njardarson, Beyond C, H, O, and, N! Analysis of the elemental composition of U.S. FDA approved drug architectures, J. Med. Chem., 2014, 57, 9764–9773 CrossRef CAS PubMed; (d) E. A. Ilardi, E. Vitaku and J. T. Njardarson, Data-mining for sulfur and fluorine: an evaluation of pharmaceuticals to reveal opportunities for drug design and discovery, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed; (e) K. A. Scott and J. T. Njardarson, Analysis of US FDA-Approved Drugs Containing Sulfur Atoms, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed.
- (a) S. Dineva, K. Uzunova, V. Pavlova, E. Filipova, K. Kalinov and T. Vekov, Comparative efficacy and safety of chlorthalidone and hydrochlorothiazide-meta-analysis, J. Hum. Hypertens., 2019, 33, 766–774 CrossRef CAS PubMed; (b) L. Lucaccioni, E. Coccolini, A. Dozza, S. L. Cantatore, A. Berardi, B. Predieri and L. Iughetti, Severe metabolic alkalosis due to diuretic treatment in a patient with distal renal tubular acidosis: a rare association, Acta Biomed., 2019, 90, 348–352 CAS; (c) J. Luo, X. Chen, C. Luo, G. Lu, L. Peng, X. Gao and Z. Zuo, Hydrochlorothiazide modulates ischemic heart failure-induced cardiac remodeling via inhibiting angiotensin II type 1 receptor pathway in rats, Cardiovasc. Ther., 2017, 35, e12246 CrossRef PubMed; (d) A. Wang, T. Hirose, Y. Ohsaki, C. Takahashi, E. Sato, I. Oba-Yabana, S. Kinugasa, Y. Muroya, S. Ito and T. Mori, Hydrochlorothiazide ameliorates polyuria caused by tolvaptan treatment of polycystic kidney disease in PCK rats, Clin. Exp. Nephrol., 2019, 23, 455–464 CrossRef CAS PubMed.
- H.-A. Ghofrani, G. Simonneau, A. M. D'Armini, P. Fedullo, L. S. Howard, X. Jaïs, D. P. Jenkins, Z.-C. Jing, M. M. Madani, N. Martin, E. Mayer, K. Papadakis, D. Richard, N. H. Kim, I. Lang, C. Kähler, M. Delcroix, Z. Bshouty, P. S. Varela, Z.-C. Jing, Y. Yang, J. Liu, G. Zhang, N. Zhang, Y. Mi, X. Zhu, P. Jansa, X. Jaïs, G. Prévot, H. Bouvaist, O. Sanchez, F. Grimminger, M. Held, H. Wilkens, S. Rosenkranz, E. Grünig, K. Karlócai, A. Temesvári, I. Edes, S. Aidietienė, S. Miliauskas, T. R. P. Zamudio, C. J. Sanchez, A. V. Noordegraaf, J. Lewczuk, P. Podolec, J. Kasprzak, T. Mularek-Kubzdela, R. Grzywna, K. Dheda, O. Moiseeva, A. Chernyavskiy, V. Shipulin, O. Barbarash, T. Martynyuk, H.-K. Kim, J.-B. Park, J. S. Lee, R. Speich, S. Ulrich, J.-D. Aubert, A. Phrommintikul, N. Jaimchariyatam, S. Sompradeekul, Z. P. Onen, G. Okumus, L. Solovey, V. Gavrysyuk, L. Howard, J. Pepke-Zaba, R. Condliffe, J. McConnell, K. Kerr, L. H. Nguyen and N. V. Pham, Macitentan for the treatment of inoperable chronic thromboembolic pulmonary hypertension (MERIT-1): results from the multicentre, phase 2, randomised, double-blind, placebo-controlled study, Lancet Respir. Med., 2017, 5, 785–794 CrossRef CAS.
- H. B. Park, Z. Wei, J. Oh, H. Xu, C. S. Kim, R. Wang, T. P. Wyche, G. Piizzi, R. A. Flavell and J. M. Crawford, Sulfamethoxazole drug stress upregulates antioxidant immunomodulatory metabolites in Escherichia coli, Nat. Microbiol., 2020, 5, 1319–1329 CrossRef CAS PubMed.
- F. A. Schutz, T. K. Choueiri, C. N. Sternberg and C. N. Pazopanib, Clinical development of a potent anti-angiogenic drug, Crit. Rev. Oncol. Hematol., 2011, 77, 163–171 CrossRef PubMed.
- B. D. C. Mapa, L. U. Araújo, N. M. Silva-Barcellos, T. G. Caldeira and J. Souza, Gliclazide: Biopharmaceutics Characteristics to Discuss the Biowaiver of Immediate and Extended Release Tablets, Appl. Sci., 2020, 10, 3836 CrossRef.
- (a) P. Mujumdar and S. A. Poulsen, Natural Product Primary Sulfonamides and Primary Sulfamates, J. Nat. Prod., 2015, 78, 1470–1477 CrossRef CAS PubMed; (b) C. Zhao, K. P. Rakesh, L. Ravidar, W. Y. Fang and H. L. Qin, Pharmaceutical and medicinal significance of sulfur (S(VI))-Containing motifs for drug discovery: A critical review, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed.
- H. X. Dai, A. F. Stepan, M. S. Plummer, Y. H. Zhang and J. Q. Yu, Divergent C-H functionalizations directed by sulfonamide pharmacophores: late-stage diversification as a tool for drug discovery, J. Am. Chem. Soc., 2011, 133, 7222–7228 CrossRef CAS PubMed.
- (a) K. B. Sloan and S. A. M. Koch, Effect of nucleophilicity and leaving group ability on the SN2 reactions of amines with (acyloxy)alkyl .alpha.-halides: a product distribution study, J. Org. Chem., 1983, 48, 635–640 CrossRef CAS; (b) P. Kovaricek and J. M. Lehn, Directional Dynamic Covalent Motion of a Carbonyl Walker on a Polyamine Track, Chem. – Eur. J., 2015, 21, 9380–9384 CrossRef CAS PubMed.
- Y. Liu, Q. Chen, C. Mou, L. Pan, X. Duan, X. Chen, H. Chen, Y. Zhao, Y. Lu, Z. Jin and Y. R. Chi, Catalytic asymmetric acetalization of carboxylic acids for access to chiral phthalidyl ester prodrugs, Nat. Commun., 2019, 10, 1675 CrossRef PubMed.
- F. Carta, A. Scozzafava and C. T. Supuran, Sulfonamides: a patent review (2008–2012), Expert Opin. Ther. Pat., 2012, 22, 747–758 CrossRef CAS PubMed.
- S. De Sarkar, S. Grimme and A. Studer, NHC catalyzed oxidations of aldehydes to esters: chemoselective acylation of alcohols in presence of amines, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef CAS PubMed.
- E. Sanchez-Larios, K. Thai, F. Bilodeau and M. Gravel, Highly enantioselective intermolecular Stetter reactions of beta-aryl acceptors: alpha-ketoester moiety as handle for activation and synthetic manipulations, Org. Lett., 2011, 13, 4942–4945 CrossRef CAS PubMed.
- (a) M. S. Kerr, J. R. Alaniz and T. Rovis, A highly enantioselective catalytic intramolecular Stetter reaction, J. Am. Chem. Soc., 2002, 124, 10298–10299 CrossRef CAS PubMed; (b) R. S. Massey, C. J. Collett, A. G. Lindsay, A. D. Smith and A. C. O'QDonoghue, Proton transfer reactions of triazol-3-ylidenes: kinetic acidities and carbon acid pKa values for twenty triazolium salts in aqueous solution, J. Am. Chem. Soc., 2012, 134, 20421–20432 CrossRef CAS PubMed; (c) J. Mahatthananchai and J. W. Bode, The effect of the N-mesityl group in NHC-catalyzed reactions, Chem. Sci., 2012, 3, 192–197 RSC; (d) C. J. Collett, R. S. Massey, O. R. Maguire, A. S. Batsanov, A. C. O'QDonoghue and A. D. Smith, Mechanistic insights into the triazolylidene-catalysed Stetter and benzoin reactions: role of the N-aryl substituent, Chem. Sci., 2013, 4, 1514–1522 RSC; (e) C. J. Collett, R. S. Massey, J. E. Taylor, O. R. Maguire, A. C. O'QDonoghue and A. D. Smith, Rate and Equilibrium Constants for the Addition of N-Heterocyclic Carbenes into Benzaldehydes: A Remarkable 2-Substituent Effect, Angew. Chem., Int. Ed., 2015, 54, 6887–6892 CrossRef CAS PubMed.
- M. S. Kerr, J. Read de Alaniz and T. Rovis, An efficient synthesis of achiral and chiral 1,2,4-triazolium salts: bench stable precursors for N-heterocyclic carbenes, J. Org. Chem., 2005, 70, 5725–5728 CrossRef CAS PubMed.
- M. S. Kerr and T. Rovis, Enantioselective synthesis of quaternary stereocenters via a catalytic asymmetric Stetter reaction, J. Am. Chem. Soc., 2004, 126, 8876–8877 CrossRef CAS PubMed.
- M. He, J. R. Struble and J. W. Bode, Highly enantioselective azadiene Diels-Alder reactions catalyzed by chiral N-heterocyclic carbenes, J. Am. Chem. Soc., 2006, 128, 8418–8420 CrossRef CAS PubMed.
- S. P. Lathrop and T. Rovis, A photoisomerization-coupled asymmetric Stetter reaction: application to the total synthesis of three diastereomers of (−)-cephalimysin A, Chem. Sci., 2013, 4, 1668–1673 RSC.
- M. Nepka, E. Perivolioti, E. Kraniotaki, L. Politi, A. Tsakris and S. Pournaras, In Vitro Bactericidal Activity of Trimethoprim-Sulfamethoxazole Alone and in Combination with Colistin against Carbapenem-Resistant Acinetobacter baumannii Clinical Isolates, Antimicrob. Agents Chemother., 2016, 60, 6903–6906 CrossRef CAS PubMed.
- G. Wang, Z. Fu and W. Huang, Access to Amide from Aldimine via Aerobic Oxidative Carbene Catalysis and LiCl as Cooperative Lewis Acid, Org. Lett., 2017, 19, 3362–3365 CrossRef CAS PubMed.
- S. Kozuch and S. Shaik, How to conceptualize catalytic cycles? The energetic span model, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2045331. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo00212k |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2021 |