DOI:
10.1039/C9LC00469F
(Paper)
Lab Chip, 2019,
19, 2500-2511
Organ-on-chips made of blood: endothelial progenitor cells from blood reconstitute vascular thromboinflammation in vessel-chips†
Received
17th May 2019
, Accepted 17th June 2019
First published on 19th June 2019
Abstract
Development of therapeutic approaches to treat vascular dysfunction and thrombosis at disease- and patient-specific levels is an exciting proposed direction in biomedical research. However, this cannot be achieved with animal preclinical models alone, and new in vitro techniques, like human organ-on-chips, currently lack inclusion of easily obtainable and phenotypically-similar human cell sources. Therefore, there is an unmet need to identify sources of patient primary cells and apply them in organ-on-chips to increase personalized mechanistic understanding of diseases and to assess drugs. In this study, we provide a proof-of-feasibility of utilizing blood outgrowth endothelial cells (BOECs) as a disease-specific primary cell source to analyze vascular inflammation and thrombosis in vascular organ-chips or “vessel-chips”. These blood-derived BOECs express several factors that confirm their endothelial identity. The vessel-chips are cultured with BOECs from healthy or diabetic patients and form an intact 3D endothelial lumen. Inflammation of the BOEC endothelium with exogenous cytokines reveals vascular dysfunction and thrombosis in vitro similar to in vivo observations. Interestingly, our study with vessel-chips also reveals that unstimulated BOECs of type 1 diabetic pigs show phenotypic behavior of the disease – high vascular dysfunction and thrombogenicity – when compared to control BOECs or normal primary endothelial cells. These results demonstrate the potential of organ-on-chips made from autologous endothelial cells obtained from blood in modeling vascular pathologies and therapeutic outcomes at a disease and patient-specific level.
Introduction
Vascular diseases are ranked amongst the leading cause of death worldwide as they are relatively poorly understood and their therapeutic approaches that exist or are in development are inadequate.1,2 These inadequacies are attributed primarily to the fact that discovery and therapeutic programs rely heavily on results from animal models which poorly predict the human pathophysiology and drug responses. In contrast, “personalized” vascular medicine has been suggested to significantly improve human healthcare. Mimicking a patient's native tissue architecture, capturing crosstalk between diseased and heterotypic cell populations, and measuring the actual functional response of such systems may provide more precise readouts of patient outcome. This can further reduce dependence on ineffective and harmful treatment strategies and bridge the treatment gaps for non-responding individuals.3,4 But achieving this objective requires the availability of physiologically-relevant in vitro models of personalized tissues and organs. In particular, there is a need for an understanding of the complex signaling mechanisms and drug responses that occur in various vascular disorders, such as diabetes and thrombosis, at a disease- and a patient-specific level as well as at a cellular, molecular and biophysical level.
Biomimetic in vitro microfluidic disease modelling platforms, such as organ-on-chips (organ-chips), have garnered significant interest recently.5,6 By mimicking the milieu of microphysiological factors like vessel microenvironments and cellular and tissue level crosstalk, these models have allowed replication of complex physiologies in vitro and have helped discern their underlying complex molecular pathways.7,8 Although these models have been able to provide dissectible analyses of several disorders, these approaches have limited precision because they rely on cell sources that are not always primary and representative of the conditions being modeled. In the context of vascular organ-chips or vessel-chips, these strategies have utilized healthy, non-disease specific cell types like human umbilical vein endothelial cells (HUVECs) or human microvascular endothelial cells (HMVECs), which limits the extent of their predictive power. Additionally, such approaches utilize exogenous factors and stimulants to create a diseased endothelium.9,10 Even though this strategy has been able to reconstitute thrombotic activity, these models might still be phenotypically dissimilar to their in vivo counterparts in cases when more complex pathophysiology may be involved. As a potential solution, the use of patient-derived cell sources can preclude the use of exogenously administered inflammatory cytokines to inflict injury and provide the most reliable source of cells for organ-on-chip technology. However, utilizing primary cells derived from patient biopsies is still not a viable option as patient tissue samples are naturally limited and often require sophisticated surgeries. Attempts at utilizing induced pluripotent stem cell derived endothelial cells (iPSC-ECs) for development of disease-specific in vitro platforms have been possible recently;11,12 however, doing so involves time-consuming procedures and highly specialized training and even then, these cells may have a significantly different gene expression of the target cell type. Interestingly, endothelial progenitor cells (EPCs) within blood circulation are easily obtainable but represent a relatively understudied cell subpopulation that can be utilized to create disease as well as patient-specific vascular models.13,14 In fact, studies with patients suffering from vascular disorders have shown increased amounts of circulating endothelial cells in the blood15,16 and therefore, we postulate that they can be harnessed as an alternative primary cell source to traditionally used endothelial cell types in in vitro disease models and organ-chips. In recent studies, research groups have utilized blood outgrowth endothelial cells (BOECs), a cell sub-population derived from patient EPCs, to examine common vascular pathophysiological states like the von Willebrand factor disease,17,18 statin treatment on arteriopathy,19 and inflammation in SCD.20,21 BOECs have also contributed to several bioengineering applications, like enhancing vascular graft revascularization and bio-prosthetics, and also in gene therapy.14,22,23 More recently, BOECs from malaria patients were shown to serve as a tool to assess the adhesion of infected erythrocytes in these patients, mediated through common inflammatory adhesion markers like ICAM-1.24 However, in prior work, BOEC monolayers were prepared in static 2D well-plate cultures,18,24 or they were cultured within extracellular matrices to investigate their angiogenic potential.25 Other progenitor endothelial cell types, like cord blood-derived endothelial cells, have been also investigated in a microfluidic system,26–28 but BOECs, which are derived more easily from venous circulation, have not been explored on 3D vessel-chips constituting a 3D lumen that is perfusable with human whole blood. As a result, none of these studies could be applied as disease models to validate functional consequences of endothelial dysfunction and blood cell adhesion. Therefore, there exists an exciting premise to use BOECs in vascular organ-chips and confirm that the application of BOECs in organ-chips may serve as a modeling platform for basic scientific investigations and drug screening purposes at a disease- and patient-specific level.
In this study, we utilized cytokine-stimulated and diabetic BOECs to create an arteriole-sized vessel-on-a-chip model of thromboinflammation. Our aim was to demonstrate that when isolated from healthy volunteer blood samples, BOECs function as mature endothelial cells within vessel-chips, similar to human primary endothelial cells.29,30 Additionally, when isolated from diabetic pigs, they exhibit several critical functions of diabetic endothelium and functional responses relative to normal controls. Our outcomes suggest that BOECs may advance the organ-chip technology and could potentially be easily deployed in preclinical research or personalized medical applications.
Materials and methods
Human blood samples
Blood from healthy adult donors was collected upon informed consent in 3.2% sodium citrate tubes (BD Biosciences). All experiments were performed according to the policies of the US Office of Human Research Protections (OHRP) and Texas A&M University Human Research Protection Program (HRPP) and approved by the Texas A&M University Institutional Review Board (IRB ID: IRB2016-0762D). Blood was used within four hours of withdrawal to prevent abnormal platelet functioning.31
Human BOEC isolation
For endothelial progenitor cell extraction, 60 mL of blood from a healthy donor was withdrawn and diluted with 1× PBS (Gibco) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The diluted blood was then gently poured over 15 mL density gradient media (Ficoll-Paque PLUS, GE Healthcare) in a 50 mL falcon tube. The tubes were then centrifuged at 400g without brake and acceleration for 35 minutes. The distinct “buffy” layer was collected and added to collagen coated cell culture flasks containing BOEC growth media (20% fetal bovine serum in EGM-2).32,33 Culture media were replaced every 36–48 hours for 2–3 weeks till BOEC colonies appeared. The colonies were then transferred to fresh culture flasks (passage 1).
:1 ratio. The diluted blood was then gently poured over 15 mL density gradient media (Ficoll-Paque PLUS, GE Healthcare) in a 50 mL falcon tube. The tubes were then centrifuged at 400g without brake and acceleration for 35 minutes. The distinct “buffy” layer was collected and added to collagen coated cell culture flasks containing BOEC growth media (20% fetal bovine serum in EGM-2).32,33 Culture media were replaced every 36–48 hours for 2–3 weeks till BOEC colonies appeared. The colonies were then transferred to fresh culture flasks (passage 1).
Porcine blood samples and BOEC isolation
All animal procedures were approved by the Baylor Scott & White Health Institutional Animal Care and Use Committee. Domestic (Yorkshire) male pigs (6 weeks old) were purchased from Real Farms (San Antonio, TX). Type 1 diabetes was induced by selective ablation of pancreatic β-cells with intravenous injection of streptozocin (STZ, Zanosar®, 200 mg kg−1 in saline) via an ear vein, as described in detail in our previous studies.34,35 The control pig was intravenously injected with saline. Fasting blood glucose levels were obtained every other day using a Bayer Contour glucometer (Bayer Corporation, Pittsburgh, PA). After 2 weeks, pigs were sedated with Telazol (4–8 mg kg−1, intramuscularly), anesthetized with 2–5% isoflurane, and intubated. The pigs were then heparinized with an intravenous administration of heparin via an ear vein (500 U kg−1). After a left thoracotomy was performed, the heart was removed and immediately placed on iced (5 °C) saline. Fifty mL of blood from diabetic pigs (fasting glucose: 300–350 mg dL−1) and control pigs (fasting glucose: 80–100 mg dL−1) was withdrawn for BOEC isolation. BOEC isolation from porcine blood samples was performed according to the method used for human blood samples. Once isolated, porcine BOECs (PBOECs) were cultured in EGM-2, with media changes every 36–48 hours.
Vessel-chip design and fabrication
Microfluidic channels were designed using SolidWorks and were subsequently patterned on silicon wafers (University Wafer Corp.) using photolithography. The microfluidic channels were then prepared using soft lithography of polydimethylsiloxane (PDMS, Dow Corning). Inlet and outlet holes were made with a 1.5 mm wide biopsy punch (Ted Pella). Each device had two independent parallel channels and the PDMS block containing the features was bonded to a PDMS coated glass slide (75 × 25 mm) using a 100 Watts plasma cleaner (Thierry Zepto, Diener Electronic). An open slip-tip syringe was connected to the channels through a curved dispensing tip (Qosina), which acted as a liquid reservoir for growth media, blood, etc. wherever required. The outlet was connected to a syringe pump (Harvard Apparatus, PHD Ultra) using 20′′ tubing (Qosina).
Device functionalization and endothelialization
The microfluidic channels were treated with oxygen plasma for 30 seconds at a power of 50 Watts prior to treatment with a 1% solution of (3-aminopropyl)-trimethoxysilane (APTES, Sigma-Aldrich) in 200 proof ethanol. After treatment for ten minutes, the channels were rinsed with 70% ethanol and 100% ethanol after which the devices were stored in a 70 °C oven for two hours. The channels were then filled with type-I rat-tail collagen (100 μg mL−1, Corning) and incubated for an hour in a 5% CO2 incubator, followed by rinsing with endothelial growth media (EGM-2, PromoCell). BOECs in the culture were seeded into the collagen coated channels and the channels were incubated upside down. After two hours, a fresh suspension of BOECs was again perfused through the channels and incubated for an additional two hours to promote cell adhesion to all the sides of the channels. Overnight perfusion of growth media was then carried out at a laminar flow rate (1 μL min−1; shear stress: 0.81 dyn cm−2; shear rate: 81 s−1) to ensure continuous supply of nutrients to the cells, also leading to cell alignment along the flow direction. For studies that required vascular activation, the endothelialized channels were treated for 18 hours with growth media spiked with TNF-α (recombinant from E. coli, Sigma) at concentrations ranging from 5–25 ng mL−1.
Live cell microscopy
For live cell culture imaging, devices seeded with BOECs and maintained under constant growth media perfusion were placed inside the incubator on a CytoSMART 2 system. Bright-field images with digital phase contrast were acquired at a 10× magnification every 15 minutes till the devices reached confluence.
Immunohistochemistry
Vessel-chips were fixed with a 4% paraformaldehyde solution (Sigma) for 15 minutes followed by permeabilization using 0.1% Triton X (Sigma-Aldrich) in BSA/DPBS for ten minutes at room temperature. To remove the non-specific binding, the channels were blocked using a 2% solution of BSA in DPBS for 30 minutes at room temperature. Mouse or rabbit antibodies against intercellular adhesion molecule-1 (ICAM-1, Invitrogen), von Willebrand factor (VWF, Invitrogen) and vascular endothelial-cadherin (VE-cadherin, Invitrogen) were added to the channels and incubated for three hours before being washed, and visualized using secondary anti-rabbit or anti-mouse fluorescent antibodies (Invitrogen) incubated for 1–2 hours at room temperature.
Barrier function and permeability assessment
Quantification of the endothelial barrier integrity in vitro was performed by measuring the gaps in confluent BOEC lumens. Briefly, confluent BOEC microchannels were fixed and stained for the junction marker (VE-cadherin), F-actin and nuclei. Following immunostaining and subsequent fluorescence microscopy, snapshots of BOEC lumens were taken and analyzed in Fiji/ImageJ. Closed loops that did not contain nuclei were regarded as gaps.36 These gap areas were summed over the compete field of view and reported as percent area coverage. For measuring permeability, we seeded endothelial cells on 24 mm tissue culture grade polycarbonate transwell inserts with 8 μm pores (Costar). Approximately 50000 cells were seeded on each transwell insert and were allowed to attain complete confluency. Once confluent, cells were either left untreated (control) or treated with growth media containing TNF-α (5–25 ng mL−1) for 18 hours. After treatment, old media were discarded and replaced with Dulbecco's modified Eagle's medium (DMEM). On top of each transwell insert, 500 μL of 1 mg mL−1 solution of 4 kDa FITC-dextran in DMEM was added. The samples were incubated for 4 hours after which 100 μL of effluent from the bottom well was isolated and added to a 96-well plate for fluorescence measurements with a plate reader (TECAN® Infinite M200PRO). The amount of fluorescence was used as a readout of permeability.
Blood perfusion
500 μL of blood pre-incubated with the FITC-conjugated anti-human CD41 antibody (10 μL mL−1 blood Invitrogen) and fluorescently labelled fibrinogen (20 μg mL−1 blood, Invitrogen) was added to the inlet reservoir. Blood was perfused through the cell laden channels at a flow rate of 15 μL min−1 which resulted in an arterial shear rate of ∼750 s−1 (ref. 37) To reinstate coagulation, a solution of 100 mM CaCl2 and 75 mM MgCl2 was mixed with blood in a 1:10 ratio prior to perfusion.38
Assessment of BOEC proliferation
Porcine BOEC proliferation was measured using the standard alamarBlue assay. Approximately 5 × 103 porcine BOECs were added to pre-treated 96-well plates and allowed to grow. After every 24 hours, 100 μL of 10% alamarBlue (Bio-Rad) in EGM-2 was added to each well containing cells. Following 2 hour incubation, fluorescence measurements were performed to assess the formation of resorufin, the colorimetric indicator of the redox reaction occurring in viable cells. Similarly, PBOEC proliferation in the vessel-chip was measured every 24 hours by adding 100 μL of 10% alamarBlue to each PBOEC-laden vessel. After 2 hour incubation, the alamarBlue solution was collected and replaced with fresh growth media. The collected effluent was then added to a 96-well plate for fluorescence measurements. Measurements were taken at 590 nm and values were reported as relative proliferation with respect to control cells.
Oxidative stress assessment
The detection of reactive oxygen species (ROS) was performed after staining cells with 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2-DCFDA; Invitrogen). A stock solution was reconstituted in molecular grade DMSO (Sigma) to a concentration of 0.5 mM and stored at −20 °C. Cells were grown to 50–75% confluence in 6-well plates. They were washed once with EGM-2. CM-H2-DCFDA was added to EGM-2 at a final concentration of 0.25 μM, and then 1 ml of the solution was added to each well. Samples were incubated for 10 minutes at 37 °C. Cells were then washed twice with ice-cold PBS and trypsin was added to detach adherent cells. EGM-2 was then added to neutralize trypsin and the cell suspension was centrifuged to finally obtain a cell pellet. The supernatant was discarded, and the pellet was resuspended in sterile PBS. Production of ROS was confirmed by the presence of the fluorescent adduct produced via the intracellular cleavage of CM-H2-DCFDA by ROS. The adduct of CM-H2-DCFDA has an excitation maximum of 495 nm and an emission maximum of 529 nm. Fluorescence was determined by measuring 10000 events per sample following excitation with a 488 nm wavelength laser and reading through a 530/30 filter.
Statistical analysis
Statistical analysis was performed using GraphPad Prism ver. 7 and comparisons between groups were made using ANOVA or Student's t-test. Differences were considered statistically significant if p < 0.05. Data are presented as mean ± standard error of the mean (SEM). Data shown are representative of at least three independent experiments.
Results and discussion
Blood outgrowth endothelial cell (BOEC) isolation and characterization
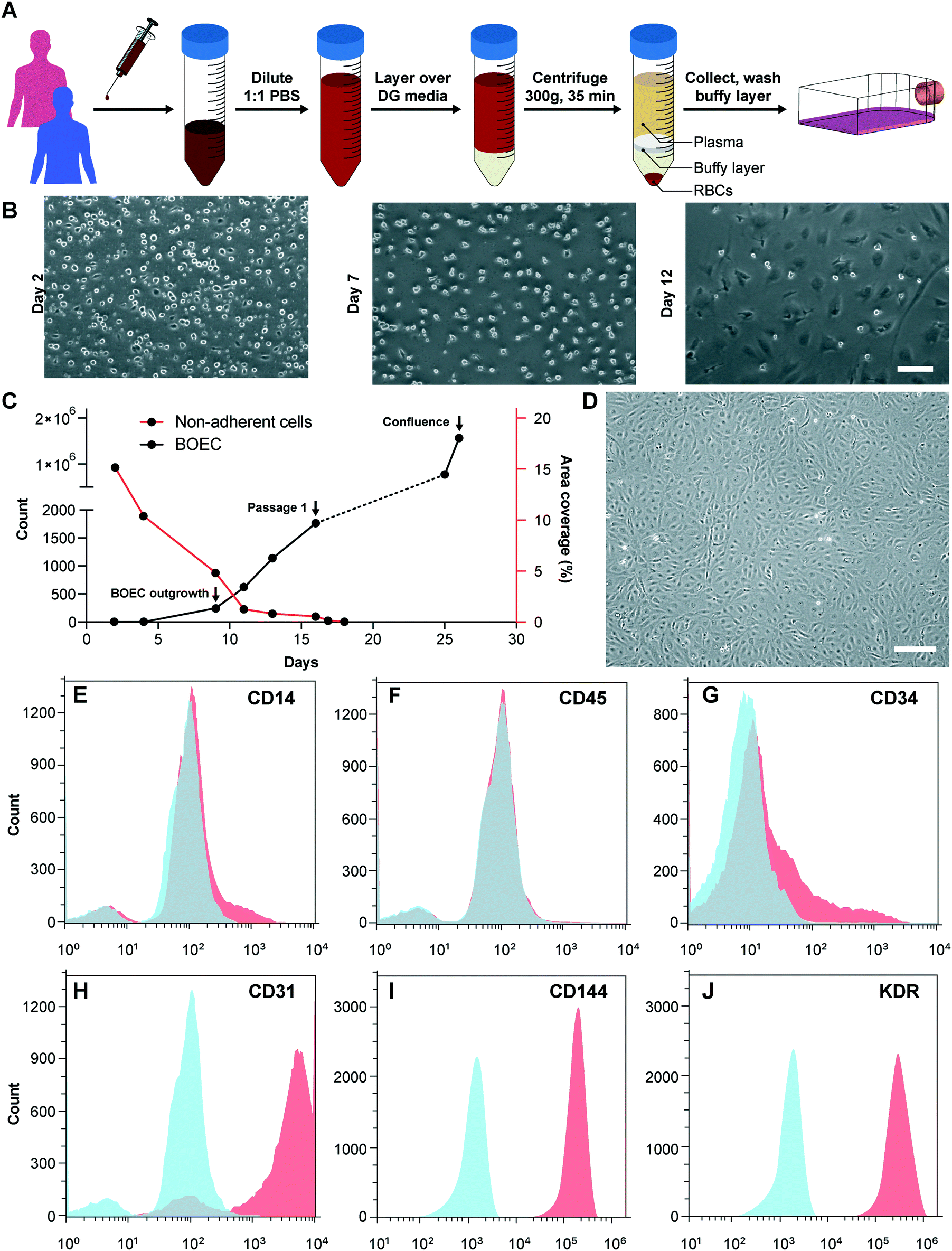
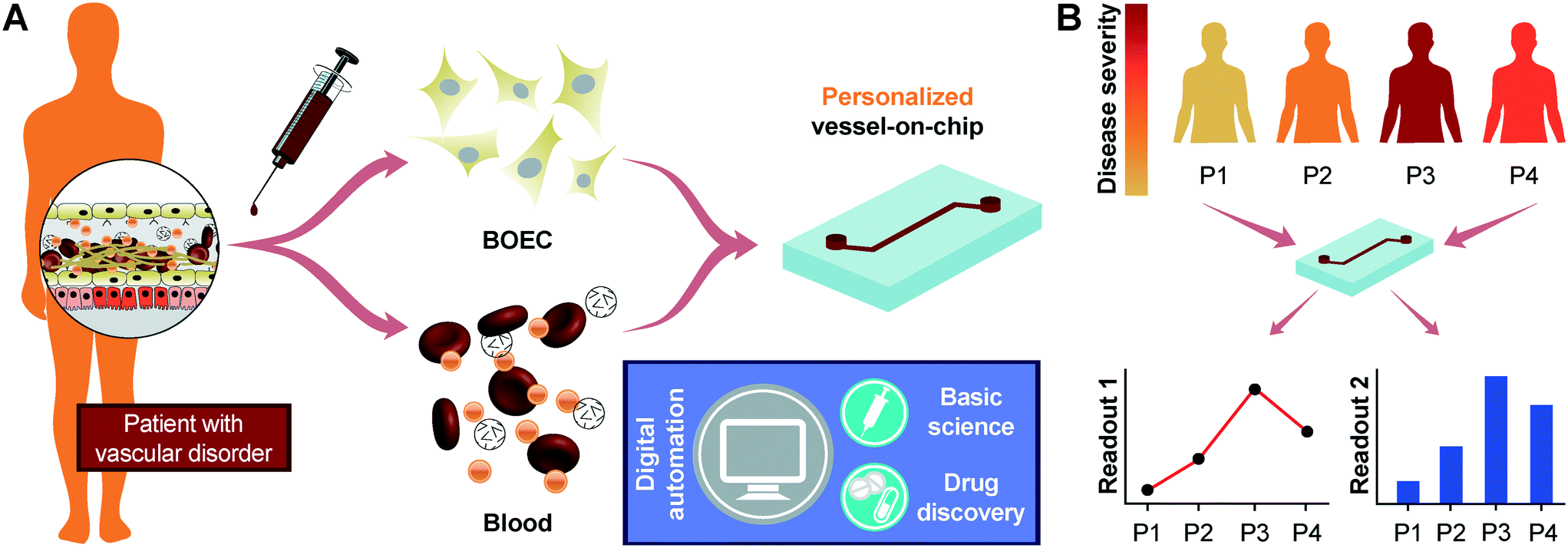
We initiated this project by first establishing the isolation strategy of BOECs from blood samples and their characterization to confirm whether these cells are feasible and appropriate for introduction into vessel-chip microfluidic devices. Isolation of BOECs from 60 mL human blood samples was achieved with a previously described protocol.32 Briefly, we isolated and washed the buffy layer twice in PBS following the density gradient centrifugation (Fig. 1A). As soon as we were able to harness the peripheral blood mononuclear cell (PBMNC) population, which typically comprises circulating immune cells and very rare circulating endothelial progenitor cells (<5 cells per mL),24 we expanded these cells in standard culture dishes pre-conditioned with type-1 rat collagen. With media changes every 48 hours, we observed that the non-adherent cells (leukocytes, macrophages, platelets, etc.) were gradually washed away (Fig. 1B). By observing these cells every 24 hours, we found that within 8–10 days after plating, BOECs began appearing and expanded into colonies (BOEC outgrowth, Fig. 1C). Subsequently, within 15–17 days, the BOEC outgrowth colonies reach beyond 1500–2000 cells after which they were transferred to a fresh T25 flask (passage 1, Fig. 1C). Within a week of subculture, BOECs were observed to expand and produce more than a million cells (confluence, Fig. 1C). Once fully confluent, these isolated BOECs displayed the classic endothelial “cobblestone” morphology in vitro (Fig. 1D) which is also exhibited by primary endothelial cells like HUVECs, reinforcing their endothelial identity. To characterize the isolated BOECs further, we measured the expression of common endothelial markers: CD31 or platelet endothelial cell adhesion marker-1 (PECAM-1), CD34 (EPC marker), CD144 (VE-cadherin), and KDR (VEGF-R2).39,40 We also assessed the expression of non-endothelial, leukocyte markers, CD14 and CD45. Analysis using flow cytometry yielded that BOECs did not express these leukocyte markers but had a strong expression of pro-endothelial surface markers, establishing the endothelial identity of these cells (Fig. 1E–J).
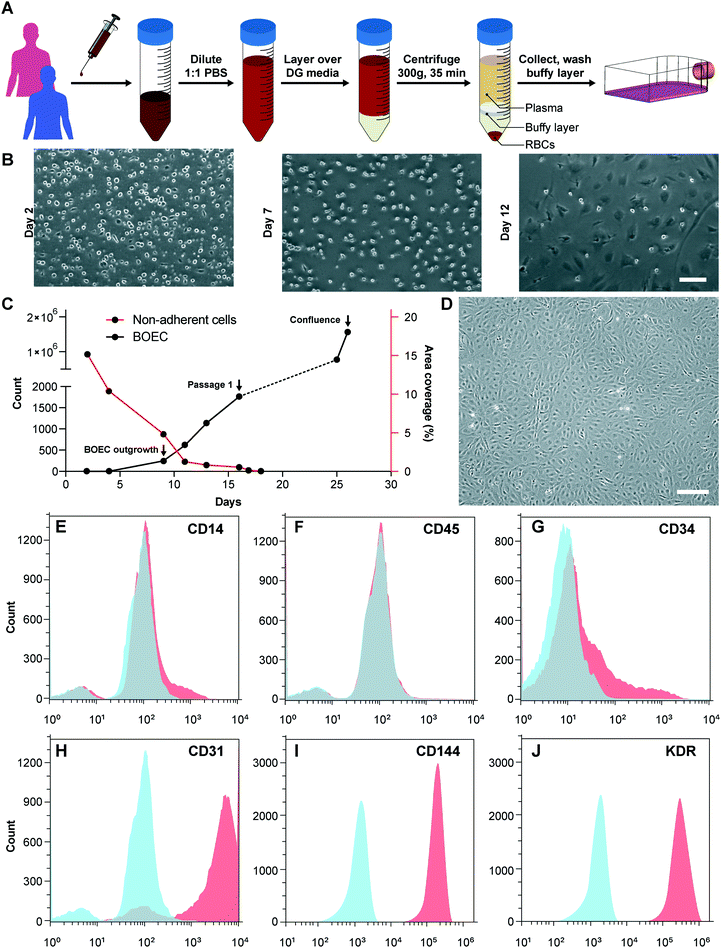 |
| | Fig. 1 Blood outgrowth endothelial cell isolation and characterization. (A) Schematic showing the BOEC isolation process. Whole blood is withdrawn in citrated tubes and diluted with PBS. The diluted blood is layered over density gradient (DG) media and centrifuged at 400g for 35 minutes after which the buffy layer is carefully collected, washed twice with PBS and finally added to culture flasks with growth media. (B) Timelapse images showing the gradual removal of non-adherent leukocytes and platelets from culture flasks seeded with cells isolated from the buffy layer (scale bar: 50 μm). (C) Representative timelapse graph showing the BOEC time of outgrowth colony appearance, subsequent subculture (passage 1) and confluence in a T25 cell culture flask (black line). Dotted line represents the growth of BOECs from passage 1 till they reach approximately 1 million cells. The red line represents the area coverage (%) of all the non-adherent cells (leukocytes, macrophages, platelets, etc.) with time. (D) Bright-field image of cultured BOECs after isolation from whole blood. BOECs exhibit the classic endothelial “cobblestone” morphology (scale bar: 200 μm). Flow cytometry analysis of surface markers, (E) CD14, (F) CD45, (G) CD34, (H) CD31, (I) CD144 and (J) KDR, present on BOECs. Red histograms represent staining with the antibody of interest; blue histograms are the relevant isotype controls. | |
Design and evaluation of the BOEC-laden vessel-on-chip
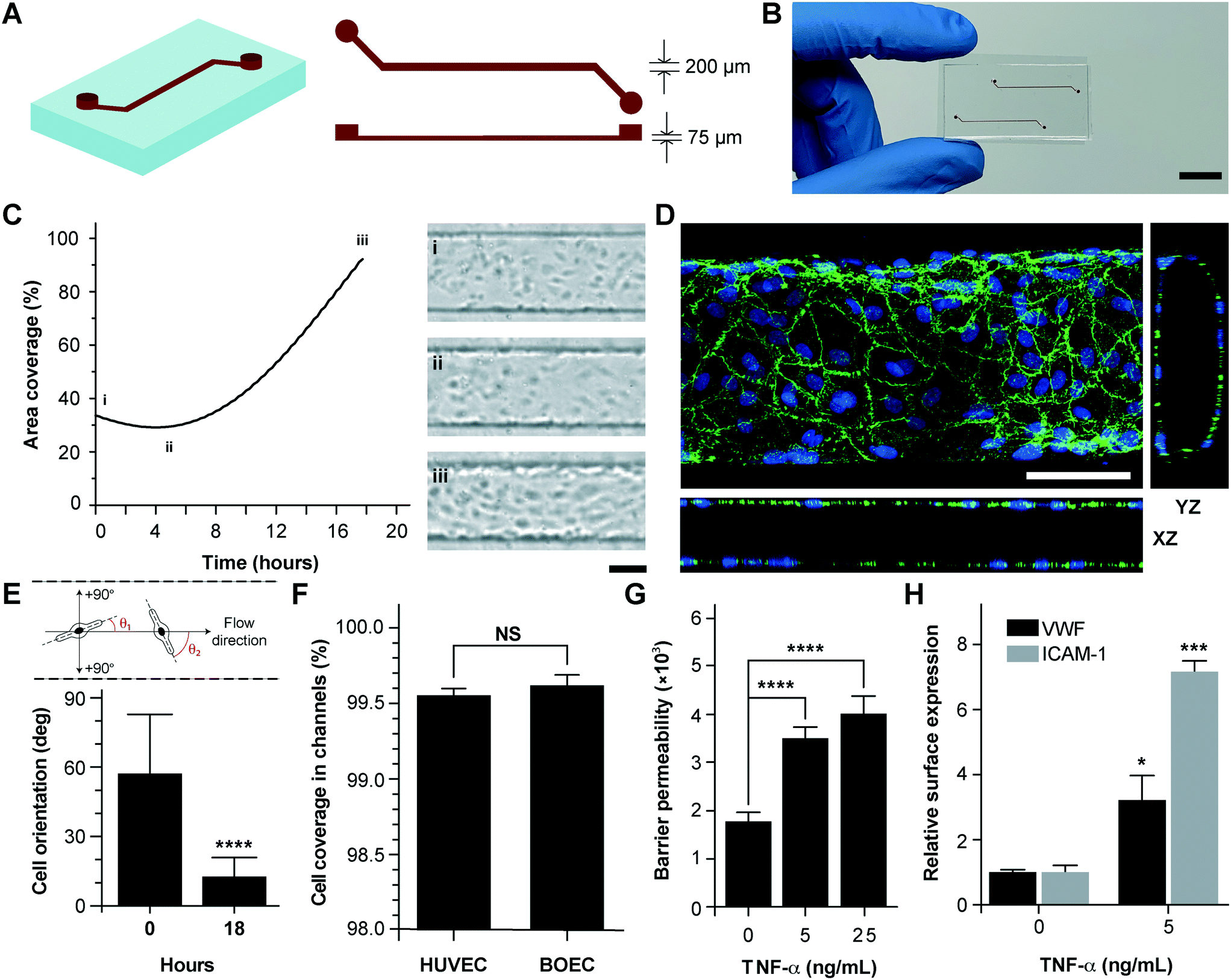
Vessel-chips are increasingly being applied as endothelialized in vitro microfluidic models of vascular research.41 In these devices, HUVECs are the most common primary endothelial cell type that are used because of their easy commercial availability, even though other cell types, such as aortic, microvascular, pulmonary and arterial endothelial cells have also been used. But incorporation of BOECs has not been initiated in such organ-chips. Therefore, we fabricated 200 μm wide, 75 μm high and 2 cm long microfluidic channels with soft lithography to mimic typical arteriolar dimensions; the hydraulic diameter of these channels roughly corresponds to 110 μm, which is the diameter of a typical human arteriole (Fig. 2A).42 Each device consisted of two similar channels parallel to each other (Fig. 2B) so that two measurements may be made consecutively when mounted on a microscope. BOECs isolated from blood samples of healthy human volunteers were introduced into collagen-coated microfluidic channels. We performed bright-field microscopy of the devices within the incubator (see the Materials and methods section) throughout the duration of culture (under flow) and observed that BOECs divided and grew within the vessel-chips (Fig. 2C, Movie S1†). At the end of culture, we observed a confluent endothelial lining of BOECs on all sides of the walls of the device through confocal microscopy, which confirmed that a lumen was formed similar to prior vessel-on-chip models8,11 (Fig. 2D, Movie S2†). We also found that after culturing BOECs in vessel-chips under continuous media perfusion, the cells first adhered to the underlying matrix at no particular preferential orientation, but by the time they reached confluence (18 hours), most of the cells within the device perfectly aligned in the axial direction parallel to the flow (Fig. 2E). This confirmed that BOECs within vessel-chips exhibit sensitivity to applied shear. Quantification of the gaps in the endothelial lumen formed by BOECs showed that they are able to cover more than 99% of the total channel area in the same manner as that typically achieved with commercially-available HUVECs (Fig. 2F). This shows that similar to HUVECs, BOECs are able to maintain their endothelial integrity in vitro with no significant junction gaps. Further, to test the quality of the BOEC-formed endothelial lumen in vitro, we assessed the barrier function of BOECs. After an overnight culture, we treated the confluent BOEC monolayers in transwell plates in the presence or absence of TNF-α (5–25 ng mL−1). After 18 hours of stimulation, we found that the TNF-α treated lumen lost their barrier maintaining capabilities, as shown by the diffusion of the fluorescent FITC-dextran across the cell barrier (Fig. 2G). This result shows that the BOEC functional response to inflammatory stimuli (activation and barrier dysfunction) is similar to that of primary endothelial cells in vitro. To further confirm the onset of vascular dysfunction, we investigated the response of BOECs to TNF-α by measuring the expression of pro-inflammatory surface adhesion markers, ICAM-1 and VWF, which mediate platelet adhesion and blood cell recruitment during thrombosis.43–46 After treatment with TNF-α, we found that BOEC-vessel-chips exhibited an increase in both ICAM-1 and VWF expression relative to the untreated endothelium (Fig. 2H), showing signs of increased endothelial activation and injury. This result further confirms that the BOEC endothelium interacts and responds to external inflammatory chemokine gradients, in a manner that has been observed both in vivo and in several human mature endothelial cells like HUVECs and HMVECs.10
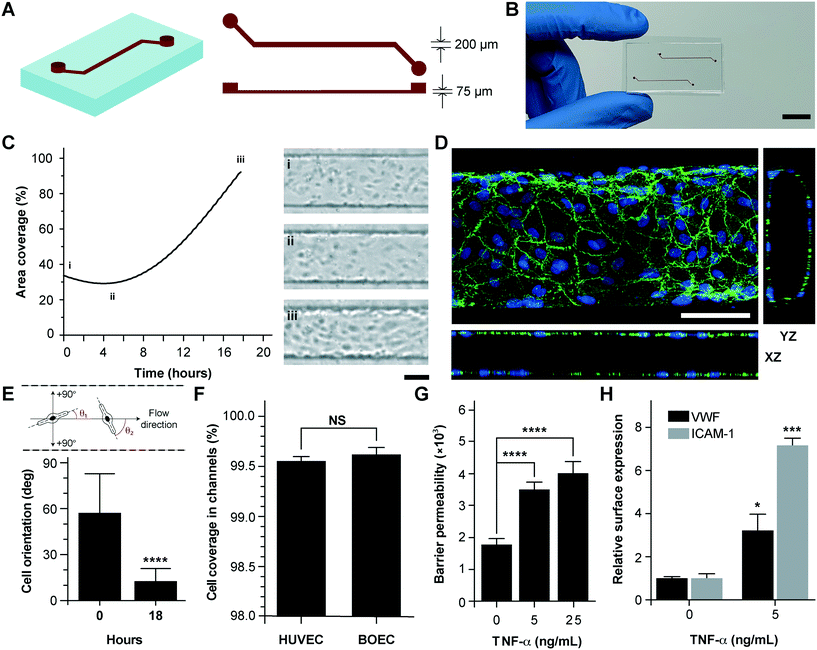 |
| | Fig. 2 Analysis of vessel-chips lined with BOECs. (A) Schematic of the arteriole-sized microchannel (vessel-chip) with an inlet, a 200 μm wide and 75 μm high straight duct, and an outlet connected to the syringe pump to draw blood through the channels. (B) Photographic representation of the vessel-chip made of polydimethylsiloxane (PDMS) containing two independent microchannels on a collagen-coated glass slide (scale bar: 10 mm). (C) Quantification of BOEC growth and spreading in microchannels with time (left); snapshots (right) show BOEC coverage at (i) seeding, (ii) initial attachment and (iii) confluence (scale bar: 100 μm). (D) Confocal micrograph showing a section of the endothelial lumen formed by BOECs in the microchannels. The orthogonal views of the endothelial lumen with the top (‘xy’), front (‘xz’) and side (‘yz’) views validate the complete coverage of BOECs on all four walls of the microchannels (green: VE-cadherin; blue: nuclei; scale bar: 100 μm). (E) Graph showing the cell orientation inside the vessel-chip. Cells are randomly oriented at the time of seeding into vessel-chips. At 18 hours, BOECs align along the flow direction. (F) Quantification of cell coverage of BOEC vs. HUVEC laden microchannels (n = 6). Nearly complete coverage was observed upon reaching confluence. (G) Endothelial barrier permeability of BOECs correlated to the diffusion of fluorescent FITC-dextran (4 kDa) through the transwell plates after treatment with TNF-α. Results demonstrate the disruption of barrier function with increasing TNF-α dosage. (H) Quantification of mean fluorescence intensity of BOECs immunostained for VWF and ICAM-1 after TNF-α treatment, normalized to untreated endothelium. NS: not significant, *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001; n = 3 for all experiments wherever not specified. | |
BOEC-vessel-chips in modeling thromboinflammatory processes
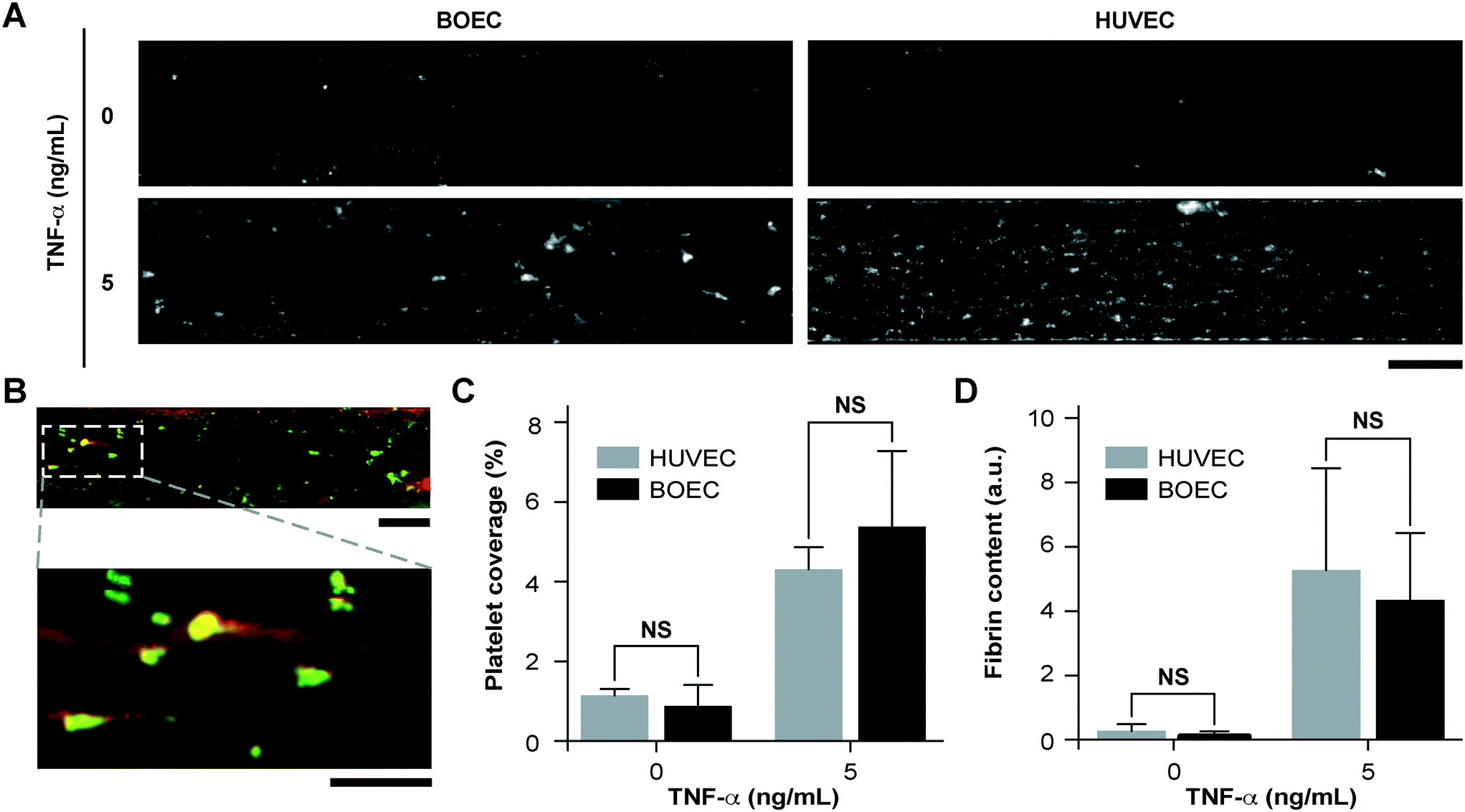
Since vascular activation results in adhesion of blood cells and thrombosis in small blood vessels, we investigated the pro-thrombotic behaviour of BOECs relative to HUVECs in the vessel-chip system. After perfusing recalcified citrated whole blood through the device at an arterial shear rate of 750 s−1 (25 dyn cm−2), we monitored the platelet adhesion to the endothelium over a period of 15 minutes.10 We treated BOEC or HUVEC laden channels with TNF-α and comparatively assessed their ability to promote platelet adhesion to the endothelial lumen, relative to those without treatment. When the endothelial lumen was left untreated and blood was perfused, we did not observe any platelet adhesion on the surface for both BOECs and HUVECs (Fig. 3A) signifying that the BOEC-covered endothelium prevents the blood cells from activating and adhering to the surface or the underlying matrix and behaves like a healthy blood vessel. But in contrast, when whole blood was perfused within vessel-on-chips pre-treated with TNF-α, we observed an increase in the platelet adhesion as well as fibrin formation. This further supports our finding that BOECs respond to the inflammatory cytokines, express adhesive factors on their surface, break their junctions, and provide an activated substrate over which platelets can adhere and initiate thrombus formation (Fig. 3A–D, Movie S3†). These events have also been observed to occur within inflamed microvessels, both in vivo, and in in vitro models utilizing HUVECs.47,48 Moreover, unlike the typical fibrillar pattern of platelet-rich thrombi formed on collagen surfaces, the thrombi formed on the inflamed BOEC endothelium exhibited a distinct “comet” or teardrop-like morphology48 (teardrop with a core and a stretched surrounding shell, Fig. 3B). These patterns have also been observed in other vessel-on-chip models containing HUVECs as well as in in vivo studies.10,49
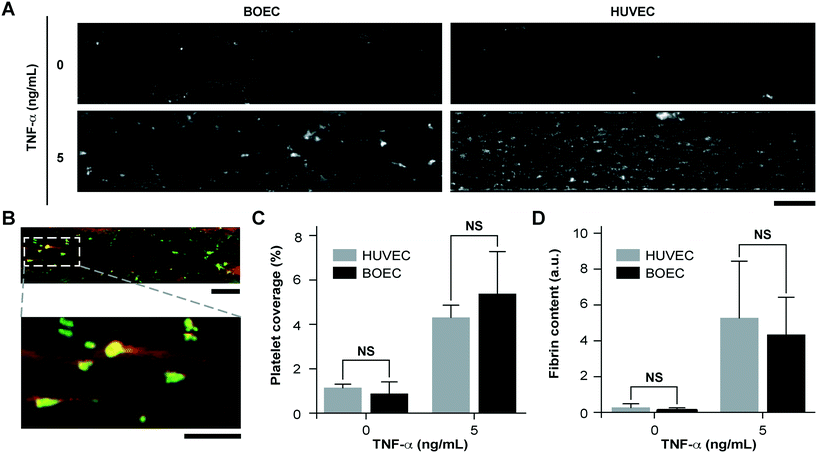 |
| | Fig. 3 Inflammatory and prothrombotic responses of TNF-α stimulated BOEC-vessel-chips. (A) Platelet adhesion micrographs of unstimulated (top) and stimulated (bottom) BOEC (left) and HUVEC (right) vessel-chips after perfusion of recalcified citrated whole blood. Platelet adhesion to the endothelial lumen increases after TNF-α treatment (scale bar: 100 μm). (B) TNF-α treatment of BOEC-vessel-chips produces “comet” shaped platelet aggregates along with fibrin formation in the surroundings (red: fibrin; green: platelets; scale bar (top): 100 μm; scale bar (bottom): 50 μm). (C) Platelet area coverage after a 15 minute perfusion of blood through the vessel-chips lined with BOECs and HUVECs with and without TNF-α treatment. The platelet coverage was not significantly different (p > 0.05) between BOECs and HUVECs for the unstimulated and stimulated endothelium. (D) Fibrin (measured with fluorescence) after blood perfusion through BOEC and HUVEC vessel-chips with and without TNF-α treatment. Fibrin formation was not significantly different (p > 0.05) between BOECs and HUVECs at different TNF-α doses. NS: not significant; n = 4 for all experiments. | |
Type-1 diabetic BOEC-vessel-chips exhibit vascular dysfunction and thrombogenicity
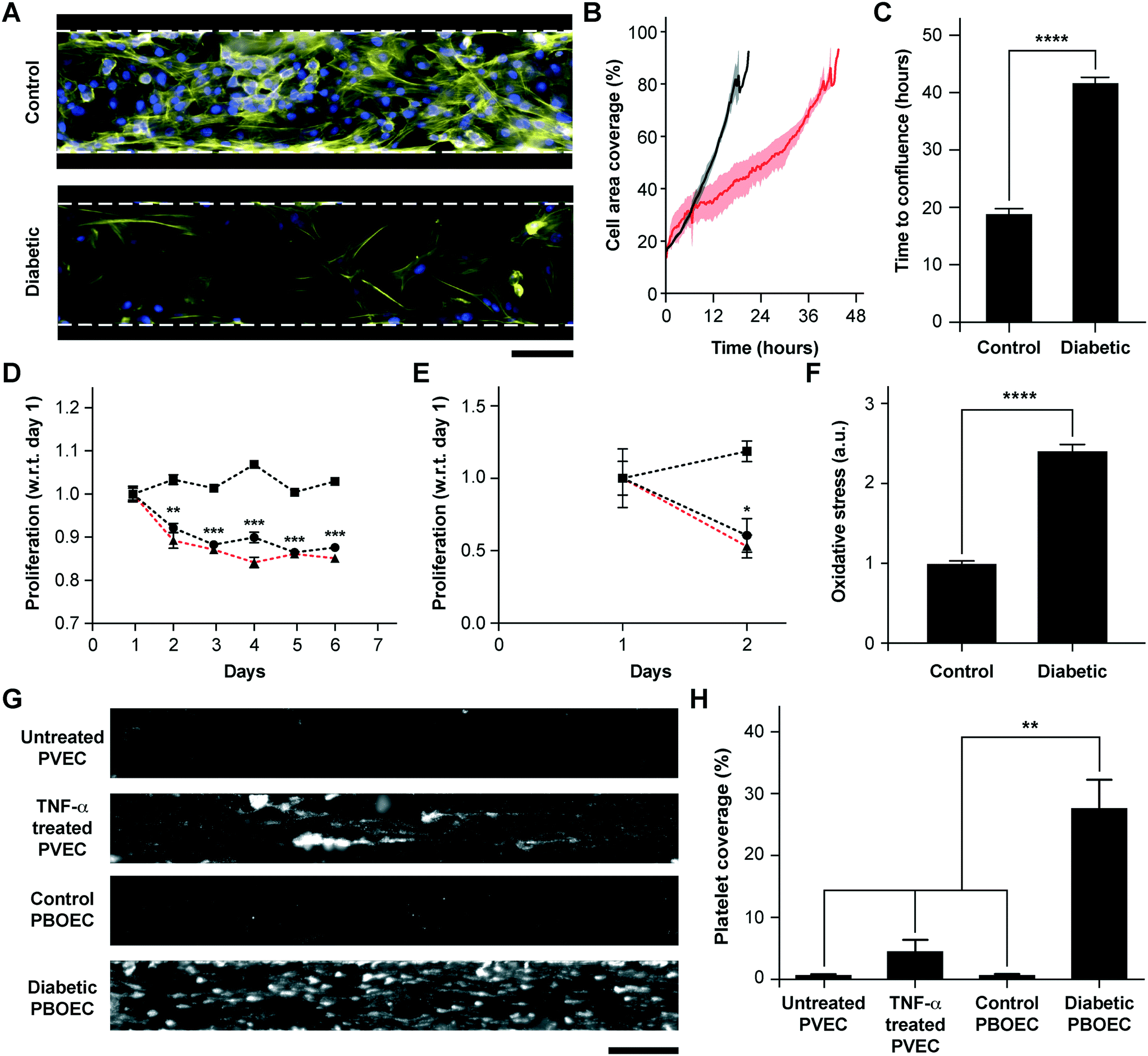
BOECs taken from healthy individuals have several functional aspects identical to HUVECs to model cytokine-stimulated vascular dysfunction and thrombosis. But we hypothesized that BOECs derived from diseased patients may reveal the in vivo disease-specific vascular dysfunction, which is not possible to model through the use of commercially-available cell types, such as HUVECs. As one example, endothelial dysfunction in type 1 diabetes has been linked with increased oxidative stress,50 reduction in endothelial progenitor cell counts,51 a significant decrease in the proliferative ability of circulating endothelial cells52 and increased vascular inflammation in vivo.53–55 Further, endothelial progenitor cells in diabetic patient circulation show a reduction in vasculogenesis and are involved in vessel formation much less than healthy controls.56 Therefore, to test the hypothesis and demonstrate that BOECs derived from diabetic hosts display nearly the same behaviour in vitro as in vivo (for example, resulting in lesser proliferative abilities and elevated thrombogenicity), we obtained fresh blood samples from pig models of type 1 diabetes mellitus and harnessed BOECs (PBOECs) with the same methods used for human blood samples (see the Materials and methods section). We found that diabetic PBOECs had a much slower rate of growth in vessel chips and after 24 hours, the diabetic PBOECs presented irregular gaps in the lumen and had a compromised barrier function, whereas PBOECs from control pigs formed a healthy intact lumen in the same time (Fig. 4A). Also, while control PBOECs were able to form a confluent lumen within 24 hours, the time required by diabetic PBOECs to form an intact lumen was significantly higher under identical culture conditions (Fig. 4B and C). Further, diabetic PBOEC cells showed a reduced proliferation rate compared to control PBOECs when cultured in well plates (Fig. 4D). Interestingly, after culturing PBOECs in the vessel-chip under constant growth media perfusion (1 μL min−1; shear stress: 0.81 dyn cm−2; shear rate: 81 s−1), the reduction in the proliferation rate of diabetic PBOECS was further amplified (Fig. 4E). The rate of proliferation of diabetic PBOECs at the end of two days was nearly half of control cells when measured within the vessel-chip (cultured under flow, Fig. 4E) as compared to nearly 90% when measured in well plates (static culture, Fig. 4C). This suggests that the diabetic vascular function is further compromised when modelled in more physiologically-relevant organ-chips. Interestingly, we were able to observe significantly increased production of reactive oxidative stress in cultured diabetic PBOECs relative to normal PBOECs (Fig. 4F), showing agreement with the clinical findings of increased oxidative stress in type 1 diabetes patients.50,57 Finally, we performed experiments where we prepared vessel-chips from seeding normal porcine primary vein endothelial cells (PVECs) and compared platelet adhesion upon whole blood flow on these chips against the ones made with diabetic BOECs. We found that when PVECs were untreated or treated with TNF-α at a typical dose (10 ng mL−1), platelet adhesion to the endothelium was significantly different from that when diabetic BOECs were used and normal endothelial cells could not exhibit the typical platelet adhesion that is expected when the endothelium is severely dysfunctional in diabetes (Fig. 4G and H). On the other hand, diabetic BOECs did show significantly different and increased platelet adhesion to the endothelium as expected to be seen in some severely diabetic patients. Therefore, the results obtained from the vessel-chip biosystem confirm that vessel-chips made from BOECs taken from diabetic porcine patients reconstitute blood cell–endothelial interactions that are representative of the disease. Notably, these easily-obtained BOECs may potentially serve as a physiologically-relevant source of endothelial cells for in vitro analysis of endothelial dysfunction in type 1 diabetes and potentially, in other vascular pathologies as well.
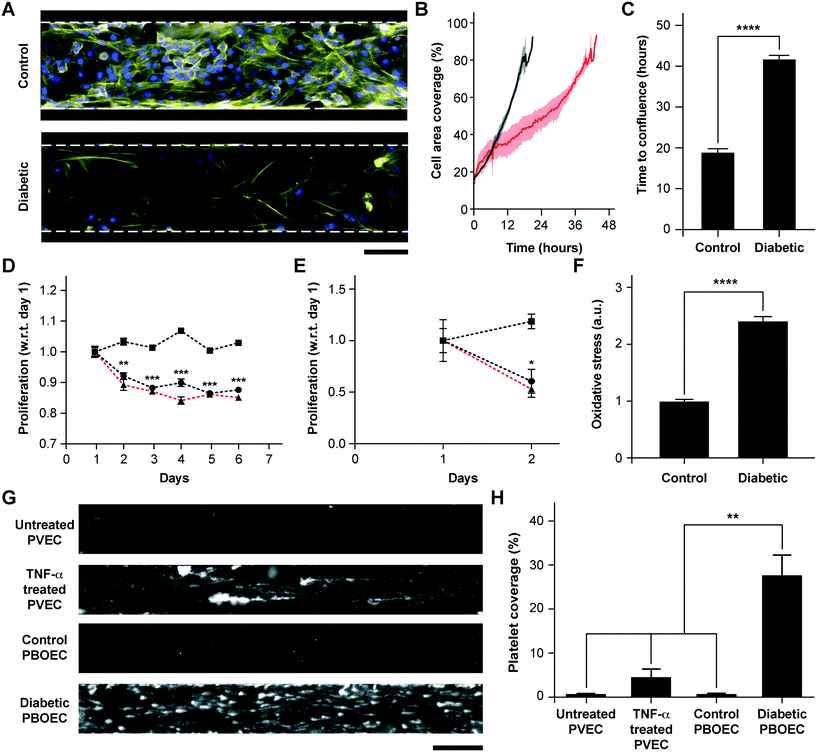 |
| | Fig. 4 Functional analysis of diabetic porcine BOECs (PBOECs) in vessel-chips. (A) Fluorescence micrographs of type 1 diabetic and control PBOECs cultured in vessel-chips for 24 hours (yellow: F-actin; blue: nuclei; scale bar: 100 μm). (B) Graph showing growth of control PBOECs (black) and diabetic PBOECs (red) with time. Solid lines represent mean and the shaded regions represent error (SEM). (C) Average time required for diabetic PBOECs to reach confluence compared to control PBOECs. Time to confluence is defined as the time when cell coverage in the vessel-chip reaches 90%. (D) Proliferation of control (square) and diabetic (circle) PBOECs in transwell plates normalized to respective day 1 values (p-values calculated versus control PBOECs on the corresponding day). Dotted line represents the ratio of proliferation of diabetic to control cells. (E) Proliferation of control (square) and diabetic (circle) PBOECs in the vessel-chip, normalized to respective day 1 values (p-values calculated versus control PBOECs on the corresponding day). Dotted line represents the ratio of proliferation of diabetic to control cells. (F) Oxidative stress measured via flow cytometry. The fluorescence reported has been normalized to the total number of cells analyzed. (G) Representative platelet adhesion micrographs of porcine primary vein endothelial cell (PVEC), TNF-α stimulated PVEC, control PBOEC and diabetic PBOEC vessel-chip devices after perfusion of whole blood (scale bar: 100 μm). (H) Platelet area coverage after 15 minute perfusion of blood in the vessel-chips. *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001; n = 3 for all experiments. | |
Conclusions
Our results show that progenitor endothelial cells circulating in blood or BOECs may serve as a primary endothelial cell source to model vascular pathology and translational outcomes through organ-on-a-chip technology. Our demonstration that BOECs can be easily isolated from patient whole blood and can be used to develop 3D lumens within vessel-chips is the first proof-of-concept that this tool offers disease-specific evaluation of thromboinflammation along with prospects of patient-specific analysis, which is not possible by the use of generic cell lines in current in vitro microfluidic platforms. As confirmed by our results, BOECs exhibit a physiologically-relevant functional response to cytokine-induced inflammation and mimic thrombosis and platelet hyperactivity similar to HUVECs. Therefore, they could be utilized as a source of endothelial cells for designing microfluidic models of thrombosis and other vascular diseases (Fig. 5A). Importantly, when we took blood samples from diabetic porcine patients, it revealed that unlike the existing vessel-chip or organ-chip models that utilize healthy primary cells that are exogenously stimulated through inflammatory cytokines, BOECs of diabetic patients are able to mimic the vascular dysfunction and thromboinflammation in vitro without any such stimulation. This novel application of BOECs in microfluidic platforms further strengthens our hypothesis that BOECs may be used for developing personalized vascular organ-chip models (Fig. 5B). This device provides more physiologically-relevant readouts by incorporating microphysiological cues that exist in vivo; for example, the exacerbation of endothelial proliferation under shear conditions from type 1 diabetic samples can be witnessed through the vessel-chip compared to that with no shear conditions. Such data cannot be obtained using 2D culture transwell plates or flow chambers. This methodology may find broader use; for example, we expect microfluidic experts who have platforms to investigate angiogenesis to explore the ability of BOECs to form capillary vessels de novo, thus opening up uncharted opportunities in tissue engineering applications. However, even though we have seen that BOECs exhibit a typical morphology, growth pattern and adhesion markers as seen in mature endothelial cells, their exact biology and phenotype still need to be identified through detailed transcriptomic profiling. To what extent can these cells be representative of the endothelial function of a disease or a patient is still relatively unknown. The faithfulness of this representation and even the amount of time BOECs spend in circulation before extraction may further vary between diseases and patients. Several other controversies still already exist in terms of the definition and characteristics of these cells, and their distinction from other progenitor cell sources is still a matter of debate.14,58–60 Although we have focused on the expression of two prominent surface adhesion ligands (ICAM-1 and VWF), additional endothelial markers might be involved in vascular-blood signalling, and a more in-depth characterization of BOECs from several disease-types and patients may be required before they may be prescribed for broader use in research and translational applications. But our results suggest that BOECs offer an alternative to other primary endothelial cells, which have been applied in personalized modelling platforms.3,61,62 Notably, BOEC isolation from whole blood is straightforward and relatively less time intensive as outgrowth colonies appear within weeks after isolation. In addition to that, studies with vascular disease patients show increased amounts of EPCs in their circulation, making them more suitable for application in organ-chips, lab-on-chips and other in vitro preclinical models.
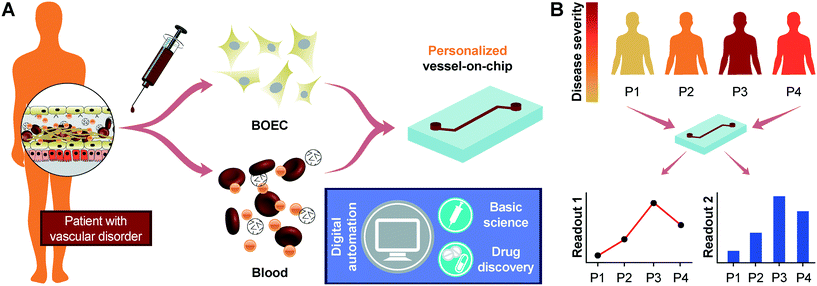 |
| | Fig. 5 Future potential of blood-derived cells to engineer “personalized” organ-chip models. (A) Schematic showing the potential of patient derived BOECs and autologous blood to engineer patient-specific vessel models that are more representative of patient distress, allow functional evaluation of disease by incorporating digital automation and overcome the predictive power of existing models. This may bridge gaps in the basic understanding of disease, ultimately leading to improved pharmaceutical research. (B) The patient-derived vessel models could also potentially provide patient-to-patient differences in disease severity, making personalized therapeutics more effective. | |
Author contributions
T. M. and A. J. designed the experiments, analysed the data and wrote the manuscript with feedback from other authors. T. M. fabricated the devices, isolated and cultured the cells and performed the experiments. S. H. T. and T. W. H. provided the blood samples from porcine models and J. M. F. provided the blood outgrowth endothelial cells. K. A. S. and A. K. G. helped set-up and analyse the cell proliferation experiments. K. A. S. and J. M. F. performed flow cytometry. N. K. R. P. helped with the initial device design.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Ms. Jo Ann Culpepper at Texas A&M University for managing the phlebotomy of healthy individuals. We thank Dr. Rola Barhoumi Mouneimne at the Texas A&M Image Analysis Laboratory for assisting with the confocal imaging. We thank Mr. David Luna and Mr. Justin Bui for fabricating the master molds with optical lithography of SU8 resist at the AggieFab Nanofabrication Facility at Texas A&M University. Research reported in this publication was supported by the NIBIB of NIH under Award Number R21EB025945 (AJ) and DP2EB026265 (AKG), the Texas A&M Triads for Transformation (T3) program by President's Excellence Fund (AJ and AKG), NEI of NIH R01EY024624 (TWH), Texas A&M Engineering Experimentation Station (TEES), and Texas A&M University.
Notes and references
- R. Gulati and R. D. Simari, Dis. Models Mech., 2009, 2, 130–137 CrossRef PubMed.
- M. A. Creager, Circulation, 2016, 133, 2593–2598 CrossRef PubMed.
- A. van den Berg, C. L. Mummery, R. Passier and A. D. van der Meer, Lab Chip, 2019, 19, 198–205 RSC.
- J. F. Dekkers, C. L. Wiegerinck, H. R. de Jonge, I. Bronsveld, H. M. Janssens, K. M. de Winter-de Groot, A. M. Brandsma, N. W. M. de Jong, M. J. C. Bijvelds, B. J. Scholte, E. E. S. Nieuwenhuis, S. van den Brink, H. Clevers, C. K. van der Ent, S. Middendorp and J. M. Beekman, Nat. Med., 2013, 19, 939–945 CrossRef CAS PubMed.
- A. Jain, R. Barrile, A. D. van der Meer, A. Mammoto, T. Mammoto, K. De Ceunynck, O. Aisiku, M. A. Otieno, C. S. Louden, G. A. Hamilton, R. Flaumenhaft and D. E. Ingber, Clin. Pharmacol. Ther., 2018, 103, 332–340 CrossRef CAS PubMed.
- D. E. Ingber, Development, 2018, 145, dev156125 CrossRef PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- Y. Sakurai, E. T. Hardy, B. Ahn, R. Tran, M. E. Fay, J. C. Ciciliano, R. G. Mannino, D. R. Myers, Y. Qiu, M. A. Carden, W. H. Baldwin, S. L. Meeks, G. E. Gilbert, S. M. Jobe and W. A. Lam, Nat. Commun., 2018, 9, 509 CrossRef PubMed.
- M. Tsai, A. Kita, J. Leach, R. Rounsevell, J. N. Huang, J. Moake, R. E. Ware, D. A. Fletcher and W. A. Lam, J. Clin. Invest., 2012, 122, 408–418 CrossRef CAS PubMed.
- A. Jain, A. D. van der Meer, A. L. Papa, R. Barrile, A. Lai, B. L. Schlechter, M. A. Otieno, C. S. Louden, G. A. Hamilton, A. D. Michelson, A. L. Frelinger, 3rd and D. E. Ingber, Biomed. Microdevices, 2016, 18, 73 CrossRef PubMed.
- P. N. Ingram, L. E. Hind, J. A. Jiminez-Torres, A. Huttenlocher and D. J. Beebe, Adv. Healthcare Mater., 2018, 7 DOI:10.1002/adhm.201700497.
- S. Sances, R. Ho, G. Vatine, D. West, A. Laperle, A. Meyer, M. Godoy, P. S. Kay, B. Mandefro, S. Hatata, C. Hinojosa, N. Wen, D. Sareen, G. A. Hamilton and C. N. Svendsen, Stem Cell Rep., 2018, 10, 1222–1236 CrossRef CAS PubMed.
- Y. Lin, D. J. Weisdorf, A. Solovey and R. P. Hebbel, J. Clin. Invest., 2000, 105, 71–77 CrossRef CAS PubMed.
- R. P. Hebbel, J. Clin. Invest., 2017, 127, 1613–1615 CrossRef PubMed.
- M. H. Strijbos, P. P. Landburg, E. Nur, T. Teerlink, F. W. Leebeek, A. W. Rijneveld, B. J. Biemond, S. Sleijfer, J. W. Gratama, A. J. Duits, J. J. Schnog and C. S. Group, Blood Cells, Mol., Dis., 2009, 43, 63–67 CrossRef CAS PubMed.
- A. Solovey, Y. Lin, P. Browne, S. Choong, E. Wayner and R. P. Hebbel, N. Engl. J. Med., 1997, 337, 1584–1590 CrossRef CAS PubMed.
- J. W. Wang, E. A. Bouwens, M. C. Pintao, J. Voorberg, H. Safdar, K. M. Valentijn, H. C. de Boer, K. Mertens, P. H. Reitsma and J. Eikenboom, Blood, 2013, 121, 2762–2772 CrossRef CAS PubMed.
- R. D. Starke, K. E. Paschalaki, C. E. Dyer, K. J. Harrison-Lavoie, J. A. Cutler, T. A. McKinnon, C. M. Millar, D. F. Cutler, M. A. Laffan and A. M. Randi, Blood, 2013, 121, 2773–2784 CrossRef CAS PubMed.
- J. Martin-Ramirez, M. G. Kok, M. Hofman, R. Bierings, E. E. Creemers, J. C. Meijers, J. Voorberg and S. J. Pinto-Sietsma, PLoS One, 2014, 9, e99890 CrossRef PubMed.
- L. Chang Milbauer, P. Wei, J. Enenstein, A. Jiang, C. A. Hillery, J. P. Scott, S. C. Nelson, V. Bodempudi, J. N. Topper, R. B. Yang, B. Hirsch, W. Pan and R. P. Hebbel, Blood, 2008, 111, 3872–3879 CrossRef PubMed.
- T. M. Sakamoto, C. Lanaro, M. C. Ozelo, V. T. Garrido, S. T. Olalla-Saad, N. Conran and F. F. Costa, Microvasc. Res., 2013, 90, 173–179 CrossRef CAS PubMed.
- P. Quaranta, S. Antonini, S. Spiga, B. Mazzanti, M. Curcio, G. Mulas, M. Diana, P. Marzola, F. Mosca and B. Longoni, PLoS One, 2014, 9, e94783 CrossRef PubMed.
- J. A. Mund, D. A. Ingram, M. C. Yoder and J. Case, Cytotherapy, 2009, 11, 103–113 CrossRef PubMed.
- G. Ecklu-Mensah, R. W. Olsen, A. Bengtsson, M. F. Ofori, L. Hviid, A. T. R. Jensen and Y. Adams, PLoS One, 2018, 13, e0204177 CrossRef PubMed.
- J. D. Stroncek, B. S. Grant, M. A. Brown, T. J. Povsic, G. A. Truskey and W. M. Reichert, Tissue Eng., Part A, 2009, 15, 3473–3486 CrossRef CAS PubMed.
- S. F. Lam, V. S. Shirure, Y. E. Chu, A. G. Soetikno and S. C. George, PLoS One, 2018, 13, e0209574 CrossRef CAS PubMed.
- M. Moya, D. Tran and S. C. George, Stem Cell Res. Ther., 2013, 4(Suppl 1), S15 CrossRef PubMed.
- M. L. Moya, Y. H. Hsu, A. P. Lee, C. C. Hughes and S. C. George, Tissue Eng., Part C, 2013, 19, 730–737 CrossRef CAS PubMed.
- D. R. Myers, Y. Sakurai, R. Tran, B. Ahn, E. T. Hardy, R. Mannino, A. Kita, M. Tsai and W. A. Lam, J. Visualized Exp., 2012, 3958, DOI:10.3791/3958.
- N. V. Menon, H. M. Tay, S. N. Wee, K. H. H. Li and H. W. Hou, Lab Chip, 2017, 17, 2960–2968 RSC.
- M. Cattaneo, C. P. Hayward, K. A. Moffat, M. T. Pugliano, Y. Liu and A. D. Michelson, J. Thromb. Haemostasis, 2009, 7, 1029 CrossRef CAS PubMed.
- M. L. Ormiston, M. R. Toshner, F. N. Kiskin, C. J. Huang, E. Groves, N. W. Morrell and A. A. Rana, J. Visualized Exp., 2015, e53384, DOI:10.3791/53384.
- J. Martin-Ramirez, M. Hofman, M. van den Biggelaar, R. P. Hebbel and J. Voorberg, Nat. Protoc., 2012, 7, 1709–1715 CrossRef CAS PubMed.
- T. W. Hein, L. B. Potts, W. Xu, J. Z. Yuen and L. Kuo, Invest. Ophthalmol. Visual Sci., 2012, 53, 7943–7949 CrossRef CAS PubMed.
- T. W. Hein, W. Xu, X. Xu and L. Kuo, Invest. Ophthalmol. Visual Sci., 2016, 57, 4333–4340 CrossRef CAS PubMed.
- V. Aragon-Sanabria, S. E. Pohler, V. J. Eswar, M. Bierowski, E. W. Gomez and C. Dong, Sci. Rep., 2017, 7, 45835 CrossRef CAS PubMed.
- T. G. Papaioannou and C. Stefanadis, Hellenic J. Cardiol., 2005, 46, 9–15 Search PubMed.
- A. Jain, A. Graveline, A. Waterhouse, A. Vernet, R. Flaumenhaft and D. E. Ingber, Nat. Commun., 2016, 7, 10176 CrossRef CAS PubMed.
- S. Fuchs, E. Dohle, M. Kolbe and C. J. Kirkpatrick, Adv. Biochem. Eng./Biotechnol., 2010, 123, 201–217 CAS.
- O. V. Halaidych, C. Freund, F. van den Hil, D. C. F. Salvatori, M. Riminucci, C. L. Mummery and V. V. Orlova, Stem Cell Rep., 2018, 10, 1642–1656 CrossRef CAS PubMed.
- K. Gold, A. K. Gaharwar and A. Jain, Biomaterials, 2019, 196, 2–17 CrossRef CAS PubMed.
-
A. J. Pappano and W. G. Wier, Cardiovascular physiology: mosby physiology monograph series, Elsevier Health Sciences, 2018 Search PubMed.
- A. Sagripanti and A. Carpi, Biomed. Pharmacother., 2000, 54, 107–111 CrossRef CAS.
- W. C. Aird, Hamostaseologie, 2015, 35, 11–16 CrossRef CAS PubMed.
- W. Hartwig, J. Werner, A. L. Warshaw, B. Antoniu, C. Fernandez-del Castillo, M. M. Gebhard, W. Uhl and M. W. Buchler, Am. J. Physiol., 2004, 287, G1194–G1199 CAS.
- A. K. Bhunia, T. Arai, G. Bulkley and S. Chatterjee, J. Biol. Chem., 1998, 273, 34349–34357 CrossRef CAS PubMed.
- S. Falati, P. Gross, G. Merrill-Skoloff, B. C. Furie and B. Furie, Nat. Med., 2002, 8, 1175–1181 CrossRef CAS PubMed.
- T. J. Stalker, J. D. Welsh, M. Tomaiuolo, J. Wu, T. V. Colace, S. L. Diamond and L. F. Brass, Blood, 2014, 124, 1824–1831 CrossRef CAS PubMed.
- Y. Qiu, B. Ahn, Y. Sakurai, C. E. Hansen, R. Tran, P. N. Mimche, R. G. Mannino, J. C. Ciciliano, T. J. Lamb, C. H. Joiner, S. F. Ofori-Acquah and W. A. Lam, Nat. Biomed. Eng., 2018, 2, 453–463 CrossRef CAS PubMed.
- V. Altabas, Internet J. Endocrinol., 2015, 2015, 848272 CAS.
- C. J. Loomans, E. J. de Koning, F. J. Staal, M. B. Rookmaaker, C. Verseyden, H. C. de Boer, M. C. Verhaar, B. Braam, T. J. Rabelink and A. J. van Zonneveld, Diabetes, 2004, 53, 195–199 CrossRef CAS PubMed.
- Y. H. Chen, S. J. Lin, F. Y. Lin, T. C. Wu, C. R. Tsao, P. H. Huang, P. L. Liu, Y. L. Chen and J. W. Chen, Diabetes, 2007, 56, 1559–1568 CrossRef CAS PubMed.
- A. C. Roberts and K. E. Porter, Diabetes Vasc. Dis. Res., 2013, 10, 472–482 CrossRef PubMed.
- A. M. Ladeia, R. R. Sampaio, M. C. Hita and L. F. Adan, World J. Diabetes, 2014, 5, 601–605 CrossRef PubMed.
- I. G. Joshua, Q. Zhang, J. C. Falcone, A. P. Bratcher, W. E. Rodriguez and S. C. Tyagi, J. Cell. Biochem., 2005, 96, 1149–1156 CrossRef CAS PubMed.
- O. M. Tepper, R. D. Galiano, J. M. Capla, C. Kalka, P. J. Gagne, G. R. Jacobowitz, J. P. Levine and G. C. Gurtner, Circulation, 2002, 106, 2781–2786 CrossRef PubMed.
- M. E. Carr, J. Diabetes Complicat., 2001, 15, 44–54 CrossRef CAS.
- K. K. Hirschi, D. A. Ingram and M. C. Yoder, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 1584–1595 CrossRef CAS PubMed.
- R. J. Medina, C. L. O'Neill, M. Sweeney, J. Guduric-Fuchs, T. A. Gardiner, D. A. Simpson and A. W. Stitt, BMC Med. Genomics, 2010, 3, 18 CrossRef PubMed.
- D. A. Ingram, L. E. Mead, H. Tanaka, V. Meade, A. Fenoglio, K. Mortell, K. Pollok, M. J. Ferkowicz, D. Gilley and M. C. Yoder, Blood, 2004, 104, 2752–2760 CrossRef CAS PubMed.
- R. Samuel, D. G. Duda, D. Fukumura and R. K. Jain, Sci. Transl. Med., 2015, 7, 309rv306 Search PubMed.
- A. Cochrane, H. J. Albers, R. Passier, C. L. Mummery, A. van den Berg, V. V. Orlova and A. D. van der Meer, Adv. Drug Delivery Rev., 2018 DOI:10.1016/j.addr.2018.06.007.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00469f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Kanwar Abhay
Singh
a,
Navaneeth K.
R. Pandian
a,
Shu-Huai
Tsai
b,
Travis W.
Hein
a,
Kanwar Abhay
Singh
a,
Navaneeth K.
R. Pandian
a,
Shu-Huai
Tsai
b,
Travis W.
Hein