
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum(IV) azido complexes undergo copper-free click reactions with alkynes†
Nicola J.
Farrer
 *a,
Gitanjali
Sharma
a,
Rachel
Sayers
a,
Evyenia
Shaili
b and
Peter J.
Sadler
b
*a,
Gitanjali
Sharma
a,
Rachel
Sayers
a,
Evyenia
Shaili
b and
Peter J.
Sadler
b
aChemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: Nicola.Farrer@chem.ox.ac.uk; Tel: +44 (0)1865 285155
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
First published on 26th February 2018
Abstract
We report our investigations into the first examples of copper-free 1,3-dipolar cycloaddition (click) reactions of electrophiles with a PtIV azido complex. The Pt-IV azido complex trans, trans, trans-[PtIV(py)2(N3)2(OH)2] (1) was reactive towards dimethyl acetylenedicarboxylate (DMAD) (2), diethyl acetylenedicarboxylate DEACD (3), N-[(1R,8S,9s)-bicyclo[6.1.0]non-4-yn-9-ylmethyloxycarbonyl]-1,8-diamino-3,6-dioxaoctane (BCN) (11) and dibenzocyclooctyne-amine (DBCO) (12) resulting in formation of the corresponding mono (a) and bis-substituted (b) complexes. Complexes of 2 undergo further reactions between the Pt centre and the carbonyl group to form 2a′ and 2b′. This is not seen for the products of the corresponding PtII azido complex trans-[Pt(py)2(N3)2] with acetylene 2. Novel complexes 2a′, 2b′, 11a and 11b have been characterised by multinuclear NMR, IR and UV-vis spectroscopy and ESI-MS. These reactions represent new synthetic routes to novel Pt(IV) complexes.
Introduction
Copper-free, azide–alkyne cycloaddition (“click”) reactions typically take place under mild conditions1,2 and provide a tantalising route to modify ligands which are coordinated to a metal centre. Since the conditions are well-tolerated by most functional groups, the reaction can be used to introduce sensitive functionality to complexes at a late stage. By avoiding the use of cytotoxic copper salts it is particularly suitable for complexes with eventual biological applications.3 Metal azido complexes have a rich cycloaddition chemistry.4–6 A range of d-block azido complexes, including those of Mn(I),7,8 Fe(III),9 Pd(II),10 Pt(II),10–15 Rh(III)16 and Au(I),17 have been reported to undergo copper-free 1,3-dipolar cycloaddition or “click” reactions with carbon–carbon and carbon-heteroatom functional groups such as alkynes, isocyanides, isonitriles, nitriles, carbon disulphides and isothiocyanates. Electron-deficient alkynes such as dimethyl acetylenedicarboxylate (DMAD) and diethyl acetylenedicarboxylate (DEACD) are relatively reactive: Mo(II),16,18 Co(III),19 Fe(III)19,20 Ru(II),20–25 Pd(II)26–28 and Ta29 azido complexes all react with DMAD. Strain-promoted azide–alkyne cycloadditions (SPAAC)30,31 are also an effective method for derivatising azido complexes.32 Click reactions between metal azides and alkynes have been used to form Pt(II) heterometallic arrays with Au(I)15 or Re/Rh;33 in the development of new catalysts (Ru)21 and metalloenzyme inhibitors (Co(III), Fe(III), Ni(II)),19 and to synthesise peptide bioconjugates (Au(I)) targeted to mitochondria.3 Azolato-bridged platinum complexes show promising anti-cancer activity,34 and 1,2,3-triazole ligands themselves have a wide-range of potential applications including in biomedical35 and materials chemistry.36Our interest in these reactions stems from our investigations of photoactivatable platinum(IV) azido anti-cancer complexes such as trans,trans,trans-[Pt(N3)2(OH)2(py)2] (1, Scheme 1). Complex 1 is inert in the absence of light, but shows potent cytotoxicity towards cancer cell lines upon irradiation with visible light.37 The mechanism of cell death may be due to the formation of a number of different species including azido radicals, nitrenes and singlet oxygen,38,39 and it is not clear whether both azido groups are necessary for the photocytotoxic effect. The synthetic route to these complexes involves oxidation from PtII to PtIV with H2O2, a step which is incompatible with a number of sensitive functional groups. The capacity for derivatising one (or both) azido groups on a PtIV centre through click chemistry therefore provides a route to a wide array of functionality, and the reaction itself is biologically compatible. It can be used to produce PtIV mono azido complexes, a relatively unexplored class of compounds. Whilst there are reports of PtII azido complexes undergoing click reactions, there are none for PtIV systems, which are anticipated to react significantly more slowly.
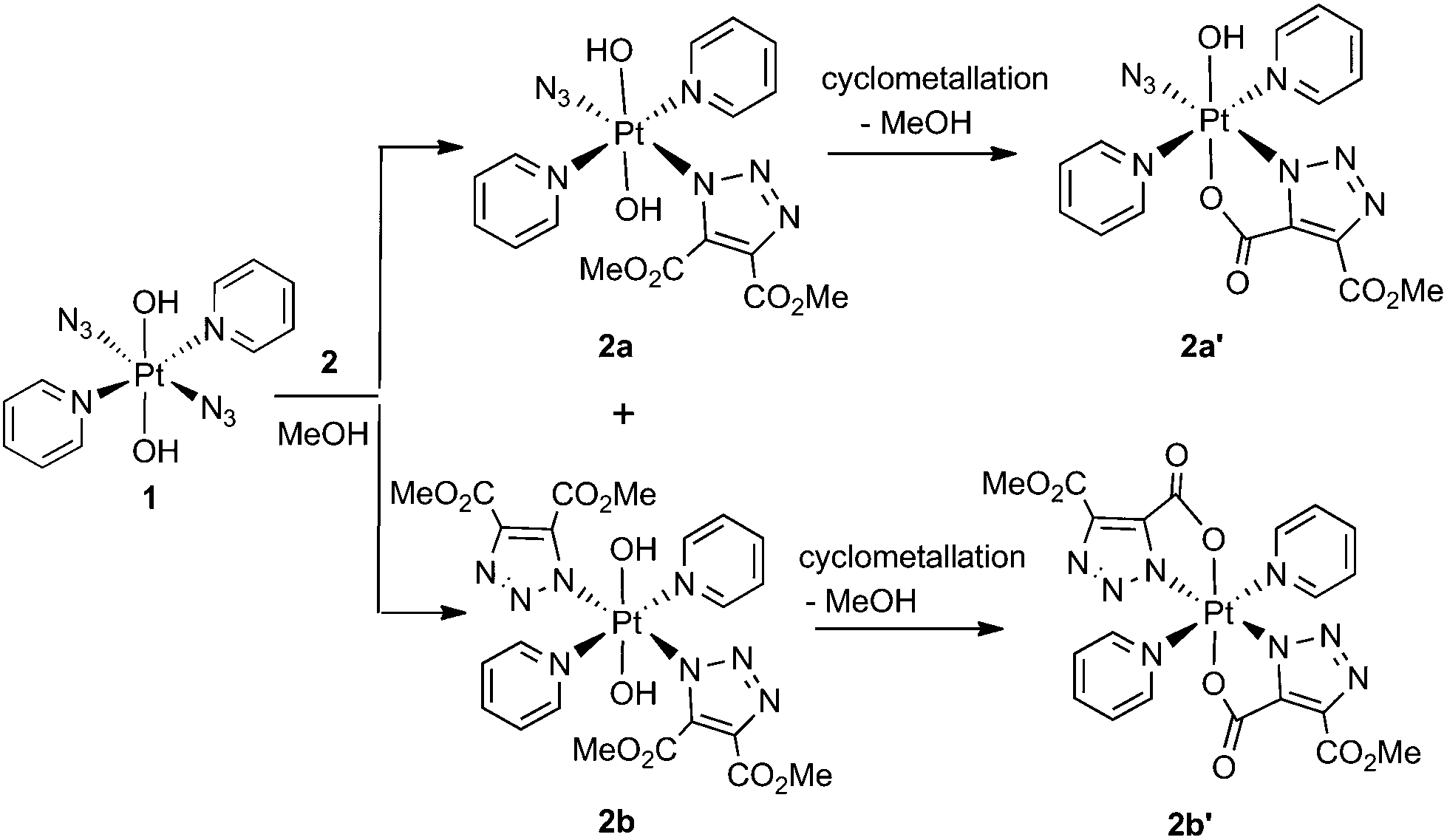 | ||
| Scheme 1 Reaction of 1 with DMAD (2). | ||
We report the results of our investigation of the reactions of trans-[PtII(N3)2(py)2] and trans,trans,trans-[PtIV(N3)2(OH)2(py)2] (1) with a range of internal and terminal alkynes (Fig. 1) including (to the best of our knowledge) the first cycloaddition reactions of a PtIV azido complex.
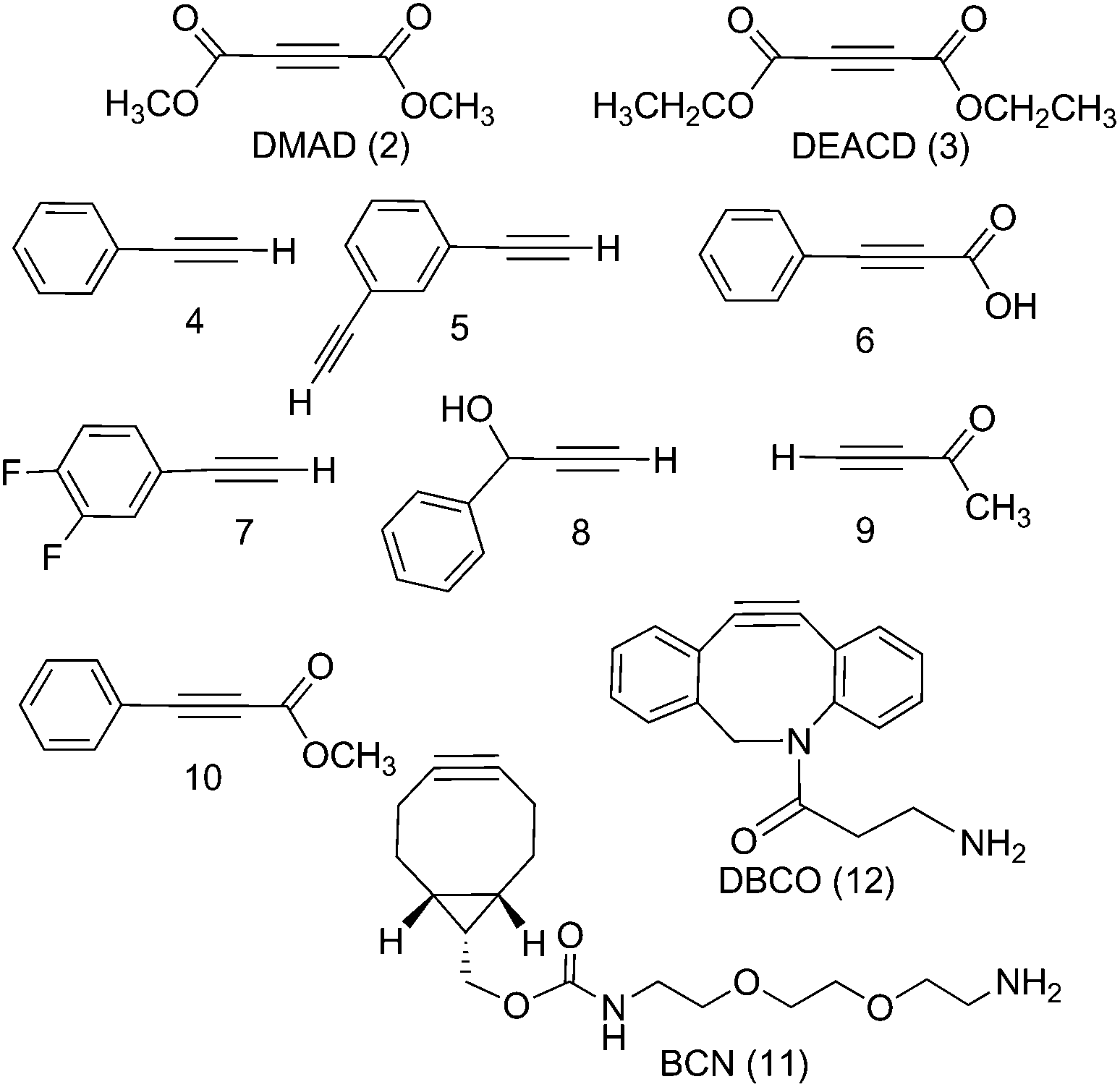 | ||
| Fig. 1 Commercially available alkynes investigated for their reactivity towards trans,trans,trans-[Pt(N3)2(OH)2(py)2] (1): DMAD (2), DEACD (3), N-[(1R,8S,9s)-bicyclo[6.1.0]non-4-yn-9-ylmethyloxycarbonyl]-1,8-diamino-3,6-dioxaoctane and BCN (11) dibenzocyclooctyne-amine, DBCO (12) were reactive, whereas phenylacetylene (4); 1,3-diethynylbenzene (5); phenylpropiolic acid (6); 3,4-difluorophenylacetylene (7) 1-phenyl-2-propyn-1-ol (8); 3-butyn-2-one (9) and 4-phenyl-3-butyne-2-one (10) were not. | ||
Results and discussion
We have previously reported a number of cis- and trans-PtIV azido complexes of the form [Pt(N3)2(R)2(amine1)(amine2)2] (where R = OH, OAc);40–42 complex 1 was chosen for cycloaddition investigations since it is readily synthesisable on a large scale, displays good solubility in water and a range of organic solvents and possesses convenient NMR spectroscopic handles. Initial experiments with the PtII precursor complex trans-[Pt(py)2(N3)2] indicated reactivity towards DMAD and DEACD which encouraged us to investigate the reactivity of complex 1.Preparation of the PtII and PtIV azido complexes
The PtII and PtIV azido complexes were synthesised as previously described.37 All reactions and manipulations were carried out so as to avoid unneccessary exposure to light.Reactivity of trans-[PtII(N3)(py)2] and 1 towards alkynes
PtII azide complexes are known to adopt various bonding modes and may also undergo a range of reactions as an alternative to cycloaddition. Ligand substitution – for example, substitution of an azide for an alkyne – instead of the anticipated cycloaddition has been reported for the complex cis-[Pt(N3)2(PPh3)2] under microwave irradiation with HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR (R = Ph, p-MeC6H4) at 100 °C.43 In the reaction of [Pd(PPh3)2(N3)2] with DMAD (the PtII derivative has not been investigated), a triazole-bridged dimer was obtained, which was recrystallised in the presence of PPh3 to give the bis substituted species [Pd(PPh3)2(triazole)2].26 In light of this product diversity, we considered a number of possible products in our investigations.
CR (R = Ph, p-MeC6H4) at 100 °C.43 In the reaction of [Pd(PPh3)2(N3)2] with DMAD (the PtII derivative has not been investigated), a triazole-bridged dimer was obtained, which was recrystallised in the presence of PPh3 to give the bis substituted species [Pd(PPh3)2(triazole)2].26 In light of this product diversity, we considered a number of possible products in our investigations.
Furthermore, for several of the alkynes further reactivity following triazole formation was possible, due to the presence of reactive ligand-based groups. We also considered the possibility of N1–N2 rearrangement of the resulting triazole: several ruthenium azides have reacted with DMAD to produce N2-triazole species,21,24,32,44 as confirmed by X-ray crystallography, however, no literature examples of monodentate PtII/PtIV triazoles could be found for us to draw comparisons with; for those PtII triazoles with chelating groups on the triazole ligand both N1 and N2 coordination has been previously observed, depending on the influence of the chelating group.45
The PtII complex trans-[Pt(N3)2(py)2] reacted with DMAD (2) in MeOH (35 °C) to give the mono (trans-[Pt(C6H6N3O4)(N3)(py)2]) and bis (trans-[Pt(C6H6N3O4)2(py)2]) substituted complexes in which both ester groups remained intact (ESI†). The 195Pt NMR resonance of the mono substituted complex trans-[Pt(C6H6N3O4)(N3)(py)2] was seen at −2219 ppm (d3-MeCN), approximately half-way between the starting material trans-[Pt(N3)2(py)2] (−2122 ppm, d6-acetone)37 and the bis triazole complex trans-[Pt(C6H6N3O4)2(py)2] (−2331 ppm, d6-acetone). The 1H NMR spectrum for the mono-substituted complex showed only one OMe environment, and no nOe correlation was observed between the pyridine protons and the OMe group. The 14N NMR spectrum of the bis substituted PtII complex revealed that the characteristic sharp Nβ and Nγ resonances (where assignment is Pt-NαNβNγ) at 230 ppm and 135 ppm were absent, consistent with loss of azido groups (Fig. S1†). Whilst a characteristic azido absorbance (2043 cm−1) was still observed for the mono substituted complex trans-[Pt(C6H6N3O4)(N3)(py)2], this IR absorbance was absent in the bis triazole.
We investigated the stability of the PtII triazole complexes in a number of different solvents; both the mono and bis triazole complexes were unstable in d6-acetone, CDCl3, and d3-MeCN, slowly converting over time to new species, such that NMR spectroscopic experiments needed to be run shortly after sample preparation.
The PtIV complex 1 showed no reactivity stirring at 35 °C, up to 7 d, with 5 eq. alkyne towards phenylacetylene (4); 1,3-diethynylbenzene (5); phenylpropiolic acid (6); 3,4-difluorophenylacetylene (7) 1-phenyl-2-propyn-1-ol (8); 3-butyn-2-one (9) and 4-phenyl-3-butyne-2-one (10). However, it was reactive in different solvents (including acetone, MeOH, EtOH, MeCN, CHCl3 and THF) towards a number of alkynes: dimethyl acetylenedicarboxylate DMAD (2), diethyl acetylenedicarboxylate DEACD (3), N-[(1R,8S,9s)-bicyclo[6.1.0]non-4-yn-9-ylmethyloxycarbonyl]-1,8-diamino-3,6-dioxaoctane, BCN (11) and dibenzocyclooctyne-amine DBCO (12) (Fig. 1).
Dimethyl acetylenedicarboxylate (DMAD, 2) and diethyl acetylenedicarboxylate (DEACD, 3) products
Complex 1 reacted with 2 in MeOH to produce the cyclometallated species 2a′ (1.1 eq. 2, 35 °C, 4 d) and 2b′ (5 eq. 2, 35 °C, 3 d), via intermediates 2a and 2b (Scheme 1).The PtIV complex 1 reacted more slowly than its PtII precursor trans-[PtII(N3)2(py)2] with 2, taking approximately twice as long under similar conditions to achieve conversion to products, as judged by 1H NMR spectroscopy. The initial complex 2a was detected by HPLC in trace amounts as the [2a + Na]+ adduct at 636.11 m/z (model 636.09 m/z) but was not isolated in sufficient quantity for further analysis. Following isolation, the cyclometallated derivative complex 2a′ was indefinitely stable in D2O.
Both 13C and 1H NMR spectra of 2a′ were consistent with attack of the axial hydroxide on the ester group and elimination of MeOH, with a sharp singlet resonance corresponding to a single methyl group (3H) being observed at 3.78 ppm. The 13C NMR spectrum for 2a′ revealed inequivalent triazole 13C resonances at 138.4 ppm and 136.1 ppm. The 195Pt NMR spectroscopic resonance in D2O was 91 ppm lower for 2a′ (873 ppm) than for 1 (964 ppm) indicating increased shielding of the Pt centre.37 The 14N NMR spectrum of 2a′ (Fig. S2†) was consistent with the proposed structure but afforded little additional structural information. The triazole 14N resonances may, like Nα in coordinated azide (Pt–Nα–Nβ–Nγ) be exceptionally broad, and/or may superimpose with either the Nβ (229.0 ppm) or the Nγ/Npy (164.6 ppm) resonances, both of which were considerably broader than for 1.46 IR spectroscopy confirmed the presence of azide in the mono substituted products, in which the Pt–N3 group gave a sharp IR absorbance ca. 2047 cm−1. Formation of bis substituted 2b′ resulted in loss of the azido IR peak, compared with the mono substituted 2a′ (Fig. S3†).47 Complex 1 also reacted with DEACD (3) to produce the corresponding cyclometallated mono (3a′) and bis (3b′) species, at a slower rate than seen for the reaction with DMAD (2).
MS/MS fragmentation studies of DMAD products 2a, 2a′, 2b and 2b′ derivatives revealed that for complexes containing non-cyclometallated triazoles (2a, 2b) these triazole ligands dissociated intact (Fig. 2(a) and S4†). For the cyclometallated complex 2a′ the triazole ligand dissociated by forming the hydroxy species Fig. 2(b); MS/MS of the bis-substituted mono-cyclometallated intermediate between 2b and 2b′ was also investigated (Fig. S5†); this fragmented through loss of both the cyclometallated and non-cyclometallated triazole ligands.
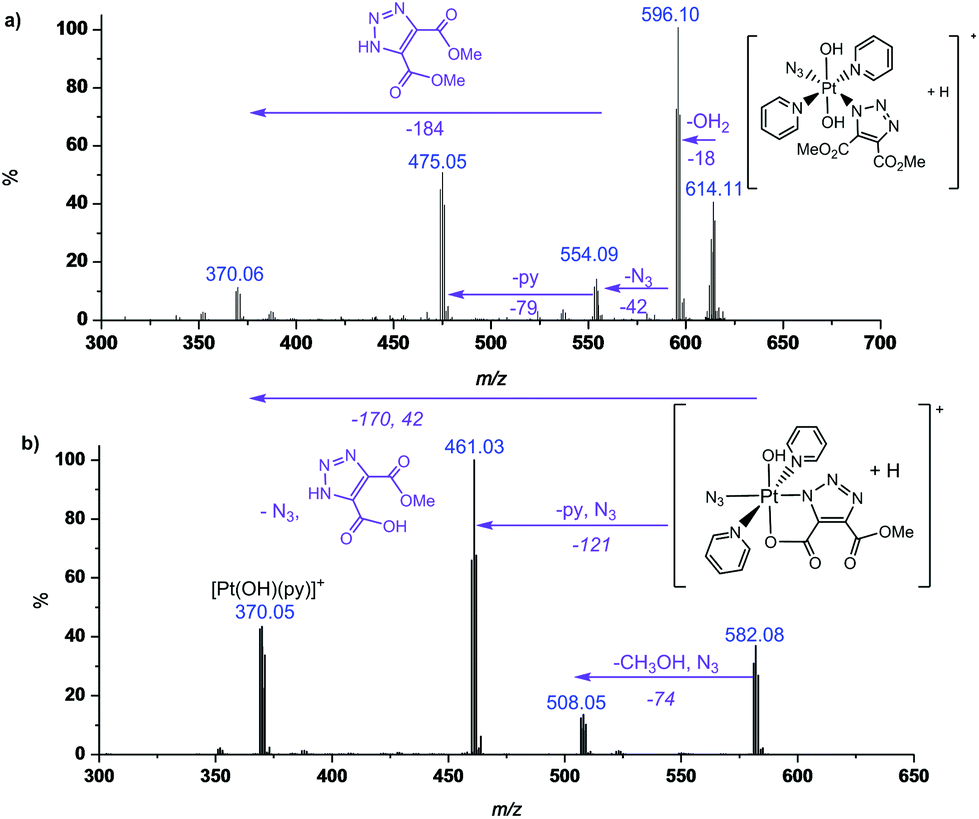 | ||
| Fig. 2 MS/MS of (a) 2a (614.11 m/z, [Pt(N3)(C6H6N3O4)(OH)2 (py)2 + H]+, model 614.10 m/z) and (b) 2a′ ([Pt(N3)(C5H3N3O4)(OH) (py)2 + H]+, model 582.08 m/z) showing fragmentation through loss of intact ligand for 2a and cyclometallated ligand for 2a′. | ||
Transesterification
We observed transesterification between the DEACD triazole ligand and the solvent when the cycloaddition reaction between 1 and 3 was carried out in d4-MeOH, replacing the remaining –OCH2CH3 group with –OCD3 (Scheme 2). This was detected by ESI-MS as the [M + Na]+ adduct [Pt(N3)(py)2(OH)(N3C3O4D3) + Na]+ at 607.08 m/z (model 607.08 m/z) (Fig. S6†). This species fragmented by MS/MS, showing loss of pyridine and N3 to give the fragment [Pt(py)(OH)(N3C3O4D3) + Na]+ at 486.04 m/z as well as further fragmentation through loss of the intact cyclised deuterated ligand, giving the [Pt(OH)(py) + Na]+ ion at 314.00 m/z.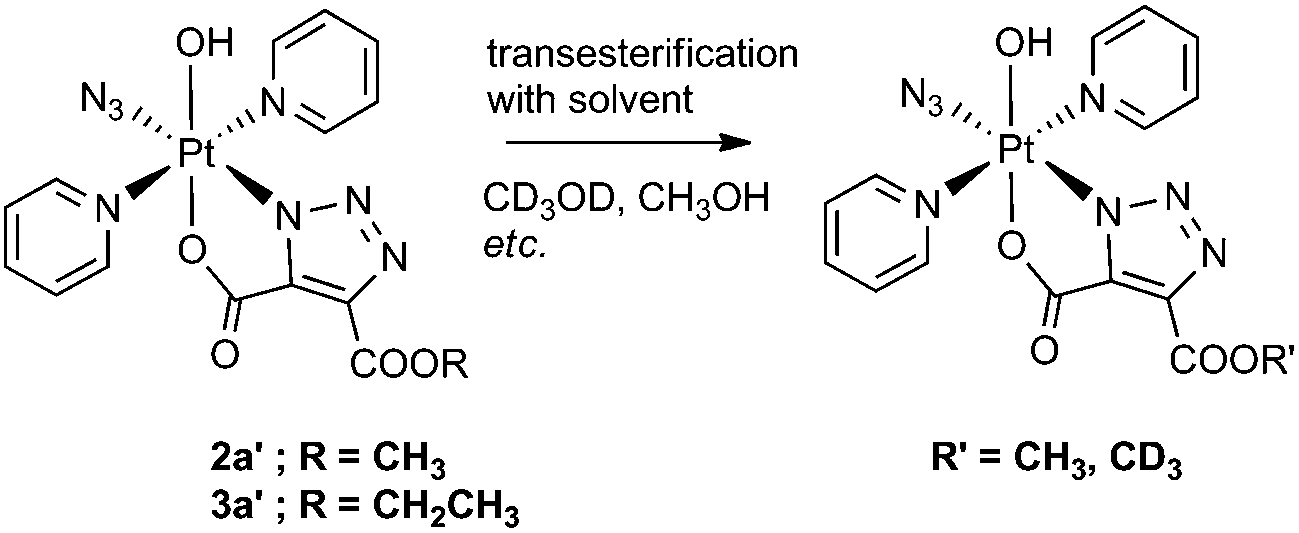 | ||
| Scheme 2 Transesterification of complexes 2a′ and 3a′ with alcoholic solvent. | ||
A similar transesterification was observed for DMAD derivatives in d4-MeOH – this could be prevented by conducting reactions and NMR spectroscopic experiments in appropriate solvents.
Subsequent reactivity
In addition to cyclometallation and transesterification, the complexes also displayed further solvent-dependent reactivity. Although D2O and d4-MeOH solutions of 2a′ were stable (with the exclusion of light) in CDCl3 a conversion from 2a′ to 2a′′ was observed. Under extended reaction times (7 d) in MeOH similar reactivity was observed; yellow 2a′ converted completely to off-white 2a′′ (insoluble in D2O). The 195Pt NMR resonance of 2a′′ in CDCl3 (764 ppm) was more shielded than for 2a′ (832 ppm) (ESI†). 14N NMR spectra of 2a′ and 2a′′ were not sufficiently diagnostic (Fig. S2†). IR N3 azide absorbances (2046 cm−1 and 2047 cm−1) were essentially the same in strength and frequency for 2a′ and 2a′′ and slightly higher than for the parent complex 1 (2032 cm−1).47 Compound 2a′′ had the same 1H NMR spectral integral ratios between pyridyl and the OCH3 group as for 2a′, indicating that the ester group was retained, but the pyridine Ho protons had become deshielded.Bis-substituted 2b′ was insoluble in D2O, MeOH and d6-acetone but soluble in MeCN and CDCl3, giving rise to a 195Pt NMR resonance at 840 ppm (CDCl3). Complex 2b′ was also unstable in CDCl3, converting over a few days to 2b′′. Complex 2b′′ exhibited inequivalent methyl 1H NMR spectroscopic environments with 3H of the singlet corresponding to the remaining OCH3 ester groups moving from 3.91 ppm to 3.88 ppm (ESI†). A new pyridyl environment was also observed, with 2HPyortho protons becoming deshielded (moving from 8.85 ppm to 8.94 ppm) and corresponding new Hm and Hp resonances overlapping with the existing resonances. Complex 2b′′ was only sparingly soluble in CDCl3, precipitating from solution over time. As with 2a′′, the dominant ESI-MS species were essentially unchanged during the transformation.
Although precedent suggests that a Pt–N1-bound triazole may isomerise to a more thermodynamically stable N2-bound complex32 we suggest that the steric requirements of the cyclometallated ring in 2a′ and 2b′ makes N1 to N2 triazole rearrangement unlikely. Further investigations are ongoing to define the precise speciation of 2a′′ and 2b′′.
BCN products
Complex 1 reacted with BCN (11) in MeOH to give mono (11a) and bis (11b) substituted products (Scheme 3). Products 11a and 11b were soluble in both H2O and MeOH. The 13C NMR spectra of 11a revealed the two alkyl carbons at 146.6 ppm and 142.4 ppm with characteristic 195Pt satellites and couplings of 27 Hz and 33 Hz, respectively (Fig. 3). This is consistent with the observation that within Pt-pyridyl systems we commonly observe 3JPtC couplings of this magnitude to Cmeta, and we typically do not observe platinum couplings to PyCortho or PyCpara. For 11a, the inequivalence of these alkyl 13C resonances could indicate N1 rather than N2 coordination of the triazole ring (Fig. 3). For 11b resonances corresponding to the two alkyl carbons were seen at 146.8 ppm and 142.1 ppm, indicating similar coordination as seen for 11a, but for 11b the 195Pt coupling on these resonances could not be resolved.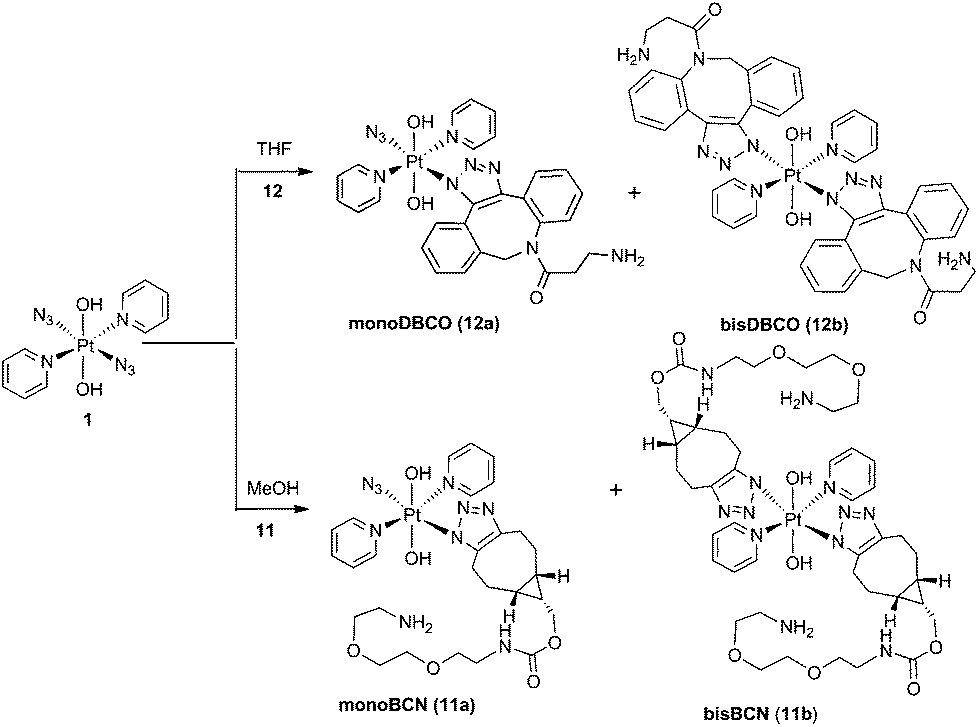 | ||
| Scheme 3 Reaction of 1 with 11 and 12. | ||
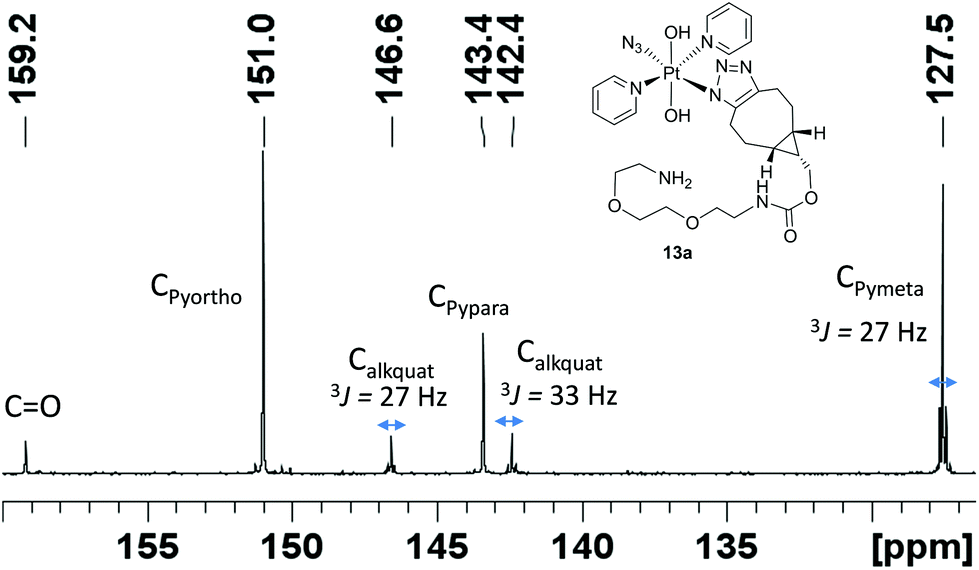 | ||
| Fig. 3 13C NMR (d4-MeOH) spectrum of 11a showing 195Pt satellites. | ||
MS/MS experiments with [mono-(11a) + H]+ (796.2 m/z) showed fragmentation through loss of the various charged (N3−, OH−, triazole etc.) and neutral (py) ligands; the triazole ligand did not readily dissociate from the [M + H]+ species to give the [M − triazole]+ fragment, but a stable fragment [Pt(OH)(py)2]+ was detected at 370.052 m/z, indicating it was possible to remove the triazole ligand in association with other ligands. MS/MS experiments with [bis-(11b) + H]+ (1120.5 m/z) gave similar fragmentation to the mono species regarding ready loss of H2O (1102.473 m/z) – a common pathway for Pt dihydroxido species (e.g.2a in Fig. 2). Complex [bis-(11b) + H]+ also showed loss of pyridine ligands to give [Pt(triazole)(triazole − H) + py]+ at 1005.423 m/z and [Pt(triazole)(triazole − H)]+ at 926.384 m/z in which it is assumed one of the amine groups of the BCN ligand provides stabilisation to the Pt centre; for both 11a and 11b species the common [Pt(triazole − H)]+ fragment was detected around 560.17 m/z.
Dibenzocyclooctyne-amine (DBCO) products
Since DBCO 12 was insoluble in MeOH, complex 1 was reacted with 12 in THF, producing both mono- (12a) and bis-(12b) substituted products (Scheme 3) which showed good agreement with HRMS isotope models (Fig. S8†). Despite concerted efforts with a range of different conditions, the compounds showed poor chromatographic resolution by HPLC and only small quantities of mono-(12a) were obtained, sufficient for ESI-MS studies.MS/MS experiments with [12a + H]+ (748.2 m/z) revealed loss of neutral molecules (H2O, py) and the residual azido ligand, and fragmentation was consistent with previous MS/MS experiments of complex 1.37 As for the BCN derivatives, the [M − triazole]+ species was not detected around 472 m/z, indicating that the triazole was relatively tightly bound.
Rates of reaction and purification of products
Compared to PdII, PtII, RuII![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32 and Au azido complexes, the rates of cycloadditions of 1 with alkynes 23 and 11 were relatively slow. Although they did proceed at room temperature, gentle heating (35 °C) was typically employed. To proceed in the absence of a catalyst, these cycloaddition reactions typically require an electron-rich azide and an electron-deficient alkyne. Electron-withdrawing groups on the alkyne lower the level of the LUMO, therefore promoting the reaction. The reactions of 1 with alkynes 2, 11, and 12 resulted in a mixture of mono and bis substituted products which in some cases then further converted to additional rearrangement species. The reactions with strained alkynes 11 and 12 proceeded significantly faster (within 24 h at 35 °C) than for the electron deficient alkyne 2 and there was no evidence of decomposition to Pt(0) or cyclometallation. Mono triazole adducts were typically pale yellow, with bis triazole adducts off-white. This was consistent with the UV-vis and IR data, and partial or total loss of the azido group. Attempts to isolate complexes in high purity using standard techniques were aided by mass-directed preparative HPLC. A range of HPLC columns under neutral (and basic – where compatible) conditions were investigated: Atlantis, Hypersil, Sunfire and X-bridge OBD. The best purification results for the complexes were obtained using mass-directed purification on a Waters X-Bridge OBD column, eluting with H2O + 0.1% NH4OH (pH 9)/MeCN + 0.1% NH4OH.
32 and Au azido complexes, the rates of cycloadditions of 1 with alkynes 23 and 11 were relatively slow. Although they did proceed at room temperature, gentle heating (35 °C) was typically employed. To proceed in the absence of a catalyst, these cycloaddition reactions typically require an electron-rich azide and an electron-deficient alkyne. Electron-withdrawing groups on the alkyne lower the level of the LUMO, therefore promoting the reaction. The reactions of 1 with alkynes 2, 11, and 12 resulted in a mixture of mono and bis substituted products which in some cases then further converted to additional rearrangement species. The reactions with strained alkynes 11 and 12 proceeded significantly faster (within 24 h at 35 °C) than for the electron deficient alkyne 2 and there was no evidence of decomposition to Pt(0) or cyclometallation. Mono triazole adducts were typically pale yellow, with bis triazole adducts off-white. This was consistent with the UV-vis and IR data, and partial or total loss of the azido group. Attempts to isolate complexes in high purity using standard techniques were aided by mass-directed preparative HPLC. A range of HPLC columns under neutral (and basic – where compatible) conditions were investigated: Atlantis, Hypersil, Sunfire and X-bridge OBD. The best purification results for the complexes were obtained using mass-directed purification on a Waters X-Bridge OBD column, eluting with H2O + 0.1% NH4OH (pH 9)/MeCN + 0.1% NH4OH.
We have previously reported DFT and TDDFT analysis of complex 1; the absorbance in the UV-vis region is dominated by 1LMCT (N3 → Pt) and mixed 1LMCT/3IL (OH → Pt, N3; IL = interligand) transitions.37 UV-vis spectroscopy of the click products were consistent with partial (mono) and complete loss (bis) of the azido absorbances, compared to the starting complex 1 (Fig. S7†) and resulted in a reduced absorption ca. 300 nm.
Experimental
General procedures
Materials. K2[PtCl4] was purchased from Precious Metals Online. HPLC-grade solvents and Millipore filtered H2O were used for the preparation and purification of compounds by HPLC. All other reagents were purchased from Sigma-Aldrich and used as received. (IM) indicates use of a nylon syringe filter (pore size 0.2 μM). All manipulations were carried out under reduced lighting and solutions were prepared stored and handled with minimal exposure to light. NMR spectroscopy. Due to the potential photosensitivity of the compounds, amberised NMR spectroscopy tubes (Goss Scientific) were used. 13C NMR: acquired on a Bruker AVII 500 MHz spectrometer equipped with a z-gradient triple resonance inverse 1H/19F(13C) TXI probe and referenced internally to residual solvent where possible or externally to TMS in CDCl3. All other NMR spectra were acquired on a Bruker AVIIIHD 500 MHz (1H: 500.13 MHz), a Bruker AVIIIHD 400 nanobay, or a Bruker AV111-600 spectrometer at 298 K and processed using Topspin 3.2. J values are quoted in Hz. All chemical shift (δ) values are given in parts per million. 1H NMR: chemical shifts were referenced to residual solvent. 195Pt NMR: chemical shifts were externally referenced to K2PtCl6 in 1.5 mM HCl in D2O (δ 0 ppm) using parameters as previously described: for spectra of PtIV species directly bonded to quadrupolar 14N, typical parameters used were d1 = 0 s, TD 2k, DE 10 μs, 256k scans.37 Data were processed with a LB of 50 Hz. 14N NMR: chemical shifts were externally referenced to [14N]NH4Cl (1.5 M) in 1 M HCl with a D2O coaxial insert and processed with a qfil baseline correction. Mass spectrometry: low resolution ESI-MS: obtained with a Waters Micromass LCT Premier XE spectrometer. HRMS: obtained with a Thermofisher Exactive Plus with a Waters Acuity UPLC system. MS/MS experiments: were performed on an Acuity UPLC in flow injection analysis mode, equipped with a Waters Xevo G25 QTOF. All MS data were processed using MassLynx 4.0. HPLC: were performed with a Waters Autopurification system, equipped with a Waters X-Bridge OBD semi-prep column (5 μm, 19 mm × 50 mm), with an injection loop of 1 ml, eluting with H2O + 0.1% NH4OH (pH 9)/MeCN + 0.1% NH4OH. The crude samples (in H2O/MeCN) were filtered (nylon, 0.2 μm) and injected in 750 μL aliquots, with mass-directed purification with an ACQUITY QDa performance mass spectrometer. UV-visible absorption spectra were acquired with a T60U Spectrometer PG Instruments Ltd using UVWin Software or the Waters HPLC. Elemental microanalyses were performed by Stephen Boyer at the London Metropolitan University.Materials and methods
Synthetic procedures
The PtII complex trans-[PtII(N3)2(py)2] and 1 were prepared in two steps from K2PtCl4, via trans-[PtIICl2(py)2]. H2O2 oxidation of trans-[PtIV(N3)2(py)2] gave trans,trans,trans-[PtIV(N3)2(OH)2(py)2 (1) which was purified by HPLC before use. Analytical data was consistent with previous reports.37 UV-Vis spectra and data concerning reactivity of trans-[PtII(N3)2(py)2] are given in the ESI.†Caution! No problems were encountered during this work, however heavy metal azides are known to be shock sensitive detonators, therefore it is essential that platinum azides compound are handled with care.
PtIV complexes
Cyclometallated trans,trans,trans-[Pt(N3)(C5H3N3O4)(OH)(py)2] (2a′). DMAD (2, 14.3 μl, 0.117 mmol) in MeOH (2 ml) was added to 1 (50 mg, 0.106 mmol) in MeOH (3 ml). The solution was stirred at 35 °C for 4 d before being placed on ice. Product 2a′ was isolated as a yellow precipitate by filtration and rinsed with cold H2O, MeOH and diethyl ether (29 mg, 0.05 mmol, 47%).
1 H NMR (400 MHz, D2O) δ: 8.71 (d, 3J1H195Pt = 25, 3JHH = 6, 4H, Ho) 8.25 (t, 3JHH = 7, 2H, Hp), 7.76 (dd, 3JHH = 7, 3JHH = 7, 4H, Hm), 3.87 (s, 3H, OMe).
195 Pt NMR (107 MHz, D2O) δ: 873 (PWHH 670 Hz).
14 N NMR (29 MHz, D2O) δ: 288.6 (N2 gas), 229.0 (Nβ), 164.6 (broad, Nγ/py). Nα not seen.
13 C NMR (126 MHz, D2O) δ: 166.6 (Cester), 161.5 (Cester), 148.9 (Cpyortho), 143.3 (Cpypara), 138.4 (Ctriazole), 136.1 (Ctriazole), 127.8 (3J13C195Pt = 25, Cpymeta), 52.9 (Calkyl).
1 H NMR (400 MHz, CDCl3) δ: 8.98 (d, 3J1H195Pt = 25, 3JHH = 6, 4H, Ho), 8.14 (t, 3JHH = 7, 2H, Hp), 7.72 (dd, 3JHH = 7, 3JHH = 7, 4H, Hm), 1.64 (br) (with peaks corresponding to 2a′′ growing in over time).
195 Pt NMR (107 MHz, CDCl3) δ: 832 (with peak at 767 ppm corresponding to 2a′′ growing in over time).
ESI-MS (MeOH, M = trans,trans,trans-[Pt(N3)(C5H3N3O4)(OH)(py)2]) m/z: 1185.12 ([2 M + Na]+ calcd 1185.14); 1163.14 ([2M + H]+ calcd 1163.15); 604.05 ([M + Na]+ calcd 604.06); 582.07 ([M + H]+ calcd 582.08).
HRMS (MeOH) m/z: 582.0808 ([M + H]+, C15H15N8O5Pt calcd 582.0766).
IR (solid) ν cm−1: 3465, 3108, 3074, 2046 (νasymN3), 1732, 1674, 1611, 1538, 1460, 1437, 1389, 1337, 1254, 1211, 1197, 1127, 1078, 1018, 810, 773, 690.
Elemental microanalysis: Calc. C15H14N8O5Pt (581.07 g mol−1): C, 30.99; H, 2.43; N, 19.27. Found: C, 31.15; H, 2.46; N, 19.10.
Bis cyclometallated bis trans-[Pt(C5H3N3O4)2(py)2] (2b′). DMAD (65.2 μl, 0.53 mmol) in MeOH (2 ml) was added dropwise to trans,trans,trans-[Pt(N3)2(OH)2(py)2] (50 mg, 0.11 mmol) in MeOH (3 ml). The solution was stirred at 35 °C for 3 days, put on ice and the resulting white compound 2b′ isolated by filtration (15.5 mg, 0.02 mmol, 21%).
1 H NMR (400 MHz, CDCl3) δ: 8.87 (d, 3J1H195Pt = 22, 3JHH = 6 Hz, 4H, Ho) 8.10 (t, 3J1H1H = 6, 2H, Hp) 7.62 (t, 3J1H1H = 6, 4H, Hm) 3.91 (s, 6H, OMe).
195 Pt NMR (107 MHz, CDCl3) δ: 840.
13 C NMR (125 MHz, CDCl3) δ: 162.7 (Cestercyclo), 160.2 (CesterOMe), 153.4, 149.4 (Co), 143.4 (Cp), 139.3 (Calkene), 132.7 (Calkene), 128.3 (Cm), 52.6 (Calkyl).
ESI-MS (MeOH) (M = trans-[Pt(C5H3N3O4)2(py)]) m/z: 1405.09 ([2 M + Na]+ calcd 1405.14); 1383.12 ([2 M + H]+ calcd 1383.15); 714.04 ([M + Na]+ calcd 714.06); 692.06 ([M + H]+ calcd 692.08).
HRMS (MeOH) m/z: 714.06298 ([M + Na]+ C20H16N8O8PtNa calcd 714.0631). IRν cm−1: 3112, 2051, 1732, 1613, 1541, 1486, 1462, 1436, 1330, 1235, 1169, 1062, 1019, 835, 812, 690.
Elemental microanalysis Calc. C20H16N8O8Pt (691.07 g mol−1): C, 34.73; H, 2.33; N, 16.21. Found: C, 34.65; H, 2.28; N, 16.14.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 151.0 (CPyortho), 146.6 (J13C195Pt = 27, Calkquat), 143.4 (CPypara), 142.4 (J13C195Pt = 33, Calkquat), 127.5 (t, 3J13C195Pt = 27, CPymeta), 72.9, 71.32, 71.27, 71.0, 63.7 (CH2OC(O)NH), 41.9, 41.7, 26.5, 25.1, 24.2, 23.5, 22.0, 21.7, 19.3. ESI-MS (MeOH) m/z (M = trans,trans,trans-[Pt(N3)(C17H28N5O4)(OH)2(py)2]): 398.63 [M + 2H]2+ calcd C27H42N10O6Pt: 398.64439, 796.25 ([M + H]+ calcd C27H41N10O6Pt: 796.2853), 819.26 ([M + Na]+ calcd C27H40N10NaO6Pt: 819.2629). HRMS (MeOH) m/z: 796.284 ([M + H]+ calcd C27H41N10O6Pt: 796.285).
O), 151.0 (CPyortho), 146.6 (J13C195Pt = 27, Calkquat), 143.4 (CPypara), 142.4 (J13C195Pt = 33, Calkquat), 127.5 (t, 3J13C195Pt = 27, CPymeta), 72.9, 71.32, 71.27, 71.0, 63.7 (CH2OC(O)NH), 41.9, 41.7, 26.5, 25.1, 24.2, 23.5, 22.0, 21.7, 19.3. ESI-MS (MeOH) m/z (M = trans,trans,trans-[Pt(N3)(C17H28N5O4)(OH)2(py)2]): 398.63 [M + 2H]2+ calcd C27H42N10O6Pt: 398.64439, 796.25 ([M + H]+ calcd C27H41N10O6Pt: 796.2853), 819.26 ([M + Na]+ calcd C27H40N10NaO6Pt: 819.2629). HRMS (MeOH) m/z: 796.284 ([M + H]+ calcd C27H41N10O6Pt: 796.285).
MS/MS (796.2) (d4-MeOH) m/z: 778.276 ([M − OH]+ C27H39N10O5Pt, calcd 778.273), 718.252 ([M + H − py]+ C22H36N9O6Pt, calcd 718.242), 657.224 ([M + H − N3, py, OH]+ C22H34N6O5Pt, calcd 657.227), 639.212 ([M − H2O2, N3, py]+, C22H32N6O4Pt, calcd 639.211), 560.174 ([M − H2O2, N3, 2py, H]+, C17H27N5O4Pt, calcd 560.172), 370.055 ([Pt(OH)(py)2]+, C10H11N2OPt calcd 370.055).
IR (MeOH-d4) cm−1: 3361 (br), 2920, 2044 (νasymN3, strong), 1695, 1613, 1543, 1457, 1264, 1211, 1104, 1077, 1020, 769, 690.
13C NMR (126 MHz, d4-MeOH) δ: 159.3 (CO), 151.3 (CPyortho), 146.8 (m), 143.7, 142.1 (m), 127.8 (3J13C195Pt = 27, CPymeta),71.5(m), 71.3(m), 71.0(m), 69.2, 63.7 (m), 43.9, 41.7(m), 26.5(m), 25.1(m), 24.3(m), 23.7 (m), 22.0(m), 21.5(m), 19.4(m).
ESI-MS (MeOH) m/z (M = trans,trans,trans-[Pt (C17H28N5O4)2(OH)2(py)2]): 560.74 ([M + 2H]2+ C44H68N12O10PtH2 calcd 560.7468), 1120.48 ([M + H]+, C44H69N12O10Pt, calcd 1120.4902). HRMS (MeOH) m/z: 1120.4933 [M + H]+, C44H69N12O10Pt, calcd 1120.4902.
MS/MS (1120.5) d4-MeOH m/z: 1102.472 ([M − OH]+, C44H67N12O9Pt, calcd 1102.484), 1067.439, 1005.423 ([M − H2O2, py, H]+ C39H60N11O8Pt, calcd 1005.430), 926.383 ([M − H2O2, 2py, H]+, C34H55N10O8Pt, calcd 926.384), 560.167 ([M − triazole, H2O2, 2py, H]+, C17H27N5O4Pt, calcd 560.172).
IR (MeOH-d4) cm−1: 3366, 2482, 2244, 2072, 1120, 973, 822.
:50 MeCN:H2O and purified by HPLC.
Monosubstituted trans,trans,trans-[Pt(N3)(C18H16N5O)(py)2(OH)2]
DBCO complex 12a was isolated as a yellow solid by HPLC (4 mg, 7% yield).ESI-MS (MeOH) m/z: (M = trans,trans,trans-[Pt(N3)(C18H16N5O)(py)2(OH)2]): 748.18 ([M + H]+, C28H29N10O3Pt calcd 748.21), 770.13 ([M + Na]+ C28H28N10O3PtNa calcd 770.19 m/z). HRMS (MeOH) m/z: 748.2067 ([M + H]+, C28H29N10O3Pt calcd 748.2066).
MS/MS (748.2) d4-MeOH m/z: 730.197 ([M − OH]+ C28H29N10O3Pt, calcd 730.197), 712.186, 688.183 ([M − N3, H2O]+, C28H27N7O2Pt calcd 688.187), 669.171 ([M − py + H]+ C23H24N9O3Pt, calcd 669.164), 609.146 ([M − N3, H2O, py]+, C23H22N6O2Pt, calcd 609.148), 590.133 ([M − 2py + H]+ C18H19N8O3Pt, calcd 590.125). IRv cm−1 (d4-MeOH): 3378(br), 2980, 2493(br), 2047 (νasymN3), 1637, 1613, 1479, 1457, 1212, 1117, 1076, 1019, 971, 765, 689.
Bis-substituted trans,trans,trans-[Pt(C18H16N5O)2(OH)2(py)2]
Complex 12b was isolated as an off-white solid by HPLC (3 mg, 4% yield).ESI-MS (MeOH) m/z: (M = trans,trans,trans-[Pt(C18H16N5O)2(OH)2 (py)2]): 1024.33 ([M + H]+ C46H44N12O4PtH, calcd 1024.33), 512.67 ([M + 2H]2+ C46H44N12O4PtH2, calcd 512.67). HRMS (MeOH) m/z: 1024.3335 ([M + H]+, C46H44N12O4PtH calcd 1024.3285). IRv cm−1 (d4-MeOH): 3346(br), 2479(br), 2216, 2071, 1120, 972, 822.
Conclusions
The reactions of azido PtIV complex 1 with acetylenes 2, 3, 11 and 12 represent the first examples of PtIV azido cycloaddition reactions. The reactions take place under mild conditions in the absence of a catalyst, enabling modification of ligands which are already coordinated to a metal centre, and providing a route to monoazido complexes. Complex 1 did not show any reactivity towards compounds 4–9; we suggest this is due to the alkynes being insufficiently electron deficient for the reaction to occur (Fig. 4).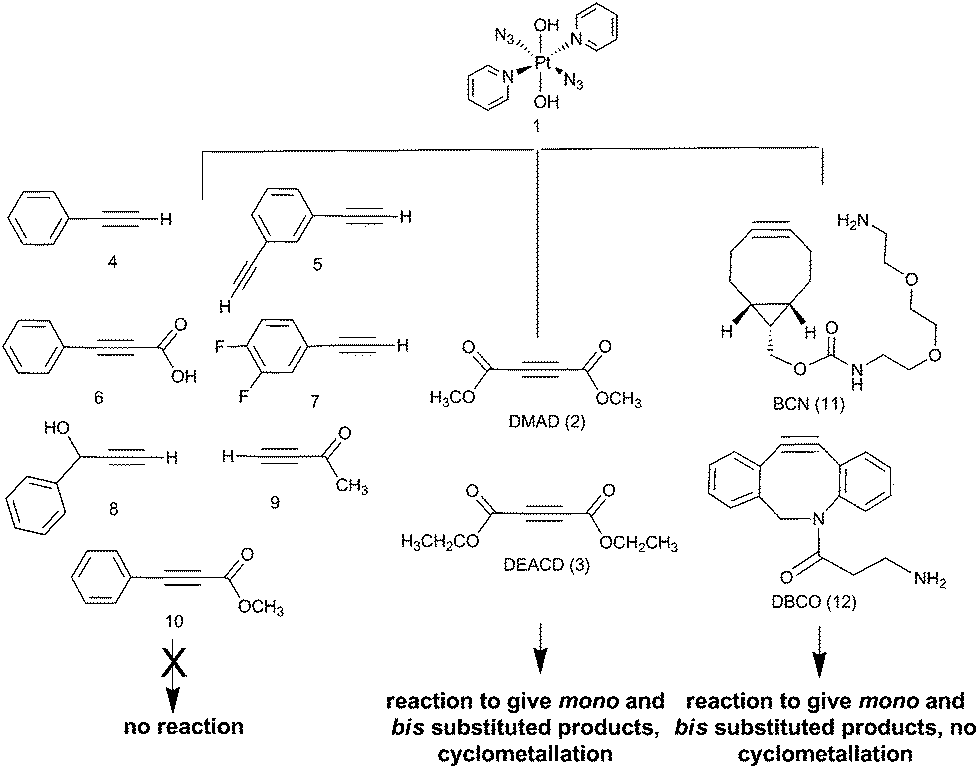 | ||
| Fig. 4 Summary of reactivity observed for complex 1. | ||
For reactions of 1 with 2 and 3 there is evidence that Pt cyclometallates with the new triazole ligand via the ester group, displacing MeOH or EtOH respectively. This reactivity is not observed for the PtII analogue trans-[Pt(N3)2(py)2]. Whilst the cyclometallated complex 2a′ was stable in D2O, in other solvents there is evidence for subsequent reactivity. In contrast, derivatives of complex 1 with cyclooctynes 11 and 12 did not show any obvious subsequent reactivity following formation of the triazole ligand.
Whilst use of electron withdrawing groups is an effective strategy to promote copper-free cycloadditions, for PtIV complexes the proximity of these reactive groups to the Pt centre can result in subsequent reactivity. Strain-promoted cycloadditions appear to be a promising alternative strategy, and we are currently investigating appropriate cyclooctynes to further investigate this chemistry.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
NF thanks Professor Stephen Faulkner for advice and support and the EPSRC (grant EP/G006792/1), the Daphne Jackson Trust, the Royal Society of Chemistry, the Medical Research Council, the University of Oxford Vice-Chancellor's Returning Carers Fund, the John Fell Fund and the Wellcome Trust (grant 201406/Z/16/Z) for financial support. This work was also supported by Cancer Research UK (CR-UK) grant number C5255/A18085, through the Cancer Research UK Oxford Centre. NF thanks Dr James Wickens for performing some of the MS/MS experiments.References
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS PubMed.
- J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182 CrossRef CAS PubMed.
- S. D. Köster, H. Alborzinia, S. Can, I. Kitanovic, S. Wölfl, R. Rubbiani, I. Ott, P. Riesterer, A. Prokop, K. Merz and N. Metzler-Nolte, Chem. Sci., 2012, 3, 2062 RSC.
- W. P. Fehlhammer and W. Beck, Z. Anorg. Allg. Chem., 2015, 641, 1599 CrossRef CAS.
- R. F. Ziolo, J. A. Thich and Z. Dori, Inorg. Chem., 1972, 11, 626 CrossRef CAS.
- H. Frühauf, Chem. Rev., 1997, 65, 4228 Search PubMed.
- J. A. K. Bauer, T. M. Becker and M. Orchin, J. Chem. Crystallogr., 2004, 34, 843 CrossRef CAS.
- L. Henry, C. Schneider, B. Mützel, P. V. Simpson, C. Nagel, K. Fucke and U. Schatzschneider, Chem. Commun., 2014, 50, 15692 RSC.
- R. Guilard, I. Perrot, A. Tabard, P. Richard, C. Lecomte, Y. H. Liu and K. M. Kadish, Inorg. Chem., 1991, 30, 21 Search PubMed.
- A. Fleischer, A. Roller, V. B. Arion, B. K. Keppler and F. Mohr, Can. J. Chem., 2009, 87, 146 CrossRef CAS.
- H. H. Lee, S. Y. Han, Y. S. Gyoung, Y. J. Kim, K. E. Lee, Y. O. Jang and S. W. Lee, Inorg. Chim. Acta, 2011, 378, 174 CrossRef CAS.
- Y. Kim, J. Han, S. Kang, S. Han and S. W. Lee, Dalton Trans., 2003, 3357 RSC.
- N. Malatesti, R. Hudson, K. Smith, H. Savoie, K. Rix, K. Welham and R. W. Boyle, Photochem. Photobiol., 2006, 82, 746 CrossRef CAS PubMed.
- L. Busetto, A. Palazzi and R. Ros, Inorg. Chim. Acta, 1975, 13, 233 CrossRef CAS.
- A. R. Powers, X. Yang, T. J. Del Castillo, I. Ghiviriga, K. A. Abboud and A. S. Veige, Dalton Trans., 2013, 14963 RSC.
- L. Waag-Hiersch, J. Mößeler and U. Schatzschneider, Eur. J. Inorg. Chem., 2017, 2017, 3024 CrossRef CAS.
- T. J. Del Castillo, S. Sarkar, K. A. Abboud and A. S. Veige, Dalton Trans., 2011, 40, 8140 RSC.
- F. C. Liu, Y. L. Lin, P. S. Yang, G. H. Lee and S. M. Peng, Organometallics, 2010, 29, 4282 CrossRef CAS.
- E. Evangelio, N. P. Rath and L. M. Mirica, Dalton Trans., 2012, 41, 7782 RSC.
- L. Busetto, F. Marchetti, S. Zacchini and V. Zanotti, Inorg. Chim. Acta, 2005, 358, 1204 CrossRef CAS.
- C. K. Chen, H. C. Tong, C. Y. C. Hsu, C. Y. Lee, Y. H. Fong, Y. S. Chuang, Y. H. Lo, Y. C. Lin and Y. Wang, Organometallics, 2009, 28, 3358 CrossRef CAS.
- K. S. Singh, C. Thöne, M. R. Kollipara, K. Sarjit and C. Tho, J. Organomet. Chem., 2005, 690, 4222 CrossRef CAS.
- K. S. Singh, K. A. Kreisel, G. P. A. Yap and M. R. Kollipara, J. Organomet. Chem., 2006, 691, 3509 CrossRef CAS.
- K. S. Singh, K. A. Kreisel, G. P. A. Yap and M. R. Kollipara, J. Coord. Chem., 2007, 60, 505 CrossRef CAS.
- K. S. Singh, V. Svitlyk and Y. Mozharivskyj, Dalton Trans., 2011, 40, 1020 RSC.
- P. H. Kreutzer, J. C. Weis, H. Bock, J. Erbe and W. Beck, Chem. Ber., 1983, 116, 2691 CrossRef CAS.
- P. Kreutzer, C. Weis, H. Boehme, T. Kemmerich, W. Beck, C. Spencer and R. Mason, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1972, 27, 745 CAS.
- J. Geisenberger, J. Erbe, J. Heidrich, U. Nagel and W. Beck, Z. Naturforsch., B: J. Chem. Sci., 1987, 42, 55 CAS.
- M. Herberhold, A. Goller and W. Milius, Z. Anorg. Allg. Chem., 2003, 629, 1162 CrossRef CAS.
- J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688 CrossRef CAS PubMed.
- M. F. Debets, S. S. Van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. Van Delft, Acc. Chem. Res., 2011, 44, 805 CrossRef CAS PubMed.
- T. Cruchter, K. Harms and E. Meggers, Chem. – Eur. J., 2013, 19, 16682 CrossRef CAS PubMed.
- M. C. Clough, P. D. Zeits, N. Bhuvanesh and J. A. Gladysz, Organometallics, 2012, 31, 5231 CrossRef CAS.
- S. Komeda, M. Lutz, A. L. Speck, M. Chikuma and J. Reedijk, Inorg. Chem., 2000, 39, 4230 CrossRef CAS PubMed.
- A. Massarotti, S. Aprile, V. Mercalli, E. Del Grosso, G. Grosa, G. Sorba and G. C. Tron, ChemMedChem, 2014, 9, 2497 CrossRef CAS PubMed.
- J. Totobenazara and A. J. Burke, Tetrahedron Lett., 2015, 56, 2853 CrossRef CAS.
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. MacKay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905 CrossRef CAS PubMed.
- Y. Zhao, N. J. Farrer, H. Li, J. S. Butler, R. J. McQuitty, A. Habtemariam, F. Wang and P. J. Sadler, Angew. Chem., Int. Ed., 2013, 52, 13633 CrossRef CAS PubMed.
- J. S. Butler, J. A. Woods, N. J. Farrer, M. E. Newton and P. J. Sadler, J. Am. Chem. Soc., 2012, 134, 16508 CrossRef CAS PubMed.
- Y. Zhao, J. A. Woods, N. J. Farrer, K. S. Robinson, J. Pracharova, J. Kasparkova, O. Novakova, H. Li, L. Salassa, A. M. Pizarro, G. J. Clarkson, L. Song, V. Brabec and P. J. Sadler, Chem. – Eur. J., 2013, 19, 9578 CrossRef CAS PubMed.
- F. S. MacKay, N. J. Farrer, L. Salassa, H.-C. Tai, R. J. Deeth, S. A. Moggach, P. A. Wood, S. Parsons and P. J. Sadler, Dalton Trans., 2009, 2315 RSC.
- N. J. Farrer, J. A. Woods, V. P. Munk, F. S. MacKay and P. J. Sadler, Chem. Res. Toxicol., 2010, 23, 413 CrossRef CAS PubMed.
- S. Mukhopadhyay, J. Lasri, M. F. C. Guedes da Silva, M. A. Januário Charmier and A. J. L. Pombeiro, Polyhedron, 2008, 27, 2883 CrossRef CAS.
- C. W. Chang and G. H. Lee, Organometallics, 2003, 22, 3107 CrossRef CAS.
- P. A. Scattergood, A. Sinopoli and P. I. P. Elliott, Coord. Chem. Rev., 2017, 350, 136 CrossRef CAS.
- N. J. Farrer, P. Gierth and P. J. Sadler, Chem. – Eur. J., 2011, 17, 12059 CrossRef CAS PubMed.
- R. R. Vernooij, T. Joshi, E. Shaili, M. Kubeil, D. R. T. Appadoo, E. I. Izgorodina, B. Graham, P. J. Sadler, B. R. Wood and L. Spiccia, Inorg. Chem., 2016, 55, 5983 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of Pt(I)I complexes, UV-vis, IR spectra, ESI-MS/MS. See DOI: 10.1039/c7dt04183g |
| This journal is © The Royal Society of Chemistry 2018 |